

Abstract
Objectives
Granulomatosis with polyangiitis (GPA) is a chronic relapsing systemic autoimmune vasculitis. Current treatment of GPA is unsatisfactory, as it relies on strong immunosuppressive regimens, with either CYC or rituximab, which reduce the immunogenicity of several vaccines and are risk factors for a severe form of COVID-19. This emphasizes the need to identify new drug targets and to develop treatment strategies with less harmful side effects. Since CD4+ effector memory T cells (TEM) play a key role in the pathogenesis of GPA, we aimed in this study to modulate CD4+TEM cell activity via Kv1.3 blockade using the specific peptide inhibiter, ShK-186.
Methods
Peripheral blood samples from 27 patients with GPA in remission and 16 age- and sex-matched healthy controls (HCs) were pre-incubated in vitro in the presence or absence of ShK-186, followed by stimulation with phorbol myristate acetate, calcium ionophore and brefeldin-A. The effect of ShK-186 on the cytokine production (IFNγ, TNFα, IL-4, IL-17, IL-21) within total and subsets of CD4+ T helper (CD4+TH) cells were assessed using flow cytometry.
Results
ShK-186 reduced the expression level of IFNγ, TNFα, IL-4, IL-17 and IL-21 in CD4+TH cells from patients with GPA in vitro. Further analysis performed on sorted CD4+T cell subsets, revealed that ShK-186 predominantly inhibited the cytokine production of CD4+TEM cells. ShK-186 treatment reduced the production of the pro-inflammatory cytokines to the level seen in CD4+ TH cells from HCs.
Conclusions
Modulation of cellular effector function by ShK-186 may constitute a novel treatment strategy for GPA with high specificity and less harmful side effects.
Keywords: granulomatosis with polyangiitis, ANCA, T cells, cytokines, Kv1.3 potassium channels
Graphical Abstract
Rheumatology key messages.
ShK-186 treatment normalized the production of the pro-inflammatory cytokines in CD4+ T helper (CD4+TH) cells from patients with GPA.
ShK-186 predominantly affects the cytokine production of CD4+ effector memory T cells (CD4+TEM) cells without impairing other CD4+T cell subsets.
Selective targeting of CD4+TEM cells by ShK-186 might have value in the treatment of patients with GPA.
Introduction
Granulomatosis with polyangiitis (GPA) is the prototype of ANCA-associated vasculitis (AAV). It is a chronic relapsing systemic autoimmune disease characterized by medium- to small-sized vessel vasculitis predominantly affecting the upper and lower respiratory tract and kidneys, which may result in life-threatening complications [1, 2]. Current treatment consists of non-specific immunosuppressive therapy, including CYC in combination with CSs [3–5]. More recently, B cell depletion therapy with rituximab has been demonstrated to be equally effective as conventional therapy in inducing disease remission [6]. Unfortunately, these immunosuppressive drugs, rituximab in particular, also reduce the immunogenicity of several vaccines and are risk factors for a severe form of COVID-19, emphasizing the need to identify novel molecular targets to develop more selective and less harmful treatment strategies [7–9].
It remains unknown how GPA develops, but accumulating evidence indicates a key role for CD4+ effector memory T cells (TEM) and their inflammatory cytokines [such as IL-17, IL-21 and IFN-gamma (IFNγ)] in the induction and progression of GPA [10–15]. Therefore, selective targeting of CD4+TEM cells without impairing other arms of cellular immunity might have value in the treatment of patients with GPA.
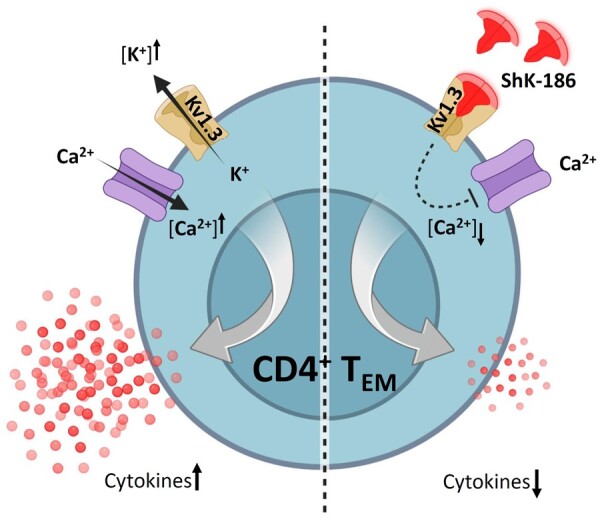
CD4+TEM cells preferentially rely on the voltage-gated potassium Kv1.3 channels to maintain their activation and effector function [16, 17]. It has been shown that Kv1.3 channels are highly expressed on CD4+TEM cells (∼1500 channels per cell), whereas naïve (CD4+TNAIVE) and central memory (CD4+TCM) CD4+T cells express lower levels of Kv1.3 channels (∼250 channels per cell) [17]. CD4+TEM cells use the Kv1.3 channels to regulate Ca2+ signalling by controlling the membrane potential. During activation, intracellular Ca2+ stores are released, leading to a depletion of these stores and an influx of extracellular Ca2+. This influx is maintained by the release of K+ through Kv1.3 channels, which helps sustain the elevated levels of Ca2+ required for optimal TEM cell activation. Blocking Kv1.3 channels can inhibit this process by preventing the release of K+, leading to a decrease in Ca2+ influx and subsequently inhibiting TEM cell activation [16]. Therefore, Kv1.3 channels may serve as an attractive target for specific immunomodulation in TEM cell–mediated chronic or autoimmune diseases. Kv1.3 channels can be selectively inhibited by synthetic analogues of a native ShK toxin isolated from the sea anemone Stichodactyla helianthus. Previous studies have demonstrated that blocking of Kv1.3 channels by ShK analogues ameliorates disease development in animal models of multiple sclerosis (MS), RA, type 1 diabetes mellitus (T1DM) and contact dermatitis, without compromising protective immune responses to acute infections [18–20]. In particular, ShK-186, a 37–amino acid synthetic analogue of ShK, has shown promising long-lasting therapeutic potential in animal models of autoimmunity, due to its high selectivity and tight binding to and slow release from the Kv1.3 channels on T cells [21]. Accordingly, we hypothesized that selective blocking of Kv1.3 channels on CD4+ TEM cells from patients with GPA, using a highly potent ShK-186 peptide, reduce their pathogenic activity through modulating their pro-inflammatory cytokine production. Selective targeting of CD4+TEM cells without impairing other arms of cellular immunity might have value in the treatment of GPA.
