

Abstract
Regioselective arene C−H bond alkylation is a powerful tool in synthetic chemistry, yet subject to many challenges. Herein, we report the meta-C−H bond alkylation of aromatics bearing N-directing groups using (hetero)aromatic epoxides as alkylating agents. This method results in complete regioselectivity on both the arene as well as the epoxide coupling partners, cleaving exclusively the benzylic C−O bond. Oxetanes, which are normally unreactive, also participate as alkylating reagents under the reaction conditions. Our mechanistic studies reveal an unexpected reversible epoxide ring opening process undergoing catalyst-controlled regioselection, as key for the observed high regioselectivities.
Subject terms: Synthetic chemistry methodology, Homogeneous catalysis, Reaction mechanisms
Regioselective arene C−H bond alkylation is a powerful tool in synthetic chemistry, yet subject to many challenges. Here, the authors report the meta-C−H bond alkylation of aromatics bearing N-directing groups using (hetero)aromatic epoxides as alkylating agents.
Introduction
Chelation-assisted transition-metal-catalyzed sp2 C−H bond alkylation has emerged as a straightforward approach to forge the fundamentally important C(sp2)−C(sp3) bond1–3. This transformation provides a precisely-controlled complementary approach to the classic Friedel-Crafts alkylation, and has accelerated the rapid assembly of structurally diverse natural products, drug scaffolds and other functional molecules from simple starting materials4,5. Alkylating reagents, such as alkyl halides, alkylboron, alkylzinc, alkyltin, and Grignard reagents as well as alkenes have been extensively utilized in C−H alkylation reactions2. While less explored, recently epoxides have received increasing attention as alkylating reagents (Fig. 1A) displaying a number of advantages6,7: 1) epoxides are stable and readily available; 2) alkylation is redox-neutral and the product incorporates all atoms from the electrophile, thus increasing atom economy; and 3) the resulting hydroxide group in the product is a useful functional handle for further diversification.
Fig. 1. Development of directed C−H alkylation enabled by epoxide and oxetane opening.
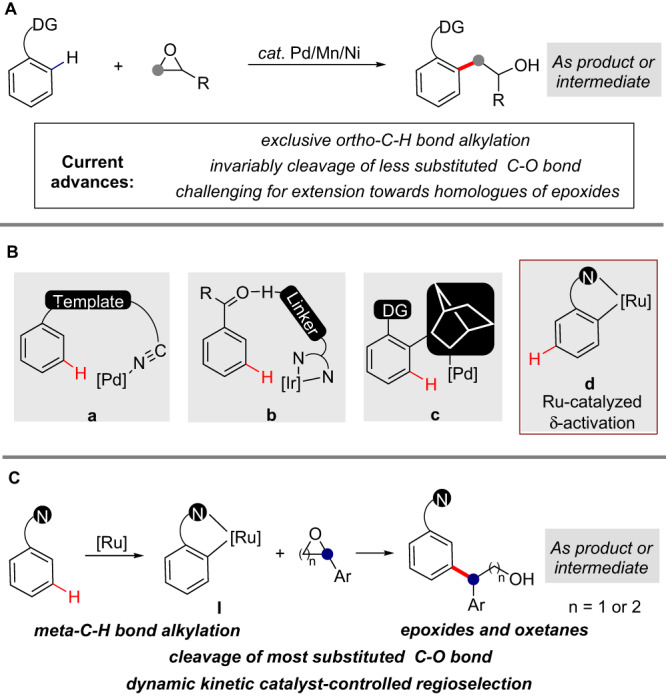
A ortho-C−H bond alkylation enabled by epoxide-opening (current advances). B Strategies for chelation-assisted meta-C−H functionalization. C Our work: meta-C−H bond alkylation with epoxides and oxetanes.
In 2015, Kanai and Kuninobu reported a seminal palladium-catalyzed aromatic ortho-C−H alkylation with terminal alkyl-epoxides8. Concurrently, the Yu group reported the ortho-C−H alkylation of benzoic acid with aliphatic epoxides to produce a variety of 3,4-dihydroisocoumarins9. Later, Hirano and Miura10 and Kuninobu11 extended such transformations by using Ni and Mn catalysis. Meanwhile, the Dong12,13 and Zhou14,15 groups reported the use of epoxides as coupling partners with aryl halides in Catellani-type processes, whereas Wang and others16–18 also significantly expanded the utility and scope of epoxide opening triggered C−H alkylation reactions (Fig. 1A). However, several key aspects have not yet been explored: firstly, in all examples to date alkylation is exclusively occurring at the ortho-C−H bond, with meta- and para-C−H bond alkylations proving elusive; Secondly, terminal or symmetric 1,2-dialkyl-substituted epoxides are predominantly used with ring opening invariably occurring at the less substituted carbon. Thirdly, the use of oxetanes or other homologs of epoxides in C−H alkylation remains a prominent challenge.
In contrast to directed sp2 ortho-C−H bond functionalization, transition-metal-catalyzed meta-C−H bond functionalization is significantly more challenging and far less developed19–24. Chelation-assisted strategies using either template-directing groups25–31, hydrogen-bonding ligands32–35 or transient norbornene mediators36–39 have been used to activate and functionalize the meta-C−H bond (Fig. 1B, a, b, c). As an alternative approach, Frost, Ackermann and others reported that arene meta-C−H functionalization can be achieved by Ru-catalysis24,40–53, proceeding via initial ortho-cycloruthenation followed by functionalization para to the ruthenium (Fig. 1B, d). This approach avoids the multi-step syntheses of ligands or templates, and provides a powerful platform for meta-alkylation of arenes bearing common nitrogen-based directing groups, with secondary or tertiary alkyl halides predominantly used as alkylating reagents. Inspired by this work, we hypothesized that meta-alkylation using epoxides as coupling partners may be achievable via Ru-catalysis, thus achieving complementary selectivity to that previously reported with epoxides. Herein, we report our latest work on meta-C−H bond alkylation with epoxides (Fig. 1C). This transformation proceeds with complete regioselectivity in both coupling partners, through exclusive cleavage of the more hindered benzylic C−O bond of (hetero)aromatic epoxides and complete meta-alkylation selectivity. It is noteworthy that the normally unreactive oxetanes, homologs of epoxides, also participate as alkylating reagents in the reaction. Our mechanistic studies reveal that an unexpected reversible ring opening of epoxide process, as well as a catalyst-controlled discrimination of reaction intermediates, are simultaneously in operation during the reaction, contributing to a dynamic kinetic regioselection, as the key to the high selectivities obtained.
