

Abstract
Mitochondria participate in a wide range of cellular processes. One essential function of mitochondria is to be a platform for antiviral signaling proteins during the innate immune response to viral infection. Recently, studies have revealed that mitochondrion-derived DNAs and RNAs are recognized as non-self molecules and act as immunogenic ligands. More importantly, the cytosolic release of these mitochondrial nucleic acids (mt-NAs) is closely associated with the pathogenesis of human diseases accompanying aberrant immune activation. The release of mitochondrial DNAs (mtDNAs) via BAX/BAK activation and/or VDAC1 oligomerization activates the innate immune response and inflammasome assembly. In addition, mitochondrial double-stranded RNAs (mt-dsRNAs) are sensed by pattern recognition receptors in the cytosol to induce type I interferon expression and initiate apoptotic programs. Notably, these cytosolic mt-NAs also mediate adipocyte differentiation and contribute to mitogenesis and mitochondrial thermogenesis. In this review, we summarize recent studies of innate immune signaling pathways regulated by mt-NAs, human diseases associated with mt-NAs, and the emerging physiological roles of mt-NAs.
Subject terms: Long non-coding RNAs, RNA metabolism, Pattern recognition receptors
Mitochondria-derived DNAs and RNAs: the hidden triggers of innate immune response
Mitochondrial nucleic acids (mt-NAs) play a crucial role in activating various innate immune signaling pathways and are associated with numerous human diseases. Downregulation of mt-NAs or their downstream receptors often alleviates interferon signature. Recent studies reveal that mt-NAs can also serve as key mediators of signaling between mitochondria and the nucleus, thereby contributing to cell physiology. In particular, studies have shown that mt-NAs can stimulate beige adipocyte development in young mice, which has potential therapeutic implications for obesity and insulin resistance. Understanding the multifaceted roles of mt-NAs will provide potential therapeutic strategies for mt-NA-mediated immune signaling regulation in pathophysiology.
This summary was initially drafted using artificial intelligence, then revised and fact-checked by the author.
Introduction
Mitochondria are semi-independent double membrane-bound organelles with their own DNA genome. The mitochondrial genome is characterized by a circular, double-stranded structure comprised of 37 genes, 13 of which encode subunits of oxidative phosphorylation (OXPHOS) complex, making mitochondria essential energy-generation centers of the cell1. In addition to ATP production, mitochondria are responsible for carrying out a wide range of cellular processes. The mitochondrial matrix stores calcium ions, which promote ATP synthesis under physiological conditions and increase mitochondrial permeability to facilitate necrosis under stress conditions2. Moreover, mitochondria generate reactive oxygen species (ROS) that can damage DNA in the nucleus and promote cellular senescence3. Last but not least, mitochondria are essential in mediating innate immune responses, serving as central hubs of cellular defense systems4.
Traditionally, the immune function of mitochondria in response to viral infection has been focused on mitochondrial antiviral signaling proteins (MAVS) located on the mitochondrial outer membrane (MOM)5. The caspase recruitment domain of MAVS interacts with oligomerized retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), such as RIG-I and melanoma differentiation-associated protein 5 (MDA5), that recognize double-stranded RNAs (dsRNAs) in the cytosol6. This interaction leads to the activation of nuclear factor-κB (NF-κB) and interferon regulatory factor 3/7 (IRF3/7), resulting in the transcription of proinflammatory cytokines and type I interferons (IFN-I)7. Therefore, by hosting MAVS on the MOM, mitochondria are the subcellular organelles in which the innate immune response by RLRs is initiated.
Recently, innate immunity to mitochondrial nucleic acids (mt-NAs) has been receiving increasing attention. The human immune system evades and protects cells from aberrant immune activation to endogenous (self) NAs through epigenetic modifications, such as DNA methylation. Indeed, the accumulation of hypomethylated DNAs in the cytosol due to mitotic defects and genomic instability trigger cyclic GMP-AMP synthase (cGAS)-dependent innate immune activation, resulting in the acquisition of autoinflammatory phenotypes8. However, mt-NAs do not undergo modifications as they are sequestered inside mitochondria away from cytosolic sensors; therefore, they do not evade immune system surveillance. As a result, these mt-NAs can be recognized and trigger an aberrant immune response when present in the cytosol. In particular, the unique circular structure of mitochondrial DNA (mtDNA) makes them sentinels of the innate immune system that recognize non-self molecules9. Moreover, the A-form helical structure of mitochondrial double-stranded RNAs (mt-dsRNAs), generated during bidirectional transcription of the circular mitochondrial genome, is recognized by dsRNA sensors and activates the IFN-I pathway. A study by Young and Attardi10 provided biochemical evidence to support the existence of long dsRNAs in human mitochondria. Recently, by utilizing J2 monoclonal antibodies that specifically recognize dsRNAs, Dhir et al. and Kim et al. independently confirmed the presence of cytosolic mt-dsRNAs11,12. More importantly, these cytosolic mt-dsRNAs are considered foreign NAs by our immune surveillance system, and therefore, mt-dsRNAs activate innate immune signaling. Interestingly, recent studies have reported the possible physiological function of cytosolic mt-NAs during beige adipocyte development, suggesting that the mt-NAs may have a function beyond triggering aberrant immune activation and potentially transmit signals from mitochondria to the nucleus13. Overall, this review summarizes the role of mt-NAs as immunogenic ligands, particularly in the development of human diseases that accompany aberrant immune activation, and provides perspectives on the emerging physiological function of mt-NAs. Understanding the multifaceted roles of mt-NAs may lead to a potential therapeutic strategy for mt-NA-mediated immune signaling regulation in pathophysiological contexts.
