

Abstract
Therapy resistance is the primary problem in treating late-stage colorectal cancer (CRC). Claudins are frequently dysregulated in cancer, and several are being investigated as novel therapeutic targets and biomarkers. We have previously demonstrated that Claudin-1 (CLDN1) expression in CRC promotes epithelial-mesenchymal transition, metastasis, and resistance to anoikis. Here, we hypothesize that CLDN1 promotes cancer stemness and chemoresistance in CRC. We found that high CLDN1 expression in CRC is associated with cancer stemness and chemoresistance signaling pathways in patient datasets, and it promotes chemoresistance both in vitro and in vivo. Using functional stemness assays, proteomics, biophysical binding assays, and patient-derived organoids, we found that CLDN1 promotes properties of cancer stemness including CD44 expression, tumor-initiating potential, and chemoresistance through a direct interaction with ephrin type-A receptor 2 (EPHA2) tyrosine kinase. This interaction is dependent on the CLDN1 PDZ-binding motif, increases EPHA2 protein expression by inhibiting its degradation, and enhances downstream AKT signaling and CD44 expression to promote stemness and chemoresistance. These results suggest CLDN1 is a viable target for pharmacological intervention and/or biomarker development.
Keywords: Colorectal cancer, chemoresistance, cancer stemness, Claudin-1, EPHA2
Graphical Abstract
INTRODUCTION
Colorectal cancer (CRC) is the second deadliest cancer in the United States with an estimated 153,020 new cases and 52,550 deaths in 2023 [1]. Prognosis varies dramatically with stage at diagnosis; localized CRC is treated surgically and has a 5-year survival rate of 91%, while the survival rate of metastatic CRC falls to 15%. Chemotherapeutic drugs are used in CRC as both adjuvant and neoadjuvant therapy [2]. However, due to intrinsic and acquired drug resistance, chemotherapy in advanced cases is largely palliative, and nearly all patients develop resistance [3]. Targeted therapies have proven effective in some cases, but their use is limited to specific molecular subtypes [4]. Understanding the mechanisms of chemoresistance and discovering new pharmaceutical targets and biomarkers for resistant tumors is crucial to improving outcomes for CRC patients.
Claudins are a large family of tight junction molecules, with 26 members expressed in humans [5]. Claudins are often dysregulated in carcinomas, and their aberrant expression and localization influence oncogenic signaling pathways including Wnt/β-catenin, Notch, MAPK, and PI3K/Akt [5-11]. Because of their tissue-specific expression, selective upregulation in cancer, and presence on the cell surface, claudins are being investigated as therapeutic targets for monoclonal antibodies, small molecule inhibitors, and immunotherapy [12]. Claudin-1 (CLDN1) is highly upregulated in CRC, and its expression promotes epithelial-mesenchymal transition (EMT), metastasis, and resistance to anoikis [6, 13, 14]. In addition, CLDN1 expression is associated with chemoresistance in ovarian, lung, and liver cancers [15-17]. It is currently unclear how claudins directly influence intracellular signaling. Dysregulation in cancer leads to increased non-junctional expression and aberrant protein-protein interactions [11]. Intracellular claudin protein-protein interactions typically occur via the C-terminal domain, which includes sites for post-translational modification and, in some family members including CLDN1, a PDZ-domain binding motif [5]. While interactions with PDZ domain-containing proteins such as ZO-1/2/3 are well documented, a more diverse interactome mediated by the C-terminal region and PDZ binding motif is now being characterized [18, 19].
Ephrin receptors are the largest class of RTKs with 14 members [20]. Upon contact with their cognate ligand, ephrin receptors participate in bidirectional signaling to modulate cell growth, migration, and differentiation [21]. Ephrin receptor signaling is highly complex and can either suppress or promote tumor growth depending on the cellular context as well as the oligomeric state of receptor complexes [22, 23]. Ligand-mediated activation tends to inhibit growth and proliferation, but overexpression of EPHA2 can lead to oncogenic, ligand-independent activation through local aggregation, oligomerization, and autophosphorylation [20]. Upon activation and autophosphorylation, EPHA2 serves as a scaffold for signaling molecules including guanine nucleotide exchange factors, src-family kinases, and the p85 subunit of phosphoinositide 3-kinase (PI3K) [20]. PI3K associates with EPHA2 through phosphorylated tyrosine residues at Y734 in the kinase domain and Y929 in the SAM domain to activate the PI3K/AKT axis [24-27]. High expression of EPHA2 in CRC is correlated with poor survival, cancer stem cell (CSC) marker expression including CD44, and therapy resistance [28, 29]. CD44 is a marker for cancer stemness in CRC, and high expression is associated with both tumor-initiating potential and resistance to therapy [30, 31].
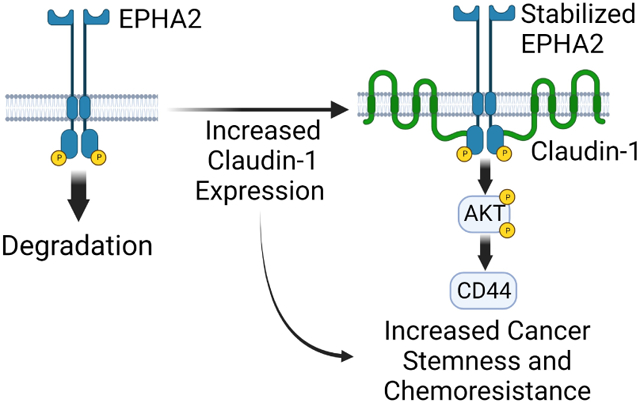
In this study, we demonstrate a direct protein-protein interaction between CLDN1 and EPHA2 via their respective intracellular domains that increases EPHA2 protein-level expression by inhibiting its degradation. Increased EPHA2 enhances downstream AKT signaling and CD44 expression to promote the cancer stemness and chemoresistance in CRC.
MATERIALS AND METHODS
Cell Lines:
The SW480, SW620, and DLD-1 cell lines were obtained from American Type Culture Collection and all experiments were conducted with low passage sub-cultured lines. All cell lines were maintained in ATCC-recommended media supplemented with 10% fetal bovine serum (R&D Systems) and 1% Pen-Strep antibiotic (Gibco). Cultures were maintained at 37°C in an atmosphere of 5% CO2. All cell lines were regularly tested for Mycoplasma infection using PCR. Additional information for antibodies and plasmids is available in Supplementary Methods.
Extreme-limiting dilution assay:
Fluorescence-activated cell sorting was used to seed live cells into 96-well ultra-low attachment plates with sphere-forming medium containing serum-free DMEM/F12 GlutaMAX (Gibco) supplemented with 20 ng/mL EGF, 20 ng/mL bFGF, B27 supplement (Gibco), N2 supplement (Gibco), and 1% Pen-Strep antibiotic (Gibco) [32]. Cells were plated in the following concentrations: 1000 cells/well x 32 wells, 100 cells/well x 32 wells, 10 cells/well x 64 wells, and 1 cell/well x 96 wells. Plates containing single cells were centrifuged at 300xg for 5 minutes and visually inspected after plating for the presence of a cell. Empty wells were excluded from analysis. Cells were cultured for 7-10 days, and wells were scored visually based on the presence of a sphere. Data was analyzed as per the method outlined by Hu and Smyth using the web-based tool located at http://bioinf.wehi.edu.au/software/elda/ [33].
