

Abstract
The Streptococcus salivarius 57.I ure cluster was organized as an operon, beginning with ureI, followed by ureABC (structural genes) and ureEFGD (accessory genes). Northern analyses revealed transcripts encompassing structural genes and transcripts containing the entire operon. A ς70-like promoter could be mapped 5′ to ureI (PureI) by primer extension analysis. The intensity of the signal increased when cells were grown at an acidic pH and was further enhanced by excess carbohydrate. To determine the function(s) of two inverted repeats located 5′ to PureI, transcriptional fusions of the full-length promoter region (PureI), or a deletion derivative (PureIΔ100), and a promoterless chloramphenicol acetyltransferase (CAT) gene were constructed and integrated into the chromosome to generate strains PureICAT and PureIΔ100CAT, respectively. CAT specific activities of PureICAT were repressed at pH 7.0 and induced at pH 5.5 and by excess carbohydrate. In PureIΔ100CAT, CAT activity was 60-fold higher than in PureICAT at pH 7.0 and pH induction was nearly eliminated, indicating that expression was negatively regulated. Thus, it was concluded that PureI was the predominant, regulated promoter and that regulation was governed by a mechanism differing markedly from other known mechanisms for bacterial urease expression.
Ureases are multisubunit enzymes requiring Ni2+ for catalytic activity. Several bacterial urease gene clusters have been isolated and characterized, and similarities in the organization of the clusters between species have been demonstrated (17, 18). Most bacterial ureases consist of three subunits, α, β, and γ, encoded by ureC, -B, and -A, respectively (18), although exceptions exist (10). The assembly of a catalytically active urease also requires ureE, -F, -G, and -D, known as accessory genes, which encode proteins required for the productive incorporation of Ni2+ into the metallocenter within the active site. In some cases, high-affinity nickel transporters, for instance, NixA of Helicobacter pylori (16), are known to be required for optimal urease activity. Additional genes, such as ureI of H. pylori and ureH and ureI of Bacillus sp. strain TB-90, have been identified (15), but the function(s) of these gene products in urease biogenesis is not well defined. However, UreH from Bacillus has homology with the high-affinity nickel transporter of Alcaligenes eutrophus, HoxN (31); thus, it has been proposed that UreH is involved in energized nickel uptake in conjunction with UreI.
The expression of bacterial urease genes is regulated by different mechanisms (7). Few known bacterial ureases are expressed constitutively, whereas most are regulated by environmental conditions. For example, urease expression in Klebsiella pneumoniae and Klebsiella aerogenes is activated only under nitrogen-limiting conditions (8, 14, 19), and expression in Proteus mirabilis is induced by urea and is mediated by the positive transcriptional regulator UreR (20). Information regarding the expression of ureases of oral bacteria is just beginning to accumulate. Among the species of oral bacteria that have been identified as ureolytic, Streptococcus salivarius is believed to be a major contributor to total oral ureolysis (25). In contrast to studies of other known mechanisms for urease expression, previous studies in our laboratory and in others demonstrated that urease expression in S. salivarius 57.I is regulated by the environmental pH (4, 27), growth rate, and carbohydrate availability (4). At a neutral pH, expression is almost completely repressed. Induction occurs when cells are grown at an acidic pH, and expression is further enhanced by excess carbohydrate and higher growth rates. Furthermore, based on the results of Northern blot analysis measuring ureC-specific mRNAs from cells grown at various pH values and in conditions with limited or excess carbohydrate, it was found that the induction is regulated, at least in part, at the transcriptional level (4).
Our initial attempts to isolate the urease genes from S. salivarius 57.I resulted in the cloning of a partial gene cluster that contained the 3′ end of ureI followed by ureABCEFGD (5). Despite the lack of the 5′ end of the cluster, Streptococcus gordonii DL1, a nonureolytic oral microorganism, expressed urease activity when harboring this partial ure cluster on a moderate-copy-number plasmid (pMC17) (Fig. 1; Table 1) and when the growth medium was supplemented with NiCl2 (5). The purpose of this work was to identify additional genes that may be involved in urease biogenesis, to investigate the transcriptional organization of the ure cluster of S. salivarius 57.I, and to explore the basis for differential expression of ure genes in response to pH and carbohydrate availability.
FIG. 1.

ure cluster and the 5′ flanking region on the S. salivarius 57.I chromosome. A restriction endonuclease map of the chromosome containing the ure cluster and the 5′ flanking region is shown on the top line. The limits of the DNA sequence shown in Fig. 2 are indicated by vertical arrows. The relative location and the direction of transcription of each ORF are indicated by horizontal arrows. The molecular mass in kilodaltons of each gene product is shown below each gene. The locations and orientations of primer pairs used in RT-PCR are indicated by arrows immediately under the restriction map. The Sau3A-XbaI region from pMC12, used as a probe for identifying pMC32, is indicated in a hatched box. The region used for the construction of pCW45 and pMC77 (see below) for integration is indicated in a shaded box within the restriction map.
TABLE 1.