Materials and methods
Study population
Twenty-seven patients with GPA in remission and 16 age-matched healthy controls (HCs) [5 males and 11 females, mean age of 60 years, range (27–77)] were included in this study. The diagnosis of GPA was established according to the definition of the Chapel Hill Consensus Conference and fulfilled the classification of the ACR [22]. Only patients with GPA without clinical signs and symptoms of active disease and considered to have complete remission of their disease, as indicated by a BVAS of 0, were included in this study [23]. All patients were PR3-ANCA positive at disease diagnosis, and 20 patients had biopsy-proven vasculitis. At the time of sampling, 18 patients were PR3-ANCA positive, as indicated by an ANCA titre of ≥1:40. The PR3-ANCA titres were measured by IIF on ethanol-fixed human granulocytes according to the standard procedure as described previously [24]. Twenty-one patients were considered to have generalized disease, and six patients were considered to have localized disease, in which the disease was confined to the upper and lower respiratory tract. None of the patients experienced an infection at the time of sampling, as indicated by a median CRP level of 5.8 mg/l. Eight of the 27 patients with GPA were treated with maintenance immunosuppressive therapy at the time of blood withdrawal. One patient with GPA received AZA, five patients with GPA received AZA in combination with prednisolone, and two patients with GPA were treated with low-dose prednisolone. Detailed clinical and laboratory characteristics of the patients are summarized in Table 1. All patients and healthy controls provided informed consent, and the local medical ethics committee of the University Medical Center Groningen approved the study.
Table 1.
Clinical and laboratory characteristics of the patients with GPA at the time of blood sampling
| GPA | |
|---|---|
| Subjects, n (% male) | 27 (44%) |
| Age, mean (range) | 61 (34–79) |
| PR3-ANCA positivea, n (% positive) | 18 (67%) |
| Localized/generalized disease, n (% generalized) | 6/21 (78%) |
| CRP (mg/l), median (range) | 5.8 (<0.3–11) |
| eGFRb ml/min/1.73 m2, median (range) | 64 (15–91) |
| Disease duration in years, median (range) | 9.5 (1.3–30.8) |
| Number of previous relapses, median (range) | 1 (0–6) |
| Non/maintenance immunosuppressive therapyc, n | 19/8 |
ANCA-positive titre ≥1:40, ANCA-negative ≤1:20.
Estimated Glomerular Filtration Rate (eGFR) was calculated using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation.
Immunosuppressive maintenance therapy: AZA, AZA + prednisolone, or prednisolone.
Sample preparation and in vitro peripheral blood stimulation
Lithium-heparinized venous blood was obtained from patients with GPA and HCs. Immediately after blood withdrawal, 2 ml of blood was mixed with 2 ml of Roswell Park Memorial Institute (RPMI) 1640 medium (Lonza, Basel, Switzerland), supplemented with 50 µg/ml Gentamicin (GIBCO, Life Technologies, Grand Island, NY, USA) and 10% foetal calf serum. The diluted blood samples were aliquoted into 5 ml polypropylene tubes (Falcon®, Corning incorporated) at 400 µl per tube. Next, the blood samples were pre-incubated in the presence or absence of ShK-186 [dose range (0.1–100 nM); Kineta Inc, Seattle, WA, USA] for 1 h at 37°C, followed by stimulation with 50 ng/ml phorbol myristate acetate (PMA; Sigma-Aldrich, St Louis, MO, USA) and 2 mM calcium ionophore (CaI, Sigma-Aldrich). The cultures were incubated for 16 h at 37°C with 5% CO2. As a negative control, one sample was kept without stimulation. To inhibit cytokine release from cells, 10 µg/ml brefeldin A (BFA; Sigma-Aldrich) was added to each sample.
IF staining of peripheral blood
After stimulation of the peripheral blood, erythrocytes were lysed using ammonium chloride, and the cells were washed in wash buffer [PBS containing 1% (w/v) BSA]. The T cells were stained with Brilliant Violet 605-conjugated anti-CD3 (Biolegend, San Diego, CA, USA), APC-eF780-conjugated anti-CD8 (eBioscience, San Diego, CA, USA), FITC-conjugated CD45RO (BD Pharmingen™, Franklin Lakes, NJ, USA) and PE-Cy7-conjugated CCR7 (BD Pharmingen™) for 15 min at room temperature. The cells were fixed with 100 µl fixation reagent A (Fix/Perm medium A, Thermo Fisher Scientific) for 15 min. After washing, the cells were resuspended in 100 µl permeabilization reagent B (Fix/Perm medium B, Thermo Fisher Scientific) and labelled with PerCP-Cy5.5-conjugated anti-IL-4 (Biolegend), APC-conjugated anti-IL-17A (eBioscience), PE-conjugated anti-IL-21 (eBioscience), Alexa Fluor®700-conjugated anti-IFNγ (BD Pharmingen™) and Pacific Blue-conjugated anti-TNFα (Biolegend) for 30 min at room temperature in the dark. Finally, the samples were washed and analysed by nine-colour flow cytometric analyses on BD™ LSR II flow cytometer. Data were collected for 5×105 events for each sample and plotted using Kaluza v1.5a (Beckman Coulter, Brea, CA, USA). Because stimulation reduces the surface expression of CD4 on T cells, CD4+T cells were identified indirectly by gating CD3-positive and CD8-negative lymphocytes. Gated CD4+T cells were further displayed as density dot plots for the evaluation of intracellular cytokine production. The unstimulated negative control sample was used to discriminate cytokine-producing from non–cytokine-producing CD4+ T cell populations.