Bioinformatic Analysis:
Stage-wise CLDN1 CRC expression data were downloaded from the University of ALabama at Birmingham CANcer data analysis portal (UALCAN) [34]. Batch-normalized PanCancer Atlas TCGA colorectal adenocarcinoma (COAD) expression data and expression correlation plots were downloaded from cBioportal [35, 36]. Genes significantly upregulated with CLDN1 were analyzed using Enrichr [37]. For Gene Set Enrichment Analysis, normalized TCGA mRNA expression data was sorted according to CLDN1 expression, and high CLDN1-expressing tumors (top 25%, n=150) and low CLDN1-expressing tumors (lowest 25%, n=150) were chosen for comparison. A false discovery rate of <0.25 was considered statistically significant according to the original manuscript [38].
Xenograft mouse model:
1.5 million SW620 control and SW620cldn1 KO cells were injected into the left and right flanks, respectively, of immunocompromised nude mice (NU/J, Jackson Laboratory). Once tumors became palpable (14 days after inoculation), mice were distributed evenly between control and treatment groups (n = 6 – 7 mice per group) based on initial tumor volume and sex. Tumors that were not palpable 14 days after inoculation (Day 1 of treatment) were excluded from analysis. Mice were injected intraperitoneally twice per week (on days 1, 4, 8 and 11) with either PBS or 10 mg/kg 5FU diluted in PBS for a total of 15 days. 10mg/kg 5FU was previously shown effective in combination with pharmacological CLDN1 inhibition in a xenograft CRC mouse model [39]. Tumor volume was measured every 2 days and calculated using the formula Volume = ½(WxWxL).
Microscale Thermophoresis (MST):
MST measurements were performed using a NanoTemper Monolith (NanoTemper Technologies) in SEC buffer. For binding experiments, the concentration of Cy3-labled CLDN1 was fixed at 0.25 μM and the EPHA2 ligand concentrations were varied from 0.05 nM to 6.5 μM. The EPHA2 ligand sub-dilutions were mixed with Cy3-labled CLDN1 and loaded into standard NT.115 capillary tubes for measurement. All experiments were conducted at room temperature using the nano-green channel, 100% excitation, high MST power; and the data were analyzed using Monolith analysis software. Binding experiments were performed in triplicate.
Patient-Derived Organoids:
Fresh tumor tissue was collected from liver biopsies of metastatic colorectal cancer under the approval of IRB 701-19-FB or from surgical resection specimens under the approval of IRB 440-16-EP. Detailed culturing methods available in supplemental material.
Statistical Analysis:
Pair-wise comparisons used Student’s t-test and multiple group comparisons used ANOVA with Tukey-Kramer post-hoc testing. A two-tailed P < 0.05 was considered statistically significant.
Additional Materials and Methods can be found in Supplementary Methods.
RESULTS
Claudin-1 expression promotes cancer stemness in CRC
Based on previous findings that CLDN1 promotes CRC, we investigated the relationship between CLDN1 expression and cancer stemness using publicly available datasets. In an analysis of The Cancer Genome Atlas (TCGA-Pan-Cancer Atlas) colorectal adenocarcinoma (COAD) cohort, CLDN1 was expressed at significantly higher levels in tumors compared to normal tissue (Figure 1A). Gene Ontology and Gene Set Enrichment Analyses (GSEA) of TCGA data revealed an upregulation of MsigDB Hallmark pathways associated with tumor progression and chemoresistance including EMT, Myc Targets, mTORC1, E2F, G2-M Checkpoint, TGFβ signaling, DNA Repair, and KRAS signaling in high CLDN1-expressing samples (Figure 1B, Sup. Figure 1A) [38, 40]. Further analysis showed that high CLDN1 expression was significantly correlated with cancer stemness pathways and the expression of colorectal CSC markers LGR5 and CD133 (PROM1) (Figure 1C, D). Receiver operating characteristic analysis of TCGA data also showed that increased CLDN1 expression was highly predictive of colon adenocarcinoma (AUC=0.99) (Sup. Figure 1B). We next analyzed gene expression datasets (GSE28702 and GSE72970) of CRC patients categorized into responders and non-responders to FOLFOX, a standard-of-care treatment combining 5-fluorouracil (5FU), leucovorin calcium, and oxaliplatin. Here we found a positive correlation between CLDN1 expression and chemoresistance (Figure 1E).
Figure 1: CLDN1 expression is associated with cancer stemness and chemoresistance in CRC datasets.

(a) CLDN1 is highly expressed in all stages of CRC (PanCancer Atlas TCGA-COAD). (b) Gene Ontology analysis of MSigDB Hallmark pathways that are significantly upregulated with CLDN1 expression in the TCGA dataset. (c) Gene Set Enrichment Analysis comparing High vs Low CLDN1-expressing TCGA tumor samples revealed a significant enrichment in cancer stemness gene expression (an FDR<0.25 is considered significant in GSEA analysis). (d) CLDN1 is significantly correlated with colorectal CSC markers LGR5 and PROM1 (CD133) in TCGA data. (e) CLDN1 was positively correlated with chemoresistance in gene expression datasets (GSE28702 and GSE72970) of CRC patient tumors categorized into responders (n=51) and non-responders (n=37) to FOLFOX. NES = Normalized Enrichment Score, FDR = False Discovery Rate, AUC = Area Under Curve, **** = p<0.0001.
Next, we developed colon cancer cell line models to investigate the relationship between CLDN1 expression, cancer stemness, and chemoresistance. SW480 and SW620 are patient-matched cell lines derived from primary and metastatic tumors, respectively [41]. SW480 cells display a more epithelial phenotype and express very low levels of CLDN1, while SW620 cells are less differentiated, more metastatic, and express high levels of CLDN1 [6]. We overexpressed HA-tagged CLDN1 in SW480 and used CRISPR cas9 to knockout CLDN1 in SW620. DLD-1 cells were chosen as a CLDN1-expressing cell line derived from a different genetic background. Here again, CLDN1 was knocked out using CRISPR cas9. We found that CLDN1 expression was positively correlated with CD44 expression, which is functionally tied to cancer stemness, tumor-initiating potential, and chemoresistance in CRC (Figure 2A) [31, 42, 43]. To test the functional relevance of CLDN1 in regulating cancer stemness, we performed standard assays including sphere formation, limiting dilution, ALDEFLUOR and side population analysis. We found a significant increase in sphere formation with CLDN1 overexpression in SW480 and a significant decrease with CLDN1 KO in both SW620 and DLD-1 (Figure 2B). The extreme limiting dilution assay (ELDA) quantifies the tumor-initiating potential of a cell population [32]. Here, we used fluorescence-activated cell sorting to seed a precise number of cells in decreasing density into ultra-low attachment plates with minimally supplemented growth medium. After an incubation of 7-10 days, we scored each well based on the presence or absence of a colony and analyzed the data according to the method detailed by Hu and Smyth [33]. Here we found a significant positive correlation between tumor-initiating cell frequency and CLDN1 expression (Figure 2C, Sup. Table 1).
Figure 2: CLDN1 promotes cancer stemness properties including tumor-initiating potential in CRC.

(a) CLDN1 expression was significantly correlated with CD44 expression in SW480 and DLD-1. In all cell lines, CLDN1 expression was positively correlated with (b) sphere-forming ability and (c) tumor-initiating potential (in vitro extreme limiting dilution assay). ELDA data available in Sup. Table 1. (d) CLDN1 expression was correlated with both ALDH activity (ALDEFLUOR assay) and side population in each cell line. C = Control, KO = Knockout. * = p<0.05, ** = p<0.01, *** = p<0.001.