Results
Optimization of the reaction conditions
We initially chose 2-phenylpyridine 1a and styrene oxide 2a as model substrates (Table 1), which would lead to important pharmacophores of 1,1-diarylalkane unit54 equipped with a hydroxide group for further product manipulation (3a). A key challenge for this design is the requirement to control regioselectivity of both the phenylpyridine and epoxide. For example, a feasible oxidative addition or nucleophilic attack onto the highly electrophilic styrene oxide6,7, would lead to the undesired ortho-alkylation product (3a’). When we used [Ru(p-cymene)Cl2]2 and Ru(p-cymene)(OPiv)2 as catalysts, both of which are predominately used in meta-alkylations40–53, together with benzoic acid (A1) as an additive to enhance the reactivity of the epoxide as well as to facilitate the ortho-C−H bond cycloruthenation step to form I55, no detectable alkylation product was formed (entries 1-2). The addition of 30% NaI, which has been shown before to facilitate reactions with epoxides56–58, also led to no reaction (entries 3-4). Gratifyingly, when cyclometalated ruthenium complex RuBnN, developed by our group59–62, was used as the catalyst, the desired product 3a was observed for the first time, albeit in low yield and accompanied with trace amount of ortho-C−H alkylation product 3a’ (entry 5). We have previously observed that RuBnN tends to lead to ortho-selective alkylations with secondary alkyl halides60. A screening of other reaction parameters resulted in no improvement, with ortho-alkylated 3a’ obtained as the major product when using methanol as solvent (entry 6). When commercially available Ru(PPh3)3Cl2 was used, both 3a and 3a’ were obtained (entry 7). The nature of the acid additive proved to be crucial to the success of this reaction (entries 8-10), with 2-ethylbutanoic acid (A4), affording 3a in 50% yield while completely suppressing the formation of 3a’ (entry 10). The carboxylic acid additive may function as a proton shuttle in the reaction by protonating the alkoxide generated after the epoxide ring opening, with the resulting carboxylate acting as a base to facilitate ortho-C−H ruthenation. A further increase in yield was gained when the reaction was run at higher concentrations (1.3 M, entry 11). Although turn-over was observed when 30% of NaI was used, a slightly higher yield of 68% of 3a was obtained when 1.0 equiv of NaI was used (entry 12), and finally 75% isolated yield was reached when reaction was run at 70 °C instead of 80 °C (entry 13). Meanwhile, similar efficiency was observed using tetrabutylammonium iodide (entry 14) whereas NaBr or NaCl provided significantly lower yields of 3a (entries 15-16).
Table 1.
Reaction optimization
| Entries | Ru-catalyst | Acid | Additive | Yield 3a / 3a’ a |
|---|---|---|---|---|
| 1 | [Ru(p-cymene)Cl2]2 | A1 | none | 0% / 0% |
| 2 | Ru(p-cymene)(OPiv)2 | A1 | none | 0% / 0% |
| 3 | [Ru(p-cymene)Cl2]2 | A1 | NaI (30 %) | 0% / 0% |
| 4 | Ru(p-cymene)(OPiv)2 | A1 | NaI (30 %) | 0% / 0% |
| 5 | RuBnN | A1 | NaI (30 %) | 27% / trace |
| 6 | RuBnN | A1 | NaI (30 %) | 19% / 25% b |
| 7 | Ru(PPh3)3Cl2 | A1 | NaI (30 %) | 15% / 5% |
| 8 | Ru(PPh3)3Cl2 | A2 | NaI (30 %) | 15% / 0% |
| 9 | Ru(PPh3)3Cl2 | A3 | NaI (30 %) | 48% / 0% |
| 10 | Ru(PPh3)3Cl2 | A4 | NaI (30 %) | 50% / 0% |
| 11 | Ru(PPh3)3Cl2 | A4 | NaI (30 %) | 62% / 0% c |
| 12 | Ru(PPh3)3Cl2 | A4 | NaI (100 %) | 68% / 0% c |
| 13 | Ru(PPh3)3Cl2 | A4 | NaI (100 %) | 75% / 0% c,d,e |
| 14 | Ru(PPh3)3Cl2 | A4 | nBu4NI (100 %) | 73% / 0% c,d |
| 15 | Ru(PPh3)3Cl2 | A4 | NaBr (100 %) | 46% / 0% c,d |
| 16 | Ru(PPh3)3Cl2 | A4 | NaCl (100 %) | 19% / 0% c,d |
aYield measured by NMR using 1,3,5-trimethoxybenzene as internal standard.
bMeOH (0.4 M of 1a) used as the solvent.
cRun at 1.3 M concentration of 1a.
dReaction was run at 70 oC.
eIsolated yield.