Isolation of CD4+ TNAIVE and CD4+ TEM cells
Peripheral blood mononuclear cells (PBMCs) of 5 HCs were used for cell sorting experiments. Cell suspensions were stained for CD3, CD8, CD45RO and CCR7. CD4+T cells were gated negatively as CD3-positive and CD8-negative cells and sorted into: CD4+TNAIVE (CD45RO–CCR7+), CD4+TCM (CD45RO+CCR7+), CD4+TEM (CD45RO+CCR7–) and CD4+TTD (CD45RO–CCR7–) cell fractions on a MoFLO Astrios sorter (Beckman Coulter). The purity of the sorted CD4+T cell subsets, as determined by a post sort analysis, was >98% for all sorted CD4+T cell subsets. From each subset, 2.5×105 cells were incubated in the presence or absence of ShK-186 [dose range (0.1–100 nM); Kineta Inc] for 1 h at 37°C, followed by stimulation with 50 ng/ml PMA (Sigma-Aldrich) and 2 mM CaI (Sigma-Aldrich) in the presence of BFA. Following incubation for 16 h at 37°C with 5% CO2, the cells were washed, premeabilized, and stained intracellularly for IL-4 (Biolegend), IL-17A, IL-21 (eBioscience), IFNγ (BD Pharmingen™) and TNFα (Biolegend). Finally, the samples were acquired on a BD™ LSR II flow cytometer (BD Biosciences), and the data was analysed using Kaluza 1.5a. Unstimulated samples were used as a negative control for setting gates to define cytokine-producing cells.
Statistical analysis
Statistical analysis was performed using GraphPad prism (GraphPad Software, San Diego, CA, USA). Data are presented as median values or mean ± SEM, as indicated. Data were analysed with the D’Agostino & Pearson omnibus normality test for Gaussian distribution. For comparison between patients with GPA and HCs, the unpaired t test was used for data with Gaussian distribution and the Mann–Whitney U test for data without Gaussian distribution. For intra-individual comparison between samples treated with or without ShK-186, the paired t test was used for data with Gaussian distribution and the Wilcoxon signed rank test for data without Gaussian distribution. Differences were considered statistically significant for 2-sided P-values ≤0.05.
Results
T cell subset distribution in peripheral blood of patients with GPA in remission
We first assessed the distribution of CD4+T cell subsets in the peripheral blood of patients with GPA in remission and HCs. CD4+T cell subsets were identified based on the surface expression of CD45RO and CCR7 and divided into CD4+TNAIVE cells (CD45RO–CCR7+), CD4+TCM (CD45RO+CCR7+), CD4+TEM cells (CD45RO+CCR7–) and CD4+ terminal differentiated cells (TTD, CD45RO–CCR7–) (Fig. 1A). We found that the percentage of circulating CD4+TEM cells in patients with GPA was significantly higher compared with that in HCs (Fig. 1B), and the percentage of circulating CD4+TNAIVE cells was significantly lower in patients with GPA compared with that in HCs, whereas the percentage of CD4+TCM cells did not differ. In addition, the percentage of circulating CD4+TTD cells was significantly higher in patients with GPA compared with that in HCs.
Figure 1.

Increased percentage of circulating CD4+ TEM cells in patients with GPA. (A) Representative flow cytometry dot plots of CD45RO and CCR7 expression to identify four CD4+ T cell subsets in the peripheral blood of a patient with GPA in remission (right plot) and a HC (left plot). (B) Percentages of CD45RO–CCR7+ (TNAIVE), CD45RO+CCR7+ (TCM), CD45RO+CCR7– (TEM) and CD45RO–CCR7– (TTD) subsets within the CD4+ T cell population in peripheral blood of patient with GPA in remission (filled squares; n = 27) and HCs (open circles; n = 16). Horizontal bar represent median percentage. **P < 0.01, ***P < 0.001 vs HCs. GPA: granulomatosis with polyangiitis; HC: healthy control; TEM: CD4+ effector memory T cells
To rule out the possibility that the increased proportion of CD4+TEM cells was influenced by the current treatment, we compared the proportions of CD4+TEM cells between patients with GPA off treatment and patients with GPA receiving immunosuppressive maintenance therapy. No significant differences were found between the treated and untreated patient groups (data not shown).
Increased intracellular pro-inflammatory T cell cytokine production in patients with GPA
Effector T cells produce pro-inflammatory cytokines (such as IL-4, IL-17, IL-21, TNFα and IFNγ) that are presumed to be involved in the disease pathogenesis of GPA. Therefore, we next analysed the pro-inflammatory cytokine profile of CD4+ T helper (CD4+TH) cells from patients with GPA and HCs. In all samples, the production of intracellular IL-4, IL-17, IL-21, TNFα and IFNγ was determined in CD4+TH cells by flow cytometry (Fig. 2A). As shown in Fig. 2, the expression of all pro-inflammatory cytokines within CD4+TH cells was significantly higher in patients with GPA compared with that in HCs.
Figure 2.