Bacterial strain and plasmids
| Strain or plasmid | Relevant phenotypea | Description | Source or reference |
|---|---|---|---|
| S. salivarius | |||
| 57.I | Ure+ | Wild-type host | 25 |
| PureICAT | CAT+ Ure+ | Wild-type chromosome containing a single copy of PureI-cat | This study |
| PureIΔ100CAT | CAT+ Ure+ | Wild-type chromosome containing a single copy of PureIΔ100-cat | This study |
| S. gordonii | |||
| DL1 | Wild-type host | 11 | |
| MC17 | Ure+ Spr | DL1 harboring pMC17 | 5 |
| E. coli DH10B | General cloning host | LTI | |
| Plasmids | |||
| pGEM-5Zf(+) | Apr | General cloning vector | Promega |
| pGEM-7Zf(+) | Apr | General cloning vector | Promega |
| pUC18 | Apr | General cloning vector | LTI |
| pSU21 | Cmr | Moderate-copy-number E. coli vector | 2 |
| pSF143 | Tcr | Integration vector for streptococcal species | 29 |
| pDL278 | Spr | Streptococcus-E. coli shuttle vector | 13 |
| pDL290 | Kmr | Low-copy-number E. coli vector | 10a |
| pCW24 | Apr Cmr | pUC18-promoterless cat | This study |
| pCW42 | Apr Cmr | pGEM-5Zf(+)-promoterless cat | This study |
| pCW45 | Tcr Cmr | pSF143-PureIcat | This study |
| pMC12 | Cmr | pSU21-ureIABCEFGDb | 5 |
| pMC17 | Spr | pDL278-ureIABCEFGDb | 5 |
| pMC23 | Cmr | pSU21-6.0-kbp XbaI fragment | This study |
| pMC32 | Cmr Kmr | pDL290-6.0-kbp XbaI fragment | This study |
| pMC63 | Apr Cmr | pCW42-PureI | This study |
| pMC68 | Apr Cmr | pCW42-PureIΔ100 + 2.0-kbp fragment | This study |
| pMC71 | Apr Cmr | pCW42-PureI + 2.0-kbp fragment | This study |
| pMC77 | Apr Tcr | pSF143-PureIΔ100-cat | This study |
Abbreviations: Ure, urease; Ap, ampicillin; Cm, chloramphenicol; Km, kanamycin; Sp, spectinomycin; Tc, tetracycline; r, resistance.
The 5′ end of ureI is incomplete.
Isolation and nucleotide sequence analysis of the 5′ portion of ureI and the flanking region from S. salivarius 57.I.
A 7.8-kbp Sau3A fragment containing the 3′ portion of ureI and complete ureABCEFGD has been previously described (5). No genes involved in urea metabolism were found within 1.5 kbp 3′ to the ure cluster, and a putative Rho-independent terminator was identified 120 bases 3′ to the stop codon of ureD, the last gene in the cluster. To obtain the complete ureI and other genes potentially involved in urease biogenesis, total chromosomal DNA was isolated from S. salivarius 57.I and digested to completion with XbaI. DNA fragments were separated on agarose gels, transferred to nitrocellulose, and hybridized at high stringency (12) to a radiolabeled Sau3A-XbaI fragment, approximately 1.0 kbp in size, which contained the 3′ portion of ureI (Fig. 1). An approximately 6.0-kbp fragment was subsequently identified (data not shown). To isolate this fragment, a subgenomic DNA library was constructed in pSU21 (2) and screened with the same probe, as described above. The resulting chimeric plasmid was designated pMC23. To maintain stably the XbaI fragment in Escherichia coli, it was necessary to subclone the insert onto a low-copy-number vector, pDL290 (10a), which generated pMC32. For convenience, all strains and plasmids used in this study are listed in Table 1.
The complete nucleotide sequences of both strands of the XbaI fragment were obtained, and the sequences of ureI and the 5′ flanking region are presented here. The ure cluster began with an open reading frame (ORF) of 513 bp, which had homology with H. pylori ureI and thus was designated ureI (Fig. 2). A putative ribosome binding site (RBS) could be found 6 bases 5′ to the predicted start codon of ureI. The ureI gene was predicted to encode a protein with an estimated molecular weight of 18,995 and a pI of 7.0. In addition, the hydropathy plot of the deduced amino acid sequences indicated that UreI was relatively hydrophobic, with six potential transmembrane domains (data not shown), suggesting that UreI could be a membrane protein.
FIG. 2.

Nucleotide and deduced amino acid sequences of the 5′ flanking region of the ure cluster. ORF3 and ureI are transcribed from the opposite DNA strands; thus, the sequence of ureI presented here is the coding strand, and the sequence of ORF3 is the noncoding strand. The locations and orientations of primers (PureIas-100 and PMC32-1) used to identify the transcriptional start site of ureI and of primers used to amplify PureI and PureIΔ100 (PureIs, PureIde12, and PureIas) are indicated by horizontal arrows. The transcriptional start site of ureI, determined by primer extension analysis, is indicated by a vertical arrow, and the corresponding −10 and −35 regions are overlined. The sequences of the inverted repeats 5′ to PureI are shaded.
Translation of the nucleotide sequences of the XbaI fragment revealed three additional ORFs located 5′ to the ure cluster. ORF1 and -2 were transcribed in the same direction as the ure cluster, and ORF3 was transcribed in the opposite direction (Fig. 1). Proposed RBSs were found in the appropriate positions 5′ to all ORFs. ORF1, 2,094 bp in size, encoded a protein with a calculated molecular weight of 78,431 and an estimated pI of 8.9. A high degree of homology was observed between ORF1 and ATP-binding cassette transporters. ORF2, located 109 bp 3′ to ORF1, was 330 bp and encoded a protein with a calculated molecular weight of 12,717 and an estimated pI of 9.6. Based on the hydropathy plot, ORF2 may encode a membrane protein; however, only low levels of similarity were observed between the ORF2 product and other known proteins. ORF3, 1,392 bp in size, was transcribed in the direction opposite to the transcription of ORF1 and -2, and it encoded a protein with a calculated molecular weight of 52,393 and an estimated pI of 9.7. Due to the proximity of these three ORFs to the ure cluster and their characteristics, the possibility of their involvement in optimal urease expression, perhaps through Ni2+ uptake, is currently under investigation.