Having found a relationship with tumor-initiating potential, we next wanted to explore the connection between CLDN1 expression and other characteristics of cancer stemness. The ALDEFLUOR and side population assays measure functional properties of cells that are linked to cancer stemness and chemoresistance. The ALDEFLUOR assay measures the activity of aldehyde dehydrogenase (ALDH) enzymes in a population of cells, and higher ALDH activity is directly linked with increased cell proliferation and chemoresistance [44, 45]. The side population assay measures a cell’s ability to efflux toxic substances through ATP-binding cassette (ABC) transporter proteins [46]. When incubated with Hoechst dye and analyzed with flow cytometry, cells with increased efflux ability form a distinct, unstained subpopulation referred to as the “side population” that exhibits increased cancer stemness [47]. CLDN1 expression was positively correlated with increased ALDH-high and side population percentage in all cell lines (Figure 2D, Sup. Figure 2). Based on these assays, we concluded that CLDN1 expression regulates properties associated with cancer stemness in colon cancer cells.
Claudin-1 expression promotes chemoresistance
Since the functional properties measured by the above assays contribute directly to chemoresistance, we next assessed CLDN1’s role in promoting resistance to 5FU. In SW480 cells, overexpression of CLDN1 led to a 4-fold increase in 5FU IC50, while CLDN1 knockout in SW620 and DLD-1 cells resulted in a 2.5 and 6-fold decrease, respectively (Figure 3A-C). Next, we tested this finding in vivo using a xenograft mouse model. SW620 control and CLDN1 KO cells were implanted into the left and right flanks, respectively, of nude mice. Once the tumors were palpable (14 days), either PBS or 10 mg/kg 5FU was injected intraperitoneally twice per week for 15 days. This dosage of 5FU was previously shown to be effective in combination with pharmacological CLDN1 inhibition [39]. This sub-clinical dose of 5FU significantly inhibited tumor growth in the CLDN1 KO xenografts (untreated = 839.9 +/− 173.8 mm3, treated = 153.7 +/− 58.0 mm3, p=0.0073) while having little effect on control tumors (untreated = 1128.3 +/− 221.9 mm3, treated = 1081.4 +/− 442.3 mm3, p=0.9230) (Figure 3D, Sup. Table 2). We confirmed the CLDN1 KO by immunohistochemistry using a CLDN1 specific antibody. Further, 5FU treated KO tumors demonstrated significantly less proliferation (Ki67) as well as increased apoptosis (cleaved Caspase-3) when compared to control, with no significant effect in 5FU treated control tumors (Figure 3E).
Figure 3: CLDN1 promotes resistance to 5-fluorouracil.

CLDN1 overexpression in SW480 (a) significantly increased the IC50 for 5FU approximately 4-fold, while knockout of CLDN1 in SW620 (b) and DLD-1 (c) cells significantly decreased it by 2.5- and 6-fold, respectively. 3000 cells/well were seeded in triplicate for each cell line in a 96-well plate. After 24 hours, cells were treated with 5FU in concentrations ranging from 10−12 – 10−2 M and incubated for 72 hours. Cell viability was measured using Prestoblue. Data represents three biological replicates. R2 > 0.90. (d) CLDN1 KO also sensitized CRC tumors to 5FU in an in vivo xenograft mouse model. SW620c and CLDN1 KO cells were injected into the left and right flank, respectively, of nude mice (n=6-7 mice per group). Mice were treated with 10mg/kg 5FU IP two times per week for 15 days. 5FU treatment significantly inhibited tumor growth in CLDN1 KO cells while having no effect on control tumors. Mean and SEM for data points available in Supplementary Table 2. (e) IHC of tumor tissues confirmed CLDN1 KO and showed significantly decreased proliferation (Ki67) and significantly increased apoptosis (cleaved Caspase-3) in 5FU-treated CLDN1 KO tumors. Five random high-power fields (10x) were taken for three representative tumors from each group, and ImageJ/FIJI was used to detect and calculate the percentage of positive cells per field. Scale bar = 100 μM. C = Control, KO = Knockout. * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001.
To further verify the role of CLDN1 in chemoresistance, we conducted a rescue experiment. For this we reintroduced either full length CLDN1 or a PDZ-binding motif deleted mutant CLDN1 construct (CLDN1ΔPDZ, aa 208-211) (Figure 4A). The PDZ binding motif mediates a variety of protein-protein interactions in claudins [19]. As previously demonstrated, this deletion did not affect subcellular localization in our model (Sup. Figure 3) [48]. We found that while overexpression of full length CLDN1 increased IC50 of SW480 cells by 3.5 to 4 fold, deletion of the PDZ-binding motif abrogated CLDN1’s ability to increase IC50 and thus 5FU resistance in SW480 cells (Figure 4B). Most importantly, reintroduction of full-length CLDN1, but not CLDN1ΔPDZ, rescued resistance to 5FU in both SW620 and DLD-1 CLDN1 KO cells (Figure 4C, D).
Figure 4: Claudin-1 expression promotes chemoresistance.

(a) Full-length CLDN1 and a mutant construct with the C-terminal PDZ-binding motif deleted (CLDN1ΔPDZ, aa 208-211). (b) When full-length and mutant transcripts were overexpressed in SW480 cells, only full-length CLDN1 induced resistance to 5FU. (c,d) Resistance to 5-FU was restored in both SW620 and DLD CLDN1 KO cells by exogenous expression of full-length CLDN1 but not CLDN1ΔPDZ. 3000 cells/well were seeded in triplicate for each cell line in a 96-well plate. After 24 hours, cells were treated with 5FU in concentrations ranging from 10−12 – 10−2 M and incubated for 72 hours. Cell viability was measured using Prestoblue. Data represents at least three biological replicates. R2 > 0.90. TM = Transmembrane domain, ECD = Extracellular domain, ICD = Intracellular domain, C = Control, KO = Knockout. * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001.
CLDN1 activates the AKT pathway to modulate CD44 expression
Next, we wanted to understand the signaling pathways involved in CLDN1-mediated cancer stemness and chemoresistance. We used an R&D Systems Human Phospho-Kinase Array to analyze the activation of 37 different kinases in our SW480 and SW620 CLDN1-manipulated cell lines (Sup. Figure 4). We found a positive correlation between CLDN1 expression and the activation of AKT and its downstream targets (Figure 5A). p-AKT (T308), p-AKT (S473), and p-PRAS40 (T246) were increased approximately 4, 13, and 4-fold, respectively, with CLDN1 overexpression in SW480 and were decreased by approximately 2, 2, and 4-fold with CLDN1 KO in SW620 when compared to controls. Activation of AKT (p-AKT S473) was confirmed with Western blot in our SW480, SW620, and DLD-1 CLDN1-manipulated cell lines (Figure 5B). In agreement with the chemoresistance data in Figure 4, we found that the increase in p-AKT and CD44 was lost with the deletion of the CLDN1 PDZ-binding motif (Figure 5C). Further, pharmacological inhibition of AKT with LY294002 (20 μM) led to a decrease in CD44 expression in SW480cldn1 but not SW480c cells (Figure 5D). Together, these results suggest that CLDN1 activates the AKT survival pathway, which in turn enhances the expression of the CRC stemness marker CD44.
Figure 5. CLDN1 activates the AKT pathway to regulate CD44 expression.