Higher percentage of intracellular cytokine production in circulating CD4+TH cells from patients with GPA. Peripheral blood from patients with GPA and HCs was stimulated with PMA and CaI and analysed with flow cytometry for intracellular IL-4, IL-17, IL-21, TNFα and IFNγ cytokine expression. (A) Percentages of IL-4+, IL-17+ IL-21+, TNFα+ and IFNγ+ CD4+TH cells from patient with GPA in remission (filled squares; n = 27) and HCs (open circles; n = 16). (B) Percentages of TNFα+IFNγ+, IFNγ+IL-17+ and IL-17+IL-21+ within CD4+TH cells from patient with GPA in remission (filled squares; n = 27) and HCs (open circles; n = 16). Horizontal bar represent median percentage. *P < 0.05, **P < 0.01, ***P < 0.001 vs HCs. CD4+TH: CD4+ T helper; CaI: calcium ionophore; GPA: granulomatosis with polyangiitis; HC: healthy control; PMA: phorbol myristate acetate
Of note, it has become evident that CD4+TH cells may produce additional cytokines in addition to their signature cytokine. For example, TH1 cells may produce IL-17 in addition to their signature cytokine IFNγ, and TH17 cells produce IL-21 in addition to their signature cytokine IL-17 [25]. We, therefore, assessed the proportion of CD4+TH cells producing three cytokines (TNFα+IFNγ+, IFNγ+IL-17+ and IL-17+IL-21+). As shown in Fig. 2B, CD4+TH cells from patients with GPA in remission produce significantly higher percentages of TNFα+IFNγ+, IFNγ+IL-17+ and IL-17+IL-21+ cytokines as compared with CD4+TH cells from HCs. Overall, these results demonstrate the pro-inflammatory nature of the CD4+TH cells in patients with GPA.
ShK-186 reduced the production of cytokines in CD4+TH cells from patients with GPA to the level seen in CD4+ TH cells from HCs
Next, we questioned whether the pro-inflammatory cytokine production could be regulated by Kv1.3 channel blockade, using the highly potent Kv1.3 peptide blocker ShK-186. To this end, we stimulated peripheral blood samples of patients with GPA in the presence and absence of ShK-186 and analysed the intracellular cytokine production of IL-4, IL-17, IL-21, TNFα and IFNγ in CD4+TH cells from patients with GPA (Supplementary Fig. S1, available at Rheumatology online). As shown in Fig. 3, addition of ShK-186 to stimulated cell cultures significantly reduced the production of IL-17, IL-21, TNFα and IFNγ in CD4+TH cells from patients with GPA. The effect of ShK-186 on the production of IL-17, IL-21, TNFα and IFNγ was dose dependent (Fig. 3B). Interestingly, the production of IL-17, IL-21, TNFα and IFNγ in CD4+TH cells was normalized to the median cytokine levels detected in HCs. Remarkably, the suppressive effect of ShK-186 on IL-4 production was less pronounced.
Figure 3.

Dose-dependent suppression of pro-inflammatory cytokines by ShK-186 in CD4+TH cells from patients with GPA. Peripheral blood from patients with GPA and HCs was stimulated with PMA and CaI with and without increasing concentrations of ShK-186. Intracellular IL-4, IL-17, IL-21, TNFα and IFNγ cytokine production in CD4+TH cells was analysed using flow cytometry. (A) Representative flow cytometry dot plots of cytokine expression within CD4+ TH cells after stimulation in the presence (lower panels) and absence (upper panels) of ShK-186 from a patient with GPA in remission. (B) Percentages of cytokine-producing CD4+ TH cells after stimulation in the presence and absence of ShK-186 from patients with GPA in remission (grey box and whiskers; n = 27). (C) Percentages of TNFα+IFNγ+, IFNγ+IL-17+ and IL-17+IL-21+ within CD4+TH cells after stimulation in the presence and absence of ShK-186 from patients with GPA in remission (grey box and whiskers; n = 27). Box-and-whiskers plots (tukey): boxes represent median values and interquartile range. Red horizontal dashed line represents median percentage of cytokine production by CD4+TH cells from HCs. *P < 0.05, **P < 0.01, ***P < 0.001 vs stimulated CD4+TH cells without ShK-186. CD4+TH: CD4+ T helper; CaI: calcium ionophore; GPA: granulomatosis with polyangiitis; HC: healthy control; phorbol myristate acetate
In addition, the percentage of CD4+TH cells producing TNFα+IFNγ+, IFNγ+IL-17+ and IL-17+IL-21+ was significantly suppressed by ShK-186 in a dose-dependent manner (Fig. 3C).
It is worth mentioning that the reduction in cytokine production upon pre-treatment with ShK-186 was not attributed to cell death, as ShK-186 does not affect the viability of CD4+ T cells (Supplementary Fig. S2, available at Rheumatology online).
ShK-186 inhibits cytokine production of CD4+ TEM cells
As described previously, CD4+TEM cells express significantly higher numbers of Kv1.3 channels on their plasma membrane compared with CD4+ TNAIVE cells and CD4+TCM cells [17, 26]. Therefore, CD4+TEM cells are the most likely target for ShK-186. To study whether Kv1.3 channel blockade by ShK-186 selectively targets cytokine production of CD4+TEM cells, we tested the effect of ShK-186 on FACS-sorted CD4+TNAIVE, CD4+TCM, CD4+TEM and CD4+TTD cells (Fig. 4A). First, we observed that the pro-inflammatory cytokine production of IL-4, IL-17 and IFNγ after in vitro stimulation was significantly increased in CD4+TEM cells compared with the other CD4+ T cell subsets (CD4+TNAIVE, CD4+TCM and CD4+TTD cells) (Fig. 4). IL-21 was significantly increased in CD4+TEM cells compared with CD4+TNAIVE and CD4+TTD cells, whereas no difference was observed compared with CD4+TCM cells. TNFα was produced by all CD4+T cell subsets, although the CD4+TEM cells showed the highest expression levels of TNFα compared with those of CD4+TNAIVE, CD4+TCM and CD4+ TTD cells. Overall, in vitro stimulation with PMA and CaI showed that CD4+TEM cells are the major producers of pro-inflammatory cytokines in comparison with other CD4+T cell subsets (Fig. 4B). Addition of ShK-186 inhibited CD4+TEM cells from producing IL-4, IL-17, TNFα and IFNγ in a dose-dependent manner, whereas the effect was less pronounced in other CD4+TH cell subsets, as their cytokine production remained almost unchanged before and after treatment (Fig. 4B). It is worth mentioning that the production of IL-4 and IL-17 by isolated CD4 TH subsets other than CD4+TEM cells were less pronounced, which could potentially impact the ability of ShK-186 to reduce cytokine production in these subsets. In contrast, the production of IL-21 was slightly affected in both CD4+TCM and CD4+TEM cells, but reached a significant decrease at a dose of 100 nM in CD4+TEM only. It should be noted that a slight reduction was observed in TNFα and IFNγ production in other CD4+TH subsets, mainly CD4+TCM cells. This might be expected, as part of TCM cells may develop to TEM cells upon stimulation, and thus express higher levels of Kv1.3 channels that can be targeted by ShK-186.
