

Abstract
Postural orthostatic tachycardia syndrome (POTS) is a chronic debilitating condition of orthostatic intolerance, predominantly affecting young females. Other than postural tachycardia, symptoms of POTS include a spectrum of non-cardiac, systemic and neuropsychiatric features. Despite the availability of widespread pharmacological and non-pharmacological therapeutic options, the management of POTS remains challenging. Exaggerated parasympathetic withdrawal and sympathetic overdrive during postural stress are principal mechanisms of postural tachycardia in POTS. Non-invasive, transcutaneous, vagus nerve stimulation (tVNS) is known to restore sympathovagal balance and is emerging as a novel therapeutic strategy in cardiovascular conditions including arrhythmias and heart failure. Furthermore, tVNS also exerts immunomodulatory and anti-inflammatory effects. This review explores the effects of tVNS on the pathophysiology of POTS and its potential as an alternative non-pharmacological option in this condition.
Keywords: Postural tachycardia, vagus nerve stimulation, parasympathetic nervous system, sympathetic nervous system, sympathovagal balance
Postural orthostatic tachycardia syndrome (POTS) is characterised by persistent symptoms (≥6 months) of orthostatic intolerance associated with an inappropriate increase in heart rate (HR; ≥30 BPM in adults and ≥40 BPM in children and adolescents) without any orthostatic hypotension (blood pressure fall >20/10 mmHg) on assuming an upright posture.[1] Other than symptoms of increased HR (palpitations, presyncope, blurry vision), the classical constellations of symptoms of POTS include gastrointestinal (nausea, gastroparesis), neuropsychiatric (mental clouding, anxiety) and systemic (fatigue, dyspnoea) symptoms.[1,2] POTS predominantly affects young females. Although the disease is not life-threatening, it is a debilitating disease and is associated with significant impairment of quality of life.
Excessive postural tachycardia in POTS results from a combination of compensatory autonomic response to haemodynamic changes and an inappropriate augmentation of sympathetic response to postural stress (Figure 1).[1,3] Conventional pharmacological and non-pharmacological therapies attempt to reduce the exaggerated HR response by expansion of intravascular volume (hydration, high sodium diet, fludrocortisone and desmopressin), reduction of venous pooling (binders, midodrine and octreotide), parasympathomimetic action (cholinesterase inhibitors), sympatholytic effects (β-blocker and α2 agonist), and inhibition of automaticity of the sinoatrial node (funny current inhibition by ivabradine).[3] Despite targeting widespread physiological derangements, the available therapies for POTS are constrained by limited efficacy and widespread cardiac and non-cardiac side-effects. Importantly, the aforementioned therapeutic strategies do not modify the non-cardiac manifestations of POTS.
Figure 1: Mechanisms of Postural Tachycardia in POTS.
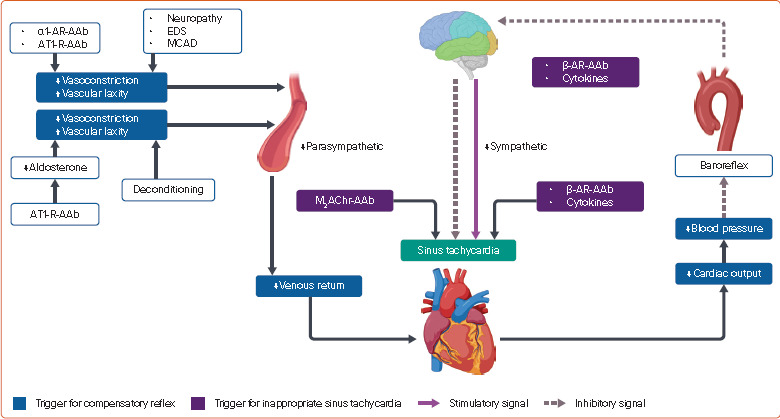
AAb = functional autoantibody; AR = adrenergic receptor; AT1-R = type 1 angiotensin II receptor; EDS = Ehlers–Danlos syndrome; M2AChR = type 2 muscarinic cholinergic receptor; MCAD = mast cell activation disorder; POTS = postural orthostatic tachycardia syndrome. Source: Created with BioRender.com.
Non-invasive transcutaneous vagus nerve stimulation (tVNS) is a novel strategy for achieving selective activation of afferent branches of the vagus nerve through electrical stimulation of part of the external ear (cymba conchae and tragus).[4] tVNS has emerged as a novel non-pharmacological approach to treat neuropsychiatric and cardiovascular disorders including AF and heart failure.[4–6] tVNS exerts parasympathomimetic and anti-adrenergic effects on the heart.[7] An anti-inflammatory effect of tVNS has also been established.[8] The current review explores the effects of tVNS on the pathophysiology of POTS and provides the rationale for its use as an alternative non-pharmacological option in this condition.
