

ABSTRACT
The development of specific, safe, and potent monoclonal antibodies (Abs) has led to novel therapeutic options for infectious disease. In addition to preventing viral infection through neutralization, Abs can clear infected cells and induce immunomodulatory functions through engagement of their crystallizable fragment (Fc) with complement proteins and Fc receptors on immune cells. Little is known about the role of Fc effector functions of neutralizing Abs in the context of encephalitic alphavirus infection. To determine the role of Fc effector function in therapeutic efficacy against Venezuelan equine encephalitis virus (VEEV), we compared the potently neutralizing anti-VEEV human IgG F5 (hF5) Ab with intact Fc function (hF5-WT) or containing the loss of function Fc mutations L234A and L235A (hF5-LALA) in the context of VEEV infection. We observed significantly reduced binding to complement and Fc receptors, as well as differential in vitro kinetics of Fc-mediated cytotoxicity for hF5-LALA compared to hF5-WT. The in vivo efficacy of hF5-LALA was comparable to hF5-WT at −24 and + 24 h post infection, with both Abs providing high levels of protection. However, when hF5-WT and hF5-LALA were administered + 48 h post infection, there was a significant decrease in the therapeutic efficacy of hF5-LALA. Together these results demonstrate that optimal therapeutic Ab treatment of VEEV, and possibly other encephalitic alphaviruses, requires neutralization paired with engagement of immune effectors via the Fc region.
KEYWORDS: Antibody dependent cell-mediated cytotoxicity (ADCC), antibody therapy, complement, complement dependent cytotoxicity (CDC), Fc effector function, Fc-engineering, neutralizing antibody, Venezuelan equine encephalitis virus (VEEV)
Introduction
The stochastic outbreaks of emerging pathogens such as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) provide a visceral sense of the urgency and importance of developing therapeutics that not only prevent virus infection, but also optimally engage the body’s immune defenses to clear infected cells.1,2 Alphaviruses are members of the Togaviridae family and represent a class of emerging pathogens that can manifest in two clinical ‘flavors’: arthritogenic (e.g., chikungunya, CHIKV; Mayaro, MAYV; Ross River virus, RRV) and encephalitic (Venezuelan equine encephalitis virus, VEEV; Eastern equine encephalitis virus; Western equine encephalitis virus). Of particular concern as a potential bioweapon is VEEV, a category B select agent that can induce febrile illness, body aches, and inflammation of the central nervous system, leading to severe neurological symptoms or even death in equines and humans.3 In addition, VEEV can be easily produced in large volumes and can spread by aerosol or transdermal inoculation (e.g., a mosquito bite).
To mediate cell entry and initiate infection, alphaviruses have two transmembrane glycoproteins E1 and E2 that form a heterodimer subunit arranged into 80 trimeric spikes on the virion. The main target of virus neutralizing antibodies (Abs) is the trimer exposed on the surface of the virion, with domain A and B of E2 as the dominant targets of neutralizing Abs.4–8 However, in infected cells the E1 and E2 proteins form a heterodimer on the surface of the cell in preparation for budding and several recent studies have identified multiple potent, pan-protective anti-alphavirus monoclonal Abs that bind the E1 protein on the surface of infected cells and prevent virion budding.5,9,10 Although several promising neutralizing and non-neutralizing monoclonal Abs have been identified, there is no approved vaccine or therapeutic to combat encephalitic alphaviruses.5,9–18
Ab-based protection against viral infections can be achieved by direct binding of Abs to viral proteins blocking virus entry (neutralization), blocking virion budding, and/or by effector functions conducted via the crystallizable fragment (Fc) of the Ab. Effector functions are mediated by interaction of the Fc with complement or Fc gamma receptors (FcγRs) expressed on immune cells, facilitating clearance of infected cells, recruitment of immune cells to reservoirs of infection, as well as other immunomodulatory functions. The three main Fc-mediated effector functions are Ab-dependent cell-mediated cytotoxicity (ADCC), Ab-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC). ADCC and ADCP occur when the Fc domain of Abs bound to viral proteins on infected cells engage with FcγRs on immune cells and either induce targeted cell death through the release of cytotoxic granules or promote the uptake of the infected cell or immune complex by phagocytosis. The Fc domain of Abs bound directly to pathogen or infected cells can also elicit CDC through activation of the complement cascade, resulting in direct, localized lysis of infected cells and recruitment of immune effector cells through their complement receptors.19,20 It was previously thought that prevention of virus cell entry through neutralization was the primary mechanism for Ab efficacy against viral infections, but recent studies are starting to demonstrate the critical role for Fc-mediated effector functions and virion egress inhibition as distinct modes of Ab-mediated protection against viral infections. For example, abrogation of Fc effector function leads to reduced serum half-life, affects bioavailability, and has been shown to diminish therapeutic efficacy in a variety of disease contexts including SARS-CoV-2, Mayaro virus, human immunodeficiency virus (HIV), Ebola virus, and Influenza.19,21–31 Even with these new findings there has been some reluctance to use Fc effector function-competent Abs as therapeutics due to the potential for FcγR-mediated antibody-dependent enhancement of infection, whereby viruses take advantage of immune complexes to enter and infect immune cells, which is a particular concern for some flaviviruses. In addition to antibody-dependent enhancement of infection, the inflammatory response activated by immune complex interactions with FcγRs may contribute to disease, so a balanced immune response is necessary for adequate protection and is critical to avoid detrimental, excessive activation.19,28,32 Neurotropic viruses, such as VEEV, pose a unique challenge in that the central nervous system is an immune-privileged site, where a measured immune response to reduce long-term sequela is vital, but an overly robust immune response may result in irreparable damage.33 Therefore, it is necessary to understand how anti-viral Abs interact with the host immune system, to ensure clearance of the viral pathogen while mitigating Ab mediated tissue damage.
Numerous studies have demonstrated the ability to engineer Abs with Fc effector gain and loss of function mutations or post-translational modifications that retain antigen-specificity.19,34–38 However, the requirement for Fc effector function may be influenced by the disease state at which efficacy is evaluated, the infection route, reservoir, and kinetics that may change across (or within) viral families. Therefore, detailed work must be done to address the prophylactic and therapeutic value of Fc effector function within the context of distinct infections. For alphaviruses, previous studies have demonstrated the value of Fc effector function for optimal protection of Abs against joint swelling caused by arthritogenic alphaviruses.5,25,26,32 A study from 2012 identified a broadly protective non-neutralizing E1 Ab against VEEV-TrD, but the role of Fc effector function was not addressed experimentally10. More recently, a pan-alphavirus E1-binding Ab required Fc effector function for prophylactic efficacy against highly pathogenic VEEV ZPC738.5 Additionally, a neutralizing and non-neutralizing Abs binding WEEV have exhibited prophylactic efficacy against lethal aerosol challenge with the pathogenic WEEV-Fleming strain, suggesting that Fc function may contribute to protection; however, Fc effector studies with these Abs have not been described17,18. In this study we demonstrate the importance of Fc effector function for E2 binding, potently neutralizing Abs in the treatment of established encephalitic alphavirus infection.
We characterize the role of Fc effector function for an E2 binding, VEEV-neutralizing human IgG1 Ab F5 (hF5),12,13,39 by evaluating wildtype hF5 Abs with intact Fc function (hF5-WT) or containing the loss of function Fc mutations L234A and L235A (hF5-LALA) in the context of VEEV infection. First, we confirmed that our WT and LALA hF5 Abs exhibit comparable trimer binding and neutralization capacity, to ensure alterations to the Fc did not affect potency. We next evaluated the ability of hF5-WT and hF5-LALA to productively engage human and murine immune response elements in vitro and assessed in vivo efficacy. Our studies reveal from a series of pre- and post-exposure dosing studies in vivo that hF5-WT and hF5-LALA can provide protection against lethal infection when delivered before and shortly after infection. However, at +48 h post infection (hpi), the hF5-LALA group exhibited a significant reduction in survival compared to the hF5-WT group, demonstrating that adequate protection and clearance requires intact Fc effector function in the context of an established VEEV infection.40,41
Results
Fc-LALA mutations do not affect ab binding or neutralization capacity
To assess the importance of Fc effector function in the context of a VEEV neutralizing Ab, we generated both a parental hF5-WT and Fc-LALA variant by engineering mutations L234A and L235A in the Fc domain of a human IgG1 construct. Although we did not expect Fc mutations to alter antigen binding, we verified intact affinity for both hF5-WT and hF5-LALA to the virulent VEEV Trinidad donkey (VEEV-TrD) E1E2 trimer and its live attenuated vaccine derivative, VEEV-TC-83 E1E2 trimer (Figure 1a,b). Despite the presence of mutations in E1E2 trimer of VEEV-TC83, we did not observe major differences in binding between hF5-WT or hF5-LALA to VEEV-TC-83 or VEEV-TrD E1E2 trimer.42 Furthermore, we performed kinetic studies using biolayer interferometry (BLI) comparing binding of the hF5-WT and hF5-LALA to VEEV-TC83 E1E2 trimer. BLI sensorgrams reveal that both hF5-WT and hF5-LALA exhibit comparably high-affinity binding with low dissociation constants, indicating alteration of the Fc region did not negatively affect antigen-binding potency (hF5-WT KD = 0.86 nM, hF5-LALA KD = 1.00 nM, Figure 1c–e). Next, we assessed neutralization capacity against VEEV-TC83 and SINV-VEEV-TrD-GFP. We found comparable neutralization capacities for the parental hF5-WT and hF5-LALA against VEEV-TC83 with IC50 values hF5-WT = 0.8101 ng/mL, and hF5-LALA = 0.1919 ng/mL (Figure 1f), and against a SINV-VEEV-TrD-GFP reporter virus in fluorescence-based neutralization assays, with IC50 values hF5-WT = 1.074 ng/mL and hF5-LALA = 1.623 ng/mL (p = 0.9617), reenforcing that binding and neutralization capacity are retained in the hF5-LALA mutant (Figure 1g).39 Of note, although hF5 was initially identified via binding affinity to inactivated TC83, the researchers complemented their initial study by demonstrating binding against several additional VEEV subtypes, including IAB, IC, ID, and IE, indicating hF5 could be a broadly neutralizing anti-VEEV therapeutic for other VEEV subtypes.13 Later work also demonstrated the efficacy of hF5-WT against virulent VEEV-TrD in vivo pre- and post-exposure (-/+24hpi), but, to our knowledge, prior to this report no binding or neutralization studies have been shown for hF5 and VEEV-TrD.39
Figure 1.