Operonic arrangement of the S. salivarius 57.I ure cluster.
To analyze the transcriptional organization of the ure cluster and potentially identify a transcript(s) that is induced at a low environmental pH, Northern blot analyses were performed with batch-grown cells at pH 7.0, 6.0, and 5.0, and ureI-, ureC-, and ureDG-specific mRNAs were examined (Fig. 1 and 3A). Total RNA was isolated according to the method of Putzer et al. (21) with modifications. Briefly, cells were cultured in brain heart infusion (BHI; Difco, Detroit, Mich.) containing 50 mM potassium phosphate buffer, pH 7.5, in BHI alone, or in BHI which had been adjusted to pH 5.5 by addition of 2 N HCl to mid-log phase, at which point the cultures were at approximately pH 7, 6, and 5, respectively. Cells were harvested, washed once with 10 mM sodium phosphate buffer, pH 7.0, and then resuspended in 1/40 of the original culture volume in 50 mM Tris–10 mM EDTA, pH 8.0. In 2.0-ml screw-cap microcentrifuge tubes, 0.5 g of glass beads (0.1-mm diameter), 500 μl of concentrated cell suspensions, 500 μl of phenol-chloroform (5:1, pH 4.7; Ambion, Austin, Tex.), and 100 μl of 10% sodium dodecyl sulfate were added. Cells were then subjected to mechanical disruption by homogenization in a Bead Beater (Biospec Products, Inc., Bartlesville, Okla.) for a total of 40 s at 4°C. The aqueous phase was first extracted with an equal volume of phenol-chloroform four to five times, followed by extraction with chloroform-isoamyl alcohol (24:1) alone. Total cellular RNA was then precipitated with 1/10 volume of 3 M Na acetate, pH 6.0, and 2 volumes of ice-cold 99% ethanol. Northern blot analysis was performed with 0.7% formaldehyde-agarose gels (22), and hybridization conditions were as described previously (1).
FIG. 3.

(A) Northern blot analysis of ure-specific messages. Ten micrograms of total cellular RNA from cultures at each pH level was probed with a ureI-specific probe (a), a ureC-specific probe (b), and a ureDG-specific probe (c). (B) PCR products generated from RT-PCR. 1% of total cDNA generated by RT-PCR from each RNA sample was amplified with specific primers (Fig. 1), and 10% of the PCR products were run on a 0.8% Tris-borate-EDTA gel. Some PCR products were generated with a primer pair specific for the ureIA intergenic region (a), and others were generated with a primer pair specific for the ureCE intergenic region (b). RT was included in some reactions (+RT), but not in control reactions which were carried out identically to the experimental samples (−RT). In other control reactions PCRs were used to amplify the target region from S. salivarius 57.I chromosomal DNA under the same conditions (57.I). The 100-bp DNA ladder was used as the molecular weight marker.
Two species, approximately 6.8 and 2.7 kb, were identified with a probe specific for ureI. The intensity of both signals increased when the culture pH became acidic, and the highest levels were observed in cells grown at pH 5.0. Furthermore, under all growth conditions, the smaller transcript was more abundant than the larger one. Extrapolating from the estimated sizes of RNA transcripts and the known sizes of the urease genes, the two transcripts presumably corresponded to ureIABCEFGD and ureIABC, respectively. When total cellular RNA was probed with a ureC-specific probe, close examination revealed four fragments, 6.8, 6.3, 2.7, and 2.2 kb, presumably corresponding to ureIABCEFGD, ureABCEFGD, ureIABC, and ureABC, respectively. However, due to degradation of the mRNA, which is commonly observed with RNA from oral streptococci, it was not possible to definitely show by Northern analysis that the transcripts could arise from two promoters. The intensities of the signals increased when the growth pH was acidic. However, it was difficult to determine whether all transcripts were enhanced at an acidic pH or only transcripts arising from the promoter 5′ to ureI (PureI). In agreement with the above results, only the larger two transcripts, 6.8 and 6.3 kb, were observed when a ureDG-specific probe was used. Although the signals were not as sharp as those seen in ureI- and ureC-specific messages, increases in intensity of signals were observed in cells grown in acidic media. We did not observe any transcripts of the appropriate size for ureEFGD, suggesting the absence of a functional promoter 5′ to the accessory genes. In addition, all attempts, including the use of primer extension analysis and promoterless reporter fusions, failed to identify functional promoters within the ureCE intergenic region.
To confirm that transcription could extend through the ureIA and ureCE intergenic regions, the presence of the larger transcripts was verified by reverse transcriptase (RT) PCR. cDNA was synthesized from 10 μg of total RNA isolated from cells grown under different conditions with Moloney murine leukemia virus RT and was further amplified by PCR with random hexamers according to standard procedures (9). Negative controls included reactions in the absence of RT. cDNAs spanning the ureIA and ureCE intergenic regions were detected by PCR with primer pairs with appropriate sequences. In both cases, PCR products with appropriate sizes (Fig. 3B) and correct sequences (data not shown) were obtained, further supporting the operonic arrangement of the ure cluster. No products were seen in control reactions that were designed for detecting chromosomal DNA contamination.