Scope of the reaction
Next, we investigated the reaction scope (Fig. 2). 2-Phenylpyridine derivatives bearing electronically diverse para-substituents efficiently participated in the reaction with 2a, delivering meta-C−H alkylation products 3a-3i in excellent yields (Fig. 2A). Functional groups such as phenolic hydroxyl 3b, alkyl ether 3d, chloride 3f, aliphatic ester 3h and internal alkyne 3i were well tolerated, leaving room for further product diversification. When 2-phenylpyridine bearing an ortho-fluoride substituent was used, the alkylation exclusively occurred at the more hindered meta-position (3j). This result is consistent with a mechanism involving formation of I, followed by a Ru-mediated para σ-activation51. Some meta-substituted phenylpyridines can give product from the reaction but only to limited degree (3k and 3l). In accordance with previous Ru-catalyzed meta-alkylation40–53, metasubstitution on the arene is not well tolerated as it forces the cyclometalation to occur on the distal ortho-position and then blocks reactivity. This reasoning is also why bis-alkylation does not occur in these reactions. Other heteroarenes, such as pyrimidine, pyrazole and non-aromatic 4,5-oxazoline were also suitable directing groups, producing 3m-3o in moderate to excellent yields. Noteworthy, a tandem reaction was discovered for 1p bearing an ester group at para position, and isochroman-1-one derivate 3p was formed in 77% yield (Fig. 2B). This tandem lactonization likely occurs via intermediate II. This type of tandem processes have been previously observed in ortho-alkylations with epoxides where the directing group itself is an electrophile8–10, while our product 3p was obtained through such tandem process occurring with a substituent within the aromatic ring. Interestingly, when using 1q bearing a para-aldehyde substituent, identical product 3p was obtained. This product may be formed through a Ru-mediated β-hydride elimination of III (Fig. 2B)63, or upon work-up in the presence of air. These results clearly highlight the unique utility of this meta-alkylation process.
Fig. 2. Substrate scopea.

A Scope of arene and directing group. B Tandem lactonization for arene substrates. C Scope of mono-substituted epoxide. D Tandem lactonization for epoxide substrare. E Scope of disubstituted epoxide. F late-stage functionalization of complex moleculars. a 1 (0.2 mmol) and 2 (0.4 mmol) were used. Yields are of isolated product; b 1 (0.2 mmol), 2 (0.8 mmol), Ru(PPh3)3Cl2 (10 mol %) and NaI (2.0 equiv) were used.
Styrene oxides bearing either electron withdrawing or donating groups at para, meta or ortho-positions also reacted smoothly to yield the corresponding meta-alkylation products in moderate to excellent yields (3q-3ad) (Fig. 2C). Moving beyond styrene oxides, the sterically hindered 1-naphthalene and 1,1’-binaphthalene substituted epoxides were converted into the desired products 3af and 3ag in reasonable to good yields. Moreover, electron rich heteroarenes, such as furan and thiophene, or electron deficient quinoline, were also tolerated in the epoxide, leading to products 3ah-3aj in moderate to good yields. Noteworthy, when styrene oxide bearing an ortho-ester substituent was used, a similar tandem alkylation-lactonization process was observed, exclusively delivering 3ak, with an isochroman-1-one unit at the meta-position (Fig. 2D).
1,2-Disubstituted epoxides have been shown to be challenging substrates on previous ortho-alkylations, leading to regioisomeric mixtures due to the difficulty to differentiate between two C−O bonds with subtle steric and electronic differences. As a result, only a handful of examples of 1,2-disubstituted epoxides have been reported, with most of them being symmetric epoxides8–18. When we tested trans−2-methyl-3-phenylepoxide as a coupling partner in this reaction (Fig. 2E), the meta-C−H alkylation regioisomer 3al was formed exclusively, resulting from cleavage of the benzylic C−O bond, albeit with a low diastereoselectivity. Identical results were obtained when using cis−2-methyl-3-phenylepoxide as the substrate. Similarly, both trans-and cis−2,3-diphenylepoxide yielded product of 3an with identical yield and diastereoselectivity. These results are in stark contrast to Miura’s work where a stereospecific ortho-C−H alkylation is observed10. Steric bulk in the epoxide could also be accommodated yielding 3am in 68% yield. Finally, indene oxide could be converted into the meta-alkylated product 3ao in 86% yield with complete regio- and excellent diastereoselectivity. These results clearly demonstrate the excellent regioselectivity control of this transformation on both arene and epoxide coupling partners.
The late-stage functionalization of biologically relevant molecules was then carried out to further showcase the utility and tolerance of the transformation (Fig. 2F). Firstly, diazepam was directly reacted with 2a under standard conditions, with the desired meta-alkylation product 3ap obtained in 82% yield, leaving the amide, imine and chloride functionalities intact. This result also highlights that imines are compatible directing group for the reaction. Benefiting from the easy accessibility of epoxides from acids, aldehyde and alkenes64, estrone, adapalene and telmisartan were derivatized to include an epoxide motif and then used in the alkylation reaction with 1a. Pleasingly, the desired complex targets 3aq-3as were produced in fair to excellent yields.
In an extension of this strategy, the use of oxetanes instead of epoxides was examined (Fig. 3). Despite their potential broad versatility as building blocks65, oxetanes have rarely been used in directed C−H alkylation66, presumably because of their less strained nature making their activation harder. Indeed, DFT calculations by Fang showed that the free energy barrier of Pd oxidative insertion into oxetane is significantly higher than that of epoxide67. We speculated that the iodide additive could lead to nucleophilic opening of oxetane, thus bypassing the high energy oxidative pathway. With a small modification to our standard conditions, 2-phenyloxetane 4 successfully alkylated 1a, affording the desired meta-alkylation product 5a in 62% yield. Interestingly, a tandem alkylation-lactonization did not occur when 1n reacted with 4, likely due to the difficulty of forming a seven-membered lactone, thus yielding 5b instead in 38% yield. Testing several substituted 2-phenyloxetanes showed that both the electron donating and electron withdrawing substitution could be well tolerated (5c-5g).
