

Abstract
Long QT syndrome (LQTS), a frequently fatal inherited arrhythmia syndrome caused by genetic variants (congenital) or drugs (acquired), affects 1 in 2,000 people worldwide. Its sentinel event is often sudden cardiac death (SCD), which makes pre-clinical diagnosis by genetic testing potentially life saving. Unfortunately, clinical experience with genetic testing has shown that it is difficult to correctly identify genetic variants as disease causing. These current deficiencies in accurately assigning pathogenicity led to the discovery of increasing numbers of rare variants classified as “variant of uncertain significance” (VUS). To overcome these challenges, new technologies such as clustered regularly interspaced short palindromic repeats (CRISPR) genome-editing can be combined with human induced pluripotent stem cell (iPSC)-derived cardiomyocytes (CMs) to provide a new approach to decipher pathogenicity of VUS and to better predict arrhythmia risk. To that end, the overarching goal of our network is to establish the utility of iPSC-based platforms to solve major clinical problems associated with LQTS by determining how to (1) differentiate pathogenic mutations from background genetic noise, (2) assess existing and novel variants associated with congenital and acquired LQTS, and (3) provide genotype- and phenotype- guided risk stratification and pharmacological management of LQTS. To achieve these goals and to further advance the use of iPSCs in disease modeling and drug discovery, our team of investigators for this Leducq Foundation Transatlantic Networks of Excellence proposal will work together to (i) improve differentiation efficiency, cellular maturation, and lineage specificity, (ii) develop new assays for high throughput cellular phenotyping, and (iii) train young investigators to clinically implement patient-specific genetic modeling.
Keywords: Long QT syndrome, arrhythmia, variant of uncertain significance, channelopathies, sudden cardiac death, precision medicine, human induced pluripotent stem cells, genome editing, prolonged QT interval
Within the universe of disorders potentially associated with significant risk for sudden cardiac death (SCD), a special place is occupied by the long QT syndrome (LQTS)1. Two types of LQTS exist: congenital LQTS (cLQTS) caused by gene mutations mostly in cardiac ion channels, and drug-induced LQTS (diLQTS). An underlying tenet that has been progressively developed and now firmly established is that LQTS can be uniquely useful in unraveling major gaps in our knowledge regarding mechanisms underlying SCD, providing unforeseen opportunities to discover novel approaches for prevention.
Over the last 45 years, there has been a tremendous progress both in the realization of the width of the clinical issues1 and in the understanding of the underlying mechanisms2. Like a series of Chinese boxes, however, many new discoveries also have unexpected ramifications that shines light in previously dark areas only to propel the curious investigator into another mystery in a seemingly endless journey of discovery. Here, we will discuss the current state and major outstanding questions in the LQTS field, followed by an overview of the joint efforts by our Consortium to address some of the major challenges in managing LQTS.
LQTS is characterized by a prolongation of the QT interval on the surface ECG, an often bizarre T wave morphology, and a predisposition to syncope, cardiac arrest, and sudden death, most commonly in conditions of increased sympathetic activity such as emotional or physical stress but can also occur at rest. The congenital form, cLQTS, occurs in at least 1:2,000 live births. However, probably because of its low penetrance and of the rather large number of genotype-positive/phenotype-negative individuals, its true prevalence is likely closer to 1:1,0003. Although 17 genes have been associated with cLQTS2 thus far, the 3 main ones, KCNQ1 (LQT1), KCNH2 (LQT2), and SCN5A (LQT3), account for approximately 75% of clinically definite LQTS, whereas the minor genes contribute an additional 5% collectively. Approximately 20% of cLQTS thus remain genetically elusive. The pillars of current therapy include the following: β-blockers (propranolol and nadolol); left cardiac sympathetic denervation, a surgical procedure that involves removal of the lower half of the left stellate ganglion with the first 3–4 thoracic ganglia; and the automatic implantable cardioverter-defibrillator (ICD). Pharmacotherapy with sodium channel blocker mexiletine has proven effective in shortening the QT interval in LQT3 patients as well as in most LQT2 patients4, 5.