It has been proposed that, after activation of the urease apoenzyme, the accessory proteins are released from the holoenzyme and are recycled for the purpose of assembling another catalytically active urease (18). Consequently, the quantity of structural proteins (UreABC) could be greater than that of accessory proteins (UreEFGD) at any given time. Results obtained with Northern analysis are consistent with the theory that transcripts containing the accessory genes are less abundant than those carrying only the structural genes. Although the molecular basis for the generation of the smaller transcripts (ureIABC and ureABC) is not clear, a strong stem-loop structure (ΔG = −34 kcal/mol) followed by a string of uridine residues was located 11 bases 3′ to the stop codon of ureC. We did not observe any transcripts with sizes corresponding to ureEFGD; thus, it is not likely that the smaller transcripts resulted from posttranscriptional processing. In preliminary half-life studies, it appears that the two smaller transcripts have half-lives roughly three times longer than those of the larger transcripts, which is not sufficient to account for the disparity in the amounts of the two sets of transcripts under fully induced conditions. Therefore, it seems likely that preferential termination occurs in the ureCE intergenic region, which gives rise to a higher proportion of ureIABC and ureABC transcripts compared with ureIABCEFGD and ureABCEFGD transcripts.
Localization of PureI.
Based on the results obtained from Northern blot analyses and the fact that ureI is the 5′-most gene in the ure cluster, it was hypothesized that PureI was differentially regulated in response to the environmental pH and carbohydrate availability. To map the putative location of PureI, the 5′ end of the transcript was determined by primer extension analysis. Total cellular RNAs from cells growing at steady state in continuous culture with a dilution rate of 0.3 h−1 (generation time ≅ 2.3 h), at pH 7.0 or 5.5 under carbohydrate-limiting conditions (10 mM fructose) or at pH 5.5 under excess carbohydrate conditions (200 mM fructose), were isolated. Cells were grown for at least 10 generations at any single set of growth parameters before cultures were defined to be at steady state, where the specific growth rate was equal to the dilution rate of the vessel (28). Two primers, containing antisense sequences of ureI, were used: primer PureIas-100, 5′-TCAAACCCAAACCACCCG-3′, and primer PMC32-1, 5′-CCCTGTACAAGCTCCAT-3′, were located 106 and 148 bases 3′ to the start codon, respectively (Fig. 2). Products extended from each reaction were analyzed on a 6% polyacrylamide gel along with a DNA sequencing reaction with the same primer. A signal, 22 bases 5′ to the translational start site of ureI (Fig. 4), was consistently observed with either primer under all growth conditions. This transcriptional initiation site could be mapped to a ς70-like promoter sequence located at an appropriate distance (Fig. 2). Furthermore, the intensity of the signal was greater with RNAs isolated from pH 5.5-grown cells than with those from pH 7.0-grown cells. The strongest signal was observed with RNAs isolated from cells grown at pH 5.5 with excess carbohydrate, suggesting that PureI is sensitive to both an acidic pH and carbohydrate concentrations.
FIG. 4.

Primer extension analysis of PureI. Total cellular RNA of S. salivarius 57.I from steady-state cultures grown in a chemostat were used. Radiolabeled primer PMC32-1 was incubated with the RNA, and the corresponding DNA was synthesized. The same primer was used to prime dideoxy sequencing reactions with plasmid pMC32 as a template. Lanes 1 and 2 show total RNA isolated from pH 7.0 and pH 5.5 cultures, respectively, with 10 mM fructose. Lane 3 shows total RNA isolated from a pH 5.5 culture with 200 mM fructose.
Based on the results of Northern blot analysis and the NiCl2-dependent urease expression in recombinant streptococcal and E. coli strains harboring ureA to -G (5), the possibility exists that there is a functional promoter 5′ to ureA (PureA). Our preliminary study using a PureA-cat fusion, in which a 400-bp fragment beginning 5′ to the translational start codon of ureA was fused to a promoterless chloramphenicol acetyltransferase (CAT) gene (cat), also demonstrated a functional but pH-unresponsive streptococcal promoter within this 400-bp region (data not shown). In addition, we found extremely low levels of urease in a strain of S. salivarius carrying a polar insertion in ureI, where the expression of the ure operon should have been derived solely from the activity of PureA (data not shown). However, all attempts to localize PureA by primer extension have failed. Multiple signals, probably due to readthrough from PureI, were observed, making it impossible to determine the location of PureA. When these observations are taken together, we cannot rule out the possibility that PureA is functional in vivo. However, it is quite clear that PureA is not the promoter used for the differential expression of urease in response to environmental signals. Thus, we focused on the analysis of the dominant promoter in the operon PureI.
Molecular analysis of the 5′ flanking region of PureI.