Mechanisms of the cytosolic release of mt-NAs
The cytosolic release of mt-NAs contributes to the activation of innate immune response systems, which in turn often leads to pathological consequences. It is important to understand how mt-NAs escape from mitochondria and are recognized by cytosolic pattern recognition receptors (PRRs). Several reports have indicated that the loss of mitochondrial integrity and compartmentalization due to mitochondrial stress could result in the cytosolic release of mt-NAs14. These mitochondrial stressors include the excessive accumulation of ROS due to an imbalance in ROS generation and antioxidant defense mechanism pathways and disruption to the mitochondrial network, resulting in mitochondria fragmentation characterized by reduced mitochondrial volume and increased mitochondrial sphericity. For example, during measles virus infection, the viral V protein sequesters the mitochondrial enzyme aminolevulinate synthase in the cytosol to disrupt the mitochondrial metabolism, which subsequently facilitates the fragmentation and degradation of the mitochondrial network, leading to the mtDNA release into the cytosol15. Similarly, herpes simplex virus type 1 infection also triggers the cytosolic efflux of mtDNAs by facilitating mitochondrial fragmentation16.
The release of mtDNAs is mediated by two types of MOM pores (Fig. 1). First, voltage-dependent anion channel 1 (VDAC1) forms oligomers and creates large pores on the MOM to allow the transportation of metabolites and mtDNAs across the mitochondrial membrane17. In this context, mtDNAs must pass through the poorly defined mitochondrial permeability transition pores (mPTPs) located at the mitochondrial inner membrane (MIM)18. mPTPs are transmembrane proteins in the MIM and responsible for the permeabilization of the MOM (MOMP)19. Normally, mPTPs are closed, but they can open under specific conditions such as during a response to increased mitochondrial Ca2+ concentration20. VDAC1 participates in the formation of mPTP complexes and triggers the opening of mPTPs under oxidative stress conditions, thereby acting as an ROS sensor17. Notably, the transcriptional activity of VDAC1 can be stimulated under starvation and oxidative stress conditions, suggesting that the release of mtDNAs might be a programmed stress response21. Moreover, during their release, mtDNAs interact with multiple VDAC1 molecules through the positively charged residues in the N-terminal domain and act as scaffolds to stabilize the VDAC1 oligomers, thereby promoting their own cytosolic efflux17. This positive feedback action is promoted under inflammatory conditions to enhance the mtDNA-mediated immune response. Indeed, inhibiting VDAC1 oligomerization alleviates inflammatory response triggered by cytosolic mtDNAs17.
Fig. 1. Efflux of mt-NAs via MOMP.

Under cellular stress, BAX and BAK are activated, leading to MOMP. MIM permeabilization and efflux of mt-NAs involve herniation through BAX/BAK macropores. The release of mt-NAs is also mediated by mPTP and VDAC1 oligomerization.
In addition to VDAC1-mediated efflux, mtDNAs can be released to the cytosol via pores created by B-cell leukemia/lymphoma 2 (BCL2)-associated X (BAX) and BCL2 antagonist/killer (BAK) proteins. In response to several cellular stressors, free cytosolic BAX proteins are recruited to the MOM and oligomerize with BAK via the exposed BH3 domain in BAK22. As the BAX/BAK oligomers assemble, macropores form, and the MIM herniates and opens into the cytosol, secreting mtDNAs, mitochondrial RNAs (mtRNAs), and other mitochondrial components23. The main difference between VDAC1 and BAX/BAK pores is that VDAC1 oligomers can form both in healthy and in stressed cells and require the opening of the mPTP opening to permeabilize the MIM. In contrast, BAX/BAK macropores form only under severe stress conditions, and their formation often leads to apoptosis23,24.