Pathophysiology of POTS
POTS, a syndrome of heterogeneous aetiologies, is linked to deconditioning from prolonged space flight or bed rest, volume dysregulation, mast cell activation disorders, autoimmunity, and hyperadrenergic state.[1–3] Irrespective of aetiology, the exaggerated orthostatic HR response in POTS is associated with three distinct pathophysiological entities, namely peripheral autonomic dysfunction, absolute hypovolaemia, and central hyperadrenergic state (Figure 2). Diverse aetiological entities ultimately converge into these three pathophysiologic mechanisms, also known as endophenotypes, and more than one endophenotype is present in many cases.[3,9]
Figure 2: Postural Haemodynamics.
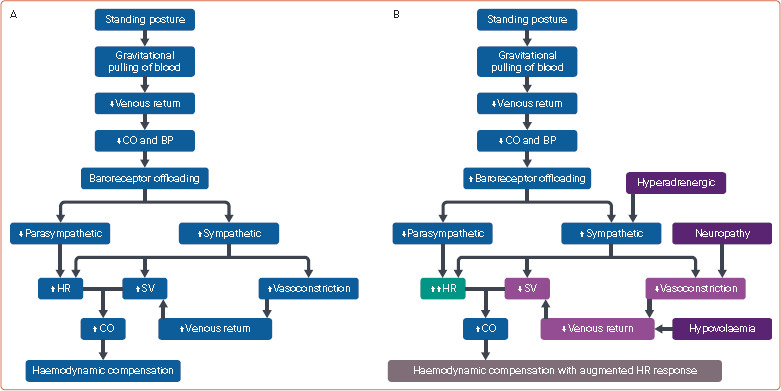
Normal postural physiology (A) and postural physiology (B) in postural orthostatic tachycardia syndrome. BP = blood pressure; CO = cardiac output; HR = heart rate; SV = stroke volume. Source: Created with BioRender.com.
In physiological conditions, standing from the recumbent position is immediately followed by the gravitational pulling of circulating blood into capacitance vessels of the lower part of the body, leading to reduced venous return and subsequent reduction of cardiac output and blood pressure (Figure 2A). The compensatory mechanisms are then activated to maintain the cardiac output. The principal early compensatory response results from offloading of aortic baroreceptors, leading to parasympathetic withdrawal and sympathetic activation. Stimulation of α1-adrenergic receptors (α1-ARs) at peripheral capacitance vessels causes vasoconstriction and an attempt to increase the venous return. Parasympathetic withdrawal increases HR. Activation of β-ARs on the sinoatrial node and ventricular myocardium is associated with an increase in HR and stroke volume, respectively. The sympathetic system also activates the renin–angiotensin–aldosterone system (RAAS) to generate angiotensin II and aldosterone, which further promotes the contraction of capacitance vessels and increases the intravascular volume. This results in increased intravascular volume, venous return and myocardial contractility, along with increased HR, with the aim of improving the cardiac output.[2,3]
In patients with POTS, due to the attenuation of one or more compensatory mechanisms, the optimal cardiac output is achieved only in the presence of a higher HR response (Figures 1 and 2B).[3] In the neuropathic endophenotype, the venous return is impaired due to inadequate constriction of blood vessels in the pelvic, splanchnic and lower extremities, leading to stasis of blood in these regions due to gravitational pooling.[3] The attenuated vasoconstriction response primarily results from the impairment of sympathetic activity in blood vessels.[10] Similarly, in Ehlers–Danlos syndrome, laxity of vascular connective tissue and coexistent autonomic dysfunction contribute to blood stasis.[11] Mast cell activation disorders also lead to inadequate venous return due to the effects of vasoactive substances released from mast cells and associated inflammatory neuropathy.[12]
A significant number of POTS patients demonstrate a state of chronic intravascular hypovolaemia.[3] Abnormal RAAS activity, characterised by inadequate aldosterone secretion by angiotensin II, is the main mechanism of hypovolaemia.[13] Chronic deconditioning is also associated with hypovolaemia.[3] However, the reduction of stroke volume in deconditioning is shown to be disproportionate to the degree of hypovolaemia, and reduced myocardial mass also contributes to suboptimal cardiac output in this condition.[14]