Sequence analysis revealed two inverted repeats, 26 and 83 bases 5′ to the −35 region of PureI, with ΔG values of −19 and −15 kcal/mol, respectively (Fig. 2). To determine whether these putative secondary structures could function as cis-acting elements in response to environmental signals, two transcriptional fusions to a promoterless cat were constructed. The promoterless cat fragment containing an E. coli RBS was purchased from Pharmacia (Piscataway, N.J.) as a HindIII fragment. To assist the cloning, sequences recognized by SalI and BamHI were included 5′ to the ATG site of the cat fragment (5′-GCG TCGACTGGATCCATGGAGAAAAAAATCACT-3′), and sequences recognized by HindIII were included in the primer which contains the antisense sequences of the 3′ end of the cat sequences (5′-CAAGGATCCAAGCTTCGACGAATT-3′). PCRs were used to amplify the cat fragment with the pair of primers described above. The PCR products were initially digested with SalI and HindIII and then cloned onto SalI- and HindIII-digested pUC18 to obtain a chimeric plasmid in which the cat fragment formed a translational fusion with lacZ. The ligation was used to transform E. coli DH10B, and selection was carried out to obtain transformants that were resistant to 50 μg of chloramphenicol per ml. The resulting plasmid was designated pCW24. To assist in the construction of pMC68 and pMC71 (see below), the HindIII site on this cat fragment was destroyed by digesting pCW24 with HindIII, followed by treatment with Klenow fragments and deoxynucleoside triphosphates. The cat fragment was then released from pCW24 by SalI digestion. The resulting fragment was subsequently cloned onto EcoRV- and SalI-digested pGEM-5Zf(+) (Promega) to generate pCW42.
Because of the proximity of the putative −35 region to the inverted repeat immediately on the 5′ side, it was difficult to design primers for the construction of a deletion derivative completely lacking both inverted repeats. Consequently, the intact promoter (PureI) and the deletion derivative lacking 1.5 inverted repeats (PureIΔ100) were amplified by PCRs with the primer pair PureIas and PureIs and the primer pair PureIas and PureIde12, respectively (Fig. 2). To facilitate the construction with the promoterless cat, a BamHI site was included immediately 5′ to the ATG site of ureI in primer PureIas, 5′-CACCTAACATGGATCCCTCCTAAG-3′. Primers PureIs (5′-GGCGACAATCAGTCCCTTAAT-3′) and PureIde12 (5′-TAAGCTTGACTAATATGTAAATG-3′), containing a HindIII site for cloning, were located 535 and 80 bases 5′ to the ATG site of ureI, respectively. The PCR products of PureI and PureIΔ100 were initially cloned onto pCRII (Invitrogen, Carlsbad, Calif.), and the nucleotide sequences were determined. The correct products were then cloned onto BamHI- and HincII-digested pCW42, selecting for resistance to chloramphenicol. Because of the lack of an alternative integration vector, a 2.0-kbp fragment (Fig. 1) located 5′ to the inverted repeats was ligated to both constructs at the HindIII site to generate pMC68 and pMC71, respectively. To facilitate the integration of the reporter constructs into the S. salivarius 57.I chromosome, the ureI promoter-cat fusions were then released from pMC68 and pMC71 and subcloned onto pSF143 (29) in E. coli, selecting for transformants resistant to 5 μg of tetracycline per ml and 50 μg of chloramphenicol per ml. The resulting plasmids were designated pCW45 and pMC77, respectively.
Both plasmids were introduced into S. salivarius by electroporation according to the guidelines of Caparon and Scott (3) with the following modifications. Briefly, an overnight culture of S. salivarius 57.I grown in Todd-Hewitt broth (Difco) and containing 0.2% yeast extract (THY) and 300 mM l-threonine was diluted 1:20 in fresh medium containing the same concentration of l-threonine. Cultures were incubated at 37°C in a 5% CO2 atmosphere until the optical density at 600 nm reached 0.2. Cells were kept on ice for 10 min prior to harvesting by centrifugation at 4°C. Cells were washed twice with an equal volume of ice-cold electroporation medium (272 mM glucose, 1 mM MgCl2 [pH 6.5]) and then resuspended in 1/15 of the original culture volume in ice-cold electroporation medium. Concentrated cell suspensions were kept on ice for at least 45 min prior to electroporation. Aliquots (40 μl) of the cells were mixed with 200 ng of plasmid DNA and then transferred into a chilled 0.1-cm Gene Pulser electroporation cuvette. Negative controls included cells with no added DNA. Electroporations were carried out at 1.8 kV, 25 μF, and 200 Ω. Cuvettes, containing cells and DNA, were kept on ice for 2 min after electroporation. The cell suspensions were subsequently recovered with 1 ml of THY plus 10 mM glucose at 37°C in a 5% CO2 atmosphere for 2 h. Cells were then concentrated and plated on BHI agar supplemented with 3 μg of tetracycline per ml. All plates were incubated at 37°C in a 5% CO2 atmosphere overnight. Colonies were usually visible in 18 to 24 h. The tetracycline-resistant transformants, strains PureICAT and PureIΔ100CAT, were further confirmed by Southern blot analysis with a tetracycline-specific probe (data not shown). Because of the nature of single-cross integration, both strains were predicted to possess a wild-type promoter 5′ to the ure cluster and a full-length promoter, or the deletion derivative, 5′ to cat. To confirm the presence and configuration of the constructs, a primer located 100 bases 3′ to the ATG and containing the antisense sequences of cat and a primer located 5′ to the HindIII site on the wild-type chromosome were used to amplify the chromosomal region of strains PureICAT and PureIΔ100CAT. The PCR products were subsequently cloned onto pCRII and subjected to sequence analysis to confirm that the junction of the promoter region and the cat gene was intact and to ensure that the deletion of the inverted repeats 5′ to cat had occurred when desired.