Fig. 3. Ring opening of 2-phenyloxetanea.

Yields are of isolated product with 1 (0.2 mmol) and 4 (0.8 mmol) were used.
Mechanistic studies
A series of experiments were carried out to investigate the reaction mechanism (Fig. 4). First, when TEMPO (2.0 equiv) was used as a radical scavenger in the standard reaction, only a trace amount of 3a was observed, with a benzylic TEMPO adduct isolated in 43% yield (Fig. 4A). This result suggests that the reaction may be operating through a radical mechanism involving the formation of a benzylic radical. Then we examined the epoxide ring opening process (Fig. 4B). No reaction was observed between epoxide 2a and NaI in the absence of Ru catalyst or acid additive (entry 1). With the addition of 5% Ru catalyst iodohydrin 7, formed by iodide opening at the less hindered C−O bond of 2a, was produced in 7%. Interestingly, iodohydrin 6, resulting from cleavage of benzylic C−O bond of 2a, was obtained in only trace amount (entry 2). This result stands in stark contrast with Doyle68 and Yang’s69 stoichiometric experiments between 2a and HI as well as NaI, respectively, where 6 was formed as the single product in excellent yields70. The addition of acid additive (A4) significantly promoted the generation of both iodohydrin 6 and 7, with 7 still the predominant regioisomer. At the same time, ester substituted products 8 and 9 were also obtained in the reaction, with 9 resulting from the cleavage of the less hindered C−O bond of 2a, as the major isomer (entry 3). Similar results were obtained when both the Ru catalyst and acid A4 were simultaneously used (entry 4). Interestingly, in the absence of NaI, acid A4 could not promote the ring opening of 2a (entry 5) by itself; However, in the presence of the Ru catalyst, 8 and 9 were obtained in high yield (entry 6). These results reveal that a number of ring opening pathways are available in the reaction conditions, with the major products resulting from the opening of the epoxide at the less hindered C−O bond. Puzzlingly, this regioselectivity is reversed compared to that observed in the alkylation products 3, which are formed with complete selectivity at the most hindered C−O bond. When 6 was reacted with 1a under the standard conditions, 3a was not detected (Fig. 4C). However, upon addition of Na2CO3 as a base, 3a was formed in 70% yield. These results are consistent with an iodide promoted epoxide opening from the benzylic C−O bond with concomitant in situ formation of a base that is required to carry out the C−H activation step. Suprisingly, when 7 was used instead in the above reaction, identical product 3a was obtained in the presence of Na2CO3 (Fig. 4D). Treatment of 6 under the reaction conditions, in the absence of arene, led to formation of regioisomer 7, as well as both 8 and 9 (Fig. 4E). Similar results were obtained when 7 was used instead of 6 (Fig. 4F). No formation of product 3a was observed when 8 or 9 were used instead of 6 and 7 in conditions analogous to those in Fig. 4C (Fig. 4G). Taken together, these results indicate a reversible ring opening of 2a is operating in the reaction. On the other hand, analogous mechanistic studies on the use of oxetanes as coupling partners suggest in that case a direct and non-reversible iodide-mediated regioselective opening of the oxetane may be responsible for the observed meta-alkylation (for details, see the Supplementary Discussion section, Mechanistic Studies of Oxetane Involved meta-Alkylation Reaction). Finally, when the standard reaction was carried out in the presence of CD3OD (300 mol %) and stopped before complete conversion, deuterium incorporation (28%) was observed at the ortho-position of the recovered starting material d5-1a, indicating that the ortho-C−H ruthenation is reversible (Fig. 4H). Meanwhile, an intermolecular kinetic isotope effect experiment revealed a relatively small KIE (kH/kD = 1.6, Fig. 4I), consistent with a non-rate determining reversible C–H cleavage71.
Fig. 4. Mechanistic studies.

A Radical trapping experiments. B Epoxide opening studies. C Testing iodohydrin 6 as the potential reaction intermediate. D Testing iodohydrin 7 as the potential reaction intermediate. E, F The experiments to prove the epoxide ring opening is a reversible process. G Testing ester substituted compounds 8 and 9 as the potential reaction intermediate. H Deuteration experiments with CD3OD. I Intermolecular kinetic isotope effect experiment. a Determined by 1HNMR analysis of crude material, and yields were reported based on NaI as the limiting reagent. b Yields were reported based on 2-ethylbutanoic acid (A4) as the limiting reagent. c Without NaI.
Based on the above results and literature reports72 a tentative mechanism is proposed (Fig. 5). With the assistance of Ru catalyst and acid additive, NaI is engaged in nucleophilic ring opening of 2a, to yield secondary benzylic iodide V and primary alkyl iodide VI. On the other hand, the aromatic substrate goes ortho-cycloruthenation to deliver the cyclometalated Ru-species I. Subsequently, I goes through single electron transfer with the more reactive intermediate V to afford a benzylic radical and a Ru(III) intermediate VII, followed by addition of the benzylic radical to intermediate VII at the para-position to the ruthenium center, to deliver the meta-alkylation product 3 and close the catalytic cycle. Meanwhile, based on results presented on Fig. 4B–F, the generation of primary alkyl iodide VI should dominate the opening of 2a in the reaction, however it is an unproductive pathway in the reaction. This is particularly unexpected since cyclometalated-Ru-species I is known to undergo facile ortho-alkylation with primary alkyl halides73,74. The absence of such by-product in our reaction (as well as the result in Fig. 4D–F) suggests that a fast-reversible equilibration of VI and V via 2a, as well as a catalyst-controlled discrimination of VI and V are simultaneously in operation, accounting for the strong regioselectivity obtained in our reaction. To the best of our knowledge, a reversible iodide-mediated epoxide ring opening dynamic kinetic process has not been reported previously in a transition metal-catalyzed coupling reaction.