A recent study demonstrated VDAC-mediated cytosolic release of mtDNAs under pyrimidine-deficient conditions25. In this context, mtDNA release was determined by the expression of mitochondrial protease YME1-like 1 ATPase (YME1L), which preserves cytosolic nucleotide pools by synthesizing pyrimidine from glutamine and limiting the accumulation of the mitochondrial pyrimidine transporter SLC25A3325. The decreased expression of YME1L resulted in the fragmentation of the mitochondrial network, thereby allowing mtDNAs to be released into the cytosol25. Cytosolic mtDNAs then activate the IFN-I response and induce the transcription of IFN-stimulated genes (ISGs)25. Notably, depletion of the YME1L protein boosted ISG expression even in BAX-/BAK-depleted cells. At the same time, treatment with VDAC inhibitor VBIT-4 impaired the induction of ISGs in YME1L-depleted cells, suggesting that mtDNAs were released from mitochondria mainly through VDAC1 pores25.
In contrast, multiple studies have shown that mtRNAs, in particular mt-dsRNAs, are released from the mitochondrial matrix and enter the cytosol via BAX/BAK macropores and MOMP. In cells deficient of polyribonucleotide nucleotidyltransferase 1 (PNPT1), an essential component of the mtRNA degradosome (mtEXO), the levels of mt-dsRNAs were increased and their cytosolic release was facilitated11. In particular, the release of mt-dsRNAs was nearly completely blocked by the downregulation of BAX and BAK11. In addition, stressors such as exogenous dsRNAs, mitochondrial dysfunction through the inhibition of ATP synthase, DNA damage, and oxidative stress can disrupt mitochondrial membrane potential to trigger the cytosolic release of mt-dsRNAs11,26. After release into the cytosol, these mt-dsRNAs are recognized by dsRNA-sensing PRRs such as MDA5, RIG-I, protein kinase R (PKR), and toll-like receptor 3 (TLR3) to trigger antiviral signaling11,12,26,27. Notably, a recent study showed that the unfolded protein response in the endoplasmic reticulum (ER) did not result in the release of mt-dsRNAs into the cytosol28, despite another study demonstrating that ER stress, exacerbated by obesity, led to mitochondrial damage in human adipocytes29.
Innate immune activation by mtDNAs under various pathological conditions
Under stress conditions, mitochondrion-derived danger-associated molecular patterns (DAMPs), such as mtDNAs, succinate, N-formyl peptides, ROS, cardiolipin, and ATP, are released from mitochondria and activate several immune response pathways30. In particular, mtDNAs function as potent agonists of PRRs when present in the cytosol or in the extracellular environment9. Herein, we describe how cytosolic mtDNAs, released in response to various pathological stressors, trigger a cascade of innate immune responses, such as cGAS-stimulator of IFN genes (STING) expression, TLR9 pathway activation, and cytosolic inflammasome formation.
cGAS-STING PATHWAY
The structure and signaling mechanisms of cGAS-STING have been extensively reviewed in recent years31,32. Structurally, cGAS contains three dsDNA-binding sites and recognizes both self- and non-self-DNA in a sequence-independent manner31. cGAS monomers interact with mtDNAs longer than 45 bp and generate stable ladder-like networks of dimers33, leading to the activation of cGAS. Upon activation, cGAS converts GTP and ATP into 2′ 3′-cyclic GMP-AMP (cGAMP), which then binds to STING located on the ER and induces the conformational change of this ER protein. Then, STING is translocated to the Golgi apparatus and subsequently activates NF-κB and IRF3 pathways33. mtDNAs can also form immunostimulatory structures known as Z-DNAs, which are non-canonical structures with a high frequency of alternating purine–pyrimidine regions34. Mitochondrial genome instability promotes Z-DNA accumulation, and Z-DNA binding protein 1 (ZBP1) stabilizes the structure and recruits cGAS to induce IFN-I signaling34. The activation of the cGAS-STING pathway via cytosolic mtDNAs has also been linked to adaptive immunity by activating macrophages35. During bacterial clearance, neutrophil extracellular trap formation results in apoptosis and the extrusion of mtDNAs into the extracellular space35. Macrophages then engulf these extracellular mtDNAs to trigger the expression of IFN-I in a cGAS-dependent mechanism, resulting in abnormal macrophage activation35.
The cGAS-STING pathway and mtDNAs are involved in the pathogenesis of several conditions and cases of organ failure. High cGAS-STING activity and IFN-I response were observed in lung samples of SARS-CoV-2-infected patients with severe tissue damage36. Pharmacological inhibition of STING alleviated lung inflammation and improved disease prognosis in a mouse model of SARS-CoV-236. The activation of the cGAS-STING pathway was also evident during liver inflammation, such as that observed in non-alcoholic fatty acid liver disease (NAFLD), hepatocellular carcinoma, cirrhosis, and viral hepatitis37. In the liver, the cGAS-STING pathway induces the progression of NAFLD to non-alcoholic steatohepatitis (NASH) in Kupffer cells (KCs)38. A study by Chen et al. showed that Sam50 gene was essential for maintaining mitochondrial membrane integrity and mtDNA stability in hepatocytes39. Liver-specific knockout of Sam50 in mice led to increased mitochondrial and cytosolic mtDNA levels, which resulted in the activation of the cGAS-STING pathway and subsequent liver inflammation39.