To be certain that the expression observed in PureIΔ100-CAT was due only to PureI and not to promoters 5′ to the deleted region, the transcriptional initiation site for the cat gene was determined in PureIΔ100CAT by primer extension analysis with a primer 100 bases 3′ to the translational start site of cat (Fig. 5). The results indicated that the transcriptional initiation site matched the initiation site used in the wild type. The expression of cat in both strains was determined in chemostat-grown cultures under different environmental conditions (Table 2). Briefly, cells were harvested and washed once with an equal volume of 10 mM Tris, pH 7.8, and then resuspended in 1/40 of the original culture volume in the same buffer. Concentrated cell suspensions were subjected to mechanical disruption in the presence of an equal volume of glass beads (0.1-mm diameter) by homogenization in a Bead Beater for a total of 2 min at 4°C. The concentration of each protein lysate was measured by using a protein assay (Bio-Rad, Hercules, Calif.), based on the method of Bradford. Bovine serum albumin served as the standard. The rates of chloramphenicol acetylation of each protein lysate were quantitated by the method of Shaw (23). Low levels of expression were observed in PureICAT grown at a neutral pH under carbohydrate-limiting conditions (Glc concentration = 20 mM). Induction, approximately eightfold increases, occurred when the culture became acidic, with the highest levels observed in cells grown at pH 5.5 under excess carbohydrate conditions (approximately 17-fold increases). This result was consistent with previous observations in which the levels of the ureC-specific mRNAs were induced to a comparable magnitude under similar growth conditions (4). Interestingly, at a neutral pH the specific activity in PureIΔ100-CAT was approximately 60-fold higher than that in PureICAT. Modest induction by an acidic pH was still observed in PureIΔ-100CAT but was never more than twofold under carbohydrate limiting conditions. No further induction by excess carbohydrate was observed in PureIΔ100CAT. These data indicated that PureI was negatively regulated and that the expression of PureICAT was derepressed at an acidic pH. The slight increase in activity by an acidic pH observed in PureIΔ100CAT under carbohydrate-limiting conditions may be due to the presence of partial sequences of the inverted repeat, with which the proposed repressor might still weakly interact.
FIG. 5.
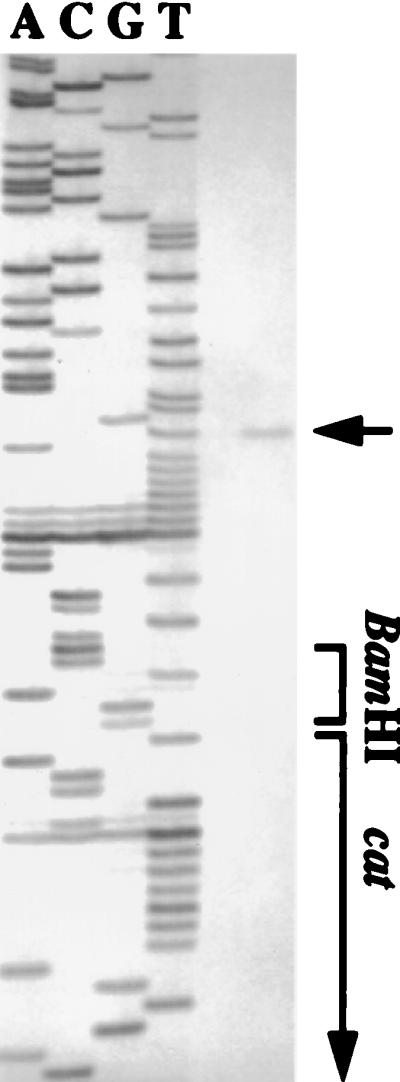
Primer extension analysis of PureIΔ100CAT. Total cellular RNA of S. salivarius PureIΔ100CAT from a steady-state culture grown at pH 5.5 with 20 mM glucose was isolated, and the radiolabeled primer cat (5′-AATGCCTCAAAATGT-3′) was used. The DNA sequences were derived from pMC71 with the same primer.
TABLE 2.
Levels of CAT specific activity in chemostat-grown S. salivarius 57.I derivatives
| Strain | CAT sp acta
|
||
|---|---|---|---|
| pH 7.0, 20 mM Glc | pH 5.5, 20 mM Glc | pH 5.5, 200 mM Glc | |
| PureICAT | 0.23 ± 0.16 | 1.93 ± 0.10 | 4.09 ± 1.17 |
| PureIΔ100CAT | 13.98 ± 0.97 | 29.88 ± 0.29 | 19.34 ± 3.18 |
Values shown are averages from three independent samples. The numbers are expressed as nanomoles of chloramphenicol acetylated per minute per milligram of protein. Negative controls were reactions carried out in the absence of chloramphenicol. Each set of reactions was performed in triplicate.
The regulation of S. salivarius 57.I urease expression by the environmental pH and carbohydrate availability, but not nitrogen availability, is distinct from previously defined urease control pathways, yet control of urease expression in this manner seems particularly well suited to the environment occupied by oral streptococci. It has been established that oral streptococci experience large and rapid changes in carbohydrate availability and pH. Moreover, it has been documented that ammonia generation from urea protects S. salivarius against lethal acidification (24). Logically, then, substantial up-regulation of urease gene expression at a low environmental pH and high carbohydrate concentrations would offer the organism a mechanism to induce synthesis of this protective system under conditions when the enzyme might be needed most for survival. High levels of ureolysis may be especially important for the survival of S. salivarius under extreme acidic conditions, as it is considered to be less aciduric than some oral streptococci and lactobacilli. On the other hand, repression of ure expression at a neutral pH could avoid overproduction of ammonia and alkalization of the environment, which can also be lethal for ureolytic organisms (6).