Fig. 5. Proposed reaction mechanism.

Proposed catalytic cycle that including a sequence of reversible epoxide ring opening and catalyst-controlled discrimination.
In conclusion, through a process co-catalyzed by Ru, a carboxylic acid and iodide, we have achieved remote meta-alkylation using epoxides as alkyl donors. In addition, unique tandem alkylation/cyclization, the employment of typically challenging substrates such as unsymmetrical 1,2-disubstituted epoxide, complex pharmaceutical compounds and previously unreactive oxetane-based alkylating partners further showcase the broad utility of this transformation. Our mechanistic studies reveal an unexpected catalyst-controlled dynamic kinetic regioselection process is responsible for the high selectivity of the reaction. We envision that this method will encourage the development of more diverse and selective epoxide ring opening reactions.
Methods
Representative procedure for Ru-catalyzed meta-C−H alkylation with epoxides
In a glove box, an oven-dried crimp-cap microwave vial equipped with a magnetic stirring bar was charged with Ru(PPh3)3Cl2 (5.0 mol %), NaI powder (1.0 equiv) and 2-ethylbutyric acid (30 mol %), then substrates 1 (0.20 mmol), epoxide 2 (2.0 equiv) and dioxane (1.3 M) were added. The vial was then capped and taken out of glovebox, stirred at 70 °C for 44 h. The reaction was then allowed to cool to room temperature and concentrated in vacuo. The residue was purified by column chromatography under the conditions noted to yield the desired product.
Supplementary information
Acknowledgements
G.-W. W. thanks the National Natural Science Foundation of China (No. 22201114), the Fundamental Research Funds for the Central Universities (lzujbky-2022-17). We gratefully acknowledge the European Research Council (ERC) for an advanced grant (RuCat, 833337) to I. L. and the Engineering and Physical Sciences Research Council (EPSRC, EP/S02011X/1) for funding to I.L.
Author contributions
G.-W.W. and I.L. conceived the project. G.-W.W. carried out initial discovery work. G.-W.W., P.-B.B. and A.D. performed optimization of the methodology and exemplification of the chemistry. G.-W.W. and I.L. secured the funding and directed the work. The manuscript was prepared by G.-W.W. and I.L. with contributions from all other authors.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Data availability
All data generated or analyzed during this study are included in this Article and the Supplementary Information. Details about materials and methods, experimental procedures, mechanistic studies, characterization data, computational details, NMR and HPLC spectra are available in the Supplementary Information. All other data are available from the corresponding author upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Gang-Wei Wang, Email: wanggw@lzu.edu.cn.
Igor Larrosa, Email: igor.larrosa@manchester.ac.uk.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-023-44219-6.
References
- 1.Dixneuf, P. H. & Doucet, H. C−H Bond activation and catalytic functionalization Vol. I and Vol. II (Springer International Publishing, Cham, 2015 and 2016).
- 2.Evano G, Theunissen C. Beyond Friedel and Crafts: directed alkylation of C−H bonds in arenes. Angew. Chem. Int. Ed. 2019;58:7202–7236. doi: 10.1002/anie.201806629. [DOI] [PubMed] [Google Scholar]
- 3.Evano G, Theunissen C. Beyond Friedel and Crafts: innate alkylation of C−H bonds in arenes. Angew. Chem., Int. Ed. 2019;58:7558–7598. doi: 10.1002/anie.201806631. [DOI] [PubMed] [Google Scholar]
- 4.Yamaguchi J, Yamaguchi AD, Itami K. C−H Bond functionalization: emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem. Int. Ed. 2012;51:8960–9009. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]
- 5.Lam NYS, Wu K, Yu J-Q. Advancing the logic of chemical synthesis: C−H activation as strategic and tactical disconnections for C−C bond construction. Angew. Chem. Int. Ed. 2021;60:15767–15790. doi: 10.1002/anie.202011901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H-H, et al. Recent advances in transition-metal-catalyzed C−H alkylation with three-membered rings. ACS Catal. 2022;12:2330–2347. doi: 10.1021/acscatal.1c05266. [DOI] [Google Scholar]
- 7.Huang C-Y, Doyle AG. The chemistry of transition metals with three-membered ring heterocycles. Chem. Rev. 2014;114:8153–8198. doi: 10.1021/cr500036t. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Kuninobu Y, Kanai M. Palladium-catalyzed oxirane opening reaction with arenes via C–H bond activation. J. Am. Chem. Soc. 2015;137:6140–6143. doi: 10.1021/jacs.5b02435. [DOI] [PubMed] [Google Scholar]
- 9.Cheng GL, Li T-J, Yu J-Q. Practical Pd(II)-catalyzed C–H alkylation with epoxides: one-step syntheses of 3,4-dihydroisocoumarins. J. Am. Chem. Soc. 2015;137:10950–10953. doi: 10.1021/jacs.5b07507. [DOI] [PubMed] [Google Scholar]