Figure 4.

ShK-186 inhibits the pro-inflammatory cytokine production of CD4+TEM cells. CD4+T cells subsets (i.e. CD4+TNAIVE, CD4+TCM, CD4+TEM and CD4+TTD cells) were isolated from peripheral blood mononuclear cells of HCs followed by stimulation with PMA and CaI with and without increasing concentrations of ShK-186. Intracellular IL-4, IL-17, IL-21, TNFα and IFNγ cytokine production in the CD4+T cell subsets was analysed using flow cytometry. (A) Representative flow cytometry dot plots of CD4+T cells subsets based on surface expression of CD45RO and CCR7 (centre dot plot), and the cytokine expression within CD4+TNAIVE cells (upper left, red), CD4+TCM cells (upper right, green), CD4+TTD cells (lower left, purple) and CD4+TEM cells (lower right, blue) after stimulation in the presence and absence of ShK-186 from a HC. (B) Percentages of intracellular cytokine production in CD4+TNAIVE cells (red symbol and line), CD4+TCM cells (green symbol and line), CD4+TTD (purple symbol and line) and CD4+TEM cells (blue symbol and line) after stimulation in the presence and absence of ShK-186 from HCs (n = 5). Data represent mean values ± SEM. ##P < 0.01 and ###P < 0.001 indicate CD4+TEM cells vs CD4+TNAIVE cells, CD4+TCM cells and CD4+TTD cells. *P < 0.05, **P < 0.01, ***P < 0.001 indicate CD4+TEM cells with vs without ShK-186. ##: IL-21 production in CD4+ TEM cells is only significantly different compared with CD4+TNAIVE and CD4+TTD cells and not significantly different compared with CD4+TCM cells. GPA: granulomatosis with polyangiitis; HC: healthy control; PMA: phorbol myristate acetate; CaI: calcium ionophore; CD4+ TCM: CD4+ central memory T cells; CD4+ TEM: CD4+ effector memory T cells; CD4+ TNAIVE: CD4+ naive T cells; CD4+ TTD: CD4+ terminal differentiated T cells
Interestingly, we observed that TNFα+IFNγ+CD4+TH cells were predominantly present within the CD4+TEM subset. Addition of ShK-186 demonstrated a significant dose-dependent inhibition of TNFα+IFNγ+ production by CD4+TEM cells compared with the other CD4+T cell subsets (Fig. 4B).
Discussion
In the present study, we show that pro-inflammatory cytokine-producing CD4+TH cells are proportionally increased in the circulation of patients with GPA in remission compared with HCs. We found that in vitro pharmacological blockade of Kv1.3 channels using ShK-186 decreased the production of pro-inflammatory cytokines, including IL-17, IL-21, TNFα and IFNγ of CD4+TH cells from patients with GPA. Importantly, ShK-186 treatment did not completely inhibit cytokine production but rather reduced the production of these pro-inflammatory cytokines to the level seen in CD4+TH cells from HCs. Furthermore, addition of ShK-186 predominantly affected cytokine production of CD4+TEM cells, with minimal effects on cytokine production by other CD4+TH cell subsets (i.e. CD4+TNAIVE, CD4+TCM and CD4+TTD cells).
Our observation that CD4+TH cells from patients with GPA display an increased pro-inflammatory cytokine profile compared with cells from HCs is consistent with previous reports demonstrating increased production of IFNγ and TNFα by peripheral blood mononuclear cells and CD4+TH cells of patients with GPA [11, 27, 28]. In addition, we and others have demonstrated that circulating IL-17– and IL-21–producing CD4+TH cells are significantly increased in patients with GPA, even in remission [13, 14, 29].
Next, we demonstrated that the increase in pro-inflammatory cytokine production in CD4+TH cells from patients with GPA can be prevented by ShK-186 treatment. These data are in line with previous reports showing that ShK-186 preferentially suppresses production of IL-2, IFNγ and TNFα from synovial T cells (mainly consisting of CD4+TEM cells) of RA patients [20]. In addition, Chi et al. have demonstrated that ShK-186 suppresses cytokine production in human T cells from whole blood [30]. Similar to our observations, these authors reported that Shk-186 was most effective in suppressing the production of IL-2, followed by IFNγ and IL-17, but had a minor effect only on IL-4 production. Interestingly, it has been shown that T cell receptor (TCR)-induced Ca2+ signalling is lower in TH2 cells than in TH1, TH17 or naïve T cells, suggesting that Kv1.3-mediated T cell activation is differently regulated not only in T cell subsets (i.e. TNAIVE, TCM and TEM) but also between different T cell phenotypes [31, 32]. This could explain the fact that blocking Kv1.3 channels using ShK-186 has a more pronounced effect on the pro-inflammatory cytokines IFNγ and IL-17 compared with IL-4.