Other than the compensatory reflex tachycardia due to the aforementioned physiological changes, an inappropriate augmentation of sinus rate is also attributed to sympathetic overactivation, inflammation and autoantibodies (Figure 1).[1] In the hyperadrenergic phenotype, the sympathetic activity is normal in the supine position. However, the sympathetic outflow is overactivated in the standing position, leading to tachycardia and sometimes hypertension.[1,3] The β-adrenergic autoantibodies may also trigger postural overactivation of the sympathetic nervous system.[15] As described later, POTS is associated with a systemic inflammatory response. Inflammation is shown to augment sympathetic outflow as well as the response of sinus node to adrenergic activation.[16,17] Autoantibodies against type 2 muscarinic cholinergic receptors (M2AChRs) and β-ARs may also contribute to inappropriate sinus tachycardia response.[18,19]
POTS and Functional Autoantibodies
Several clinical observations suggest that autoimmune pathophysiology plays a role in POTS. These observations include disease onset following viral infections or vaccination, family history of systemic autoimmune disorders, and association with peripheral neuropathy of autoimmune origin.[2] The presence of autoantibodies against common G protein-coupled receptors (GPCRs) including α1-AR, β1-AR, β2-AR, M2AChRs and type 1 angiotensin 2 receptor (AT1-R) has been demonstrated in POTS (Figure 3).[15,18–21] The autoantibodies bind to the extracellular loop between the fourth and fifth membrane-spanning segments of GPCR (ECL2, extracellular loop 2), a site that is different from the true ligand binding site.[18] Unlike other autoantibodies that invoke immune-mediated cell damage, the antibodies against GPCR induce functional response inside the cell. In the absence of true ligands, the antibodies induce an agonistic response, whereas in the presence of true ligands, the antibodies either enhance (allosteric activation) or reduce (partial agonist) the functional response of true ligand (Figure 3).[18] Antibodies against α1-AR, AT1-R and M2AChR act as partial agonists and β1-AR and β2-AR antibodies are allosteric activators.[15,18,19,21] Following orthostatic challenge, the α1-AR and AT1-R antibodies attenuate the vasoconstriction response of catecholamines and angiotensin II whereas M2AChR and β1-AR antibodies exaggerate the HR-accelerating effects of parasympathetic withdrawal and sympathetic stimulation, respectively.[18,[19,21] In addition to activating the β-ARs on the sinus node, the autoantibodies against β-AR can also increase sympathetic outflow.[15] Although the role of AT1-R antibodies on volume regulation has not been tested, they can potentially attenuate aldosterone synthesis in response to RAAS activation and may promote the state of intravascular hypovolaemia.[22] Recently, there was a surge in the incidence of POTS after the COVID-19 pandemic. Functional antibodies against different GPCRs have also been demonstrated in the serum of symptomatic post-COVID-19 patients.[23] Interestingly, detection of functional antibodies by conventional ELISA may be erroneous.[24]
Figure 3: G Protein Receptor-coupled Functional Autoantibodies in POTS.
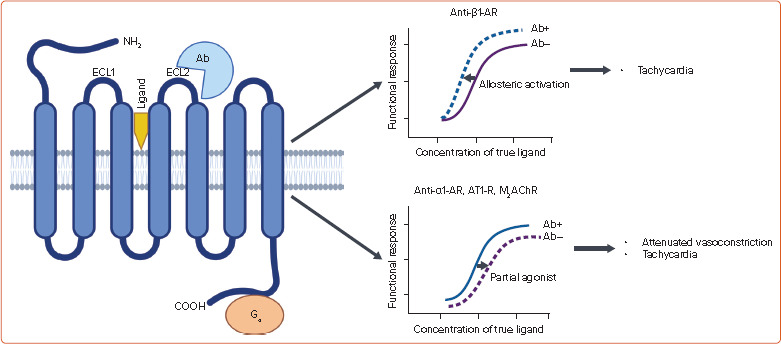
Ab = functional antibody (+ and −, presence or absence of autoantibodies, respectively); AR = adrenergic receptor; AT1-R = type 1 angiotensin II receptor; COOH = carboxy terminal of peptide chain; ECL = extracellular loop; NH2 = amino terminal of peptide chain; M2AChR = type 2 muscarinic cholinergic receptor; POTS = postural orthostatic tachycardia syndrome. True ligands represent catecholamines, angiotensin II, and acetylcholine. Source: Created with BioRender.com
Autonomic Remodelling in POTS
Autonomic remodelling in POTS is responsible for an inappropriate augmentation of HR response that is out of proportion to physiological reflex tachycardia. The characteristic autonomic abnormalities include attenuated parasympathetic response and augmented sympathetic activity on the sinoatrial node.[2,3,18,19] Interestingly, autonomic remodelling in POTS is not only limited to the sinoatrial node: autonomic remodelling with subsequent electrophysiological changes is also noted in the atrium and ventricle.[25] However, the consequences of atrial and ventricular autonomic remodelling are still to be determined.