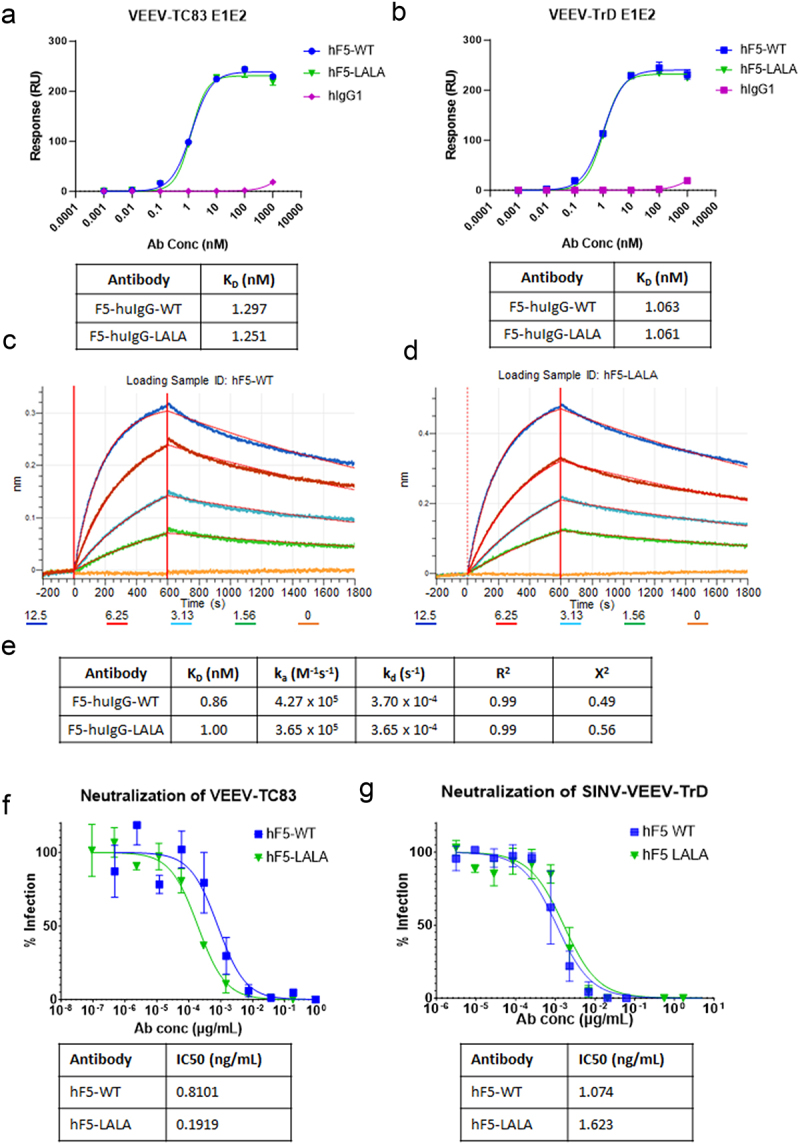
hF5-WT and hF5-LALA binding and neutralization potency. hF5-WT, hF5-LALA mutant, and human IgG isotype control binding to immobilized, recombinant VEEV-TC83 E1E2 trimer (a), and VEEV-TrD E1E2 trimer (b) mean and standard deviation (SD) from experiments performed in duplicate on gyrolabs immunoassay platform. BLI kinetic sensorgrams for hF5-WT (c) and hF5-LALA (d) against recombinant VEEV-TC83 E1E2 trimer. Table with equilibrium dissociation constant (KD), on-rate (ka), and Off-rate (kd) calculated from BLI kinetics experiments (e). TC83 neutralization and calculated IC50 values for hF5-WT and hF5-LALA calculated from plaque neutralization assay (PNA) experiments performed in triplicate (f). SINV-TrD GFP neutralization and calculated IC50 values for hF5-WT and hF5-LALA calculated from focus forming unit neutralization experiments performed in triplicate (g). Representative data shown for one of three independent experiments.
hF5-LALA mutant displays a loss in productive engagement with complement
The classical complement pathway is initiated by Ab-bound pathogen or virus-infected cells interacting with the first subcomponent of the C1 complex, C1q, via the Ab Fc domain. The productive engagement of C1q with Ab causes activation of a series of proteolytic cleavage events and release of inflammation-inducing and immune cell-recruiting cleavage products. Ultimately, deposition and insertion of complement components onto and into the cell membrane facilitate phagocytosis by responding immune cells, and/or direct lysis via the membrane attack complex. To determine the impact of the LALA mutation on the hF5 Ab, we characterized the ability of hF5-WT or hF5-LALA to bind mouse and human complement component, C1q. Although hF5-WT bound comparably well with both murine and human orthologs of C1q from normal mouse (NMS) and normal human serum (NHS), there was a significant decrease in C1q binding for hF5-LALA in comparison to hF5-WT over a range of serum concentrations (p < .003 for 2.8 to 75 µg/ml) with a 5.1-fold (NMS) and 6.2-fold (NHS) reduction in binding at 2.8 µg/ml (Figure 2a,b). Since the complement system functions in a series of successive proteolytic cleavages, we further assessed the productivity of C1q engagement by assaying the level of downstream complement fixation. As expected, hF5-WT demonstrated comparable levels of murine and human C3 fixation, signifying successful activation of the complement pathway. As with C1q, there was a significant decrease in complement fixation for hF5-LALA in comparison to hF5-WT over a range of NMS serum concentrations (p < .02 for 0.3 to 8.33 µg/ml) and NHS serum concentrations (p < .001 for 2.8 to 75 µg/ml) (Figure 2c,d). Of note, C3 fixation via hF5-WT and NHS serum peaked at 8.33 ug/ml (A450 = 2.24), but hF5-LALA reached a maximum A450 = 1.74 at the highest concentration of Ab (75ug/ml). While hF5-LALA mutant showed a substantial reduction in C3 fixation, complete ablation was not observed, suggesting that there may still be some complement activation with the LALA mutation at high concentrations of Ab. These findings are noteworthy, since little is known about the effect of Fc mutations on cross-species receptor–ligand interactions and downstream effector functions. Some previous reports demonstrate that LALA mutants abolish C1q binding and CDC43–45 while others suggest an additional mutation is required to completely abrogate CDC activity.32,35,46,47 These referenced studies were performed with human, primate, and rabbit serum; therefore, to our knowledge, a hIgG1 Fc-LALA mutation has not previously been described as compromising the interaction with mouse serum-derived C1q and C3.
Figure 2.
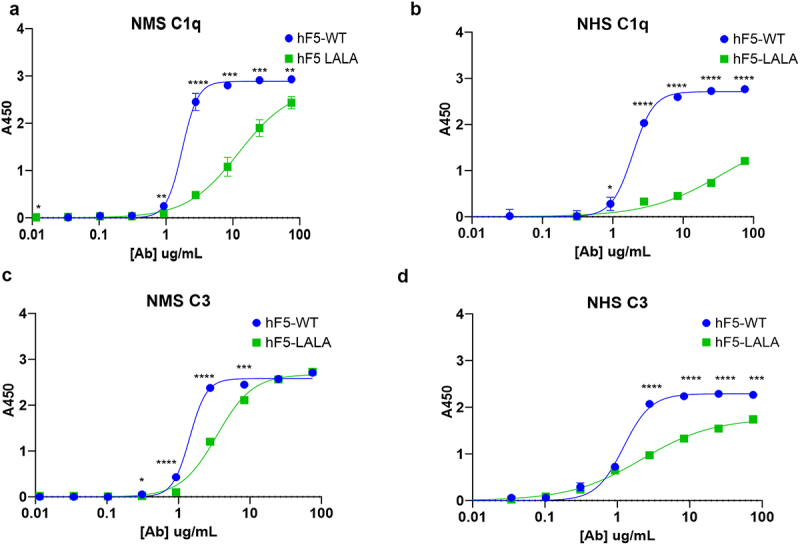
Complement binding and fixation with hF5-WT and hF5-LALA. hF5-WT and hF5-LALA mutant ab binding complement (C1q) from normal mouse serum (NMS) and normal human serum (NHS), (panel a and b, respectively). hF5-WT and hF5-LALA ab complement C3 fixation in the presence of NMS and NHS (panels c and d, respectively.) hF5-WT ab binds C1q in mouse and human sera, while hF5-LALA exhibits reduced binding to C1q and C3 fixation in human and mouse sera. Data are from experimental conditions performed in triplicate, and the error bars represent the standard deviation from the mean. P values considered significant if < .05 (*), or very significant if < .01 (**), < .001 (***), or < .0001 (****).
Although ELISA-based detection of binding to C1q and fixation of C3 informs successful initiation of the complement cascade, it fails to provide cell-based, functional data. Also, reporter endpoint assays, though useful for a single timepoint may obscure the subtle differences in kinetic interactions in the context of an infection over time.48,49 Therefore, we developed a real time (RT)-CDC assay to monitor the impact of Ab-dependent complement activation on cell viability in the context of VEEV infection over time (Figure 3). Uninfected cells and VEEV-TC83 infected cells treated with isotype control Ab demonstrated no cytotoxicity up to 20 hpi (Figure 3b,c). As expected, the VEEV-TC83-infected cells treated with hF5-WT and complement containing murine serum and human serum displayed rapid onset of cytotoxic efficacy within 4 h and two h of serum spikes, respectively. The hF5-LALA variant displayed a striking reduction in CDC activity over time compared with hF5-WT in the presence of NMS (p < .0001) and NHS (p = .0013) (Figure 3b,c). Cytotoxicity associated with infection alone (isotype control Ab) started at 20 hpi (human) to 27 hpi (murine) and increased over time. Images of all wells were captured hourly for the first 24 hpi, every 3 h until 30 hpi, with a final timepoint taken at 48 hpi based on established viral replication kinetics of VEEV-TC83. Images of infected, hF5-WT treated NHS wells at 24 hpi illustrate a robust cytotoxic effect with easily visualized Hoechst+ Propidium Iodide+, dead cells (Figures 3d and S1-S6 Videos). Interestingly, infected hF5-LALA plus NHS-treated wells exhibit comparably potent cytotoxicity by 24 hpi, indicating a single timepoint or endpoint evaluation of LALA-complement interactions may obscure more subtle loss of function phenotypic effects (Figures 3f and S1-S6 Videos).
Figure 3.
Real-time complement dependent cytotoxicity of hF5-WT and hF5-LALA. Schematic diagram illustrating RT-CDC assay conditions (a). Percent cell viability of Vero cells over the course of VEEV-TC83 infection for 48 h with hF5-WT and hF5-LALA, wells spiked with mouse (b) or human (c) sera at 8hpi (red arrows). % viability = [(number of blue cells – number of red cells)/number of blue cells] *100. Blue signal indicates Hoechst+, red and blue indicates Hoechst+ propidium iodide+. Images of NMS and NHS assays are taken from timecourse videos that span 9-25hpi (hours post infection). All image snapshots are taken at 24hpi. NMS treated hF5-WT (D, S1 video), hIgg1 (E, S2 video), and hF5-LALA (F, S3 video). NHS treated hF5-WT (G, S4 video), hIgg1 (H, S5 video), and hF5-LALA (I, S6 video). CDC activity was pronounced in hF5-WT treated wells with both mouse and human sera, whereas hF5-LALA mutant treatment with mouse or human sera showed significantly reduced cytotoxicity. In CDC-NMS assay, hIgg1 v hF5-WT p < 0.0001, hF5-WT v hF5-LALA p < 0.0001, hIgg1 v hF5-LALA p = 0.0001. In CDC-NHS assay, hIgg1 v hF5-WT and hF5-LALA p < 0.0001, hF5-WT v hF5-LALA p = 0.0013, by paired t-test. Data shown is representative of three or more experiments, each with three technical replicates (b and c), quantified using CellInsight C×7High Content Analysis software. Images (d-i) taken at 10X magnification on a CellInsight C×7High Content imaging platform.