Despite significant advances in clinical management, the care of almost 1/3 of cLQTS patients is often plagued by inconclusive clinical genetic testing due to variants of uncertain significance (VUS). VUSs are emerging as a significant challenge in clinical genetics due to the increasing use of genetic testing coupled with a paucity of experimental platforms that can rapidly and reliably establish the functional significance of genetic variants. Computational methods fare poorly in their ability to distinguish pathogenic from benign variants6. Adding to the complexity, different variants in the same gene can give rise to a multitude of diseases. For example, variants in SCN5A have been linked to LQTS3, Brugada syndrome, idiopathic ventricular fibrillation, sick sinus syndrome, and even familial DCM6. Hence, the presence of a VUS not only leads to questions as to whether or not it is pathogenic, but also to what disease it may be associated with. Thus, there is a strong need for better diagnostic platforms, ideally of human origin, that can validate the functional significance of genetic variants and accurately assess responses to therapeutic agents.
The elegance of iPSC technology is exemplified by the fact that patient-derived iPSC-CMs can serve as a disease model in a dish by retaining the genetic makeup of the patient and exhibiting the phenotypic features of the disease in vitro. Although CRISPR editing of existing iPSCs now makes it possible to rapidly establish variant pathogenicity in vitro using patient-independent iPSC-CM models7, to accurately model a patient’s clinical phenotype in a dish with respect to the effect of modifier alleles, patient-derived iPSC-CMs remain indispensable. In the future, both platforms could be integrated into genetic testing clinics where patients who have undergone DNA sequencing and are found to have a VUS in LQTS susceptibility genes could be given functional annotation of the variant within a few months, or a shorter period of time using patient-independent iPSC models, to guide the patient’s prognosis and management7, 8.
A large number of drugs can prolong cardiac repolarization thus inducing diLQTS and consequent life-threatening ventricular arrhythmias called torsades de pointes (TdP). The typical cause is the inhibition of the inward rectifying potassium channel (hERG or human ether-a-go-go related gene encoded by KCNH29). Importantly, the risk of developing diLQTS and TdP varies markedly between individuals; for cLQTS, there is a need for better diagnostic platforms to understand molecular mechanisms underlying this susceptibility to diLQTS and evaluate individual patient’s risks.
The main purpose of our Leducq Foundation Transatlantic Networks of Excellence is to fill in the crucial knowledge gaps plaguing the interpretation of rare variants by adopting, optimizing, and standardizing patient- and disease-specific iPSC-based platforms to better our understanding of arrhythmia risks in LQTS. Our primary goals include: (i) differentiation of pathogenic mutations from background genetic noise (i.e., assign functional significance to VUS), which will help us decipher mechanisms underlying patient-specific phenotypic variations; (ii) assessment of novel variants associated with diLQTS; and (iii) providing genotype- and phenotype-guided risk stratification and pharmacological management of LQTS.
To accomplish these goals, we have assembled an international multidisciplinary team of leading experts in the fields of clinical cardiology, electrophysiology, stem cell biology, genomics, pharmacogenomics bioinformatics, and tissue engineering, all working on projects towards realizing the full potential of precision medicine. Our team is led by Joseph C. Wu from Stanford University (North American coordinator), who studies disease mechanisms using iPSC-CMs, next generation sequencing (NGS), genome editing, and high-throughput (HT) screening methods; and Peter Schwartz from IRCCS Istituto Auxologico Italiano (European coordinator), who has pioneered genotype-phenotype correlation for LQTS “modifier” genes, diLQTS, and drug therapy for LQTS. Our network members include Shinya Yamanaka (2012 Nobel Prize in Medicine & Physiology) and Yoshinori Yoshida (Kyoto, Japan), expert in iPSC generation and iPSC-CM differentiation; Lior Gepstein (Haifa, Israel), expert in iPSC electrophysiology and arrhythmia; Jean-Sébastien Hulot (Inserm, Paris), an expert in precision medicine and using iPSCs for studying diLQTS; and Bjorn Knollmann (Vanderbilt University, USA), clinical pharmacologist and expert in cellular electrophysiology and excitation-contraction coupling of iPSC-CMs and drug repurposing studies. Raymond L. Woosley (USA), Minoru Horie (Japan), and Stefan Kääb (Germany), all experts in LQTS science, will serve as scientific advisors. This team will work together to adopt, optimize, and standardize patient- and disease-specific iPSC-based platforms for improving our understanding of arrhythmia risk associated with LQTS.