a) R&D Systems Proteome Profiler Human Phospho-Kinase Array analysis of the AKT pathway in SW480cldn1 and SW620cldn1 KO vs. respective controls. (b) Confirmation of AKT activation (p-S473) in SW480, SW620, and DLD-1 CLDN1-manipulated cells with p-AKT densitometry. (c) Full-length CLDN1 but not the CLDN1ΔPDZ mutant increased both CD44 expression and AKT activation (p-S473). (d) Inhibition of AKT using PI3K inhibitor LY294002 significantly reduced CD44 expression in SW480 CLDN1 overexpressing cells. Cells were incubated overnight in serum-free medium and treated for 4 hours with 20 μM LY294002 followed by 30 minutes incubation with FBS. Data represents at least three biological replicates. C = Control, KO = Knockout. * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001.
CLDN1 physically associates with EPHA2 and regulates its expression
Although we found a connection between CLDN1 expression and AKT activation, it was still unclear how CLDN1 altered AKT signaling. Since CLDN1 is not enzymatically active, we hypothesized that it must be interacting with another protein to affect signaling. We used a panel of inhibitors towards ERK, EPHA2, SRC, and EGFR (all known regulators of the AKT pathway in CRC) and found that only EPHA2 and EGFR inhibition reduced p-AKT in SW480 CLDN1-expressing cells (Sup. Figure 5). We next used co-immunoprecipitation (co-IP) of CLDN1 in SW620 cells followed by mass spectrometry shotgun proteomics to identify interacting partners (data not shown). Among the interacting proteins, we found EPHA2, an upstream regulator of AKT identified in our inhibitor panel and a known promotor of cancer stemness and chemoresistance in CRC [28, 29]. This interaction was confirmed through co-IP in SW620, DLD-1, and SW480cldn1-HA cells (Figure 6A). Interestingly, the association with EPHA2 was not found in SW480 cells expressing CLDN1ΔPDZ-HA. We confirmed that the PDZ-binding motif is required for the CLDN1/EPHA2 interaction in HEK cells co-transfected with EPHA2-FLAG and either CLDN1-HA or CLDN1ΔPDZ-HA, suggesting that this finding is not specific to CRC cell lines or a result of differences in endogenous EPHA2 or CLDN1 expression (Sup. Figure 6A). To test this interaction directly, we recombinantly expressed and purified CLDN1 and three EPHA2 constructs: WT, EPHA2ΔN (extracellular and transmembrane domains deleted), and EPHA2ΔC (intracellular and transmembrane domains deleted). We then measured their ability to interact using microscale thermophoresis (MST). We found that CLDN1 binds with 150.04 nM affinity to EPHA2 WT and 125.31 nM affinity to EPHA2ΔN, while EPHA2ΔC bound to CLDN1 with 4-fold decreased affinity (581.7 nM) (Figure 6B). These results confirm that CLDN1 and EPHA2 can directly interact via their intracellular domains.
Figure 6. CLDN1 associates with EPHA2 and regulates its expression.

(a) EPHA2 co-immunoprecipitated with CLDN1 in all cell lines, and the association was dependent on the CLDN1 PDZ-binding motif. (b) Microscale thermophoresis confirmed a direct interaction between CLDN1 and EPHA2 intracellular domains. WT and intracellular region of EPHA2 (EPHA2ΔN) displayed ~4-fold higher binding affinity than EPHA2 extracellular region (EPHA2ΔC). (c) CLDN1 expression positively correlated with EPHA2, p-EPHA2 S897, and p-EPHA2 Y588 protein expression in SW480, SW620, and DLD-1 control and CLDN1-manipulated cell lines. (d) Full-length CLDN1 but not CLDN1ΔPDZ increased EPHA2 expression in SW480 cells. (e) Western blot analysis of nine patient-derived CRC organoids including one patient-matched primary and metastatic tumor pair showed a positive correlation between CLDN1, EPHA2, and CD44. (f) Cycloheximide (10 μM) inhibition of protein synthesis revealed a significant increase in EPHA2 stability in full-length CLDN1 but not CLDN1ΔPDZ expressing SW480 cells when compared to control. Stars denote significance of SW480cldn1 vs. SW480c, and hashmarks refer to SW480cldn1 vs. SW480cldn1ΔPDZ. (g) 12hr chloroquine (100 μM) treatment but not MG132 (10 μM) significantly increased EPHA2 protein in SW480c and SW480cldn1ΔPDZ but not SW480cldn1 cells. Data represents at least three biological replicates. C = Control, KO = Knockout, PT = Primary tumor, Met = metastasis, CHX = Cycloheximide, MG = MG132, CQ = Chloroquine */# = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001.
We next tested the status of EPHA2 expression and activation in our cell lines. Along with increased CD44 and AKT activation, CLDN1 expression was correlated with increased total EPHA2, p-EPHA2 Y588, and p-EPHA2 S897 expression (Figure 6C, Sup. Figure 6B). Additionally, total and phospho-EPHA2 expression increased with full-length but not CLDN1ΔPDZ (Figure 6D). Based on this data, we postulated that the expression of both total and phosphorylated EPHA2 was increased as a result of an interaction with CLDN1 via the CLDN1 PDZ-binding motif.
We further investigated this relationship in patient-derived organoids. Nine organoid lines were derived from CRC tumors of eight patients (Figure 6E). Two lines were developed from the primary and metastatic tumors of the same patient (denoted as P1 (PT) and P1 (Met) in Figure 6E). When we compared high CLDN1-expressing (P1-P4) with low CLDN1-expressing (P5-P8) organoids, we found a positive correlation with CD44 and both total and phospho-EPHA2 (S897 and Y588) (Sup. Figure 6C).
To test whether EPHA2 expression was increased on a transcriptional or post-transcriptional level, we measured mRNA expression using qRT-PCR and found that EPHA2 transcription was not significantly affected by CLDN1 expression (Sup. Figure 6D). Next, we tested EPHA2 protein stability by inhibiting protein synthesis with 10 μg/mL cycloheximide in SW480 control, CLDN1, and CLDN1ΔPDZ cell lines for multiple time points between 0 and 32 hours (Figure 6F). Here we found that the expression of full-length CLDN1 significantly delayed the degradation of EPHA2 for up to 24 hours when compared to either control or CLDN1ΔPDZ-expressing cells. We verified this finding by treating cells with MG132 (10 μM) or chloroquine (100 μM) for 12hrs to block the proteosomal and lysosomal degradation pathways, respectively. Chloroquine significantly increased EPHA2 expression in SW480c and CLDN1ΔPDZ cells but not in cells expressing full-length CLDN1, suggesting that EPHA2 is more rapidly degraded through the lysosomal pathway in these cell lines (Figure 6G).
Together, these findings suggest a direct association between CLDN1 and EPHA2 via their intracellular domains that inhibits EPHA2 degradation and increases its overall protein expression level.