Using our RT-CDC assay, we determined that there was a 2 (NHS) to 4-h (NMS) delay in hF5-LALA-mediated CDC activity initiation compared to hF5-WT. The pronounced 20% reduction in cytotoxicity at 30 hpi in the hF5-LALA compared with hF5-WT conditions demonstrate compelling evidence that hIgG1-LALA variants fail to efficiently engage mouse complement, even in this closed system. By examining the kinetics of cytotoxicity between complement and hF5-WT versus hF5-LALA, we gain insight into the significant reduction in CDC activity with LALA in mouse and human systems. These results demonstrate the value of this assay to capture rapid Ab-induced CDC activity, as well as differences in CDC kinetics observed with Abs of differing functionality.
hF5-LALA mutant exhibits loss of functional binding to Fcγ receptors
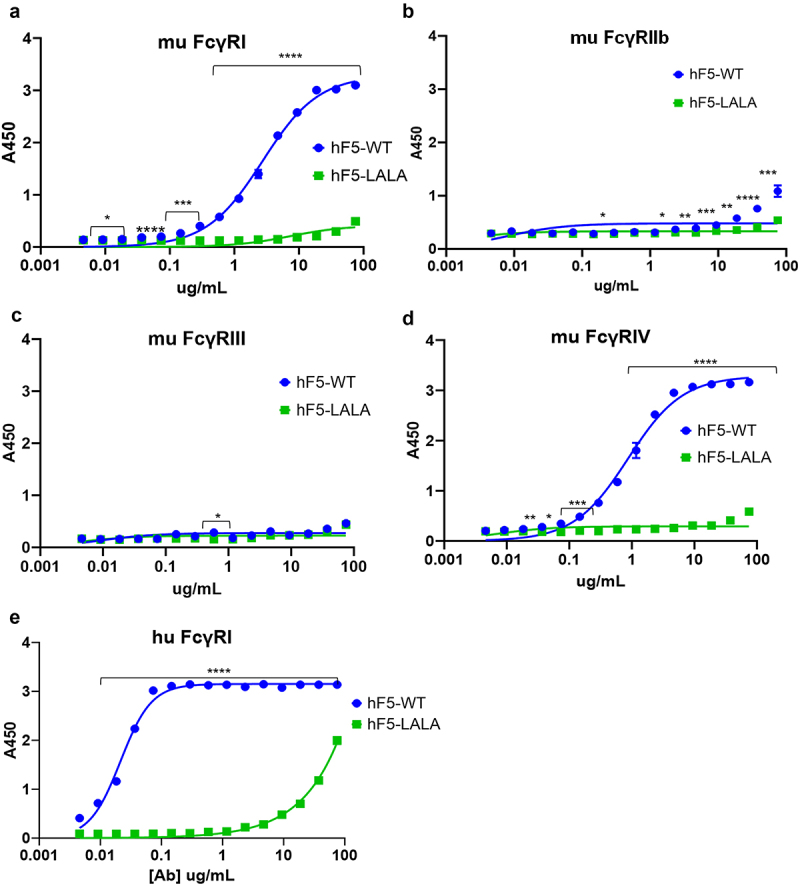
Previous reports have suggested that human IgG1 is functionally equivalent to murine IgG2a, with similar binding affinity for murine FcγR and similar effects on immune activation.36,50–52 To determine the impact of the LALA mutation on hF5 IgG1 FcγR engagement, we assessed binding of hF5-WT and hF5-LALA to four recombinant murine FcγRs and 6 human FcγRs (Figures 4 and S1). As expected hF5-WT had high affinity for murine FcγRI and IV (Figure 4a,d), but we observed little binding to murine FcγRIIb, and unlike previous reports we did not observe binding of hF5-WT to FcγRIII (Figure 4b,c), which has been shown by Dekkers et al. using surface plasmon resonance (SPR) measurements to have the lowest affinity to hIgG1 (KD hIgG1 for mFcγRI = 0.035 µM, mFcγRIIb = 1.1µM, mFcγRIII = 9.3µM, and mFcγRIV = 0.28µM).51,52 Based on the markedly low-affinity interactions between hIgG1 and murine FcγRIIb and III that we and others have observed, we expect the weak affinity of these interactions goes beyond the limit of our ELISA-based detection method. As expected, on the other hand, the hF5-WT human IgG1 isotype demonstrated the greatest affinity for human FcγRI, little binding to hFcγRIIA and IIIA, and no binding of human FcγRIIB or IIIB (Figures 4e and S1).37,53 Importantly, there was little to no detectable binding of hF5-LALA mutant to all murine and human FcγRs by ELISA (Figures 4a–e and S1).
Figure 4.
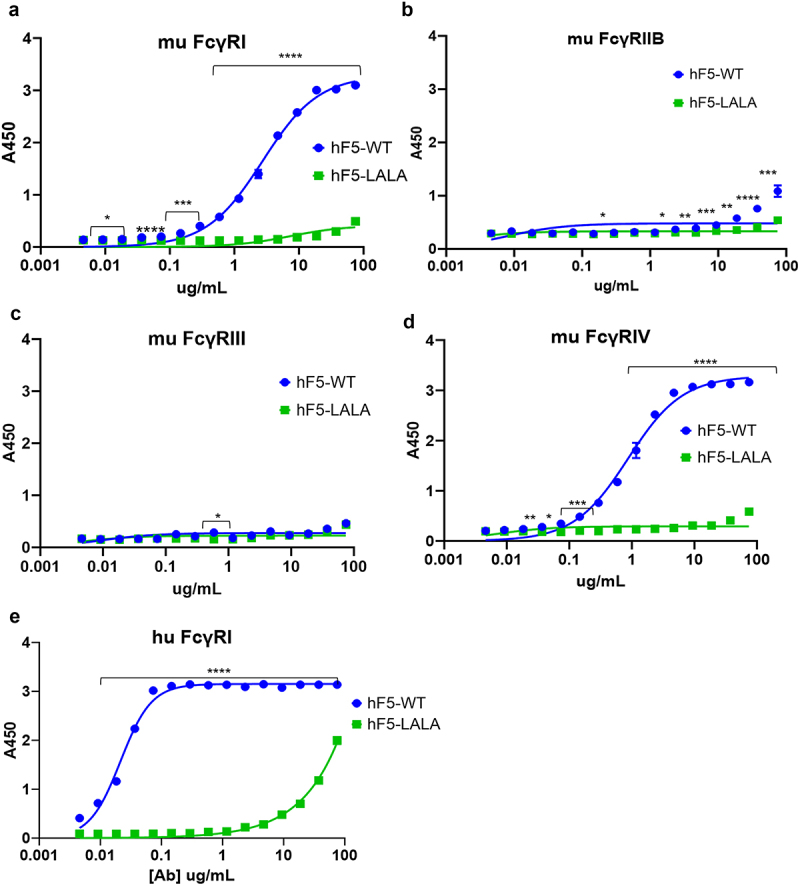
FcγR binding with hF5-WT and hF5-LALA. hF5-WT and hF5-LALA mutant Ab binding murine FcγRs I (a), IIb (b), III (c), and IV (d). hF5-WT and hF5-LALA Ab binding to human FcγRI (e). hF5-WT Ab binds to both mouse and human FcγR I orthologs, and mouse FcγRIV, but fails to bind to murine FcγR IIb and III, while the hF5-LALA mutant exhibits abrogated binding to all murine FcγRs and substantially reduced binding to human FcγRI. Data are from experimental conditions performed in triplicate, and the error bars represent the standard deviation from the mean. P values considered significant if < .05 (*), or very significant if < 0.01 (**), <.001 (***), or < .0001 (****).
Next, we determined how the altered binding affinity of the LALA mutant would impact cytotoxicity using a novel assay for real time observation of ADCC (RT-ADCC). We used murine RAW264.7 macrophages, which express mFcγRI/IIb/III and IV, and human THP-1 macrophages, which express hFcγRI and II.54,55 We exposed VEEV-TC83-infected cells to hF5-WT, hF5-LALA, or isotype control Ab, spiked in activated murine or human macrophages 8hpi, and assessed cell death over time (Figure 5). hF5-WT facilitated significant and rapid ADCC activity with both human (hF5-WT v hIgG1 p = .0004) and murine (hF5-WT v hIgG1 p < .0001) macrophages, indicating productive interaction with FcγRs. Importantly, the LALA mutant exhibited a complete loss of specific ADCC activity (human: hF5-WT v hF5-LALA p = .0006; mouse: hF5-WT v hF5-LALA p < .0001) (Figure 5a,b). In the human (THP1) assay, the hF5-LALA variant exhibited significantly reduced cytotoxic activity compared with hF5-WT, with activity like the isotype control (Figure 5c). The changes in cell viability over time in the isotype control recapitulate the expected cell death kinetics from VEEV-TC83 infection in vitro and indicate a lack of ADCC-specific cell killing by macrophages in the absence of virus-specific Ab (Figures 5b,c and S7-S12 Videos). Representative images taken at 24 hpi illustrate the high level of ADCC-mediated death by the number of Hoechst and propidium iodide double positive cells in the hF5-WT-treated wells compared with hF5-LALA and isotype control (Figures 5d–f and S7-S12 Videos). Taken together, the complete loss of hF5-LALA binding to murine FcγRI and IV, and human FcγRI, combined with the abrogation of Fc-mediated functional engagement to drive ADCC activity, confirms a cross-species loss of Fc effector function.
Figure 5.