Our network member, Shinya Yamanaka, received the 2012 Nobel Prize in Physiology or Medicine for his groundbreaking work on generating iPSCs directly from patients10. Since his landmark discovery, our understanding of novel pathological mechanisms has progressed remarkably11 with new drugs originating from iPSC screens now emerging, including the first clinical trial using iPSC-retinal pigment epithelial cells for macular degeneration12. Importantly, the use of iPSCs allows researchers to understand disease mechanisms at a personalized level13, potentially offering an ideal model for better identification and management of LQTS patients who are susceptible to SCD.
In addition to scientific and clinical discovery into LQTS therapeutic strategies, training and education of emerging investigators is an essential goal of our network. We will train young investigators to clinically implement the latest iPSC technology and genetic modeling. Our proposal also represents the first concerted effort to create an international biorepository of standardized, high-quality iPSC lines from USA, Italy, France, Israel, and Japan (Figure 1).
Figure 1. An overview of our Leducq consortium grant.
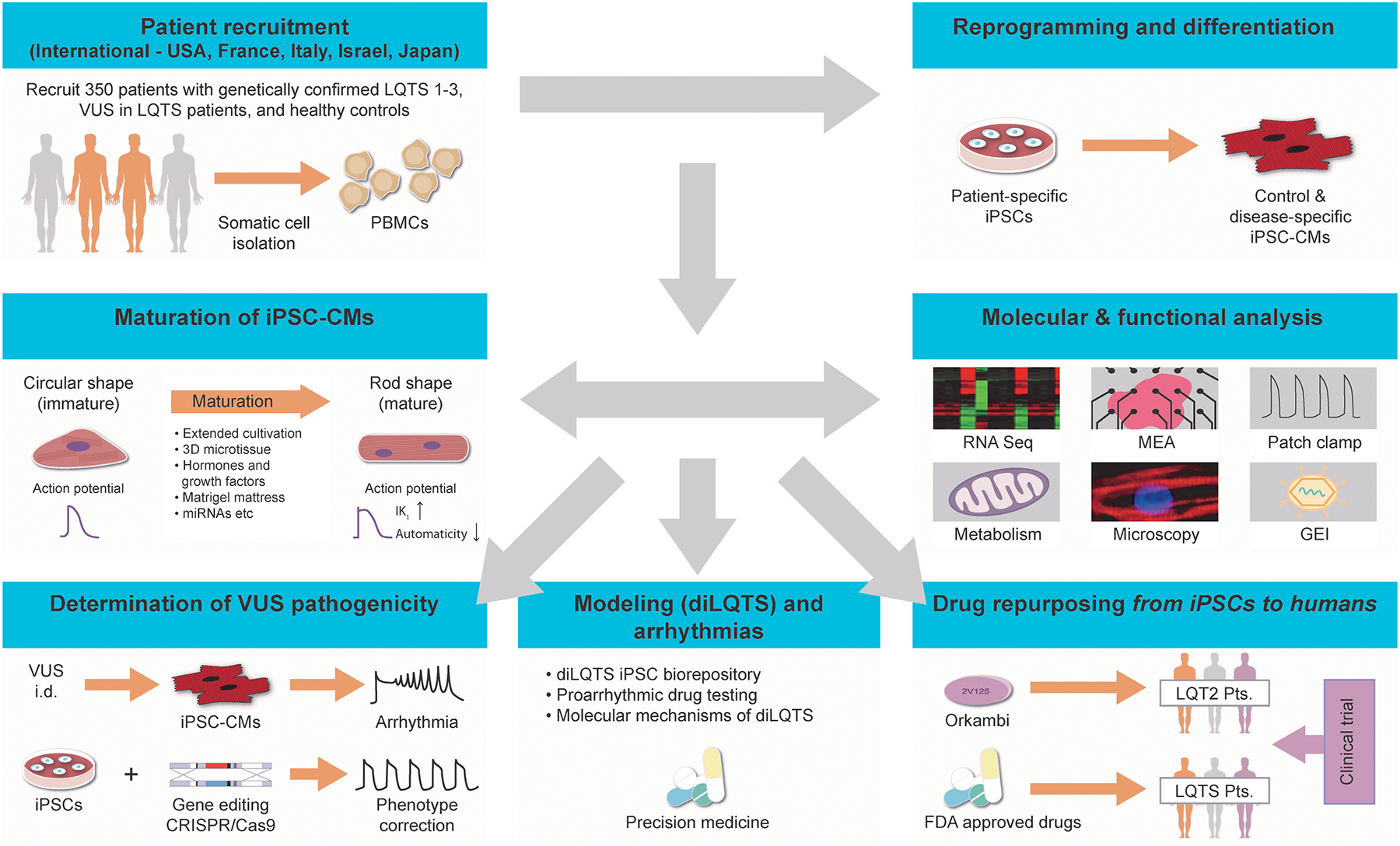
Network members include Wu, Schwartz, Yamanaka, Yoshida, Gepstein, Hulot and Knollmann. Our network will recruit patients internationally from cardiology clinics in USA, France, Italy, Israel and Japan to generate their iPSC-CMs, all of which will undergo complete molecular and functional characterization. Patient-specific iPSC-CMs will then be used to assign functional significance of rare genetic variants, model diLQTS and arrhythmia, and repurpose drugs for treating LQTS patients. These studies will help identify novel pathological mechanisms that will improve drug therapy and advance personalized medicine for LQTS.