- 10.Xu SB, Takamatsu K, Hirano K, Miura M. Nickel-catalyzed stereospecific C−H coupling of benzamides with epoxides. Angew. Chem. Int. Ed. 2018;57:11797–11801. doi: 10.1002/anie.201807664. [DOI] [PubMed] [Google Scholar]
- 11.Sueki S, Wang ZJ, Kuninobu Y. Manganese and borane-mediated synthesis of isobenzofuranones from aromatic esters and oxiranes via C−H bond activation. Org. Lett. 2016;18:304–307. doi: 10.1021/acs.orglett.5b03474. [DOI] [PubMed] [Google Scholar]
- 12.Li R, Dong G. Direct annulation between aryl iodides and epoxides through palladium/norbornene cooperative catalysis. Angew. Chem. Int. Ed. 2018;57:1697–1701. doi: 10.1002/anie.201712393. [DOI] [PubMed] [Google Scholar]
- 13.Li R, Liu F, Dong G. Palladium-catalyzed asymmetric annulation between aryl iodides and racemic epoxides using a chiral norbornene cocatalyst. Org. Chem. Front. 2018;5:3108–3112. doi: 10.1039/C8QO00808F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng H-G, et al. Epoxides as alkylating reagents for the catellani reaction. Angew. Chem. Int. Ed. 2018;57:3444–3448. doi: 10.1002/anie.201800573. [DOI] [PubMed] [Google Scholar]
- 15.Cheng H-G, et al. A concise total synthesis of (−)‐Berkelic acid. Angew. Chem. Int. Ed. 2021;60:5141–5146. doi: 10.1002/anie.202014660. [DOI] [PubMed] [Google Scholar]
- 16.Wang HH, et al. Pd(II)-catalyzed annulation reactions of epoxides with benzamides to synthesize isoquinolones. Org. Lett. 2021;23:863–868. doi: 10.1021/acs.orglett.0c04097. [DOI] [PubMed] [Google Scholar]
- 17.Thombal RS, Feoktistova T, González-Montiel GA, Cheong PH-Y, Lee YR. Palladium-catalyzed synthesis of β-hydroxy compounds via a strained 6,4-palladacycle from directed C−H activation of anilines and C−O insertion of epoxides. Chem. Sci. 2020;11:7260–7265. doi: 10.1039/D0SC01462A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li D-D, Niu L-F, Ju Z-Y, Xu ZH, Wu CZ. Palladium-catalyzed C(sp2)−H bond alkylation of ketoximes by using the ring-opening of epoxides. Eur. J. Org. Chem. 2016;18:3090–3096. doi: 10.1002/ejoc.201600335. [DOI] [Google Scholar]
- 19.Sinha SK, et al. Toolbox for distal C–H bond functionalizations in organic molecules. Chem. Rev. 2022;122:5682–5841. doi: 10.1021/acs.chemrev.1c00220. [DOI] [PubMed] [Google Scholar]
- 20.Dutta U, Maiti S, Bhattacharya T, Maiti D. Arene diversification through distal C(sp2)−H functionalization. Science. 2021;372:eabd5992. doi: 10.1126/science.abd5992. [DOI] [PubMed] [Google Scholar]
- 21.Meng G, et al. Achieving site-selectivity for C–H activation processes based on distance and geometry: a carpenter’s approach. J. Am. Chem. Soc. 2020;142:10571–10591. doi: 10.1021/jacs.0c04074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, Dong G. Palladium/norbornene cooperative catalysis. Chem. Rev. 2019;119:7478–7528. doi: 10.1021/acs.chemrev.9b00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mihai MT, Genov GR, Phipps RJ. Access to the meta position of arenes through transition metal catalyzed C–H bond functionalization: a focus on metals other than palladium. Chem. Soc. Rev. 2018;47:149–171. doi: 10.1039/C7CS00637C. [DOI] [PubMed] [Google Scholar]
- 24.Leitch JA, Frost CG. Ruthenium-catalyzed σ-activation for remote meta-selective C−H functionalization. Chem. Soc. Rev. 2017;46:7145–7153. doi: 10.1039/C7CS00496F. [DOI] [PubMed] [Google Scholar]
- 25.Tang R-Y, Li G, Yu J-Q. Conformation-induced remote meta-C–H activation of amines. Nature. 2014;507:215–220. doi: 10.1038/nature12963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bag S, Jayarajan R, Mondal R, Maiti D. Template-assisted meta-C−H alkylation and alkenylation of arenes. Angew. Chem. Int. Ed. 2017;56:3182–3186. doi: 10.1002/anie.201611360. [DOI] [PubMed] [Google Scholar]
- 27.Li S, Cai L, Ji H, Yang L, Li G. Pd (II)-catalysed meta-C–H functionalizations of benzoic acid derivatives. Nat. Commun. 2016;7:10443–10450. doi: 10.1038/ncomms10443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi H, Herron AN, Shao Y, Shao Q, Yu J-Q. Enantioselective remote meta-C–H arylation and alkylation via a chiral transient mediator. Nature. 2018;558:581–585. doi: 10.1038/s41586-018-0220-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bag S, et al. Palladium-catalyzed meta-C–H allylation of arenes: a unique combination of a pyrimidine-based template and hexafluoroisopropanol. J. Am. Chem. Soc. 2020;142:12453–12466. doi: 10.1021/jacs.0c05223. [DOI] [PubMed] [Google Scholar]
- 30.Saha A, et al. Photoinduced regioselective olefination of arenes at proximal and distal sites. J. Am. Chem. Soc. 2022;144:1929–1940. doi: 10.1021/jacs.1c12311. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Z, Tanaka K, Yu J-Q. Remote site-selective C–H activation directed by a catalytic bifunctional template. Nature. 2017;543:538–542. doi: 10.1038/nature21418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuninobu Y, Ida H, Nishi M, Kanai M. A meta-selective C–H borylation directed by a secondary interaction between ligand and substrate. Nat. Chem. 2015;7:712–717. doi: 10.1038/nchem.2322. [DOI] [PubMed] [Google Scholar]