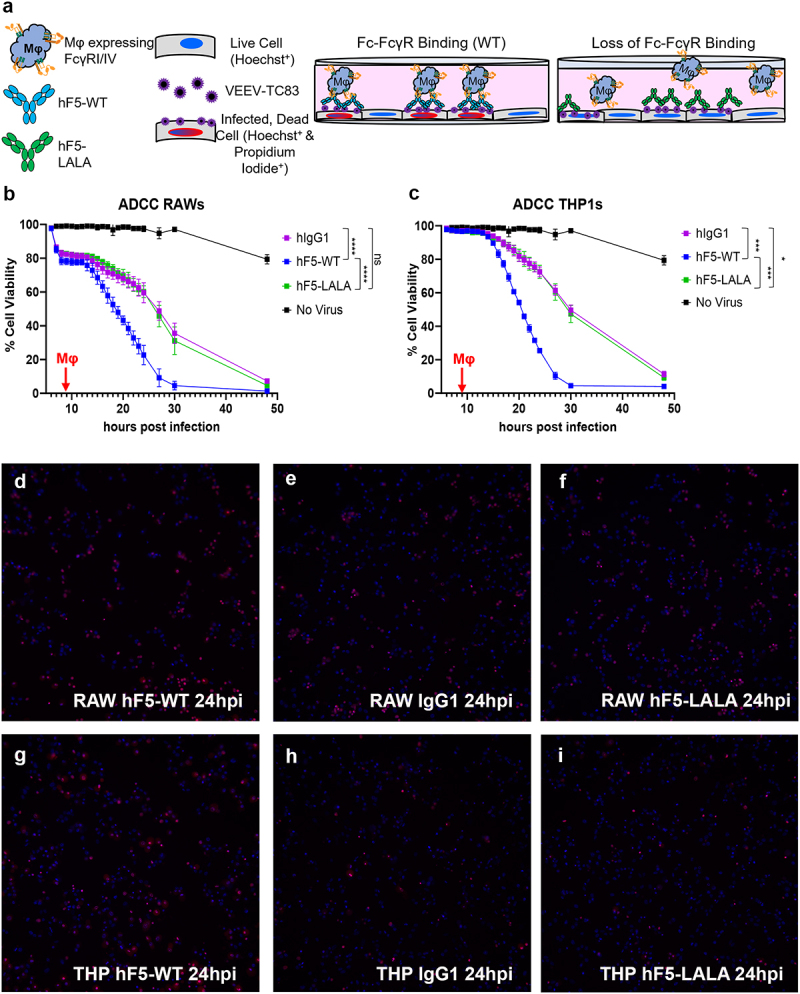
Real-Time Ab-Dependent-Cell-mediated-Cytotoxicity of hF5-WT and hF5-LALA. Schematic diagram illustrating RT-ADCC assay conditions (a). Percent cell viability of Vero cells over the course of VEEV-TC83 infection for 48h with hF5-WT and hF5-LALA, wells spiked with RAW mouse (b) or THP1 human (c) macrophages (Mφ) at 8hpi (red arrows). % Viability = [(number of blue cells – number of red cells)/ number of blue cells] *100. Blue signal indicates Hoechst+, red and blue indicates Hoechst+ propidium iodide+. Images of murine RAW macrophage-treated timecourse taken at 24hpi (hours post infection). hF5-WT (d, S7 Video), hIgG1 (e, S8 Video), and hF5-LALA (f, S9 Video). Images of human THP1 macrophage-treated timecourse taken at 24hpi (hours post infection). hF5-WT (g, S10 Video), hIgG1 (h, S11 Video), and hF5-LALA (i, S12 Video). ADCC activity was pronounced in hF5-WT treated wells with both mouse and human Mφ, whereas hF5-LALA mutant treatment with mouse or human Mφ completely abrogated ADCC; hF5-LALA cytotoxicity follows VEEV-TC83 infection cytopathic kinetics. In ADCC-RAWs assay, hIgG1 v hF5-WT p < .0001, hF5-WT v p < .0001, hIgG1 v hF5-LALA was not significant. In ADCC-THP1 assay, hIgG1 v hF5-WT p = .0004, and hF5-WT v hF5-LALA p = .0006, hIgG1 v hF5-LALA p = .0138 by paired t-test. Data shown is representative of three or more experiments, each with three technical replicates (B and C), quantified using CellInsight CX7 High Content Analysis software. Images (D-I) taken at 10X magnification on a CellInsight CX7 High Content Imaging platform.
hF5 fc effector mutants exhibit reduced therapeutic efficacy against VEEV-TC83 in vivo
hF5-WT, hF5-LALA, or human isotype control Abs were used in a series of pre- and post-exposure treatment studies using VEEV-TC83 intranasal infection in C3H/HeN mice41 to understand the importance of Fc effector function in the context of VEEV infection (Figure 6). Animals dosed with hF5-WT at −24 h, +24 h, and + 48 hpi exhibited little or no severe neurological symptoms and had a consistently high survival rate, across experiments (90% n = 20, 70% n = 20 and 85.7% n = 35, respectively: Figure 6b–d). However, a significant reduction in efficacy (50% survival), and development of neurological symptoms by 4 days post infection (dpi) was observed when hF5-WT was given at + 72 hpi (n = 10; Figure 5e).
Figure 6.
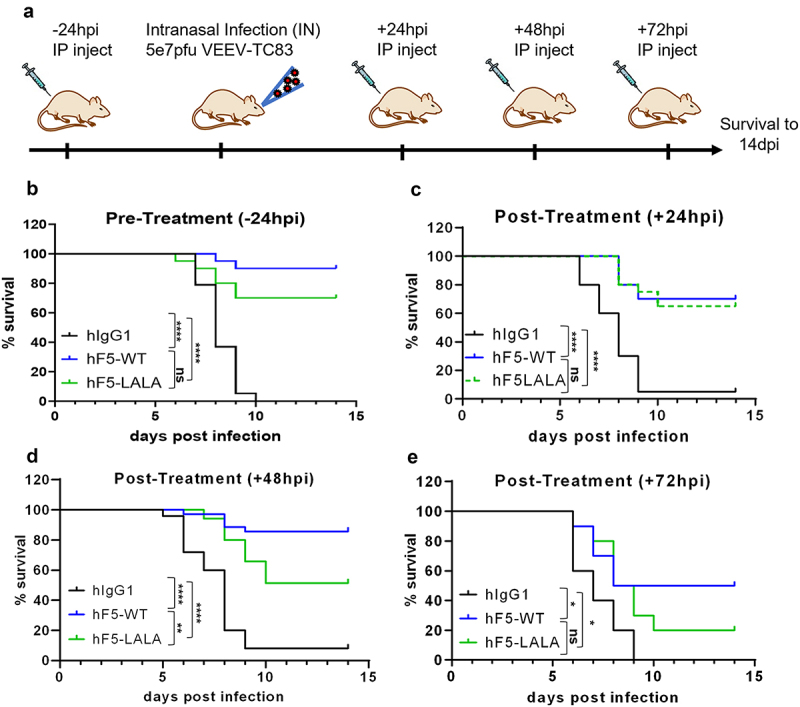
in vivo efficacy of hF5-WT and hF5-LALA dosed prophylactically and therapeutically. Schematic of dosing and infection timeline through 14 dpi (days post infection) survival (a). Prophylaxis study involved a -24-h pre-infection 100ug dose of Ab (b), while therapeutic studies included +24hpi (c), +48-h (d), and +72-h (e) post-exposure 100ug dose. Animals were infected intranasally (IN) with 5e7pfu VEEV-TC83, and survival was measured to 14dpi. Pre-treatment (-24hpi) and early exposure (+24hpi) animals receiving hF5-WT or hF5-LALA Ab showed comparable survival rates, while in the +48hpi post-exposure context, hF5-LALA-treated animals did exhibit a significant reduction in survival. By +72hpi both hF5-WT and hF5-LALA -treated individuals showed similarly high degrees of mortality, indicating a diminishing therapeutic benefit regardless of Fc effector function, in the context of an established VEEV infection. For the -24hpi (hours post infection) study hIgG1 v. hF5-WT ****p< .0001, hIgG1 v. hF5-LALA ****p < .0001, hF5 v. hF5-LALA was not significant (p = .1034). For the +24hpi study hIgG1 v. hF5-WT ****p< .0001, hIgG1 v. hF5-LALA ****p< .0001, hF5-WT v. hF5-LALA was not significant (p = .7818). For the +48hpi study hIgG1 v. hF5-WT ****p< .0001, hIgG1 v. hF5-LALA ****p< .0001, hF5-WT v. hF5-LALA **p= .0036. For the +72hpi study hIgG1 v. hF5-WT *p = .0190, hIgG1 v. hF5-LALA *p = .0343, hF5-WT v. hF5-LALA was not significant (p= .3751), by Log-rank Mantel-Cox test. In -24hpi and +24hpi studies n=20; in +48hpi studies n=35; and in +72hpi study n=10. Data shown represent at least two experiments, except for the +72hpi study, performed once.
The hF5-LALA-treated animals at −24 hpi showed slightly decreased survival rates (70%, n = 20) compared with the −24 hpi hF5-WT group (90%, n = 20), although the difference was not statistically significant (Figure 6b). Interestingly, mice treated with hF5-LALA at + 24 hpi also provided therapeutic protection (65%, n = 20) comparable to the hF5-WT group (70%, n = 20, Figure 6c), indicating the potent neutralizing activity of the hF5 Ab does not require Fc effector function to protect early in infection. In contrast, the hF5-LALA +48 hpi treatment group, showed a significant reduction in therapeutic protection compared with the hF5-WT group (hF5-WT 85.7%, hF5-LALA 51.4%, **p = .0036, n = 35), suggesting an important role for Fc effector function in VEEV clearance and survival (Figure 6d). The hF5-LALA +72hpi group also developed neurological signs by 4 dpi and diminished survival compared with hF5-WT (WT 50%, LALA 20%, n = 10), although the differences were not statistically significant (p = .3751). As expected, animals that succumbed to VEEV infection exhibited neurological symptoms, including altered gait causing circling, lethargy, weakness, and in later stages of disease showed hyperactivity and even seizures, indicating encephalitis. These data indicate that within 48hpi there is a window of opportunity for Ab therapeutics to mediate peripheral virus clearance and prevent the development of VEEV-induced encephalitis, but this effective clearance at + 48 hpi is contingent on intact Fc effector function.
Discussion
Abs and Ab cocktails are promising prophylactic and therapeutic treatments for many emerging and reemerging infectious diseases. While most studies focus on the ability of Abs to potently neutralize the virus, several recent studies have shown that the ability of Abs to engage innate and adaptive immune responses is vital for in vivo protection from influenza, SARS-CoV-2, flaviviruses, alphaviruses, and filoviruses.5,21,24–27,31,32,56–60 To fully protect the host from viral infections, Abs form immune complexes, engage complement and FcγRs on immune cells to boost neutralization, facilitate clearance of infected cells, opsonize virions, enhance antigen presentation, and regulate inflammation.20,32 The delicate balance between immune effector function, antimicrobial factors and their regulatory structures via Fc engagement highlights an opportunity to improve therapeutic outcomes for pathogen-specific disease states.