In this study, we have completed the cloning and sequence analysis of the ure operon. The operon began with ureI, followed by the structural genes, ureABC, and then the accessory genes, ureEFGD. The function of UreI is not well defined, although it is clearly not required for urease biogenesis, since heterologous expression of the S. salivarius ureA to -D in streptococcal hosts yields a urease enzyme indistinguishable from that of the parent (5). Notably, we have investigated the possibility that UreI could be involved in a regulatory circuit governing transcription of ure genes in response to pH. Using a strain containing a polar insertion in ureI, we found that pH responsiveness of PureI remained identical to that of the wild type (data not shown), strongly suggesting that the expression of ureI is not autogenously regulated, nor is its gene product required for pH-dependent regulation.
The expression of both CAT and urease was induced by an acidic pH and excess carbohydrate in strain PureICAT, in which the transcription of both the ure operon and cat was driven by a full-length PureI. However, the magnitude of increases in CAT level was substantially less than that of urease. The differences are likely attributable to the observation that the half-lives of CAT and of urease in oral streptococci are dramatically different. It has been shown that the S. salivarius urease is extremely stable in vivo (26); thus, the activity observed with each growth condition would reflect the combination of the amount of stable mRNA and the accumulation of a stable enzyme. On the other hand, the turnover rate of CAT in oral streptococci appears to be relatively high (30); hence, the level of CAT specific activities would be more closely related to the level of transcription initiation. Consistent with this, the magnitudes of increases in the levels of transcription initiated by PureI measured by primer extension (Fig. 4) and in the levels of ureC-specific messages quantitated by slot blot analysis (4) most closely parallel the increases in CAT specific activity in response to an acidic pH and excess carbohydrate. It should also be noted that the level of expression observed in PureICAT under acidic pH and excess carbohydrate conditions never reached the level observed in PureIΔ100CAT. It is possible that other factors may influence the expression of PureIΔ100CAT, which may have been reflected in the apparent higher strength of PureIΔ100. However, it is more likely that PureI is not completely derepressed under the conditions examined and that perhaps more-acidic conditions are needed to fully derepress the operon.
In summary, we have established that the urease genes of S. salivarius constitute an operon, have identified a promoter that is sensitive to both the environmental pH and carbohydrate availability, and have determined that the induction of urease expression by an acidic pH and excess carbohydrate was negatively regulated. Unlike reports of enteric bacterial ureases, which are either constitutively expressed or regulated by an activator, this is the first report to demonstrate negative control of urease expression. Efforts to isolate and characterize the trans-acting factors which bind near PureI are under way.
Nucleotide sequence accession numbers.
The sequence of ureI was submitted to GenBank and assigned accession no. AF042344. The sequences of ORF1, -2, and -3 were assigned GenBank accession no. AF043280, AF043281, and AF043282, respectively.
ACKNOWLEDGMENTS
We thank R. G. Quivey and K. A. Clancy for critical review of the manuscript.
This study was supported by PHS grant DE10362 from the National Institute for Dental Research to R.A.B.
REFERENCES
- 1.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current protocols in molecular biology. New York, N.Y: John Wiley and Sons; 1989. [Google Scholar]
- 2.Bartolomé B, Jubete Y, Martinez E, dela Cruz F. Construction and properties of a family of pACYC184-derived cloning vectors compatible with pBR322. Gene. 1991;102:75–78. doi: 10.1016/0378-1119(91)90541-i. [DOI] [PubMed] [Google Scholar]
- 3.Caparon M G, Scott J R. Genetic manipulation of pathogenic streptococci. Methods Enzymol. 1991;204:580–581. doi: 10.1016/0076-6879(91)04028-m. [DOI] [PubMed] [Google Scholar]
- 4.Chen Y M, Burne R A. Analysis of Streptococcus salivarius urease expression using continuous chemostat culture. FEMS Microbiol Lett. 1996;135:223–229. doi: 10.1111/j.1574-6968.1996.tb07993.x. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y M, Clancy K A, Burne R A. Streptococcus salivarius urease: genetic and biochemical characterization and expression in a dental plaque streptococcus. Infect Immun. 1996;64:585–592. doi: 10.1128/iai.64.2.585-592.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clyne M, Labigne A, Drumm B. Helicobacter pylori requires an acidic environment to survive in the presence of urea. Infect Immun. 1995;63:1669–1673. doi: 10.1128/iai.63.5.1669-1673.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins C M, D’Orazio S E F. Bacterial ureases: structure, regulation of expression and role in pathogenesis. Mol Microbiol. 1993;9:907–913. doi: 10.1111/j.1365-2958.1993.tb01220.x. [DOI] [PubMed] [Google Scholar]
- 8.Friedrich B, Magasanik B. Urease of Klebsiella aerogenes: control of its synthesis by glutamine synthetase. J Bacteriol. 1977;131:446–452. doi: 10.1128/jb.131.2.446-452.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawasaki E S. Amplification of RNA. In: Innis M A, Gelfand D H, Sninsky J J, White T J, editors. PCR protocols. San Diego, Calif: Academic Press, Inc.; 1990. pp. 21–27. [Google Scholar]
- 10.Labigne A, Cussac V, Courcoux P. Shuttle cloning and nucleotide sequences of Helicobacter pylori genes responsible for urease activity. J Bacteriol. 1991;173:1920–1931. doi: 10.1128/jb.173.6.1920-1931.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10a.LeBlanc, D. J. Unpublished data.