EPHA2 regulates AKT in CLDN1-expressing CRC cells to promote cancer stemness and chemoresistance
While our initial inhibitor screen suggested that EPHA2 regulates AKT in our system, EPHA2 signaling is complex and context dependent [21]. EPHA2 can function as both an upstream regulator and downstream substrate of AKT [20, 24]. Upon activation of EPHA2, phosphorylated tyrosine residues can serve as docking sites for the p85 subunit PI3K to activate AKT signaling [24-27]. AKT can also phosphorylate EPHA2 at S897, which is known to promote invasiveness and migration in cancer [24]
To understand this relationship in our CRC models, we used both pharmacological and genetic approaches. The EPHA2-specific inhibitor ALW-II-41-27 has shown promise as a therapeutic agent and inhibits downstream AKT activation in both CRC and hepatocellular carcinoma [28, 49]. EPHA2 inhibition decreased both p-AKT and CD44 expression in our SW480 CLDN1-expressing cells (Figure 7A). We next used CRISPR cas9 to knockout EPHA2 in both SW480cldn1 and SW620 cells. Similar to the inhibitor, we observed a significant decrease in both p-AKT and CD44 expression (Figure 7B, Sup. Figure 6E). Next, we wanted to test the functional effects of EPHA2 knockout. We found that EPHA2 KO in SW480 CLDN1-expressing cells significantly decreased tumor-initiating potential in an in vitro ELDA, and resistance to 5FU was significantly decreased based on in vitro IC50 measurements (Figure 7C & D, Sup. Table 3). Finally, we reanalyzed the CRC patient dataset of responders and non-responders to FOLFOX and found that increased EPHA2 expression was, like CLDN1, correlated with chemoresistance. (Figure 7E). This concordance is further evidence of a functional relationship between these two proteins in CRC therapy resistance. Together, these findings establish EPHA2 as a CLDN1-interacting protein that regulates cancer stemness to promote a tumorigenic and chemoresistant phenotype in CLDN1-expressing CRC.
Figure 7. EPHA2 regulates AKT in CLDN1-expressing CRC cells to promote CD44 expression, tumor-initiating potential, and chemoresistance.

(a) Inhibition of EPHA2 with ALW-II-41-27 reduced AKT activation (p-S473) in both SW480 control and CLDN1 overexpressing cells and significantly decreased CD44 only in SW480cldn1 cells. Cells were incubated overnight in serum-free medium and treated for 4 hours with 1 μM inhibitor followed by 30 minutes incubation with FBS. (b) EPHA2 KO in SW480 CLDN1-expressing cells reduced AKT activation (p-S473) and CD44 expression. (c, d) EPHA2 KO in SW480cldn1 cells reduced both tumor-initiating potential and resistance to 5FU. ELDA data available in Sup. Table 3. (e) EPHA2 expression was positively correlated with chemoresistance in CRC patient datasets (GSE28702 and GSE72970) categorized into responders (n=51) and non-responders (n=37) to FOLFOX. C = Control, KO = Knockout, EPH = EPHA2. * = p<0.05, ** = p<0.01, *** = p<0.001.
DISCUSSION
These results support our initial hypothesis and detail a novel mechanism through which CLDN1 directly interacts with EPHA2 to enhance AKT signaling and promote properties of cancer stemness including increased CD44 expression, tumor-initiating potential, and chemoresistance. Claudins are known to modulate oncogenic pathways, but the biggest gap in our understanding has been the physical mechanism mediating these effects. To our knowledge, this is the first study to establish a direct mechanism through which a claudin regulates a receptor tyrosine kinase to promote cancer stemness and chemoresistance in CRC.
In previous studies, CLDN1 expression increases in CRC with exposure to both 5FU and oxaliplatin, and subsequent CLDN1 knockdown restores sensitivity, indicating a role in acquired resistance [50, 51]. CLDN1 is also associated with therapy resistance in other cancers. In breast cancer, scRNA-seq identified a subpopulation of CD44highCLDN1high cells that exhibited high phenotypic plasticity and were intrinsically resistant to endocrine therapy [52]. In ovarian cancer, CLDN1 expression was correlated with chemoresistance (carboplatin and paclitaxel) and cancer stem cell properties (CD44/CD133 expression and sphere-forming ability), as well as shorter overall survival in recurrent, chemoresistant tumors [15]. In non-small cell lung cancer, CLDN1 expression promoted resistance to both cisplatin and doxorubicin [16, 53]. In liver cancer, 5FU-resistant cell lines showed increased CLDN1 expression, and CLDN1 silencing re-sensitized cells to 5FU [17]. Here, we have shown that high CLDN1 is associated with cancer stemness and FOLFOX resistance in patient datasets. We then established that CLDN1 promotes properties of cancer stemness including increased CD44 expression, tumor-initiating potential, ALDH activity, side population, and resistance to 5FU both in vitro and in vivo. We demonstrated that CLDN1 expression induced AKT activation, a known driver of multidrug resistance and cancer stemness [54, 55]. In agreement with our findings, other studies have found that AKT activation specifically promotes CD44 expression in breast, cervical, and colorectal cancer cells [56, 57]. Together, these results suggest that CLDN1 expression plays a causal role in intrinsic chemoresistance and increased tumor-initiating potential in CRC through AKT activation.
Our lab has previously demonstrated CLDN1-mediated AKT activation in both CRC cell and mouse models; however, the connection between CLDN1 and AKT activation was unclear [58, 59]. Our results show that CLDN1 directly interacts with EPHA2 via its PDZ-binding motif and increases EPHA2 protein-level expression by inhibiting its degradation. A previous study suggested that CLDN4 and EPHA2 interact via their extracellular domains [60]. Our results in CRC and HEK cells along with in vitro biophysical binding assays clearly show that CLDN1 and EPHA2 interact via their intracellular domains. The increase in EPHA2 as a result of this interaction enhances AKT activation, thus bridging the gap between increased CLDN1 expression and oncogenic signaling. It was previously shown that ligand-mediated EPHA2 signaling negatively regulates AKT activation and migration in glioblastoma and prostate cancer cells [24]. While ligand-mediated signaling tends to suppress tumor growth, high expression of EPHA2 results in oncogenic ligand-independent activation and subsequent PI3K/AKT activation [20, 61]. In our system, both pharmacological and genetic EPHA2 targeting decreased p-AKT, indicating that EPHA2 is operating as an upstream activator of AKT signaling. This agrees with previous findings in both CRC and HCC [28, 49]. It is currently unclear exactly how the CLDN1/EPHA2 interaction inhibits EPHA2 degradation. Interactions with membrane-associated proteins can inhibit EGFR and ERBB2 internalization and degradation, and CLDN1 may be playing a similar role with EPHA2 [62, 63]. Interestingly, the intracellular region of EPHA2 contains a SAM domain that interacts with protein regulators of receptor stability including phosphatases [64]. Whether these regulators are involved in CLDN1-mediated EphA2 stabilization remains to be determined. Future studies will address this question.
Both CLDN1 and EPHA2 are being developed as therapy targets. Claudins are recognized as promising target molecules because they are present on the cell surface, exhibit tissue-specific expression, and can selectively increase during carcinogenesis [12]. Our lab has designed a first-generation CLDN1 inhibitor that decreases AKT signaling and shows promise in CRC mouse models [39]. Identifying the PDZ-binding motif as necessary for CLDN1’s oncogenic effects will guide further inhibitor development. Anti-CLDN1 monoclonal antibodies are being investigated as therapies for both HCC and CRC [11, 51, 65]. Mislocalized, non-junctional claudins present in cancer cells display extracellular epitopes that are masked in intact junctional complexes, preventing off-target binding in healthy tissue [66, 67]. In HCC cells, an anti-CLDN1 mAb modulated the CLDN1 interactome, including a 3-fold decrease in CLDN1/EPHA2 interaction, and a significant decrease in AKT activation [67]. EPHA2 has shown promise as a therapeutic target in multiple cancers [68]. In CRC, high EPHA2 expression promotes invasion and is correlated with poor prognosis [29]. EPHA2 overexpression has been linked to acquired resistance to EGFR inhibitors in CRC, and the EPHA2 inhibitor ALW-II-41-27 restored sensitivity to cetuximab in vivo [28, 69]. Our results confirm that EPHA2 overexpression in CRC enhances tumor-initiating potential and resistance to 5FU. Future studies will detail the impact of these CLDN1 and EPHA2-based therapies on the CLDN1/EPHA2 interaction.