Most of the detailed studies characterizing the role of Fc effector function during alphavirus infection have been performed with Abs against the arthritic alphaviruses. However, the requirement for Fc effector function within these studies is variable, with some Abs requiring Fc effector function for optimal prophylactic efficacy24,25 for therapeutic efficacy (+24-72hpi).26,61 and others not requiring Fc effector function for optimal protection from disease.32,62–64 These studies demonstrate that the contribution of Fc effector function to optimal Ab efficacy is affected by the virus targeted, its ability to escape neutralization, the Ab epitope, dose, and timing of administration. Therefore, detailed analysis is needed to characterize the influence of these factors on the contribution of Fc effector function to therapeutic efficacy in the context of viral infection to develop optimized Abs as potential therapies. Although a recent study determined that a pan-protective non-neutralizing anti-alphavirus Ab (targeting E1) requires effector functions for optimal prophylactic efficacy against VEEV infection in vivo,5 this is the first study to investigate the role of Fc effector function for potently neutralizing (E2-binding) Abs at different stages of encephalitic alphavirus infection.32
Given the sustained affinity in our hF5-WT and hF5-LALA Abs for both VEEV-TC83 and VEEV-TrD E1E2 trimer (Figure 1a,b), and effective neutralization of VEEV-TC83 and SINV-TrD-GFP viruses (Figure 1f,g), we utilized the VEEV-TC83 model of infection for constructing our in vitro and in vivo system for validation and characterization of anti-VEEV Fc-engineered mutant antibodies. Importantly, the attenuated VEEV-TC83 strain may be handled at a lower bio-containment level (BSL-2), compared with the WT VEEV-TrD (BSL-3), permitting more comprehensive assessment of Ab-Fc-mediated immune functions in the context of infection. TC83 challenge in C3H/HeN mice represents an acute lethal infection via intranasal inoculation, with 90–100% of animals succumbing to overwhelming meningoencephalitis within 7–10 days (Figure 6).41 Although viral replication kinetics and ensuing neurovirulence are delayed in the VEEV-TC83 infection model compared with WT VEEV-TrD infection, we can draw parallels between disease progression parameters, neuropathology, and lethality, that reenforce VEEV-TC83 as a highly informative model of VEEV infection.
In this study we investigated the impact of Fc effector function on the efficacy of a potent neutralizing anti-E2 VEEV Ab. We evaluated the ability of the human F5-WT and hF5-LALA (L234A and L235A) to bind human and murine orthologs of complement, verifying that the LALA mutation indeed decreases binding to C1q and fixation of C3, thereby significantly reducing Ab-mediated CDC in vitro, in agreement with other studies.43–45 We also confirmed that hF5-WT IgG1 is capable of robustly binding to mouse FcγR orthologs I and IV, demonstrating similar immune effector activating potential to murine isotype IgG2a. However, our hIgG1 LALA-Fc mutant Ab showed complete loss of binding to mFcγR I and IV, as well as abrogation of ADCC in vitro. Importantly, some reports evaluating the LALA mutations for loss of function have historically shown varying results depending on the specificity of the Ab, the isotype, and the species of host Ab relative to the species of Fc receptor in an ELISA or bioassay.27,44,46 Wilkinson and colleagues performed a highly comprehensive assessment of a series of Fc mutations in 3 different human IgG1 antibodies comparing their binding affinity to the WT affinities of each Ab for a panel of human FcγRs. Interestingly, they found LALA mutations confer an 80% reduction in binding to human FcγRI, but achieve complete abrogation of binding between hIgG1-LALA and mouse FcγRI.
We developed comprehensive in vitro validation studies for characterizing the loss of productive Fc region engagement using two novel, high-resolution, real-time assays. Using our RT assays, the hF5-LALA mutant displayed a significant reduction in Fc-mediated CDC over time, an effect that may not be readily apparent using traditional, single timepoint analysis methods. The limited scope of such methods may further explain the inconsistent reports regarding L234A and L235A Fc mutations and their effect on CDC and Fc effector functions.48,49,65 In addition, our RT-ADCC assay illustrates a striking abrogation of functional, Fc-directed cell death with the hF5-LALA in comparison to hF5-WT over the course of VEEV-TC83 infection. Traditional luciferase reporter cell line assays rely on single endpoint analysis, which may obscure differential Fc effector function capacities. The comprehensive view of in vitro RT-CDC and ADCC activity in these assays provides valuable, detailed insight into Fc-engineered Ab characterization, thus facilitating thorough evaluation of the impact of gain and loss of function mutations on effector function capacity. Importantly, these assays are adaptable for a variety of viruses, Ab isotypes, and effector cells. Our findings demonstrate the importance of careful characterization of novel therapeutics, considering that changes in mutations can affect cross-species reactivity, which may confound in vivo data, when left unaddressed. It will be important going forward to use high resolution, time course-based methods to thoroughly evaluate the Fc effector function of novel gain and loss of function mutant Abs to accurately reflect differences in effector function capacity.
Finally, we determined the impact of Fc effector function on the efficacy of hF5 at different timepoints prior to and after lethal VEEV-TC83 challenge in vivo. Our pre-exposure (−24 hpi) and early exposure (+24 hpi) dosage groups displayed similar degrees of protection for both hF5-WT and hF5-LALA, indicating that Fc effector function is not required for protection at these disease stages. This is likely due in large part to the high neutralizing capability of E2-targeting hF5 and that the peripheral VEEV infection is not fully established.40,41 By + 48 hpi, however, when virus titers are at peak levels in all tissues except the brain, hF5-WT continues to provide protection, while hF5-LALA exhibits a significant drop in therapeutic capacity (Figure 6d).40,41 Our results demonstrate that Fc-mediated clearance is required for therapeutic efficacy and prevention of VEEV-induced neuropathogenesis and subsequent mortality.
While potent neutralization alone may be sufficient for prophylactic and therapeutic protection at early timepoints post-infection, our data suggest that optimal treatment of an established infection requires Fc effector functions for clearance and protection from lethal challenge and neuropathogenic disease. Although we did not delineate the exact Fc effector function(s) required for in vivo protection + 48 hpi, the in vitro assays performed showed a decrease in complement activation and abrogation in binding of mouse FcγRI and IV for hF5-LALA in comparison to hF5-WT. Other studies also suggest a role for complement activation in VEEV clearance, where the use of C3−/− mice demonstrated impaired survival during VEEV infection compared with C3+/+ infected controls.66 In addition to Fc-mediated activation of the complement pathway for virus and infected cell clearance, productive engagement of Ab Fc with activating FcγRs such as human FcγRI and mouse FcγRI and IV (largely expressed on macrophages, DCs, and neutrophils) help to mobilize a comprehensive innate and adaptive immune response.19,32,36 Recent reports implicate macrophages as important players for effective killing and clearance of VEEV infected cells, a function which is greatly hampered in our LALA mutant Ab, based on our in vitro data.67 It is therefore vital to improve our understanding of the immune mechanisms required for clearance of VEEV at different stages of infection to better inform the design of therapeutic strategies both in the periphery and ultimately with active blood-brain barrier-penetrating functionality to treat neurotropic infection without causing immune-mediated neuropathology.
Materials and methods
Ab and recombinant protein engineering
Sequences for hF5-WT and hF5-LALA mutants were designed in gene blocks and cloned into pSF-CMV vectors.12 Production of recombinant Abs was achieved using the ExpiCHO system (Thermo Fisher Cat # A29133) according to the manufacturer’s ‘Max Titer’ protocol. Briefly, Ab-containing plasmids for heavy and light chain were transfected into CHO-S cells, cultured at 32o C and 5% CO2 shaking in flasks. Cells were harvested by centrifugation at 4000 × g at 4°C for 20 minutes. Clarified supernatant was applied to a HiTrap PrismA column (GE Cat # 17549851) equilibrated in 20 mM sodium phosphate (pH 7.4), 150 mM NaCl, and partially purified protein was eluted with 100 mM sodium citrate (pH 3.0) and immediately neutralized with 1 M Tris (pH 9.0). Fractions containing protein were pooled, concentrated, and subsequently purified by size exclusion chromatography on a Superdex 200 increase 10/300 GL column (Cytiva, Cat # 28990944) equilibrated in PBS. Protein was concentrated, flash frozen in liquid nitrogen, and stored at −80°C.
pMTBip-TC83-E1E2 and pMTBip-TrD-E1E2 were constructed as follows and was adapted from other work to express recombinant E1-E2 heterodimer from Sindbis virus.68 A double-stranded DNA fragment was commercially obtained (IDT) encoding the following TC83 or TrD structural protein coding sequences, E3 (1-59), E2 (1-344), and E11−384). A Strep-tag II sequence (GGGSWSHPQFEKGGGG) was present in between the coding sequence for E2 and E1 in addition to a C-terminal hexa-histidine tag followed by a Avi site specific biotinylation sequence. Trans-membrane regions of E2 and E1 were not included to produce soluble protein. This DNA fragment was assembled with agarose purified pMT/Bip/V5-HisA vector (Thermo), restriction digested with BglII and XbaI, and treated with phosphatase rSAP (NEB) using NEBuilder HiFi DNA master mix (NEB).
VEEV-TC83 E1E2 trimer was produced using a Drosophila S2 expression system. A Stable S2 cell line that expressed VEEV-TC83 E1E2 protein under an inducible promoter was generated as described in the technical literature provided by Gibco (Drosophila Schneider 2 (S2) Cells User Guide). In brief, S2 cells were cultured in complete Schneider’s Drosophila Medium (10% heat inactivated fetal bovine serum (FBS), 1% penicillin streptomycin). S2 cells were transfected using a CaCl2 protocol. Cells were transfected at 3.0 × 106 cells/mL with 19 μg of pMTBip-TC83-E1E2 and 1 μg pCoPuro in a 35 mm plate as described in the manufacturers protocol for 18 h at 28°C. Cells were washed and media exchanged, then allowed to grow in the absence of selection agent for 48 h. Cells were then expanded in the presence of 7 μg/mL puromycin. Stable polyclonal S2 cells were expanded and adapted to EX-CELL 420 serum-free media (Sigma) supplemented with 0.1% Pluronic F-68 over several passages with shaking. Protein expression was induced at a cell density reached 6 × 106 cells/mL with 500 μM CuSO4. Protein was allowed to express for 5 days after which the supernatant containing recombinant E1-E2 protein was harvested. Cells were centrifuged at 4,000 × g for 30 minutes at 4°C. Supernatant was then clarified by 0.45 μm filter prior to application to a 5 mL HITrap TALON Crude (Cytiva) equilibrated with 50 mM sodium phosphate 300 mM NaCl. The column was washed extensively with buffer followed by washing with equilibration buffer with 3 mM and 5 mM imidazole. The E1-E2 complex was eluted from the column with equilibration buffer supplemented with 150 mM imidazole. Protein was concentrated and applied to Superdex 200 Increase 10/300 GL (Cytiva) equilibrated with phosphate-buffered saline (PBS). Fractions containing E1-E2 complex were concentrated, aliquoted, and snap frozen with liquid N2. Protein was stored at −80°C.
pSF-CMV human and mouse Fc receptors were constructed as follows. A double-stranded DNA fragment was commercially obtained (IDT) encoding huFcγI (Met1-Pro288), huFcγIIa (Met1-Ile218), huFcγIIb (Met1-Pro217), huFcγIIINA1 (Met1-Ser200), huFcγIIINA2 (Met1-Ser200), muFcγI (Met1-Pro297), muFcγII (Met1–217), muFcγIII (Met1-Thr215), muFcγIV (met1-Glu203) each followed by TEV protease cleavage site, and a 10× His tag. This DNA fragment was assembled with agarose-purified pSF-CMV vector, restriction digested with EcoRI and BamHI, and treated with phosphatase rSAP (NEB) using NEBuilder HiFi DNA master mix (NEB).