To date, the majority of iPSC studies have utilized a mixed population of iPSC-CMs (ventricular, atrial, and nodal cells) that display an early-stage fetal-like phenotype. To model LQTS and other channelopathies, maturation of cardiomyocytes is essential. Recapitulating physiological conditions of adult human heart and its microenvironment in our iPSC platform remains a major goal of our study. This will allow us to efficiently characterize pathogenic mutations and screen drugs that can eventually alleviate fatal consequences of LQTS and help improve quality of life for the patients. Five tentative hallmarks that represent common denominators of adult human myocardium are the following: (1) cellular alignment-driven anisotropy, (2) efficient energy conversion (requiring oxidative metabolism), (3) excitation-contraction coupling, (4) achieving electrophysiological property similar to adult-cardiomyocytes with increased conduction velocity, and (5) positive force-frequency relationship. We seek to uncover novel mechanisms driving these hallmarks in iPSC-CMs that will lead to more precise disease drug modeling and drug discovery process.
We also plan to devise several strategies to reduce the heterogeneity of iPSC-CMs and augment their maturation. These include: (i) derivation of chamber-specific CMs (focusing on ventricular CMs); (ii) using multiple strategies to induce maturation; (iii) utilization of multicellular preparations that tend to decrease intercellular variability; and (iv) development of high-throughput cellular phenotyping strategies to average out residual heterogeneities. To induce iPSC-CM maturation, a number of approaches have been used, including cultivation for extended periods, 3-D and mechanical conditioning, rapid electrical stimulation, enhanced IK1 expression, hormonal stimulation (thyroid hormones, IGF-1 and glucocorticoids), Matrigel-based mattress approach, and ectopic overexpression of miRNAs. Recently, our network member Knollmann used these strategies to promote t-tubule structural and functional maturation.14 Over the next 5 years, we plan to further mature iPSC-CMs by integrating the aforementioned strategies together with newer approaches such as synthetic miRNA switches for efficient detection and purification of cell populations, results of which were published recently by our network members Yoshida and Yamanaka.15 Notably, our network member Gepstein was among the pioneers to generate LQT2 patient-specific iPSC-CMs16. Finally, we plan to adopt optogenetic sensors such as genetically encoded voltage and calcium indicators (GEVIs and GECIs, respectively) for high-throughput cellular phenotyping, and optogenetic actuators (light-sensitive proteins such as channelrhodopsin-2) for contact-free pacing17 that will allow standardized analysis across all network sites.