Finally, our analysis of patient datasets suggests that high expression of both CLDN1 and EPHA2 correlate with resistance to FOLFOX, suggesting a possible biomarker for therapy selection. Current treatment guidelines for metastatic CRC recommend initial three-drug combination therapy with 5FU/leucovorin (LV), oxaliplatin, and irinotecan; however, for patients who cannot tolerate the increased toxicity of this treatment, a two-drug combination of 5FU/LV with either oxaliplatin (FOLFOX) or irinotecan (FOLFIRI) is used [70]. The choice of therapy is generally driven by their different side-effect profiles, with FOLFOX being more popular in the United States [71]. With no accepted rationale for choosing initial therapy, the overall response rate for either two-drug combination is low (FOLFOX 53%, FOLFIRI 39%), and new biomarkers are needed to prescribe the most effective therapeutic approach for each patient [71]. A recent study found a correlation between acquired oxaliplatin resistance and high CLDN1 expression in CRC [51]. In that study, an anti-CLDN1 antibody drug conjugate significantly potentiated the effectiveness of oxaliplatin in vivo, suggesting a therapeutic strategy for overcoming oxaliplatin resistance. Future studies will investigate the role of CLDN1 expression in resistance to other treatment regimens as well as strategies for overcoming chemoresistance using anti-CLDN1 based therapy.
CONCLUSION
Our study presents a novel CLDN1-EPHA2-AKT-CD44 pathway that promotes cancer stemness and chemoresistance in CRC. In this pathway, CLDN1 interacts directly with EPHA2 via its C-terminal PDZ-binding motif to increase EPHA2 expression and promote downstream AKT signaling and CD44 expression. This interaction offers new opportunities for targeted therapy and may provide a possible biomarker for therapy selection in CRC.
Supplementary Material
Acknowledgement:
This study was supported by BX002086 & CX002228 (VA merit), CA250383 (NIH/NCI) to PD, T32CA009476 (NIH/NCI) to MP, CA22287 (NIH/NCI) to KWF, and DK124095 & BX002761 (VA merit) to ABS.
Footnotes
Conflict of Interest: None to declare.
References
- [1].Society AC, Cancer Facts & Figures 2022, American Cancer Society, Atlanta, GA, 2022. [Google Scholar]
- [2].Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, Starling N, Colorectal cancer, Lancet, 375 (2010) 1030–1047. [DOI] [PubMed] [Google Scholar]
- [3].Hu T, Li Z, Gao C-Y, Cho CH, Mechanisms of drug resistance in colon cancer and its therapeutic strategies, World journal of gastroenterology, 22 (2016) 6876–6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Xie Y-H, Chen Y-X, Fang J-Y, Comprehensive review of targeted therapy for colorectal cancer, Signal Transduction and Targeted Therapy, 5 (2020) 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Günzel D, Yu ASL, Claudins and the Modulation of Tight Junction Permeability, Physiological Reviews, 93 (2013) 525–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dhawan P, Singh AB, Deane NG, No Y, Shiou SR, Schmidt C, Neff J, Washington MK, Beauchamp RD, Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer, J Clin Invest, 115 (2005) 1765–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pope JL, Bhat AA, Sharma A, Ahmad R, Krishnan M, Washington MK, Beauchamp RD, Singh AB, Dhawan P, Claudin-1 regulates intestinal epithelial homeostasis through the modulation of Notch-signalling, Gut, 63 (2014) 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gowrikumar S, Ahmad R, Uppada SB, Washington MK, Shi C, Singh AB, Dhawan P, Upregulated claudin-1 expression promotes colitis-associated cancer by promoting β-catenin phosphorylation and activation in Notch/p-AKT-dependent manner, Oncogene, 38 (2019) 5321–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Li J, Context-Dependent Roles of Claudins in Tumorigenesis, Frontiers in oncology, 11 (2021) 676781–676781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Singh AB, Sharma A, Dhawan P, Claudin family of proteins and cancer: an overview, J Oncol, 2010 (2010) 541957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Roehlen N, Muller M, Nehme Z, Crouchet E, Jühling F, Del Zompo F, Cherradi S, Duong FHT, Almeida N, Saviano A, Fernández-Vaquero M, Riedl T, El Saghire H, Durand SC, Ponsolles C, Oudot MA, Martin R, Brignon N, Felli E, Pessaux P, Lallement A, Davidson I, Bandiera S, Thumann C, Marchand P, Moll S, Nicolay B, Bardeesy N, Hoshida Y, Heikenwälder M, Iacone R, Toso A, Meyer M, Elson G, Schweighoffer T, Teixeira G, Zeisel MB, Laquerriere P, Lupberger J, Schuster C, Mailly L, Baumert TF, Treatment of HCC with Claudin-1 specific antibodies suppresses carcinogenic signaling and reprograms the tumor microenvironment, J Hepatol, (2022). [DOI] [PubMed] [Google Scholar]
- [12].Li J, Targeting claudins in cancer: diagnosis, prognosis and therapy, Am J Cancer Res, 11 (2021) 3406–3424. [PMC free article] [PubMed] [Google Scholar]
- [13].Singh AB, Sharma A, Dhawan P, Claudin-1 expression confers resistance to anoikis in colon cancer cells in a Src-dependent manner, Carcinogenesis, 33 (2012) 2538–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bhat AA, Ahmad R, Uppada SB, Singh AB, Dhawan P, Claudin-1 promotes TNF-alpha-induced epithelial-mesenchymal transition and migration in colorectal adenocarcinoma cells, Exp Cell Res, 349 (2016) 119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Visco ZR, Sfakianos G, Grenier C, Boudreau M-H, Simpson S, Rodriguez I, Whitaker R, Yao DY, Berchuck A, Murphy SK, Huang Z, Epigenetic Regulation of Claudin-1 in the Development of Ovarian Cancer Recurrence and Drug Resistance, Frontiers in Oncology, 11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhao Z, Li J, Jiang Y, Xu W, Li X, Jing W, CLDN1 Increases Drug Resistance of Non-Small Cell Lung Cancer by Activating Autophagy via Up-Regulation of ULK1 Phosphorylation, Med Sci Monit, 23 (2017) 2906–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tong H, Li T, Qiu W, Zhu Z, Claudin-1 silencing increases sensitivity of liver cancer HepG2 cells to 5-fluorouracil by inhibiting autophagy, Oncol Lett, 18 (2019) 5709–5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang Y, Yeh S, Appleton BA, Held HA, Kausalya PJ, Phua DCY, Lee Wong W, Lasky LA, Wiesmann C, Hunziker W, Sidhu SS, Convergent and Divergent Ligand Specificity among PDZ Domains of the LAP and Zonula Occludens (ZO) Families *, Journal of Biological Chemistry, 281 (2006) 22299–22311. [DOI] [PubMed] [Google Scholar]
- [19].Suarez-Artiles L, Breiderhoff T, Girardello R, Gonschior H, Rodius S, Lesur A, Reimer U, Ramberger E, Perez-Hernandez D, Müller D, Mertins P, Dittmar G, Pan-claudin family interactome analysis reveals shared and specific interactions, Cell Reports, 41 (2022). [DOI] [PubMed] [Google Scholar]