Expression of human and mouse Fc receptors was performed in Expi293F cells (Thermo, A14527) in 50–100 ml transfections at 1 μg DNA/mL. Transfections were fed 18–22 h later according to the manufacturer’s specifications and harvested 6 days post-transfection by centrifugation at 4000 ×g for 20 min and filtering through a 0.22 μm filter (Nalgene). The filtered supernatant was run on first a HisTrap Excel 5 ml Column (Cytiva) equilibrated in 20 mM phosphate, 300 mM NaCl buffer at pH 7.4 and subsequently through a Superdex 200 Increase 10/300 GL SEC column (GE) equilibrated in PBS. Purified proteins were concentrated using 10 kDa MWCO concentrators (Amicon) and quantified using the DC assay (Biorad).
VEEV E1E2 antigen-binding assay
Purified Avi tagged VEEV E1E2 proteins were biotinylated using the BirA biotin-protein ligase reaction kit (Avidity) according to manufacturer instructions, and free biotin removed on a Zeba Spin desalting column (Thermo Fisher). Antigen binding assays were conducted on a Gyrolabs Explore immunoassay system. Assays were performed on a Bioaffy 1000HC CD, with a capture antigen (VEEV E1E2) concentration of 1 uM. Concentration curves for hF5-WT and hF5-LALA were prepared as a 10-fold dilution series with a starting concentration of 1 uM. AffiniPure F(ab’)₂ Fragment Goat Anti-Human IgG (H+L) conjugated with Alexa 467 (Jackson ImmunoResearch) was used for detection at a concentration of 10 nM. Antigen and antibody dilutions were made in Rexxip A buffer, with Detection reagent diluted in Rexxip F buffer (Gyros Protein Technologies). Detection was performed at 0.1% and 1% PMT. Binding curves were analyzed in Graphpad prism, using a Specific Binding with Hill slope non-linear fit analysis function.
Biolayer interferometry
Kinetic parameters for each anti-VEEV IgG antibody were determined using an OctetⓇ RED384 (Sartorius). Each IgG antibody was immobilized on anti-human Fc sensors in 10 mM phosphate (pH 7.4), 300 mM NaCl, 1 mg/mL bovine serum albumin (BSA), 0.02% NP-40. VEEV TC83 E1E2 was used as the analyte and sensorgrams were fit to 1:1 global kinetics.
Complement ELISAs
NUNC 384-well plates (Thermo Fisher Cat # 464718) were coated with 25 µL analyte (hF5-WT or hF5-LALA, at 75 µg/mL in a 3-fold dilution series) overnight in amine labeling buffer (100 mM NaCO3, 150 mM NaCl, pH 8.3 at 25°C) at 4°C. Plates were washed with 0.05% Tween20 (v/v) in 1X PBS and commercial protein-free block (Thermo Fisher Cat #37572) was added. Plates were incubated for 2 h at room temperature on a tabletop shaker. Plates were washed and 25 µL/well of 3% NMS or NHS (Complement Tech Cat # NHS and NMS) in PBS was added for 1 h at room temperature. Plates were washed and primary anti-human C1q or anti-human C3 (Abcam Cat #s ab11861 and ab11862) was diluted 1:1000 and added at 25 µL/well. Abs were incubated at room temperature for 1 h with shaking. Plates were washed and an anti-rat horseradish peroxidase (HRP) secondary Ab (Thermo Fisher Cat # 31470) was diluted to 1:5,000 or 1:2,000 for C1q and C3 plates, respectively, and added for 1 hour at room temperature with shaking. Plates were washed and developed using 25 µl/well 1-Step Ultra TMB-ELISA (Thermo Fisher Cat # 34029), and the development arrested using an equal volume of 2 M H2SO4. Absorbance readings were taken using the Tecan SPARK at 450 nm and plotted using Prism (GraphPad).
FcγR ELISAs
NUNC 384-well plates (Thermo Fisher Cat # 464718) were coated with human or mouse FcγRs in amine labeling buffer (100 mM NaCO3, 150 mM NaCl, pH 8.3 at 25°C) overnight at 4°C. Plates were washed with 0.05% Tween20 (v/v) in 1X PBS and blocked using 0.5% BSA (w/v) and 0.05% Tween20 in 1X PBS for 2 h at room temperature with vigorous shaking. Blocking solution was washed off as done previously, and serially diluted hF5-WT and hF5-LALA primary Abs were added to the plates and incubated for 2 h at room temperature with shaking. Primary Abs were washed off, and secondary Ab (Goat anti-human IgG H+L F(ab’)2-HRP; Sigma Cat # AQ112P; 1:2000) was added for 1 h at room temperature with shaking. Plates were washed and developed using 25 µl/well 1-Step Ultra TMB-ELISA (Thermo Fisher), and the development arrested using an equal volume of 2 M H2SO4. Absorbance readings were taken using the Tecan SPARK at 450 nm and plotted using Prism (GraphPad).
In vitro fc effector function assays
Vero (African green monkey kidney, (ATCC CCL-81) cells were cultured to 70% confluence and infected (MOI = 1) with VEEV-TC83 in 96-well plates. At 4 h post infection (hpi), wells were dosed with hF5-WT, LALA, or isotype control (BioXcell Cat #BE0297) Abs (0.6 µg/mL). Cells were counterstained with Hoechst 33,342 (Thermo Scientific Cat #62249) and Propidium Iodide (Invitrogen Cat #699050) to identify live and dead cells, respectively. For RT-CDC, at 8 hpi NHS or NMS was added (10% final concentration) to each well. For RT-ADCC, at 8 hpi, RAW264.7 (ATCC TIB-71) mouse or THP1 (ATCC TIB-202) human macrophages [stimulated for 48 h with 10 ng/mL LPS (Sigma Cat # L3024), then allowed to rest overnight] were added in a 1:3 effector: target cell ratio to each well. Time course imaging using a Thermo Fisher Scientific C×7 microscope was performed for 48 hpi to gauge cell viability over time. Data analysis was performed using the C×7CellInsight High Content Screening and Prism software, with paired t-test.
Virus stock and plaque assays
VEEV-TC83 was obtained through the National Institutes of Health (NIH) Biodefense and Emerging Infections Research Resources Repository, NIAID, NIH (NR-63). TC83 viral stocks were propagated using cultured Vero cells grown to be 70% confluent in alphaMEM complemented with 10% FBS and 1% penicillin/streptomycin (P/S) (Gibco), incubated at 37°C in 5%CO2. The supernatant was collected 48 h post infection and cleared of debris by centrifugation (1000 × g for 10 min). Viral supernatants were then purified by ultracentrifugation, resuspended in PBS; then aliquots were stored at −80°C, and titers were determined by plaque assay.
The SINV-VEEV (TrD) GFP chimera was constructed from the genetic backbone of SINV originating from the addgene plasmid pSin REP5-GFP-GluR2 (https://www.addgene.org/24003/). The cargo gene (GFP-GluR2) was removed by restriction enzyme digest with XbaI and SphI, to generate a backbone containing the SINV non-structural coding genes and cis-acting RNA elements required for replication and transcription of the sub genomic RNA (encoding the structural genes). Synthetic DNA was designed containing the structural gene region of VEEV TrD (NCBI Reference Sequence: NC_075022.1) with an insertion of EGFP followed by a T2A self-cleaving peptide sequence in frame between the VEEV TrD capsid and E3 genes and ordered from Twist Biosciences as clonal genes in the pTwist amp high copy vector. The structural gene insert, including > 25 bp terminal overlaps with the SINV backbone were PCR amplified out of the cloning vector, and assembled with the SINV non-structural backbone by NEB Hi-Fi assembly. Final constructs contain constructs contain nt 1–7647 of the SINV genome (5’ UTR, non-structural genes, and sub-genomic RNA promoter); nt 7647–8471 (825 nt) capsid sequence (VEEV TrD); nt 8472–9260 (789nt) linker-EGFP-T2A cleavage site; nt 9261–12,203 (2943 nt) E3, E2, 6K, and E1 glycoprotein sequence (VEEV TrD) and 12,215–12,524 (310nt) SINV 3’ UTR followed by a poly(A). After purification and conformation of plasmid sequences by enzymatic digest and Sanger sequencing, DNA was linearized by digestion with NotI, and infectious RNA transcripts were generated by in vitro transcription with the mMessage mMachine SP6 transcription kit (Thermo Fisher). Virus was rescued after transfection of BHK-21 cells, cultured in DMEM complemented with 10% FBS, 10% Triptose Phosphate Broth, and 1% P/S (Gibco), with infectious RNA using Lipofectamine MessengerMax transfection reagent (Thermo Fisher). Cells were monitored for GFP expression and visible CPE, and virus containing supernatant was collected at day 3 post transfection. SINV-VEEV (TrD) GFP was propagated using Vero cells grown to be 70% confluent in alphaMEM complemented with 10% FBS and 1% P/S (Gibco), incubated at 37°C in 5%CO2. The supernatant was collected 48 h post infection and cleared of debris by centrifugation (1000 × g for 10 min).
For plaque assays (PA) and plaque neutralization assays (PNA), Vero cells were cultured to 70% confluence in alphaMEM complemented with 10% FBS and 1% P/S. For PA titers, Virus stocks or infected brain homogenates were diluted with alphaMEM in a 10-fold dilution series using 12-well plates and inoculating with 200 µL per well, for 30 min at 37°C, 5%CO2. For PNAs, diluted virus was mixed with 10-fold dilutions of hF5-WT, LALA, or isotype control Ab for 1 h and then used to inoculate Veros with 200 µL per well for 30 min, as above, followed by addition of 1.5 mL 0.5% agarose and 2XMEM overlay to each well. Assays were stopped at 38hpi. Agarose plugs were removed, cells were washed once with PBS, and then stained with 2% paraformaldehyde (PFA) with 0.25% crystal violet. Wells were rinsed twice with DI water and plaques were counted.
For focus forming unit neutralization assays performed with GFP reporter virus, Vero cells were cultured to 70% confluence in alphaMEM complemented with 10% FBS and 1% P/S in a 384-well black clear bottom tissue culture plate (Corning Costar). Diluted virus was mixed with 3-fold dilutions of hF5-WT, LALA, or isotype control Ab for 1 hour and then used to inoculate Veros with 30 µL per well. Assay plates were incubated for 48 h at 37°C, 5% CO2, and fixed with 4% PFA containing Hoechst nuclear co-stain. Cells were washed gently 1× with PBS and imaged on a TECAN Spark Cyto plate reader. Total cell number (Blue) and infected cell number (green) were quantified for each well, and data were normalized to 100% infection (no antibody) and 0% infection (no virus) control samples.