Attenuation of resting sympathovagal imbalance with attenuation of parasympathetic activity is suggested by decreased HR variability (HRV), along with a reduced high-frequency (HF) component and an increased low-frequency (LF) to HF ratio (LF/HF ratio) of HRV in POTS patients compared with those without POTS.[26,27] Following standing posture, immediate reduction of the HF component of HRV is a physiological phenomenon. However, the reduction is more profound in POTS patients, suggesting an exaggerated parasympathetic withdrawal. Similarly, a higher increase in LF/HF ratio following standing in POTS patients suggests significant sympathovagal imbalance.[28,29] Unlike in the control population, a significant rise in the LF component of HRV and muscle sympathetic nerve activity (MSNA) with standing and hypotension suggests augmented sympathetic outflow following haemodynamic challenges in POTS patients.[28,29]
Unlike consistent attenuation of resting parasympathetic tone, supine sympathetic activity is reported to be normal, reduced or increased in POTS patients.[28,30–32] The heterogeneous aetiology may explain the conflicting findings on resting sympathetic activity. Although the supine sympathetic tone is principally unaffected in POTS patients, decreased MSNA may suggest a neuropathic form, while a high MSNA and LF component of HRV may suggest hyperadrenergic POTS. A discordance between vascular and cardiac sympathetic response following orthostatic stress, a subtle increase in MSNA with a profound increase in LF component, is also reported in neuropathic POTS.[30]
Transcutaneous Vagus Nerve Stimulation in POTS
tVNS in POTS: The Mechanistic Concept
As described previously, the blunted postural parasympathetic activity and exaggerated sympathetic activity are the primary mechanisms of postural tachycardia in POTS. Hence, potentiation of parasympathetic response and inhibition of sympathetic drive may work as an effective therapeutic strategy in POTS. Consistent with this notion, cholinesterase inhibitors (ChEIs) and β-blockers are used for pharmacological management of POTS.[3] However, resting bradycardia is a common concern with these approaches. The generalised parasympathomimetic effects of ChEIs are also an issue. Moreover, in the setting of reduced myocardial mass and hypovolaemia, inhibition of myocardial contraction by β-blocker may provoke postural hypotension in vulnerable patients.[33]
tVNS provides a unique, non-pharmacological opportunity to restore sympathovagal balance by electrical stimulation of the auricular branch of the vagus, a selective afferent branch (Figure 4).[34] Selective recruitment of afferent vagus fibres with low-level VNS and tVNS leads to activation of the sensory nucleus of the vagus (nucleus tractus solitarius or NTS). The NTS subsequently transmits stimulatory signals to the motor nucleus of the vagus and caudal ventrolateral medulla (CVLM). Tonic discharge from neurons in the rostral ventrolateral medulla (RVLM) is responsible for the activation of sympathetic motor nuclei in the spinal cord. However, upon activation, the CVLM sends inhibitory signals to the RVLM, leading to inhibition of the sympathetic system. In contrast, activation of the motor nuclei of the vagus leads to activation of the parasympathetic system. The parasympathomimetic and sympatholytic effects of tVNS have been demonstrated in both preclinical and clinical studies. In patients with heart failure, 1 hour of tVNS was associated with significant improvement of HRV parameters (increased HRV, increased HF component, and decreased LF and LF/HF ratio), which suggests an increase in cardiac parasympathetic activity and attenuation of sympathetic activity with improvement of sympathovagal balance.[35] Similar outcomes have been observed with intermittent tVNS treatment (1 hour daily) given for an extended duration.[6,36] Likewise, the decrease in both the frequency and incidence of MSNA during tVNS in healthy volunteers implies a reduction in sympathetic outflow.[37] Additionally, suppression of stellate ganglion nerve activity through VNS has been reported in a canine model of AF.[38] Importantly, clinical studies did not demonstrate significant resting bradyarrhythmia, hypotension or systemic parasympathomimetic effects with tVNS.[6] Hence, there is a mechanistic plausibility of correction of autonomic dysfunction with tVNS in POTS without significant side-effects of available pharmacotherapy (Figure 5).
Figure 4: Mechanisms of Action of Transcutaneous Vagus Nerve Stimulation.
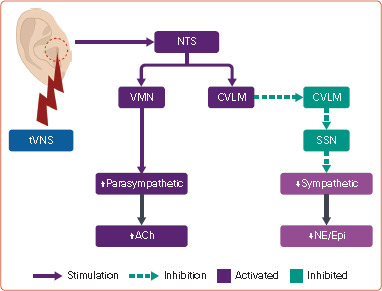
ABV = auricular branch of vagus; ACh = acetylcholine; CVLM = caudal ventrolateral medulla; Epi = epinephrine; NE = norepinephrine; NTS = nucleus tractus solitarius; RVLM = rostral ventrolateral medulla; SSN = sympathetic spinal cord nuclei (intermediolateral cell column of the thoracolumbar spinal cord); tVNS = transcutaneous vagus nerve stimulation; VMN = vagus motor nuclei (nucleus accumbens and dorsal motor nucleus of vagus). Source: Created with BioRender.com.
Figure 5: Mechanisms of Symptom Improvement with tVNS in POTS.
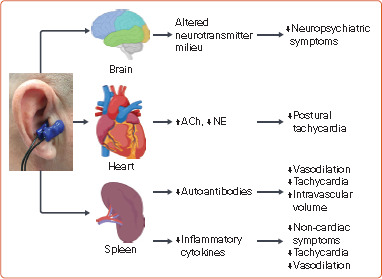
ACh = acetylcholine; NE = norepinephrine; POTS = postural orthostatic tachycardia syndrome; tVNS = transcutaneous vagus nerve stimulation. Source: Created with BioRender.com.