In addition to its effect on T cells, we have previously explored the anti-inflammatory effect of ShK-186 on B cells. We found that ShK-186 modulates the effector functions of B cells of patients with GPA in vitro by reducing the production of ANCAs and pro-inflammatory cytokines [33]. Thus, utilizing a Kv1.3-based therapy in GPA would represent a significant improvement over existing therapies. In addition, ShK-186 may also be beneficial in the treatment of COVID-19, as it has the potential to mitigate the production of pro-inflammatory cytokines and thus suppresses the cytokine storms occurring in COVID-19 patients [34].
Using sorted CD4+T cell subsets, we observed that cytokine production is most effectively suppressed by ShK-186 in CD4+TEM cells. This can be explained by the fact that activation of T cells has differential effects on the expression of potassium channels in different T cells subsets. CD4+TNAIVE and CD4+TCM cells preferentially upregulate the Ca2+-activated potassium KCa3.1 channel, while CD4+TEM cells preferentially increase their Kv1.3 expression [17]. This switch in channel expression significantly affects the responsiveness of T cell subsets to Kv1.3 and KCa3.1 blockers, CD4+TEM cells being highly sensitive to Kv1.3 channel blockers and CD4+TNAIVE/TCM cells being more sensitive to KCa3.1 channel blockers.
In addition, ShK analogues have shown similar effects on rat T cells in various immune-mediated inflammatory disease models. In these studies, ShK analogues showed efficacy in preventing and ameliorating acute experimental autoimmune encephalomyelitis (EAE, a model of multiple sclerosis) and pristine-induced arthritis in rats [20, 35]. Moreover, in a rat model of anti- GBM GN, the majority of CD4+T cells infiltrating the kidney were Kv1.3high TEM cells [36]. Rats treated with a Kv1.3 blocker developed less proteinuria and had fewer crescentic glomeruli than rats treated with placebo. ShK-186 may therefore be useful in the treatment of autoimmune kidney disease like GPA.
In addition to the ShK-186, several other types of Kv1.3 blockers have been developed and studied. For instance, ShK-related peptides derived from parasitic worms, such as AcK1 and BmK1, have been shown to block Kv1.3 channels and suppress TEM cell responses in vitro and in vivo [37]. Other scorpion venom–derived peptides, including HsTX1 and Imk, have been also identified as potent Kv1.3 channel blockers, and have demonstrated effectiveness in controlling arthritis and reducing the severity of experimental autoimmune encephalomyelitis, respectively [38, 39]. Additionally, Vm24, a peptide derived from the scorpion venom, has shown high selectivity and potency in blocking Kv1.3 channels and impairing the synthesis and secretion of Th-cell cytokines in response to TCR engagement [40]. These Kv1.3 blockers are currently being investigated as potential therapeutic agents for autoimmune diseases.
Compared with other Kv1.3 blockers, ShK-186 has several advantages. One of the key advantages of ShK-186 over other Kv1.3 blockers is its high selectivity. It has been shown to be >100-fold more selective for Kv1.3 compared with other potassium channels [19]. Another unique aspect of ShK-186 is its tight binding to the Kv1.3 channels and long duration of action, which may allow for less frequent dosing. The unique pharmacokinetic profile of ShK-186 may make it a promising therapeutic option for the treatment of autoimmune diseases.
It is worth noting that in addition to CD4+TEM cells, Kv1.3 channels are also expressed in various tissues in the body, including the kidney, liver and the CNS. Therefore, one may argue that toxic side effects are a potential concern when using Kv1.3 channel blockers. However, Kv1.3 blockers (especially the ShK analogues) have been shown to have an excellent safety profile in animal models [19, 20, 41]. ShK-186 was reported to exhibit no perceptible in vitro toxicity, was negative in the Ames test, and had no effect on cardiac parameters [19]. Furthermore, repeated subcutaneous administration of ShK-186 in rats did not cause clinical toxicity, as evidenced by normal blood cell counts and serum chemistry parameters, and no signs of histopathological changes in various tissues [19, 20]. Moreover, in vivo studies have demonstrated that the efficacy of ShK186 can be achieved without general immunosuppression [41]. In rats, administration of ShK-186 did not compromise the protective immune response to acute viral (influenza) or bacterial (Chlamydia) infections at pharmacological doses that did ameliorate autoimmune diseases [41]. Importantly, ShK-186 has completed phase 1 b trial in psoriasis patients, showing the blocker is well tolerated and improves psoriatic skin lesions by inhibiting T cell mediators of inflammation [42].
Therapies targeting CD4+TEM cells via blocking Kv1.3 channels may have an advantage over current therapies in GPA, because CD4+TNAIVE and CD4+TCM would escape the inhibition by ShK-186. Leaving TNAIVE and TCM CD4+T cells unimpaired. patients with GPA treated with ShK-186 would therefore be able to preserve protective immune responses against most pathogenic challenges. On the other hand, a potential disadvantage of Kv1.3 blockade is that it likely suppresses all CD4+TEM cells, thereby affecting immune responses against chronic infections. However, as demonstrated here, it may be possible to titrate ShK-186 to a dose at which it normalizes, but does not completely suppress, CD4+TEM cell responses. Moreover, the Kv1.3 blocker is reversible, and therapy could be paused in the event of an acute infection, unlike current treatments in GPA (i.e. CYC, high-dose CSs and rituximab), which take several months to subside.
In this study, blood samples from patients with GPA in remission were evaluated for the effect of ShK-186, rather than blood samples from those with active disease. We have previously shown that during active disease CD4+TEM cells appear to migrate towards inflamed tissues [12]. Analysis of the effect of ShK-186 on circulating CD4+TH cells in patients with GPA with active disease will exclude cells that have migrated to inflamed tissue, which are (probably) the most relevant cells. Therefore, studying samples from patients in remission seems more relevant for this analysis. However, it should be noted that our findings may not fully reflect the effect of ShK-186 on TEM cells during disease progression, and therefore future studies should aim to include samples from patients with active disease to better understand the potency of ShK-186 on CD4+ TEM cells in various stages of the disease. Future in vivo studies should also consider the impact of ShK-186 on PR3-specific T cells and its impact on protective immunity.