In vivo challenge studies
All animal work was performed in accordance with protocols approved by the Lawrence Livermore National Laboratory Institutional Animal Care and Use Committee (IACUC # 275). Experiments were conducted under ABSL2 containment, with C3H/HeN females aged 4–8 weeks (Charles River). Each animal received 5e7pfu VEEV-TC83 by intranasal inoculation while under isoflurane-induced anesthesia. Animals were dosed with Abs either −24 h prior to or + 24, +48, or +72 h post infection via intraperitoneal injection (4 mg/kg), n = 20 (−24 hpi, +24 hpi) and n = 35 (+48 hpi) across two experiments, and n = 10 (+72 hpi). Mice were checked daily for neurological symptoms and survival for 14 dpi. Survival data were analyzed using a Log-rank Mantel-Cox test in GraphPad Prism.
Supplementary Material
Acknowledgments
This work was supported by the Defense Threat Reduction Agency [contract HDTRA140027, project CB10489, PI: Harmon]. Sandia National Laboratories is a multimission laboratory managed and operated by National Technology & Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International Inc., for the U.S. Department of Energy’s National Nuclear Security Administration under contract DE-NA0003525. All work performed at Lawrence Livermore National Laboratory is performed under the auspices of the U.S. Department of Energy under Contract DE-AC52-07NA27344. This paper describes objective technical results and analysis. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government.
Funding Statement
This work was supported by the Defense Threat Reduction Agency [contract HDTRA140027, project CB10489, PI: Harmon].
Abbreviations
- Ab
Antibody
- ADCC
Antibody-dependent cell-mediated cytotoxicity,
- ADCP
Antibody-dependent cellular phagocytosis
- BSA
Bovine serum albumin
- CDC
Complement-dependent cytotoxicity
- Fc
Crystallizable fragment
- FcγRs
Fc gamma receptors
- GFP
Green fluorescent protein
- hF5
VEEV-neutralizing human IgG1 antibody F5
- hF5-LALA
Human antibody hF5 with loss of function Fc mutations L234A and L235A
- hF5-WT
Wildtype human antibody F5 with intact Fc function
- hpi
hours post infection
- HRP
Horseradish peroxidase
- IgG
Immunoglobulin G
- NHS
normal human serum
- NMS
normal mouse serum
- PFA
paraformaldehyde
- VEEV
Venezuelan Equine Encephalitis Virus
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2023.2297451
References
- 1.Steele K, Twenhafel N.. Review paper: pathology of animal models of alphavirus encephalitis. Vet Pathol. 2010;47(5):790–16. doi: 10.1177/0300985810372508. [DOI] [PubMed] [Google Scholar]
- 2.Zacks M, Paessler S. Encephalitic alphaviruses. Vet Microbiol. 2010;140:281–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weaver SC, Ferro C, Barrera R, Boshell J, Navarro JC. Venezuelan equine encephalitis. Annu Rev Entomol. 2004;49(1):141–74. doi: 10.1146/annurev.ento.49.061802.123422. [DOI] [PubMed] [Google Scholar]
- 4.Jose J, Snyder JE, Kuhn RJ. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 2009;4(7):837–56. doi: 10.2217/fmb.09.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim AS, Kafai NM, Winkler ES, Gilliland TC Jr., Cottle EL, Earnest JT, Jethva PN, Kaplonek P, Shah AP, Fong RH, et al. Pan-protective anti-alphavirus human antibodies target a conserved E1 protein epitope. Cell. 2021;184(17):4414–29.e19. doi: 10.1016/j.cell.2021.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leung JY, Ng MM, Chu JJ. Replication of alphaviruses: a review on the entry process of alphaviruses into cells. Adv Virol. 2011;2011:249640. doi: 10.1155/2011/249640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li L, Jose J, Xiang Y, Kuhn RJ, Rossmann MG. Structural changes of envelope proteins during alphavirus fusion. Nature. 2010;468(7324):705–8. doi: 10.1038/nature09546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanchez-San Martin C, Liu CY, Kielian M. Dealing with low pH: entry and exit of alphaviruses and flaviviruses. Trends Microbiol. 2009;17(11):514–21. doi: 10.1016/j.tim.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williamson LE, Reeder KM, Bailey K, Tran MH, Roy V, Fouch ME, Kose N, Trivette A, Nargi RS, Winkler ES, et al. Therapeutic alphavirus cross-reactive E1 human antibodies inhibit viral egress. Cell. 2021;184(17):4430–46.e22. doi: 10.1016/j.cell.2021.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rülker T, Voß L, Thullier P, LM OB, Pelat T, Perkins SD, Langermann C, Schirrmann T, Dübel S, Marschall H-J, et al. Isolation and characterisation of a human-like antibody fragment (scFv) that inactivates VEEV in vitro and in vivo. PLoS ONE. 2012;7(5):e37242. doi: 10.1371/journal.pone.0037242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porta J, Jose J, Roehrig JT, Blair CD, Kuhn RJ, Rossmann MG, Dermody TS. Locking and blocking the viral landscape of an alphavirus with neutralizing antibodies. J Virol. 2014;88(17):9616–23. doi: 10.1128/JVI.01286-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunt AR, Frederickson S, Hinkel C, Bowdish KS, Roehrig JT. A humanized murine monoclonal antibody protects mice either before or after challenge with virulent Venezuelan equine encephalomyelitis virus. J Gen Virol. 2006;87(Pt 9):2467–76. doi: 10.1099/vir.0.81925-0. [DOI] [PubMed] [Google Scholar]
- 13.Hunt AR, Frederickson S, Maruyama T, Roehrig JT, Blair CD, Weaver SC. The first human epitope map of the alphaviral E1 and E2 proteins reveals a new E2 epitope with significant virus neutralizing activity. PLoS neglected tropical diseases. PLoS Negl Trop Dis. 2010;4(7):e739. doi: 10.1371/journal.pntd.0000739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kafai NM, Williamson LE, Binshtein E, Sukupolvi-Petty S, Gardner CL, Liu J, Mackin S, Kim AS, Kose N, Carnahan RH, et al. Neutralizing antibodies protect mice against Venezuelan equine encephalitis virus aerosol challenge. J Exp Med. 2022;219(4):e20212532. doi: 10.1084/jem.20212532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodchild S, O’Brien L, Steven J, Muller M, Lanning O, Logue C, D’Elia RV, Phillpotts RJ, Perkins SD. A humanised murine monoclonal antibody with broad serogroup specificity protects mice from challenge with Venezuelan equine encephalitis virus. Antiviral Res. 2011;90(1):1–8. doi: 10.1016/j.antiviral.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 16.Kim AS, Diamond MS. A molecular understanding of alphavirus entry and antibody protection. Nat Rev Microbiol. 2023;21(6):396–407. doi: 10.1038/s41579-022-00825-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hülseweh B, Rülker T, Pelat T, Langermann C, Frenzel A, Schirrmann T, Dübel S, Thullier P, Hust M. Human-like antibodies neutralizing Western equine encephalitis virus. MAbs. 2014;6(3):718–27. doi: 10.4161/mabs.28170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burke CW, Froude JW, Miethe S, Hülseweh B, Hust M, Glass PJ. Human-like neutralizing antibodies protect mice from aerosol exposure with western equine encephalitis virus. Viruses. 2018;10(4):147. doi: 10.3390/v10040147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Erp EA, Luytjes W, Ferwerda G, van Kasteren PB. Fc-mediated antibody effector functions during respiratory syncytial virus infection and disease. Front Immunol. 2019;10(548):548. doi: 10.3389/fimmu.2019.00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu LL, Suscovich TJ, Fortune SM, Alter G. Beyond binding: antibody effector functions in infectious diseases. Nat Rev Immunol. 2017;18(1):46. doi: 10.1038/nri.2017.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saphire EO, Schendel SL, Fusco ML, Gangavarapu K, Gunn BM, Wec AZ, Halfmann PJ, Brannan JM, Herbert AS, Qiu X, et al. Systematic analysis of monoclonal antibodies against ebola virus GP defines features that contribute to protection. Cell. 2018;174(4):938–52.e13. doi: 10.1016/j.cell.2018.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dias AG, Atyeo C, Loos C, Montoya M, Roy V, Bos S, Narvekar P, Singh T, Katzelnick LC, Kuan G, et al. Antibody fc characteristics and effector functions correlate with protection from symptomatic dengue virus type 3 infection. Sci Transl Med. 2022;14(651):eabm3151. doi: 10.1126/scitranslmed.abm3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DiLillo DJ, Ravetch JV. Fc-Receptor Interactions Regulate Both Cytotoxic and Immunomodulatory Therapeutic Antibody Effector Functions. Cancer Immunol Res. 2015;3(7):704. doi: 10.1158/2326-6066.CIR-15-0120. [DOI] [PubMed] [Google Scholar]
- 24.Earnest JT, Basore K, Roy V, Bailey AL, Wang D, Alter G, Fremont DH, Diamond MS. Neutralizing antibodies against mayaro virus require fc effector functions for protective activity. J Exp Med. 2019;216(10):2282–301. doi: 10.1084/jem.20190736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Earnest JT, Holmes AC, Basore K, Mack M, Fremont DH, Diamond MS. The mechanistic basis of protection by non-neutralizing anti-alphavirus antibodies. Cell Rep. 2021;35(1):108962. doi: 10.1016/j.celrep.2021.108962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fox JM, Roy V, Gunn BM, Huang L, Edeling MA, Mack M, Fremont DH, Doranz BJ, Johnson S, Alter G, et al. Optimal therapeutic activity of monoclonal antibodies against chikungunya virus requires Fc-FcγR interaction on monocytes. Sci Immunol. 2019;4(32):eaav5062. doi: 10.1126/sciimmunol.aav5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hessell AJ, Hangartner L, Hunter M, Havenith CE, Beurskens FJ, Bakker JM, Lanigan CMS, Landucci G, Forthal DN, Parren PWHI, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. 2007;449(7158):101–4. doi: 10.1038/nature06106. [DOI] [PubMed] [Google Scholar]
- 28.Nasser R, Pelegrin M, Michaud HA, Plays M, Piechaczyk M, Gros L. Long-lasting protective antiviral immunity induced by passive immunotherapies requires both neutralizing and effector functions of the administered monoclonal antibody. J Virol. 2010;84(19):10169–81. doi: 10.1128/JVI.00568-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang A, Stacey HD, D’Agostino MR, Tugg Y, Marzok A, Miller MS. Beyond neutralization: Fc-dependent antibody effector functions in SARS-CoV-2 infection. Nat Rev Immunol. 2022;23(6):381–96. doi: 10.1038/s41577-022-00813-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winkler ES, Gilchuk P, Yu J, Bailey AL, Chen RE, Chong Z, Zost SJ, Jang H, Huang Y, Allen JD, et al. Human neutralizing antibodies against SARS-CoV-2 require intact Fc effector functions for optimal therapeutic protection. Cell. 2021;184(7):1804–20.e16. doi: 10.1016/j.cell.2021.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vanderven HA, Liu L, Ana-Sosa-Batiz F, Nguyen THO, Wan Y, Wines B, Hogarth PM, Tilmanis D, Reynaldi A, Parsons MS, et al. Fc functional antibodies in humans with severe H7N9 and seasonal influenza. JCI Insight. 2017;2(13). doi: 10.1172/jci.insight.92750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keeler SP, Fox JM. Requirement of Fc-Fc gamma receptor interaction for antibody-based protection against emerging virus infections. Viruses [Internet]. 2021;13(6):1037. doi: 10.3390/v13061037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuller JP, Stavenhagen JB, Teeling JL. New roles for fc receptors in neurodegeneration-the impact on immunotherapy for Alzheimer’s disease. Front Neurosci. 2014;8:235. doi: 10.3389/fnins.2014.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Mathieu M, Brezski RJ. IgG fc engineering to modulate antibody effector functions. Protein Cell. 2018;9(1):63–73. doi: 10.1007/s13238-017-0473-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Idusogie EE, Presta LG, Gazzano-Santoro H, Totpal K, Wong PY, Ultsch M, Meng YG, Mulkerrin MG. Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 fc. J Immunol. 2000;164(8):4178–84. doi: 10.4049/jimmunol.164.8.4178. [DOI] [PubMed] [Google Scholar]