Stimulation of the efferent vagus nerve by tVNS has also been shown to attenuate systemic inflammation.[6,36] The immunomodulatory effect of the parasympathetic system in the spleen is known to be responsible for the anti-inflammatory activity of tVNS.[8] A state of systemic inflammatory response with the rise of inflammatory cytokines is described in POTS.[1,2,39,40] The exact role of inflammation in POTS is still unclear. However, inflammatory cytokines may participate in multiple pathophysiological processes including attenuation of vasoconstriction, potentiation of central sympathetic flow, and promotion of inappropriate sinus tachycardia.[1,41,42] Inflammatory cytokines may also play important roles in the pathophysiology of non-cardiac symptoms such as depression and fatigue. Hence, the anti-inflammatory effect of tVNS may help to resolve cardiac and non-cardiac symptoms (Figure 5).
tVNS is used to treat neuropsychiatric conditions including depression and post-traumatic stress disorders.[4,43] Functional brain imaging studies suggest tVNS-induced altered activation patterns in several brain regions.[4] Other than the anti-inflammatory effect, the beneficial effects of tVNS may also result from the alteration of the neurotransmitter milieu in the brain.[4,44,45] This pleiotropic effect may help to alleviate neuropsychiatric symptoms of POTS (Figure 5).
tVNS and POTS: Experimental Evidence
The POTS phenotype is demonstrated in rabbits following immunisation with peptides from adrenergic receptors (α1- and β1-ARs) or muscarinic cholinergic receptors (M2AChRs). Six weeks after immunisation, animals developed autoantibodies against respective GPCRs (α1- and β1-ARs or M2AChRs) and had a greater degree of postural increase in HR without significant changes in blood pressure during the tilt test.[46,47] HRV analysis showed reduced HF components along with increased LF and LF/HF ratio in the immunised animals compared with preimmune values.
In the M2AChR autoantibody model, daily 30-minute tVNS for 2 weeks was associated with significant suppression of postural tachycardia and associated inflammation.[46] tVNS for 4 weeks also ameliorated postural tachycardia in the autoantibody-induced hyperadrenergic POTS model (immunisation with α1- and β1-AR proteins).[48] HRV analysis following tVNS showed an improvement of cardiac sympathovagal balance, as indicated by an increase in the HF component and a decrease in the LF component and LF/HF ratio following orthostatic stress.[46,48] The cholinomimetic effect of tVNS was also demonstrated by an increase in ACh secretion and augmentation of the HR-lowering action of the M2AChR agonist.[46,48] A concurrent mitigation of systemic inflammation by tVNS with subsequent reduction of serum inflammatory cytokines was reported in both studies.
tVNS and POTS: Clinical Evidence
Case reports and small clinical studies demonstrated modulation of POTS pathophysiology and symptoms by invasive and non-invasive VNS.[45] There are anecdotal reports of improvement of POTS symptoms following invasive VNS for refractory epilepsy.[49,50] In a sham control study, short-term transcutaneous auricular vagal nerve stimulation during orthostatic challenge was associated with an increase in the HF component of HRV, which was translated into an increase in tilt time and a tendency toward reduction of orthostatic change of HR (supine HR–standing HR).[51] Interestingly, the beneficial effect was achieved only with low-level stimulation (high-frequency power <200 ms[2]). In another open-label clinical trial, 4 hours of tVNS daily at the cymba conchae region for 14 days was associated with a significant reduction in orthostatic intolerance and gastrointestinal symptoms in POTS patients.[45] A tendency of reduced HR following standing posture was also noted in the tVNS group.
The recently published sham-controlled, double-blind, randomised clinical trial by our group showed that 1-hour tVNS (20 Hz, 1 mA below the discomfort threshold) for 2 months is associated with significant improvement of postural tachycardia and a tendency towards improved autonomic symptoms in patients with POTS.[52] Postural changes in HRV parameters (increased LF, decreased HF, and increased LF/HF ratio) were attenuated by tVNS, suggesting attenuation of sympathetic response and improvement of parasympathetic response and sympathovagal balance. tVNS was also associated with a reduction in levels of functional antibodies against α1-AR and β1-AR, and proinflammatory cytokine tumour necrosis factor-α (TNF-α). More importantly, no patient in the tVNS group developed blood pressure reduction during the postural test. Despite the mitigation of compensatory postural sympathetic hyperactivation, the absence of postural hypotension in the tVNS group suggests that the beneficial effects of tVNS in POTS are beyond the modulation of autonomic balance. By attenuating the α1-AR antibodies and inflammation, tVNS may improve vasoconstriction and venous return following orthostatic stress. Mitigation of inflammation and reduction of anti-β1-AR antibodies may help in alleviating central sympathetic outflow and inappropriate tachycardia response. Although the effect of tVNS on AT1-R autoantibodies has not been evaluated in published experimental and clinical studies, a reduction of AT1-R antibodies may potentially improve the vascular contraction and intravascular volume and thus the venous return following standing posture. Currently, a few other studies are also evaluating the role of tVNS in POTS (NCT04632134, NCT03124355), and dysautonomia syndromes associated with Ehlers–Danlos syndrome (NCT05212129) and COVID-19 (NCT05630040, NCT05421208).
It should be noted that the physiological effects of tVNS are dependent on the parameters of stimulation including pulse width, frequency and amplitude of the electric current. In addition, although tVNS has been shown to exert a ‘memory effect’, the optimal duration of stimulation has not been determined. These shortcomings notwithstanding, current evidence suggests that further investigation of tVNS as a non-pharmacological therapeutic option in POTS is warranted.