The Expert Consensus Statement18 on management of inherited arrhythmias requires the presence of an “unequivocally” pathogenic mutation in LQTS susceptibility genes as Class I criterion for LQTS diagnosis. However, up to 50% of rare variants cannot be classified as “unequivocally” pathogenic due to inadequate evidence and are instead labeled as “variant of uncertain significance”. This creates a major clinical dilemma for patients and physicians, as a VUS cannot be used in clinical decision-making.19 This problem is further complicated by the high variability of phenotypic expression in LQTS patients. Even family members carrying the same mutation may exhibit different QT intervals and clinical manifestations. Under such circumstances, patient-specific electrophysiological characterization of these rare variants to assign pathogenicity is crucial not only for accurate diagnosis but also for appropriate clinical management. Hence, we aim to establish the utility of iPSC-CMs in distinguishing causal from benign rare genetic variants to improve diagnosis and management of LQTS. We will optimize and validate iPSC-based platforms to decipher pathogenicity of VUSs in LQTS families. For each family, iPSC-CMs will be generated from VUS carriers and VUS-negative family members. VUS pathogenicity will be determined by verifying whether hallmark LQTS features such as action potential duration (APD) prolongation, early afterdepolarizations (EADs), or triggered arrhythmias (EADs that give rise to triggered action potentials), manifested in LQTS iPSC-CMs as isolated premature beats or even multiple sequential triggered action potentials,16 are present in VUS-positive iPSC-CMs. Using CRISPR genome editing, variant pathogenicity will be established based on whether the introduction of VUS in healthy iPSC-CMs recapitulates the pathogenic phenotype or if correction of the VUS in patient iPSC-CMs normalizes the aberrant cellular phenotype20, 21. Finally, we will determine if the candidate variant alters the density of respective ionic current. We anticipate the reclassification of VUSs into “benign” vs “causal” will benefit not only for VUS carriers, but also for extended family members who have not been screened or genetically tested. Whenever a “VUS” is proven pathogenic, it will enter our drug screening and clinical trial studies.
Certain individuals have a higher propensity to develop prolonged QT interval and consequently life-threatening ventricular arrhythmias in response to drugs that alter cardiac ion currents, notably the IKr. The risk of arrhythmia can be further elevated in the presence of (i) acquired factors (e.g., ionic disturbances) and (ii) genetic factors (e.g., underlying mutations)9, 22. Network member Hulot recently demonstrated in vitro the potential of patient-specific iPSC-CMs to recapitulate the patient’s in vivo predilection to diLQTS23. Hence, we will utilize patient-specific iPSC-CMs to better understand mechanisms of diLQTS, and predict the arrhythmogenic risk of drugs at both the cellular level (drug screening assays) and the patient level (precision medicine). We will recruit and generate iPSCs from patients (i) diagnosed as carriers of mutations in one of the 17 cLQTS genes or of functional polymorphisms recognized as being often present in diLQTS (i.e., KCNE1-D85N) (referred as genotype positive)22, 24, or (ii) without any LQTS mutations (genotype negative) and acquired conditions explaining arrhythmias (e.g., hypokalemia, bradycardia, hypothermia, heart failure, etc.). We will assess whether iPSC-CMs from our diLQTS biorepository exhibit a higher propensity to develop arrhythmias under pharmacological stimulation for typical drugs that block IKr (e.g., sotalol, dofetilide, disopyramide, ibutilide, and quinidine). Finally, we will elucidate factors responsible for the heterogeneity of drug responses in iPSC-CMs from patients of different ethnicities and from different clones and batches of differentiation. Furthermore, to study the mechanisms leading to differential susceptibility of iPSC-CMs to diLQTS, we recently performed a transcriptomic profile of iPSC-CMs using RNA-seq23. Using a prior knowledge-based approach, we identified potential risk markers mechanistically connected to QT prolongation in iPSC-CMs derived from high risk patients, including 4 up-regulated (DLG2, KCNE4, PTRF, and HTR2C) and 1 down-regulated (CAMKV) genes. These genes encode proteins that act as direct interactors of ion channels, suggesting that diLQTS is favored by changes in the regulation of the cardiac repolarization machinery (i.e., channel trafficking and scaffolding), which in turn limits the cardiac repolarization reserve. To test this hypothesis, we will analyze expression of new candidate genes from our diLQTS iPSC biorepository. We will compare the results observed in genotype-positive vs. negative patients. We will then study their functional effect by modulating their expression and by targeting candidates using genome editing.
Recently, Schwartz and colleagues in an encouraging study demonstrated the potential of Lumacaftor (LUM) + Ivacaftor in significantly shortening the QTc in two LQT2 patients with a trafficking defect. These clinical results recapitulate in vitro results with LUM, a drug already in clinical use for cystic fibrosis: LUM rescued the pathological phenotype of class 2 LQT2 iPSC-CMs derived from same two LQT2 patients not protected by β-blockers25. This proof-of-concept result illustrates the potential for mechanism of action studies using iPSC-CMs to identify novel therapies for LQTS patients. Importantly, the safety and pharmacokinetic profile of LUM are already known, making it a suitable first candidate for translating our findings from bench to bedside. LUM is marketed clinically as Orkambi (LUM plus ivacaftor, an enhancer of the CFTR protein function). In our network, we will implement a two-stage approach to confirm the hypothesis that LQT2 iPSC-CMs can be used to predict clinical response to Orkambi. We will first identify KCNH2 mutations with a channel trafficking defect from the total 160 KCNH2 mutations present in our Milan database, followed by testing Orkambi in iPSC-CMs using our published protocols2 and comparing in vitro results to clinical effects in the patients themselves.