In conclusion, the data presented here demonstrate that the Kv1.3 blocker ShK-186 suppresses pro-inflammatory cytokine production in CD4+TH cells from patients with GPA, and predominantly affects the cytokine production of CD4+ TEM cells. Importantly, ShK-186 treatment reduced the production of the pro-inflammatory cytokines to the level seen in CD4+TH cells from HCs. These findings support the potential of selective Kv1.3 blockade as a therapeutic strategy for patients with GPA.
Supplementary Material
Contributor Information
Lucas L Lintermans, Department of Rheumatology and Clinical Immunology, University of Groningen, University Medical Center Groningen, Groningen, The Netherlands.
Coen A Stegeman, Department of Internal Medicine, Division of Nephrology, University of Groningen, University Medical Center Groningen, Groningen, The Netherlands.
Ernesto J Muñoz-Elías, Prometheus Biosciences, San Diego, CA, USA.
Eric J Tarcha, Kineta Inc, Seattle, WA, USA.
Shawn P Iadonato, Kineta Inc, Seattle, WA, USA.
Abraham Rutgers, Department of Rheumatology and Clinical Immunology, University of Groningen, University Medical Center Groningen, Groningen, The Netherlands.
Peter Heeringa, Department of Pathology and Medical Biology, University of Groningen, University Medical Center Groningen, Groningen, The Netherlands.
Wayel H Abdulahad, Department of Rheumatology and Clinical Immunology, University of Groningen, University Medical Center Groningen, Groningen, The Netherlands; Department of Pathology and Medical Biology, University of Groningen, University Medical Center Groningen, Groningen, The Netherlands.
Supplementary material
Supplementary material is available at Rheumatology online.
Data availability
The data underlying this article will be shared on reasonable request to the corresponding author.
Contribution statement
All authors contributed to the concept and design. L.L. performed the experiments, statistical analysis, drafted the manuscript, and contributed to interpretation of the data. W.A. and P.H. contributed to interpretation of the data and critically revised the manuscript. A.R. and C.S. contributed to inclusion of patients with GPA, and assessed and participated in the interpretation of clinical data, and critical revision of the manuscript. E.M.-E., E.T. and S.I. critically revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the Dutch Arthritis foundation (Reumafonds project number 12–2-407).
Disclosure statement: The authors have declared no conflicts of interest.
References
- 1. Hilhorst M, van Paassen P, Tervaert JWC.. Proteinase 3-ANCA vasculitis versus myeloperoxidase-ANCA vasculitis. J Am Soc Nephrol 2015;26:2314–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jennette JC, Falk RJ.. Pathogenesis of antineutrophil cytoplasmic autoantibody–mediated disease. Nat Rev Rheumatol 2014;10:463–73. [DOI] [PubMed] [Google Scholar]
- 3. Biedroń G, Włudarczyk A, Wawrzycka-Adamczyk K. et al. Treatment and its side effects in ANCA-associated vasculitides – study based on POLVAS registry data. Adv Med Sci 2020;65:156–62. [DOI] [PubMed] [Google Scholar]
- 4. Jain K, Jawa P, Derebail VK, Falk RJ.. Treatment updates in antineutrophil cytoplasmic autoantibodies (ANCA) vasculitis. Kidney360 2021;2:763–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schönermarck U, Gross WL, de Groot K.. Treatment of ANCA-associated vasculitis. Nat Rev Nephrol 2014;10:25–36. [DOI] [PubMed] [Google Scholar]
- 6. Stone JH, Merkel PA, Spiera R. et al. ; RAVE-ITN Research Group. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. New Engl J Med 2010;363:221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guilpain P, le Bihan C, Foulongne V. et al. Rituximab for granulomatosis with polyangiitis in the pandemic of covid-19: lessons from a case with severe pneumonia. Ann Rheum Dis 2021;80:e10. [DOI] [PubMed] [Google Scholar]
- 8. Schulze-Koops H, Krueger K, Vallbracht I, Hasseli R, Skapenko A.. Increased risk for severe COVID-19 in patients with inflammatory rheumatic diseases treated with rituximab. Ann Rheum Dis 2021;80:e67. [DOI] [PubMed] [Google Scholar]
- 9. Deepak P, Kim W, Paley MA. et al. Effect of immunosuppression on the immunogenicity of mRNA vaccines to SARS-CoV-2. Ann Intern Med 2021;174:1572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abdulahad WH, van der Geld YM, Stegeman CA, Kallenberg CGM.. Persistent expansion of CD4+ effector memory T cells in Wegener’s granulomatosis. Kidney Int 2006;70:938–47. [DOI] [PubMed] [Google Scholar]
- 11. Komocsi A, Lamprecht P, Csernok E. et al. Peripheral blood and granuloma CD4+CD28– T cells are a major source of interferon-γ and tumor necrosis factor-α in Wegener’s granulomatosis. Am J Pathol 2002;160:1717–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abdulahad WH, Kallenberg CGM, Limburg PC, Stegeman CA.. Urinary CD4+ effector memory T cells reflect renal disease activity in antineutrophil cytoplasmic antibody–associated vasculitis. Arthritis Rheum 2009;60:2830–8. [DOI] [PubMed] [Google Scholar]
- 13. Abdulahad WH, Stegeman CA, Limburg PC, Kallenberg CGM.. Skewed distribution of Th17 lymphocytes in patients with Wegener’s granulomatosis in remission. Arthritis Rheum 2008;58:2196–205. [DOI] [PubMed] [Google Scholar]