- [20].Liang L-Y, Patel O, Janes PW, Murphy JM, Lucet IS, Eph receptor signalling: from catalytic to non-catalytic functions, Oncogene, 38 (2019) 6567–6584. [DOI] [PubMed] [Google Scholar]
- [21].Pasquale EB, Eph receptors and ephrins in cancer: bidirectional signalling and beyond, Nature Reviews Cancer, 10 (2010) 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Singh DR, Kanvinde P, King C, Pasquale EB, Hristova K, The EphA2 receptor is activated through induction of distinct, ligand-dependent oligomeric structures, Communications Biology, 1 (2018) 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zapata-Mercado E, Biener G, McKenzie DM, Wimley WC, Pasquale EB, Raicu V, Hristova K, The efficacy of receptor tyrosine kinase EphA2 autophosphorylation increases with EphA2 oligomer size, Journal of Biological Chemistry, 298 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, Sloan AE, Cohen ML, Wang B, EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt, Cancer Cell, 16 (2009) 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fang WB, Brantley-Sieders DM, Hwang Y, Ham AJ, Chen J, Identification and functional analysis of phosphorylated tyrosine residues within EphA2 receptor tyrosine kinase, J Biol Chem, 283 (2008) 16017–16026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Subbarayal P, Karunakaran K, Winkler AC, Rother M, Gonzalez E, Meyer TF, Rudel T, EphrinA2 receptor (EphA2) is an invasion and intracellular signaling receptor for Chlamydia trachomatis, PLoS Pathog, 11 (2015) e1004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pandey A, Lazar DF, Saltiel AR, Dixit VM, Activation of the Eck receptor protein tyrosine kinase stimulates phosphatidylinositol 3-kinase activity, J Biol Chem, 269 (1994) 30154–30157. [PubMed] [Google Scholar]
- [28].Martini G, Cardone C, Vitiello PP, Belli V, Napolitano S, Troiani T, Ciardiello D, Della Corte CM, Morgillo F, Matrone N, Sforza V, Papaccio G, Desiderio V, Paul MC, Moreno-Viedma V, Normanno N, Rachiglio AM, Tirino V, Maiello E, Latiano TP, Rizzi D, Signoriello G, Sibilia M, Ciardiello F, Martinelli E, EPHA2 Is a Predictive Biomarker of Resistance and a Potential Therapeutic Target for Improving Antiepidermal Growth Factor Receptor Therapy in Colorectal Cancer, Mol Cancer Ther, 18 (2019) 845–855. [DOI] [PubMed] [Google Scholar]
- [29].Dunne PD, Dasgupta S, Blayney JK, McArt DG, Redmond KL, Weir JA, Bradley CA, Sasazuki T, Shirasawa S, Wang T, Srivastava S, Ong CW, Arthur K, Salto-Tellez M, Wilson RH, Johnston PG, Van Schaeybroeck S, EphA2 Expression Is a Key Driver of Migration and Invasion and a Poor Prognostic Marker in Colorectal Cancer, Clin Cancer Res, 22 (2016) 230–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen C, Zhao S, Karnad A, Freeman JW, The biology and role of CD44 in cancer progression: therapeutic implications, J Hematol Oncol, 11 (2018) 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Primeaux M, Gowrikumar S, Dhawan P, Role of CD44 isoforms in epithelial-mesenchymal plasticity and metastasis, Clin Exp Metastasis, 39 (2022) 391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Agro L, O'Brien C, In vitro and in vivo Limiting Dilution Assay for Colorectal Cancer, Bio-protocol, 5 (2015) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hu Y, Smyth GK, ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays, J Immunol Methods, 347 (2009) 70–78. [DOI] [PubMed] [Google Scholar]
- [34].Chandrashekar DS, Karthikeyan SK, Korla PK, Patel H, Shovon AR, Athar M, Netto GJ, Qin ZS, Kumar S, Manne U, Creighton CJ, Varambally S, UALCAN: An update to the integrated cancer data analysis platform, Neoplasia, 25 (2022) 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N, The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data, Cancer Discov, 2 (2012) 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N, Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal, Sci Signal, 6 (2013) pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, Jeon M, Ma'ayan A, Gene Set Knowledge Discovery with Enrichr, Curr Protoc, 1 (2021) e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles, Proc Natl Acad Sci U S A, 102 (2005) 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Fatima I, Uppada JP, Chhonker YS, Gowrikumar S, Barman S, Roy S, Tolentino KT, Palermo N, Natarajan A, Beauchamp DR, Vecchio A, Murry DJ, Singh AB, Hopkins CR, Dhawan P, Identification and characterization of a first-generation inhibitor of claudin-1 in colon cancer progression and metastasis, Biomed Pharmacother, 159 (2023) 114255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chen L, Tian B, Liu W, Liang H, You Y, Molecular Biomarker of Drug Resistance Developed From Patient-Derived Organoids Predicts Survival of Colorectal Cancer Patients, Front Oncol, 12 (2022) 855674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ghosh D, Yu H, Tan XF, Lim TK, Zubaidah RM, Tan HT, Chung MCM, Lin Q, Identification of Key Players for Colorectal Cancer Metastasis by iTRAQ Quantitative Proteomics Profiling of Isogenic SW480 and SW620 Cell Lines, Journal of Proteome Research, 10 (2011) 4373–4387. [DOI] [PubMed] [Google Scholar]
- [42].Wang Z, Tang Y, Xie L, Huang A, Xue C, Gu Z, Wang K, Zong S, The Prognostic and Clinical Value of CD44 in Colorectal Cancer: A Meta-Analysis, Front Oncol, 9 (2019) 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chen C, Zhao S, Karnad A, Freeman JW, The biology and role of CD44 in cancer progression: therapeutic implications, Journal of Hematology & Oncology, 11 (2018) 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Moreb JS, Ucar D, Han S, Amory JK, Goldstein AS, Ostmark B, Chang LJ, The enzymatic activity of human aldehyde dehydrogenases 1A2 and 2 (ALDH1A2 and ALDH2) is detected by Aldefluor, inhibited by diethylaminobenzaldehyde and has significant effects on cell proliferation and drug resistance, Chem Biol Interact, 195 (2012) 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhou L, Sheng D, Wang D, Ma W, Deng Q, Deng L, Liu S, Identification of cancer-type specific expression patterns for active aldehyde dehydrogenase (ALDH) isoforms in ALDEFLUOR assay, Cell Biol Toxicol, 35 (2019) 161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA, Brenner MK, A distinct “side population” of cells with high drug efflux capacity in human tumor cells, Proceedings of the National Academy of Sciences, 101 (2004) 14228–14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Shimoda M, Ota M, Okada Y, Isolation of Cancer Stem Cells by Side Population Method, Methods Mol Biol, 1692 (2018) 49–59. [DOI] [PubMed] [Google Scholar]
- [48].Rüffer C, Gerke V, The C-terminal cytoplasmic tail of claudins 1 and 5 but not its PDZ-binding motif is required for apical localization at epithelial and endothelial tight junctions, Eur J Cell Biol, 83 (2004) 135–144. [DOI] [PubMed] [Google Scholar]