To summarize, this viewpoint highlights some of the critical challenges in the identification, risk-stratification, and clinical management of a potentially fatal, but highly treatable genetic disorder such as LQTS. The systematic approach that we outline here will help develop and validate new iPSC-based platforms to solve major intractable clinical problems associated with LQTS and to improve our understanding of the underlying mechanisms.
Acknowledgment
We are grateful for the support by our LQTS patients and by Leducq Foundation 18CVD05. We thank Antonio Lucena-Cacace and Massimiliano Gnecchi for their contribution to this Viewpoint We thank Amy Thomas for her assistance with the figure.
Footnotes
Disclosure
JCW is a co-founder of Khloris Biosciences but has no competing interests, as the work presented here is completely independent. SY is a scientific advisor of iPS Academia Japan without salary. YY owns stock in iPS Portal. All other authors have no disclosures.
References
- 1.Schwartz PJ and Ackerman MJ. The long QT syndrome: a transatlantic clinical approach to diagnosis and therapy. Eur Heart J. 2013;34:3109–16. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz PJ, Ackerman MJ, George AL Jr. and Wilde AAM. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol. 2013;62:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, Gabbarini F, Goulene K, Insolia R, Mannarino S, Mosca F, Nespoli L, Rimini A, Rosati E, Salice P and Spazzolini C. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roden DM. Clinical practice. Long-QT syndrome. N Engl J Med. 2008;358:169–76. [DOI] [PubMed] [Google Scholar]
- 5.Bos JM, Crotti L, Rohatgi RK, Castelletti S, Dagradi F, Schwartz PJ and Ackerman MJ. Mexiletine shortens the QT interval in patients with potassium channel-mediated type 2 long QT syndrome. Circ Arrhythm Electrophysiol. 2019;12:e007280. [DOI] [PubMed] [Google Scholar]
- 6.Musunuru K, Sheikh F, Gupta RM, Houser SR, Maher KO, Milan DJ, Terzic A, Wu JC, American Heart Association Council on Functional G, Translational B, Council on Cardiovascular Disease in the Y, Council on C and Stroke N. Induced pluripotent stem cells for cardiovascular disease modeling and precision medicine: A scientific statement from the American Heart Association. Circ Genom Precis Med. 2018;11:e000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chavali NV, Kryshtal DO, Parikh SS, Wang L, Glazer AM, Blackwell DJ, Kroncke BM, Shoemaker MB and Knollmann BC. Patient-independent human induced pluripotent stem cell model: A new tool for rapid determination of genetic variant pathogenicity in long QT syndrome. Heart Rhythm. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sallam K, Li Y, Sager PT, Houser SR and Wu JC. Finding the rhythm of sudden cardiac death: new opportunities using induced pluripotent stem cell-derived cardiomyocytes. Circ Res. 2015;116:1989–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garg P, Garg V, Shrestha R, Sanguinetti MC, Kamp TJ and Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes as models for cardiac channelopathies: A primer for non-electrophysiologists. Circ Res. 2018;123:224–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K and Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. [DOI] [PubMed] [Google Scholar]
- 11.Shi Y, Inoue H, Wu JC and Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 2016;16:115–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mandai M, Watanabe A, Kurimoto Y, Hirami Y, Morinaga C, Daimon T, Fujihara M, Akimaru H, Sakai N, Shibata Y, Terada M, Nomiya Y, Tanishima S, Nakamura M, Kamao H, Sugita S, Onishi A, Ito T, Fujita K, Kawamata S, Go MJ, Shinohara C, Hata K-i, Sawada M, Yamamoto M, Ohta S, Ohara Y, Yoshida K, Kuwahara J, Kitano Y, Amano N, Umekage M, Kitaoka F, Tanaka A, Okada C, Takasu N, Ogawa S, Yamanaka S and Takahashi M. Autologous Induced stem-cell–derived retinal cells for macular degeneration. New England Journal of Medicine. 2017;376:1038–1046. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Sallam K, Schwartz PJ and Wu JC. Patient-specific induced pluripotent stem cell-based disease model for pathogenesis studies and clinical pharmacotherapy. Circ Arrhythm Electrophysiol. 2017;10:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parikh SS, Blackwell DJ, Gomez-Hurtado N, Frisk M, Wang L, Kim K, Dahl CP, Fiane A, Tonnessen T, Kryshtal DO, Louch WE and Knollmann BC. Thyroid and glucocorticoid hormones promote functional T-tubule development in human-induced pluripotent stem cell-derived cardiomyocytes. Circ Res. 2017;121:1323–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miki K, Endo K, Takahashi S, Funakoshi S, Takei I, Katayama S, Toyoda T, Kotaka M, Takaki T, Umeda M, Okubo C, Nishikawa M, Oishi A, Narita M, Miyashita I, Asano K, Hayashi K, Osafune K, Yamanaka S, Saito H and Yoshida Y. Efficient detection and purification of cell populations using synthetic microRNA switches. Cell Stem Cell. 2015;16:699–711. [DOI] [PubMed] [Google Scholar]
- 16.Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, Boulos M and Gepstein L. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–9. [DOI] [PubMed] [Google Scholar]
- 17.Nussinovitch U and Gepstein L. Optogenetics for in vivo cardiac pacing and resynchronization therapies. Nature Biotechnology. 2015;33:750–4. [DOI] [PubMed] [Google Scholar]
- 18.Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G and Tracy C. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10:1932–63. [DOI] [PubMed] [Google Scholar]
- 19.Giudicessi JR, Roden DM, Wilde AAM and Ackerman MJ. Classification and reporting of potentially proarrhythmic common genetic variation in long QT syndrome genetic testing. Circulation. 2018;137:619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garg P, Oikonomopoulos A, Chen H, Li Y, Lam CK, Sallam K, Perez M, Lux RL, Sanguinetti MC and Wu JC. Genome editing of induced pluripotent stem cells to decipher cardiac channelopathy variant. J Am Coll Cardiol. 2018;72:62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Liang P, Lan F, Wu H, Lisowski L, Gu M, Hu S, Kay MA, Urnov FD, Shinnawi R, Gold JD, Gepstein L and Wu JC. Genome editing of isogenic human induced pluripotent stem cells recapitulates long QT phenotype for drug testing. J Am Coll Cardiol. 2014;64:451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Itoh H, Crotti L, Aiba T, Spazzolini C, Denjoy I, Fressart V, Hayashi K, Nakajima T, Ohno S, Makiyama T, Wu J, Hasegawa K, Mastantuono E, Dagradi F, Pedrazzini M, Yamagishi M, Berthet M, Murakami Y, Shimizu W, Guicheney P, Schwartz PJ and Horie M. The genetics underlying acquired long QT syndrome: impact for genetic screening. Eur Heart J. 2016;37:1456–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stillitano F, Hansen J, Kong CW, Karakikes I, Funck-Brentano C, Geng L, Scott S, Reynier S, Wu M, Valogne Y, Desseaux C, Salem JE, Jeziorowska D, Zahr N, Li R, Iyengar R, Hajjar RJ and Hulot JS. Modeling susceptibility to drug-induced long QT with a panel of subject-specific induced pluripotent stem cells. Elife. 2017;6:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaab S, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, Bezzina CR, Schulze-Bahr E, Guicheney P, Bishopric NH, Myerburg RJ, Schott JJ, Pfeufer A, Beckmann BM, Martens E, Zhang T, Stallmeyer B, Zumhagen S, Denjoy I, Bardai A, Van Gelder IC, Jamshidi Y, Dalageorgou C, Marshall V, Jeffery S, Shakir S, Camm AJ, Steinbeck G, Perz S, Lichtner P, Meitinger T, Peters A, Wichmann HE, Ingram C, Bradford Y, Carter S, Norris K, Ritchie MD, George AL Jr. and Roden DM. A large candidate gene survey identifies the KCNE1 D85N polymorphism as a possible modulator of drug-induced torsades de pointes. Circ Cardiovasc Genet. 2012;5:91–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwartz PJ, Gnecchi M, Dagradi F, Castelletti S, Parati G, Spazzolini C, Sala L and Crotti L. From patient-specific induced pluripotent stem cells to clinical translation in long QT syndrome Type 2. Eur Heart J. 2019;40:1832–1836. [DOI] [PubMed] [Google Scholar]