- 14. Abdulahad WH, Lepse N, Stegeman CA. et al. Increased frequency of circulating IL-21 producing Th-cells in patients with granulomatosis with polyangiitis (GPA). Arthritis Res Ther 2013;15:R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lintermans LL, Stegeman CA, Heeringa P, Abdulahad WH.. T cells in vascular inflammatory diseases. Front Immunol 2014;5:504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cahalan MD, Chandy KG.. The functional network of ion channels in T lymphocytes. Immunol Rev 2009;231:59–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wulff H, Calabresi PA, Allie R. et al. The voltage-gated Kv1.3 K+ channel in effector memory T cells as new target for MS. J Clin Invest 2003;111:1703–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beeton C, Barbaria J, Giraud P. et al. Selective blocking of voltage-gated K+ channels improves experimental autoimmune encephalomyelitis and inhibits T cell activation. J Immunol 2001;166:936–44. [DOI] [PubMed] [Google Scholar]
- 19. Beeton C, Pennington MW, Wulff H. et al. Targeting effector memory T cells with a selective peptide inhibitor of Kv1.3 channels for therapy of autoimmune diseases. Mol Pharmacol 2005;67:1369–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beeton C, Wulff H, Standifer NE. et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc Natl Acad Sci USA 2006;103:17414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tarcha EJ, Chi V, Muñoz-Elías EJ. et al. Durable pharmacological responses from the peptide ShK-186, a specific Kv1.3 channel inhibitor that suppresses T cell mediators of autoimmune disease. J Pharmacol Exp Ther 2012;342:642–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jennette JC, Falk RJ, Bacon PA. et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65:1–11. [DOI] [PubMed] [Google Scholar]
- 23. Leavitt RY, Fauci AS, Bloch DA. et al. The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis Rheum 1990;33:1101–7. [DOI] [PubMed] [Google Scholar]
- 24. Luqmani RA, Bacon PA, Moots RJ. et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. 1994;87:671–8. [PubMed] [Google Scholar]
- 25. Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S.. Defining the human T helper 17 cell phenotype. Trends Immunol 2012;33:505–12. [DOI] [PubMed] [Google Scholar]
- 26. Chandy KG, Wulff H, Beeton C. et al. K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci 2004;25:280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lúdvíksson BR, Sneller MC, Chua KS. et al. Active Wegener’s granulomatosis is associated with HLA-DR+ CD4+ T cells exhibiting an unbalanced Th1-type T cell cytokine pattern: reversal with IL-10. J Immunol 1998;160:3602–9. [PubMed] [Google Scholar]
- 28. Csernok E, Trabandt A, Müller A. et al. Cytokine profiles in Wegener’s granulomatosis: predominance of type 1 (Th1) in the granulomatous inflammation. Arthritis Rheum 1999;42:742–50. [DOI] [PubMed] [Google Scholar]
- 29. Nogueira E, Hamour S, Sawant D. et al. Serum IL-17 and IL-23 levels and autoantigen-specific Th17 cells are elevated in patients with ANCA-associated vasculitis. Nephrol Dial Transplant 2010;25:2209–17. [DOI] [PubMed] [Google Scholar]
- 30. Chi V, Pennington MW, Norton RS. et al. Development of a sea anemone toxin as an immunomodulator for therapy of autoimmune diseases. Toxicon 2012;59:529–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sloan-Lancaster J, Steinberg TH, Allen PM.. Selective loss of the calcium ion signaling pathway in T cells maturing toward a T helper 2 phenotype. J Immunol 1997;159:1160–8. [PubMed] [Google Scholar]
- 32. Weber KS, Miller MJ, Allen PM.. Th17 cells exhibit a distinct calcium profile from Th1 and Th2 cells and have Th1-like motility and NF-AT nuclear localization. J Immunol 2008;180:1442–50. [DOI] [PubMed] [Google Scholar]
- 33. Land J, Lintermans LL, Stegeman CA. et al. Kv1.3 channel blockade modulates the effector function of B cells in granulomatosis with polyangiitis. Front Immunol 2017;8:1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kazama I. Targeting lymphocyte Kv1.3-channels to suppress cytokine storm in severe COVID-19: can it be a novel therapeutic strategy? Drug Discov Ther 2020;14:143–4. [DOI] [PubMed] [Google Scholar]
- 35. Beeton C, Wulff H, Barbaria J. et al. Selective blockade of T lymphocyte K+ channels ameliorates experimental autoimmune encephalomyelitis, a model for multiple sclerosis. Proc Natl Acad Sci USA 2001;98:13942–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hyodo T, Oda T, Kikuchi Y. et al. Voltage-gated potassium channel Kv1.3 blocker as a potential treatment for rat anti-glomerular basement membrane glomerulonephritis. Am J Physiol Renal Physiol 2010;299:F1258–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chhabra S, Chang SC, Nguyen HM. et al. Kv1.3 channel‐blocking immunomodulatory peptides from parasitic worms: implications for autoimmune diseases. FASEB J 2014;28:3952–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tanner MR, Tajhya RB, Huq R. et al. Prolonged immunomodulation in inflammatory arthritis using the selective Kv1.3 channel blocker HsTX1[R14A] and its PEGylated analog. Clin Immunol 2017;180:45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yuan XL, Zhao YP, Huang J. et al. A Kv1.3 channel-specific blocker alleviates neurological impairment through inhibiting T-cell activation in experimental autoimmune encephalomyelitis. CNS Neurosci Ther 2018;24:967–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Veytia-Bucheli JI, Jiménez-Vargas JM, Melchy-Pérez EI. et al. Kv1.3 channel blockade with the Vm24 scorpion toxin attenuates the CD4+ effector memory T cell response to TCR stimulation. Cell Commun Signal 2018;16:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Matheu MP, Beeton C, Garcia A. et al. Imaging of effector memory T cells during a delayed-type hypersensitivity reaction and suppression by Kv1.3 channel block. Immunity 2008;29:602–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tarcha EJ, Olsen CM, Probst P. et al. Safety and pharmacodynamics of dalazatide, a Kv1.3 channel inhibitor, in the treatment of plaque psoriasis: a randomized phase 1b trial. PLoS One 2017;12:e0180762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author.