Conclusion
Considering the potential impact of non-invasive vagal stimulation on various key targets of POTS, such as sympathovagal imbalance, inflammation and autoantibody production, tVNS holds promise in altering the underlying disease mechanisms and mitigating the tachycardia response. In addition, the pleiotropic effects of tVNS may provide further benefits by alleviating non-cardiac and neuropsychiatric symptoms. The existing evidence suggests that chronic tVNS is well tolerated and, unlike pharmacotherapies, does not carry any major side-effects. Preliminary clinical data suggest that tVNS decreased postural tachycardia in patients with POTS compared with sham stimulation. Further evaluation of tVNS in randomised, sham-controlled clinical trials, adequately powered to detect clinical outcomes, is warranted.
Clinical Perspective
Exaggerated parasympathetic withdrawal and sympathetic overdrive during postural stress are principal mechanisms of postural tachycardia in postural orthostatic tachycardia syndrome (POTS).
Non-invasive autonomic modulation using low-level tragus stimulation holds promise in altering the underlying disease mechanisms and mitigating the tachycardia response in patients with POTS, by decreasing anti-adrenergic autoantibodies, improving cardiac autonomic function and decreasing serum inflammatory cytokines.
Future studies should correlate the use of autonomic modulation to improved patient-reported outcomes.
Funding Statement
NIH/NHLBI R01HL161008 to SS. PC is supported by the George Mines Travelling Fellowship from the Canadian Heart Rhythm Society.
References
- 1.Vernino S, Bourne KM, Stiles LE et al. Postural orthostatic tachycardia syndrome (POTS): state of the science and clinical care from a 2019 National Institutes of Health Expert Consensus Meeting – Part 1. Auton Neurosci. 2021;235:102828. doi: 10.1016/j.autneu.2021.102828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bryarly M, Phillips LT, Fu Q et al. Postural orthostatic tachycardia syndrome: JACC focus seminar. J Am Coll Cardiol. 2019;73:1207–28. doi: 10.1016/j.jacc.2018.11.059. [DOI] [PubMed] [Google Scholar]
- 3.Mar PL, Raj SR. Postural orthostatic tachycardia syndrome: mechanisms and new therapies. Annu Rev Med. 2020;71:235–48. doi: 10.1146/annurev-med-041818-011630. [DOI] [PubMed] [Google Scholar]
- 4.Farmer AD, Strzelczyk A, Finisguerra A et al. International consensus based review and recommendations for minimum reporting standards in research on transcutaneous vagus nerve stimulation (version 2020). Front Hum Neurosci. 2020;14:568051. doi: 10.3389/fnhum.2020.568051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang Y, Po SS, Amil F, Dasari TW. Non-invasive low-level tragus stimulation in cardiovascular diseases. Arrhythm Electrophysiol Rev. 2020;9:40–6. doi: 10.15420/aer.2020.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stavrakis S, Stoner JA, Humphrey MB et al. TREAT AF (Transcutaneous Electrical Vagus Nerve Stimulation to Suppress Atrial Fibrillation): a randomized clinical trial. JACC Clin Electrophysiol. 2020;6:282–91. doi: 10.1016/j.jacep.2019.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sha Y, Scherlag BJ, Yu L et al. Low-level right vagal stimulation: anticholinergic and antiadrenergic effects. J Cardiovasc Electrophysiol. 2011;22:1147–53. doi: 10.1111/j.1540-8167.2011.02070.x. [DOI] [PubMed] [Google Scholar]
- 8.Pavlov VA, Tracey KJ. The cholinergic anti-inflammatory pathway. Brain Behav Immun. 2005;19:493–9. doi: 10.1016/j.bbi.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 9.Raj SR, Robertson D. Moving from the present to the future of postural tachycardia syndrome: what we need. Auton Neurosci. 2018;215:126–8. doi: 10.1016/j.autneu.2018.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Streeten DH. Pathogenesis of hyperadrenergic orthostatic hypotension. Evidence of disordered venous innervation exclusively in the lower limbs. J Clin Invest. 1990;86:1582–8. doi: 10.1172/JCI114878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roma M, Marden CL, De Wandele I et al. Postural tachycardia syndrome and other forms of orthostatic intolerance in Ehlers–Danlos syndrome. Auton Neurosci. 2018;215:89–96. doi: 10.1016/j.autneu.2018.02.006. [DOI] [PubMed] [Google Scholar]
- 12.Doherty TA, White AA. Postural orthostatic tachycardia syndrome and the potential role of mast cell activation. Auton Neurosci. 2018;215:83–8. doi: 10.1016/j.autneu.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Zhang ZY, Qian LL, Wang RX. Molecular mechanisms underlying renin-angiotensin-aldosterone system mediated regulation of BK channels. Front Physiol. 2017;8:698. doi: 10.3389/fphys.2017.00698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine BD, Zuckerman JH, Pawelczyk JA. Cardiac atrophy after bed-rest deconditioning: a nonneural mechanism for orthostatic intolerance. Circulation. 1997;96:517–25. doi: 10.1161/01.cir.96.2.517. [DOI] [PubMed] [Google Scholar]