- 33.Davis HJ, Mihai MT, Phipps RJ. Ion pair-directed regiocontrol in transition-metal catalysis: a meta-selective C–H borylation of aromatic quaternary ammonium salts. J. Am. Chem. Soc. 2016;138:12759–12762. doi: 10.1021/jacs.6b08164. [DOI] [PubMed] [Google Scholar]
- 34.Neel AJ, Hilton MJ, Sigman MS, Toste FD. Exploiting non-covalent π interactions for catalyst design. Nature. 2017;543:637–646. doi: 10.1038/nature21701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoque ME, Bisht R, Haldar C, Chattopadhyay B. Noncovalent interactions in Ir-catalyzed C–H activation: L-shaped ligand for para-selective borylation of aromatic esters. J. Am. Chem. Soc. 2017;139:7745–7748. doi: 10.1021/jacs.7b04490. [DOI] [PubMed] [Google Scholar]
- 36.Wang X-C, et al. Ligand-enabled meta-C–H activation using a transient mediator. Nature. 2015;519:334–338. doi: 10.1038/nature14214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye J, Lautens M. Palladium-catalysed norbornene-mediated C–H functionalization of arenes. Nat. Chem. 2015;7:863–870. doi: 10.1038/nchem.2372. [DOI] [PubMed] [Google Scholar]
- 38.Dong Z, Wang J, Dong G. Simple amine-directed meta-selective C–H arylation via Pd/norbornene. Catal. J. Am. Chem. Soc. 2015;137:5887–5890. doi: 10.1021/jacs.5b02809. [DOI] [PubMed] [Google Scholar]
- 39.Ca’ ND, Fontana M, Motti E, Catellani M. Pd/Norbornene: a winning combination for selective aromatic functionalization via C–H bond activation. Acc. Chem. Res. 2016;49:1389–1400. doi: 10.1021/acs.accounts.6b00165. [DOI] [PubMed] [Google Scholar]
- 40.Saidi O, et al. Ruthenium-catalyzed meta sulfonation of 2-phenylpyridines. J. Am. Chem. Soc. 2011;133:19298–19301. doi: 10.1021/ja208286b. [DOI] [PubMed] [Google Scholar]
- 41.Hofmann N, Ackermann L. meta-Selective C−H bond alkylation with secondary alkyl halides. J. Am. Chem. Soc. 2013;135:5877–5884. doi: 10.1021/ja401466y. [DOI] [PubMed] [Google Scholar]
- 42.Li J, et al. N-Acyl amino acid ligands for ruthenium (II)-catalyzed meta-C−H tert-alkylation with removable auxiliaries. J. Am. Chem. Soc. 2015;137:13894–13901. doi: 10.1021/jacs.5b08435. [DOI] [PubMed] [Google Scholar]
- 43.Paterson AJ, John-Campbell SS, Mahon MF, Press NJ, Frost CG. Catalytic meta-selective C–H functionalization to construct quaternary carbon centres. Chem. Commun. 2015;51:12807–12810. doi: 10.1039/C5CC03951G. [DOI] [PubMed] [Google Scholar]
- 44.Yu Q, Hu L, Wang Y, Zheng S, Huang J. Directed meta-selective bromination of arenes with ruthenium catalysts. Angew. Chem. Int. Ed. 2015;54:15284–15288. doi: 10.1002/anie.201507100. [DOI] [PubMed] [Google Scholar]
- 45.Paterson AJ, et al. α-Halo carbonyls enable meta selective primary, secondary and tertiary C–H alkylations by ruthenium catalysis. Org. Biomol. Chem. 2017;15:5993–6000. doi: 10.1039/C7OB01192J. [DOI] [PubMed] [Google Scholar]
- 46.Ruan Z, et al. Ruthenium(II)-catalyzed meta C−H mono-and difluoromethylations by phosphine/carboxylate cooperation. Angew. Chem. Int. Ed. 2017;56:2045–2049. doi: 10.1002/anie.201611595. [DOI] [PubMed] [Google Scholar]
- 47.Korvorapun K, Kuniyil R, Ackermann L. Late-stage diversification by selectivity switch in meta-C−H activation: evidence for singlet stabilization. ACS Catal. 2020;10:435–440. doi: 10.1021/acscatal.9b04592. [DOI] [Google Scholar]
- 48.Li J, et al. Ruthenium(II)-catalyzed remote C−H alkylations as a versatile platform to meta-decorated arenes. Nat. Commun. 2017;8:15430–15438. doi: 10.1038/ncomms15430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leitch JA, McMullin CL, Mahon MF, Bhonoah Y, Frost CG. Remote C6-selective ruthenium-catalyzed C–H alkylation of indole derivatives via σ-activation. ACS Catal. 2017;7:2616–2623. doi: 10.1021/acscatal.7b00038. [DOI] [Google Scholar]
- 50.Wang X-G, et al. Three-component ruthenium-catalyzed direct meta-selective C–H activation of arenes: a new approach to the alkylarylation of alkenes. J. Am. Chem. Soc. 2019;141:13914–13922. doi: 10.1021/jacs.9b06608. [DOI] [PubMed] [Google Scholar]
- 51.Sagadevan A, Greaney M. F. meta-Selective C−H activation of arenes at room temperature using visible light: dual-function ruthenium catalysis. Angew. Chem., Int. Ed. 2019;58:9826–9830. doi: 10.1002/anie.201904288. [DOI] [PubMed] [Google Scholar]
- 52.Gou X-Y, et al. Ruthenium-catalyzed radical cyclization/meta-selective C–H alkylation of arenes via σ-activation strategy. ACS Catal. 2021;11:4263–4270. doi: 10.1021/acscatal.1c00359. [DOI] [Google Scholar]
- 53.Chen S, Yuan B, Wang Y, Ackermann L. Ruthenium-catalyzed remote difunctionalization of nonactivated alkenes for double meta-C(sp2)H/C-6(sp3)H functionalization. Angew. Chem. Int. Ed. 2023;62:e202301168. doi: 10.1002/anie.202301168. [DOI] [PubMed] [Google Scholar]
- 54.Ameen D, Snape TJ. Chiral 1, 1-diaryl compounds as important pharmacophores. Med. Chem. Commun. 2013;4:893–907. doi: 10.1039/c3md00088e. [DOI] [Google Scholar]