- 11.LeBlanc D J, Hassell F P. Transformation of Streptococcus sanguis Challis by plasmid deoxyribonucleic acid from Streptococcus faecalis. J Bacteriol. 1976;128:347–355. doi: 10.1128/jb.128.1.347-355.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LeBlanc D J, Lee L N. Characterization of two tetracycline resistance determinants in Streptococcus faecalis JH1. J Bacteriol. 1982;150:835–843. doi: 10.1128/jb.150.2.835-843.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LeBlanc D J, Lee L N, Abu-Al-Jaibat A. Molecular, genetic and functional analysis of the basic replicon of pVA380-1, a plasmid of oral streptococcal origin. Plasmid. 1992;28:130–145. doi: 10.1016/0147-619x(92)90044-b. [DOI] [PubMed] [Google Scholar]
- 14.Macaluso A, Best E A, Bender R A. Role of the nac gene product in the nitrogen regulation of some NTR-regulated operons of Klebsiella aerogenes. J Bacteriol. 1990;172:7249–7255. doi: 10.1128/jb.172.12.7249-7255.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maeda M, Hidaka M, Nakamura A, Masaki H, Uozumi T. Cloning, sequencing, and expression of thermophilic Bacillus sp. strain TB-90 urease gene complex in Escherichia coli. J Bacteriol. 1994;176:432–442. doi: 10.1128/jb.176.2.432-442.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mobley H L T, Garner R M, Bauerfeld P. Helicobacter pylori nickel-transport gene nixA: synthesis of catalytically active urease in Escherichia coli independent of growth conditions. Mol Microbiol. 1995;16:97–109. doi: 10.1111/j.1365-2958.1995.tb02395.x. [DOI] [PubMed] [Google Scholar]
- 17.Mobley H L T, Hausinger R P. Microbial ureases: significance, regulation, and molecular characterization. Microbiol Rev. 1989;53:85–108. doi: 10.1128/mr.53.1.85-108.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mobley H L T, Island M D, Hausinger R P. Molecular biology of microbial ureases. Microbiol Rev. 1995;59:451–480. doi: 10.1128/mr.59.3.451-480.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mulrooney S B, Pankratz H S, Hausinger R P. Regulation of gene expression and cellular localization of cloned Klebsiella aerogenes (K. pneumoniae) urease. J Gen Microbiol. 1989;135:1769–1776. doi: 10.1099/00221287-135-6-1769. [DOI] [PubMed] [Google Scholar]
- 20.Nicholson E B, Concaugh E A, Foxall P A, Island M D, Mobley H L T. Proteus mirabilis urease: transcriptional regulation by UreR. J Bacteriol. 1993;175:465–473. doi: 10.1128/jb.175.2.465-473.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Putzer H, Gendron N, Grunberg-Manago M. Co-ordinate expression of two threonyl-tRNA synthetase genes in Bacillus subtilis: control by transcriptional antitermination involving a conserved regulatory sequence. EMBO J. 1992;11:3117–3127. doi: 10.1002/j.1460-2075.1992.tb05384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 23.Shaw W V. Chloramphenicol acetyltransferase activity from chloramphenicol-resistant bacteria. Methods Enzymol. 1979;43:737–755. doi: 10.1016/0076-6879(75)43141-x. [DOI] [PubMed] [Google Scholar]
- 24.Sissons C H, Hancock E M. Urease activity in Streptococcus salivarius at low pH. Arch Oral Biol. 1993;38:507–516. doi: 10.1016/0003-9969(93)90187-q. [DOI] [PubMed] [Google Scholar]
- 25.Sissons C H, Hancock E M, Perinpanayagam H E R, Cutress T W. The bacteria responsible for ureolysis in artificial dental plaque. Arch Oral Biol. 1988;33:727–734. doi: 10.1016/0003-9969(88)90006-4. [DOI] [PubMed] [Google Scholar]
- 26.Sissons C H, Perinpanayagam H E R, Hancock E M. Processes involved in the regulation of urease levels in Streptococcus salivarius by pH. Oral Microbiol Immunol. 1992;7:159–164. [PubMed] [Google Scholar]
- 27.Sissons C H, Perinpanayagam H E R, Hancock E M, Cutress T W. pH regulation of urease levels in Streptococcus salivarius. J Dent Res. 1990;69:1131–1137. doi: 10.1177/00220345900690050301. [DOI] [PubMed] [Google Scholar]
- 28.Stafford K. Continuous fermentation. In: Demain A L, Solomon N A, editors. Manual of industrial microbiology and biotechnology. Washington, D.C: American Society for Microbiology; 1986. pp. 137–151. [Google Scholar]
- 29.Tao L, LeBlanc D J, Ferretti J J. Novel streptococcal-integration shuttle vectors for gene cloning and inactivation. Gene. 1992;120:105–110. doi: 10.1016/0378-1119(92)90016-i. [DOI] [PubMed] [Google Scholar]
- 30.Wexler D L, Hudson M C, Burne R A. Streptococcus mutans fructosyltransferase (ftf) and glucosyltransferase (gtfBC) operon fusion strains in continuous culture. Infect Immun. 1993;61:1259–1267. doi: 10.1128/iai.61.4.1259-1267.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolfram L, Friedrich B, Eitinger T. The Alcaligenes eutrophus protein HoxN mediates nickel transport in Escherichia coli. J Bacteriol. 1995;177:1840–1843. doi: 10.1128/jb.177.7.1840-1843.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]