- 15.Fedorowski A, Li H, Yu X et al. Antiadrenergic autoimmunity in postural tachycardia syndrome. Europace. 2017;19:1211–9. doi: 10.1093/europace/euw154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mayuga KA, Fedorowski A, Ricci F et al. Sinus tachycardia: a multidisciplinary expert focused review. Circ Arrhythm Electrophysiol. 2022;15:e007960. doi: 10.1161/CIRCEP.121.007960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pongratz G, Straub RH. The sympathetic nervous response in inflammation. Arthritis Res Ther. 2014;16:504. doi: 10.1186/s13075-014-0504-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Yu X, Liles C et al. Autoimmune basis for postural tachycardia syndrome. J Am Heart Assoc. 2014;3:e000755. doi: 10.1161/JAHA.113.000755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Zhang G, Forsythe E et al. Implications of antimuscarinic autoantibodies in postural tachycardia syndrome. J Cardiovasc Transl Res. 2022;15:438–40. doi: 10.1007/s12265-021-10167-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Badiudeen T, Forsythe EA, Bennett G et al. A functional cell-based bioassay for assessing adrenergic autoantibody activity in postural tachycardia syndrome. J Transl Autoimmun. 2019;2:100006. doi: 10.1016/j.jtauto.2019.100006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu X, Li H, Murphy TA et al. Angiotensin II type 1 receptor autoantibodies in postural tachycardia syndrome. J Am Heart Assoc. 2018;7:e008351. doi: 10.1161/JAHA.117.008351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mustafa HI, Garland EM, Biaggioni I et al. Abnormalities of angiotensin regulation in postural tachycardia syndrome. Heart Rhythm. 2011;8:422–8. doi: 10.1016/j.hrthm.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wallukat G, Hohberger B, Wenzel K et al. Functional autoantibodies against G-protein coupled receptors in patients with persistent Long-COVID-19 symptoms. J Transl Autoimmun. 2021;4:100100. doi: 10.1016/j.jtauto.2021.100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall J, Bourne KM, Vernino S et al. Detection of G protein-coupled receptor autoantibodies in postural orthostatic tachycardia syndrome using standard methodology. Circulation. 2022;146:613–22. doi: 10.1161/CIRCULATIONAHA.122.059971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malik V, Nguyen MT, Seeley MC et al. Abnormal cardiac remodeling in postural orthostatic tachycardia syndrome: further insights into its cardiac origin. JACC Clin Electrophysiol. 2022;8:1044–6. doi: 10.1016/j.jacep.2022.02.012. [DOI] [PubMed] [Google Scholar]
- 26.Baker J, Racosta JM, Balint B, Kimpinski K. Utility of time and frequency domain parameters of heart rate variability in the context of autonomic disorders characterized by orthostatic dysfunction. J Clin Neurophysiol. 2018;35:123–9. doi: 10.1097/WNP.0000000000000452. [DOI] [PubMed] [Google Scholar]
- 27.Baker J, Racosta JM, Kimpinski K. Comparison of heart rate variability parameters to the autonomic reflex screen in postural orthostatic tachycardia syndrome and neurogenic orthostatic hypotension. J Clin Neurophysiol. 2018;35:115–22. doi: 10.1097/WNP.0000000000000436. [DOI] [PubMed] [Google Scholar]
- 28.Orjatsalo M, Alakuijala A, Partinen M. Heart rate variability in head-up tilt tests in adolescent postural tachycardia syndrome patients. Front Neurosci. 2020;14:725. doi: 10.3389/fnins.2020.00725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swai J, Hu Z, Zhao X et al. Heart rate and heart rate variability comparison between postural orthostatic tachycardia syndrome versus healthy participants; a systematic review and meta-analysis. BMC Cardiovasc Disord. 2019;19:320. doi: 10.1186/s12872-019-01298-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furlan R, Jacob G, Snell M et al. Chronic orthostatic intolerance: a disorder with discordant cardiac and vascular sympathetic control. Circulation. 1998;98:2154–9. doi: 10.1161/01.cir.98.20.2154. [DOI] [PubMed] [Google Scholar]
- 31.Jacob G, Diedrich L, Sato K et al. Vagal and sympathetic function in neuropathic postural tachycardia syndrome. Hypertension. 2019;73:1087–96. doi: 10.1161/HYPERTENSIONAHA.118.11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muenter Swift N, Charkoudian N, Dotson RM et al. Baroreflex control of muscle sympathetic nerve activity in postural orthostatic tachycardia syndrome. Am J Physiol Heart Circ Physiol. 2005;289:H1226–33. doi: 10.1152/ajpheart.01243.2004. [DOI] [PubMed] [Google Scholar]