- 36.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520–. doi: 10.3389/fimmu.2014.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bournazos S, Ravetch JV. Fcγ receptor function and the design of vaccination strategies. Immunity. 2017;47(2):224–33. doi: 10.1016/j.immuni.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lofano G, Gorman MJ, Yousif AS, Yu W-H, Fox JM, Dugast A-S, Ackerman ME, Suscovich TJ, Weiner J, Barouch D, et al. Antigen-specific antibody fc glycosylation enhances humoral immunity via the recruitment of complement. Sci Immunol. 2018;3(26):eaat7796. doi: 10.1126/sciimmunol.aat7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hunt AR, Bowen RA, Frederickson S, Maruyama T, Roehrig JT, Blair CD. Treatment of mice with human monoclonal antibody 24h after lethal aerosol challenge with virulent Venezuelan equine encephalitis virus prevents disease but not infection. Virology. 2011;414(2):146–52. doi: 10.1016/j.virol.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cain MD, Salimi H, Gong Y, Yang L, Hamilton SL, Heffernan JR, Hou J, Miller MJ, Klein RS. Virus entry and replication in the brain precedes blood-brain barrier disruption during intranasal alphavirus infection. J Neuroimmunol. 2017;308:118–30. doi: 10.1016/j.jneuroim.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Julander JG, Skirpstunas R, Siddharthan V, Shafer K, Hoopes JD, Smee DF, Morrey JD. C3H/HeN mouse model for the evaluation of antiviral agents for the treatment of Venezuelan equine encephalitis virus infection. Antiviral Res. 2008;78(3):230–41. doi: 10.1016/j.antiviral.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kinney RM, Chang GJ, Tsuchiya KR, Sneider JM, Roehrig JT, Woodward TM, Trent DW. Attenuation of Venezuelan equine encephalitis virus strain TC-83 is encoded by the 5’-noncoding region and the E2 envelope glycoprotein. J Virol. 1993;67(3):1269–77. doi: 10.1128/jvi.67.3.1269-1277.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hezareh M, Hessell AJ, Jensen RC, van de Winkel JGJ, Parren PWHI. Effector function activities of a panel of mutants of a broadly neutralizing antibody against human immunodeficiency virus Type 1. J Virol. 2001;75(24):12161–68. doi: 10.1128/JVI.75.24.12161-12168.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilkinson I, Anderson S, Fry J, Julien LA, Neville D, Qureshi O, Watts G, Hale G. Fc-engineered antibodies with immune effector functions completely abolished. PLoS ONE. 2021;16(12):e0260954. doi: 10.1371/journal.pone.0260954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu D, Alegre M-L, Varga SS, Rothermel AL, Collins AM, Pulito VL, Hanna LS, Dolan KP, Parren PWHI, Bluestone JA, et al. In vitro characterization of five humanized OKT3 effector function variant antibodies. Cell Immunol. 2000;200(1):16–26. doi: 10.1006/cimm.2000.1617. [DOI] [PubMed] [Google Scholar]
- 46.Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umaña P, Mössner E, et al. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel. 2016;29(10):457–66. doi: 10.1093/protein/gzw040. [DOI] [PubMed] [Google Scholar]
- 47.Lo M, Kim HS, Tong RK, Bainbridge TW, Vernes J-M, Zhang Y, Lin YL, Chung S, Dennis MS, Zuchero YJY, et al. Effector-attenuating substitutions that maintain antibody stability and reduce toxicity in mice. J Biol Chem. 2017;292(9):3900–8. doi: 10.1074/jbc.M116.767749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karimi MA, Lee E, Bachmann MH, Salicioni AM, Behrens EM, Kambayashi T, Baldwin, CL. Measuring cytotoxicity by bioluminescence imaging outperforms the standard chromium-51 release assay. PLoS One [Internet]. 2014;9(2):e89357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uccellini MB, Aslam S, Liu STH, Alam F, García-Sastre A. Development of a macrophage-based ADCC assay. Vaccines (Basel). 2021;9(6):660. doi: 10.3390/vaccines9060660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arduin E, Arora S, Bamert PR, Kuiper T, Popp S, Geisse S, Grau R, Calzascia T, Zenke G, Kovarik J, et al. Highly reduced binding to high and low affinity mouse Fc gamma receptors by L234A/L235A and N297A Fc mutations engineered into mouse IgG2a. Mol Immunol. 2015;63(2):456–63. doi: 10.1016/j.molimm.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 51.Dekkers G, Bentlage AEH, Stegmann TC, Howie HL, Lissenberg-Thunnissen S, Zimring J, Rispens T, Vidarsson G. Affinity of human IgG subclasses to mouse Fc gamma receptors. MAbs. 2017;9(5):767–73. doi: 10.1080/19420862.2017.1323159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Overdijk MB, Verploegen S, Ortiz Buijsse A, Vink T, Leusen JHW, Bleeker WK, Parren PWHI. Crosstalk between human IgG isotypes and murine effector cells. J Immunol. 2012;189(7):3430–38. doi: 10.4049/jimmunol.1200356. [DOI] [PubMed] [Google Scholar]
- 53.Nimmerjahn F, Ravetch JV. Divergent Immunoglobulin G Subclass Activity Through Selective Fc Receptor Binding. Sci. 2005;310(5753):1510–2. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]
- 54.Forrester MA, Wassall HJ, Hall LS, Cao H, Wilson HM, Barker RN, Vickers MA. Similarities and differences in surface receptor expression by THP-1 monocytes and differentiated macrophages polarized using seven different conditioning regimens. Cell Immunol. 2018;332:58–76. doi: 10.1016/j.cellimm.2018.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Verma A, Ngundi MM, Meade BD, De Pascalis R, Elkins KL, Burns DL. Analysis of the Fc gamma receptor-dependent component of neutralization measured by anthrax toxin neutralization assays. Clin Vaccine Immunol. 2009;16(10):1405–12. doi: 10.1128/CVI.00194-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gunn BM, Yu WH, Karim MM, Brannan JM, Herbert AS, Wec AZ, Halfmann PJ, Fusco ML, Schendel SL, Gangavarapu K, et al. A role for fc function in therapeutic monoclonal antibody-mediated protection against ebola virus. Cell Host & Microbe. 2018;24(2):221–33.e5. doi: 10.1016/j.chom.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bailey MJ, Duehr J, Dulin H, Broecker F, Brown JA, Arumemi FO, Bermúdez González MC, Leyva-Grado VH, Evans MJ, Simon V, et al. Human antibodies targeting Zika virus NS1 provide protection against disease in a mouse model. Nat Commun. 2018;9(1):4560. doi: 10.1038/s41467-018-07008-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chung KM, Nybakken GE, Thompson BS, Engle MJ, Marri A, Fremont DH, Diamond MS. Antibodies against west nile virus nonstructural protein NS1 prevent lethal infection through Fc γ receptor-dependent and -independent mechanisms. J Virol. 2006;80(3):1340–51. doi: 10.1128/JVI.80.3.1340-1351.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.DiLillo DJ, Tan GS, Palese P, Ravetch JV. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcgammaR interactions for protection against influenza virus in vivo. Nat Med. 2014;20(2):143–51. doi: 10.1038/nm.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu L, Liu X, Ye X, Su W, Zhang X, Deng W, Luo J, Xiang M, Guo W, Zhang S, et al. Monoclonal antibodies against zika virus NS1 protein confer protection via Fcγ receptor-dependent and -independent pathways. mBio. 2021;12(1):10–128. doi: 10.1128/mBio.03179-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pal P, Dowd KA, Brien JD, Edeling MA, Gorlatov S, Johnson S, Lee I, Akahata W, Nabel GJ, Richter MKS, et al. Development of a highly protective combination monoclonal antibody therapy against Chikungunya virus. PLoS Pathog. 2013;9(4):e1003312. doi: 10.1371/journal.ppat.1003312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou QF, Fox JM, Earnest JT, Ng T-S, Kim AS, Fibriansah G, Kostyuchenko VA, Shi J, Shu B, Diamond MS, et al. Structural basis of Chikungunya virus inhibition by monoclonal antibodies. Proc Natl Acad Sci USA. 2020;117(44):27637–45. doi: 10.1073/pnas.2008051117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Powell LA, Miller A, Fox JM, Kose N, Klose T, Kim AS, Bombardi R, Tennekoon RN, Dharshan de Silva A, Carnahan RH, et al. Human mAbs broadly protect against arthritogenic alphaviruses by recognizing conserved elements of the Mxra8 receptor-binding site. Cell Host & Microbe. 2020;28(5):699–711. e7. doi: 10.1016/j.chom.2020.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fox JM, Huang L, Tahan S, Powell LA, Crowe JE Jr., Wang D, Diamond MS. A cross-reactive antibody protects against Ross River virus musculoskeletal disease despite rapid neutralization escape in mice. PLoS Pathog. 2020;16(8):e1008743. doi: 10.1371/journal.ppat.1008743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parekh BS, Berger E, Sibley S, Cahya S, Xiao L, LaCerte MA, Vaillancourt P, Wooden S, Gately D. Development and validation of an antibody-dependent cell-mediated cytotoxicity-reporter gene assay. MAbs. 2012;4(3):310–18. doi: 10.4161/mabs.19873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brooke CB, Schäfer A, Matsushima GK, White LJ, Johnston RE. Early activation of the host complement system is required to restrict central nervous system invasion and limit neuropathology during Venezuelan equine encephalitis virus infection. J Gen Virol. 2012;93(Pt 4):797–806. doi: 10.1099/vir.0.038281-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sharma A, Knollmann-Ritschel B. Current understanding of the molecular basis of venezuelan equine encephalitis virus pathogenesis and vaccine development. Viruses. 2019;11(2):164. doi: 10.3390/v11020164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li L, Jose J, Xiang Y, Kuhn RJ, Rossman MG. Structural changes of envelope proteins during alphavirus fusion. Nature. 2010;468(7324):705–8. doi: 10.1038/nature09546. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.