- [49].Wang H, Hou W, Perera A, Bettler C, Beach JR, Ding X, Li J, Denning MF, Dhanarajan A, Cotler SJ, Joyce C, Yin J, Ahmed F, Roberts LR, Qiu W, Targeting EphA2 suppresses hepatocellular carcinoma initiation and progression by dual inhibition of JAK1/STAT3 and AKT signaling, Cell Rep, 34 (2021) 108765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gowrikumar S, Primeaux M, Pravoverov K, Wu C, Szeglin BC, Sauve CG, Thapa I, Bastola D, Chen XS, Smith JJ, Singh AB, Dhawan P, A Claudin-Based Molecular Signature Identifies High-Risk, Chemoresistant Colorectal Cancer Patients, Cells, 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cherradi S, Garambois V, Marines J, Andrade AF, Fauvre A, Morand O, Fargal M, Mancouri F, Ayrolles-Torro A, Vezzo-Vié N, Jarlier M, Loussaint G, Huvelle S, Joubert N, Mazard T, Gongora C, Pourquier P, Boissière-Michot F, Rio MD, Improving the response to oxaliplatin by targeting chemotherapy-induced CLDN1 in resistant metastatic colorectal cancer cells, Cell & Bioscience, 13 (2023) 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hong SP, Chan TE, Lombardo Y, Corleone G, Rotmensz N, Bravaccini S, Rocca A, Pruneri G, McEwen KR, Coombes RC, Barozzi I, Magnani L, Single-cell transcriptomics reveals multi-step adaptations to endocrine therapy, Nature Communications, 10 (2019) 3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Akizuki R, Maruhashi R, Eguchi H, Kitabatake K, Tsukimoto M, Furuta T, Matsunaga T, Endo S, Ikari A, Decrease in paracellular permeability and chemosensitivity to doxorubicin by claudin-1 in spheroid culture models of human lung adenocarcinoma A549 cells, Biochim Biophys Acta Mol Cell Res, 1865 (2018) 769–780. [DOI] [PubMed] [Google Scholar]
- [54].Liu R, Chen Y, Liu G, Li C, Song Y, Cao Z, Li W, Hu J, Lu C, Liu Y, PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers, Cell death & disease, 11 (2020) 797–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Xia P, Xu XY, PI3K/Akt/mTOR signaling pathway in cancer stem cells: from basic research to clinical application, Am J Cancer Res, 5 (2015) 1602–1609. [PMC free article] [PubMed] [Google Scholar]
- [56].Li J, Zhou BP, Activation of β-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters, BMC Cancer, 11 (2011) 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Niu T, Zhu J, Dong L, Yuan P, Zhang L, Liu D, Inorganic pyrophosphatase 1 activates the phosphatidylinositol 3-kinase/Akt signaling to promote tumorigenicity and stemness properties in colorectal cancer, Cellular Signalling, 108 (2023) 110693. [DOI] [PubMed] [Google Scholar]
- [58].Gowrikumar S, Ahmad R, Uppada SB, Washington MK, Shi C, Singh AB, Dhawan P, Upregulated claudin-1 expression promotes colitis-associated cancer by promoting beta-catenin phosphorylation and activation in Notch/p-AKT-dependent manner, Oncogene, 38 (2019) 5321–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Singh AB, Sharma A, Smith JJ, Krishnan M, Chen X, Eschrich S, Washington MK, Yeatman TJ, Beauchamp RD, Dhawan P, Claudin-1 up-regulates the repressor ZEB-1 to inhibit E-cadherin expression in colon cancer cells, Gastroenterology, 141 (2011) 2140–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tanaka M, Kamata R, Sakai R, EphA2 phosphorylates the cytoplasmic tail of Claudin-4 and mediates paracellular permeability, J Biol Chem, 280 (2005) 42375–42382. [DOI] [PubMed] [Google Scholar]
- [61].Fang WB, Brantley-Sieders DM, Hwang Y, Ham A-JL, Chen J, Identification and functional analysis of phosphorylated tyrosine residues within EphA2 receptor tyrosine kinase, The Journal of biological chemistry, 283 (2008) 16017–16026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Borg J-P, Marchetto S, Le Bivic A, Ollendorff V, Jaulin-Bastard F, Saito H, Fournier E, Adélaïde J, Margolis B, Birnbaum D, ERBIN: a basolateral PDZ protein that interacts with the mammalian ERBB2/HER2 receptor, Nature Cell Biology, 2 (2000) 407–414. [DOI] [PubMed] [Google Scholar]
- [63].Lazar CS, Cresson CM, Lauffenburger DA, Gill GN, The Na+/H+ Exchanger Regulatory Factor Stabilizes Epidermal Growth Factor Receptors at the Cell Surface, Molecular Biology of the Cell, 15 (2004) 5470–5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Mercurio FA, Vincenzi M, Leone M, Hunting for Novel Routes in Anticancer Drug Discovery: Peptides against Sam-Sam Interactions, Int J Mol Sci, 23 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Cherradi S, Ayrolles-Torro A, Vezzo-Vié N, Gueguinou N, Denis V, Combes E, Boissière F, Busson M, Canterel-Thouennon L, Mollevi C, Pugnière M, Bibeau F, Ychou M, Martineau P, Gongora C, Del Rio M, Antibody targeting of claudin-1 as a potential colorectal cancer therapy, J Exp Clin Cancer Res, 36 (2017) 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Eichner M, Augustin C, Fromm A, Piontek A, Walther W, Bücker R, Fromm M, Krause G, Schulzke J-D, Günzel D, Piontek J, In Colon Epithelia, Clostridium perfringens Enterotoxin Causes Focal Leaks by Targeting Claudins Which are Apically Accessible Due to Tight Junction Derangement, The Journal of Infectious Diseases, 217 (2018) 147–157. [DOI] [PubMed] [Google Scholar]
- [67].Roehlen N, Saviano A, El Saghire H, Crouchet E, Nehme Z, Del Zompo F, Jühling F, Oudot MA, Durand SC, Duong FHT, Cherradi S, Gonzalez Motos V, Almeida N, Ponsolles C, Heydmann L, Ostyn T, Lallement A, Pessaux P, Felli E, Cavalli A, Sgrignani J, Thumann C, Koutsopoulos O, Fuchs BC, Hoshida Y, Hofmann M, Vyberg M, Viuff BM, Galsgaard ED, Elson G, Toso A, Meyer M, Iacone R, Schweighoffer T, Teixeira G, Moll S, De Vito C, Roskams T, Davidson I, Heide D, Heikenwälder M, Zeisel MB, Lupberger J, Mailly L, Schuster C, Baumert TF, A monoclonal antibody targeting nonjunctional claudin-1 inhibits fibrosis in patient-derived models by modulating cell plasticity, Sci Transl Med, 14 (2022) eabj4221. [DOI] [PubMed] [Google Scholar]
- [68].Xiao T, Xiao Y, Wang W, Tang YY, Xiao Z, Su M, Targeting EphA2 in cancer, Journal of Hematology & Oncology, 13 (2020) 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Torlot L, Jarzab A, Albert J, Pók-Udvari Á, Stahler A, Holch JW, Gerlinger M, Heinemann V, Klauschen F, Kirchner T, Kumbrink J, Küster B, Jung A, Proteomics uncover EPHA2 as a potential novel therapeutic target in colorectal cancer cell lines with acquired cetuximab resistance, Journal of Cancer Research and Clinical Oncology, 149 (2023) 669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Clark JS, Hanna K Systemic Therapy for Nonoperable Metastatic Colorectal Cancer: Selecting the Initial Therapeutic Approach, in: Goldberg RM (Ed.) UpToDate, UpToDate, Waltham, MA. (Accessed on May 3, 2023). [Google Scholar]
- [71].Neugut AI, Lin A, Raab GT, Hillyer GC, Keller D, O'Neil DS, Accordino MK, Kiran RP, Wright J, Hershman DL, FOLFOX and FOLFIRI Use in Stage IV Colon Cancer: Analysis of SEER-Medicare Data, Clin Colorectal Cancer, 18 (2019) 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.