- 55.Ackermann L. Carboxylate-assisted transition-metal-catalyzed C−H bond functionalizations: mechanism and scope. Chem. Rev. 2011;111:1315–1345. doi: 10.1021/cr100412j. [DOI] [PubMed] [Google Scholar]
- 56.Lau SH, et al. Ni/Photoredox-catalyzed enantioselective cross-electrophile coupling of styrene oxides with aryl iodides. J. Am. Chem. Soc. 2021;143:15873–15881. doi: 10.1021/jacs.1c08105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parasram M, Shields BJ, Ahmad O, Knauber T, Doyle AG. Regioselective cross-electrophile coupling of epoxides and (hetero)aryl iodides via Ni/Ti/photoredox catalysis. ACS Catal. 2020;10:5821–5827. doi: 10.1021/acscatal.0c01199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teng S, Tessensohn ME, Webster RD, Zhou JS. Palladium-catalyzed intermolecular Heck-type reaction of epoxides. ACS Catal. 2018;8:7439–7444. doi: 10.1021/acscatal.8b02029. [DOI] [Google Scholar]
- 59.Simonetti M, Cannas DM, Just-Baringo X, Vitorica-Yrezabal IJ, Larrosa I. Cyclometallated ruthenium catalyst enables late-stage directed arylation of pharmaceuticals. Nat. Chem. 2018;10:724–731. doi: 10.1038/s41557-018-0062-3. [DOI] [PubMed] [Google Scholar]
- 60.Wang G-W, Wheatley M, Simonetti M, Cannas DM, Larrosa I. Cyclometalated ruthenium catalyst enables ortho-selective C−H alkylation with secondary alkyl bromides. Chem. 2020;6:1459–1468. doi: 10.1016/j.chempr.2020.04.006. [DOI] [Google Scholar]
- 61.Wheatley M, et al. Ru-catalyzed room-temperature alkylation and late-stage alkylation of arenes with primary alkyl bromides. Chem. Catal. 2021;1:691–703. doi: 10.1016/j.checat.2021.05.008. [DOI] [Google Scholar]
- 62.Hogg A, et al. Ruthenium-catalyzed mono selective C–H methylation and d3-methylation of arenes. JACS Au. 2022;2:2529–2538. doi: 10.1021/jacsau.2c00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hamid MHSA, et al. Ruthenium-catalyzed N-alkylation of amines and sulfonamides using borrowing hydrogen methodology. J. Am. Chem. Soc. 2009;131:1766–1774. doi: 10.1021/ja807323a. [DOI] [PubMed] [Google Scholar]
- 64.Yudin. A. K. Aziridines and Epoxides in Organic Synthesis (Wiley-VCH, Weinheim, 2006).
- 65.Bull JA, Croft RA, Davis OA, Doran R, Morgan KF. Oxetanes: recent advances in synthesis, reactivity, and medicinal chemistry. Chem. Rev. 2016;116:12150–12233. doi: 10.1021/acs.chemrev.6b00274. [DOI] [PubMed] [Google Scholar]
- 66.Xu S, Takamatsu K, Hirano K, Miura M. Synthesis of seven-membered benzolactones by nickel-catalyzed C-H coupling of benzamides with oxetanes. Chem. Eur. J. 2019;25:9400–9404. doi: 10.1002/chem.201900543. [DOI] [PubMed] [Google Scholar]
- 67.Lian B, Zhang L, Li S-J, Zhang L-L, Fang D-C. PdIV Species mediation in PdII-catalyzed direct alkylation of arenes with oxiranes: a DFT study. J. Org. Chem. 2018;83:3142–3148. doi: 10.1021/acs.joc.7b03236. [DOI] [PubMed] [Google Scholar]
- 68.Lau SH, et al. Ni/ Photoredox-catalyzed enantioselective cross-electrophile coupling of styrene oxides with aryl iodides. J. Am. Chem. Soc. 2021;143:15873–15881. doi: 10.1021/jacs.1c08105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luo X, et al. CuX Dual catalysis: construction of oxazolo[2,3-b][1,3]oxazines via a tandem CuAAC/ring cleavage/[4+2+3] annulation reaction. Org. Lett. 2022;24:7300–7304. doi: 10.1021/acs.orglett.2c02705. [DOI] [PubMed] [Google Scholar]
- 70.Zhao Y, Weix DJ. Nickel-catalyzed regiodivergent opening of epoxides with aryl halides: Co-catalysis controls regioselectivity. J. Am. Chem. Soc. 2014;136:48–51. doi: 10.1021/ja410704d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Simmons EM, Hartwig JF. On the interpretation of deuterium kinetic isotope effects in C−H bond functionalizations by transition-metal complexes. Angew. Chem. Int. Ed. 2012;51:3066–3072. doi: 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
- 72.Chen X, et al. Close-shell reductive elimination versus open-shell radical coupling for site-selective ruthenium-catalyzed C−H activations by computation and experiments. Angew. Chem. Int. Ed. 2023;62:e202302021. doi: 10.1002/anie.202302021. [DOI] [PubMed] [Google Scholar]
- 73.Ackermann L, Novák P, Vicente R, Hofmann N. Ruthenium-catalyzed regioselective direct alkylation of arenes with unactivated alkyl halides through C−H bond cleavage. Angew. Chem. Int. Ed. 2009;48:6045–6048. doi: 10.1002/anie.200902458. [DOI] [PubMed] [Google Scholar]
- 74.Ackermann L, Hofmann N, Vicente R. Carboxylate-assisted ruthenium-catalyzed direct alkylations of ketimines. Org. Lett. 2011;13:1875–1877. doi: 10.1021/ol200366n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this Article and the Supplementary Information. Details about materials and methods, experimental procedures, mechanistic studies, characterization data, computational details, NMR and HPLC spectra are available in the Supplementary Information. All other data are available from the corresponding author upon request.