- 33.Bhanu C, Nimmons D, Petersen I et al. Drug-induced orthostatic hypotension: a systematic review and meta-analysis of randomised controlled trials. PLoS Med. 2021;18:e1003821. doi: 10.1371/journal.pmed.1003821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deuchars SA, Lall VK, Clancy J et al. Mechanisms underpinning sympathetic nervous activity and its modulation using transcutaneous vagus nerve stimulation. Exp Physiol. 2018;103:326–31. doi: 10.1113/EP086433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tran N, Asad Z, Elkholey K et al. Autonomic neuromodulation acutely ameliorates left ventricular strain in humans. J Cardiovasc Transl Res. 2019;12:221–30. doi: 10.1007/s12265-018-9853-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stavrakis S, Elkholey K, Morris L et al. Neuromodulation of inflammation to treat heart failure with preserved ejection fraction: a pilot randomized clinical trial. J Am Heart Assoc. 2022;11:e023582. doi: 10.1161/JAHA.121.023582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clancy JA, Mary DA, Witte KK et al. Non-invasive vagus nerve stimulation in healthy humans reduces sympathetic nerve activity. Brain Stimul. 2014;7:871–7. doi: 10.1016/j.brs.2014.07.031. [DOI] [PubMed] [Google Scholar]
- 38.Shen MJ, Shinohara T, Park HW et al. Continuous low-level vagus nerve stimulation reduces stellate ganglion nerve activity and paroxysmal atrial tachyarrhythmias in ambulatory canines. Circulation. 2011;123:2204–12. doi: 10.1161/CIRCULATIONAHA.111.018028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gunning WT 3rd, Stepkowski SM, Kramer PM et al. Inflammatory biomarkers in postural orthostatic tachycardia syndrome with elevated G-protein-coupled receptor autoantibodies. J Clin Med. 2021;10:623. doi: 10.3390/jcm10040623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johansson M, Yan H, Welinder C et al. Plasma proteomic profiling in postural orthostatic tachycardia syndrome (POTS) reveals new disease pathways. Sci Rep. 2022;12:20051. doi: 10.1038/s41598-022-24729-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kennedy RH, Silver R. In: Neuroscience in the 21st Century. Pfaff DW, Volkow ND, editors. New York:: Springer; 2016. Neuroimmune signaling: cytokines and the CNS. pp. 1–41. [Google Scholar]
- 42.Vila E, Salaices M. Cytokines and vascular reactivity in resistance arteries. Am J Physiol Heart Circ Physiol. 2005;288:H1016–21. doi: 10.1152/ajpheart.00779.2004. [DOI] [PubMed] [Google Scholar]
- 43.Breit S, Kupferberg A, Rogler G, Hasler G. Vagus nerve as modulator of the brain–gut axis in psychiatric and inflammatory disorders. Front Psychiatry. 2018;9:44. doi: 10.3389/fpsyt.2018.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conway CR, Xiong W. The mechanism of action of vagus nerve stimulation in treatment-resistant depression: current conceptualizations. Psychiatr Clin North Am. 2018;41:395–407. doi: 10.1016/j.psc.2018.04.005. [DOI] [PubMed] [Google Scholar]
- 45.Diedrich A, Urechie V, Shiffer D et al. Transdermal auricular vagus stimulation for the treatment of postural tachycardia syndrome. Auton Neurosci. 2021;236:102886. doi: 10.1016/j.autneu.2021.102886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deng J, Li H, Guo Y et al. Transcutaneous vagus nerve stimulation attenuates autoantibody-mediated cardiovagal dysfunction and inflammation in a rabbit model of postural tachycardia syndrome. J Interv Card Electrophysiol. 2023;66:291–300. doi: 10.1007/s10840-022-01144-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, Zhang G, Zhou L et al. Adrenergic autoantibody-induced postural tachycardia syndrome in rabbits. J Am Heart Assoc. 2019;8:e013006. doi: 10.1161/JAHA.119.013006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo Y, Li H, Deng J et al. Low-level tragus stimulation improves autoantibody-induced hyperadrenergic postural tachycardia syndrome in rabbits. Heart Rhythm. 2023;4:127–33. doi: 10.1016/j.hroo.2022.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lankford J, Numan M, Adejumo R et al. Vagal nerve stimulation in autonomic dysfunction: a case study. Auton Neurosci Basic Clin. 2015;192:83. doi: 10.1016/j.autneu.2015.07.094. [DOI] [Google Scholar]
- 50.Petelin Gadze Z, Bujan Kovac A, Adamec I et al. Vagal nerve stimulation is beneficial in postural orthostatic tachycardia syndrome and epilepsy. Seizure. 2018;57:11–3. doi: 10.1016/j.seizure.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 51.Diedrich A, Okamoto L, Black B et al. Abstract P387: sub-perception transdermal vagal stimulation in postural tachycardia syndrome. Hypertension. 2018;72((Suppl 1)):AP387. doi: 10.1161/hyp.72.suppl_1.P387. [DOI] [Google Scholar]
- 52.Stavrakis S, Chakraborty P, Farhat K Noninvasive vagus nerve stimulation in postural tachycardia syndrome: a randomized clinical trial. JACC Clin Electrophysiol. 2023. epub ahead of press. [DOI] [PMC free article] [PubMed]