In cases of acute kidney injury (AKI), a nephrotoxic reagent induced mtDNA leakage into the cytosol and activated cGAS-STING signaling, ultimately resulting in renal tubular inflammation40. Indeed, STING deficiency ameliorated the kidney damage and inflammation induced by nephrotoxic cisplatin40. In addition, macrophages from rheumatoid arthritis (RA) patients expressed tumor necrosis factor (TNF), which inhibited PTEN-induced putative kinase 1 (PINK1)-mediated mitophagy and altered mitochondrial function to elevate cytosolic mtDNA levels41. mtDNA-induced cGAS-STING pathway activation has also been implicated in multiple neurodegenerative disorders, notably amyotrophic lateral sclerosis (ALS), a condition characterized by the cytosolic accumulation of TAR DNA-binding protein 43 (TDP-43)42. In neurons of ALS patients, TDP-43 invades mitochondria, which results in the release of mtDNAs via mPTPs and subsequent neuroinflammation42. Consistent with this mechanism, the level of cGAS signaling-related metabolites is significantly upregulated in the spinal cords of ALS patients42.
TLR9 PATHWAY
The cytosolic and extracellular mtDNAs can be sensed by TLR9, which is an immune cell (neutrophils, macrophages, and dendritic cells)-specific receptor located in the ER43. TLR9 directly senses hypomethylated CpG motifs in early endosomes43. As CpG motifs of endogenous mtDNAs are unmethylated, mtDNAs function as strong agonists for TLR9. When bound to mtDNAs, TLR9 recruits myeloid differentiation factor 88 (MyD88) and forms a signaling complex with interleukin-1 (IL-1) receptor-associated kinases (IRAKs) and tumor necrosis factor receptor-associated factor 6 (TRAF6) as well as with IRF7, transforming growth factor-β-activated kinase 1 (TAK1)-NF-κB inhibitor IκB (IκKBα) or TAK1-mitogen-activated protein kinase 1 (MAPK1) to promote proinflammatory gene expression via the activation of the transcription factor IRF7, NF-κB or activating protein-1 (AP-1)44. During SARS-CoV-2 infection, the mitochondrial network is impaired, and the released mtDNAs activated TLR9 signaling in endothelial cells, which exacerbates disease progression45. The release of mtDNAs also plays a critical role in the severity of SARS-CoV-2 infection and the initiation of sterile systemic inflammatory response syndrome by activating the TLR9/NF-κB pathway, resulting in the overproduction of proinflammatory cytokines45,46.
Cytosolic Inflammasomes
Finally, cytosolic mtDNAs contribute to the activation of inflammasomes, such as nucleotide-binding and oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) and absent in melanoma 2 (AIM2), which facilitate the secretion of proinflammatory cytokines9. NLRP3 is a cytosolic PRR that is activated in response to a broad range of pathogen-derived and endogenous agents. In response to DAMPs, NLRP3 undergoes conformational changes to expose its central nucleotide-binding and oligomerization domain, promoting its oligomerization. AIM2 is another cytosolic receptor that recognizes dsDNAs and activates the inflammasome cascade. Extracellular dsDNAs released from necrotic cells are engulfed and recognized in cells by the AIM2 inflammasome, which drives pyroptosis and proteolytic maturation of the proinflammatory cytokines such as IL-18 and IL-1β47. Thus, cytosolic mtDNAs are ideal endogenous agonists that trigger NLRP3/AIM2 inflammasomes during necrosis or under cellular stress conditions. Once NLRP3/AIM2 inflammasomes are assembled, pro-caspase-1 is autocleaved, and the activated caspase drives the maturation and secretion of proinflammatory cytokines48. NLRP3/AIM2 inflammasome-activated caspase-1 also cleaves Gasdermin D (GSDMD), releasing the N-fragment of GSDMD, which in turn forms cell membrane pores to facilitate pyroptosis49.
The activation of cytosolic inflammasomes plays a critical role in regulating disease pathogenesis, such as acute myocardial infarction, systemic lupus erythematosus (SLE), inflammatory bowel disease such as Crohn’s disease, and bacterial infections50. Moreover, the increased production of IL-18 and IL-1β mediated by inflammasomes contributes to the onset and progression of RA, type II diabetes, and various neurodegenerative disorders, including Alzheimer’s disease (AD) and ALS51. The levels of AIM2 inflammasome-related proteins in the liver and those of IL-18 and IL-1β in serum were elevated in high-fat diet (HFD)-induced NAFLD model mice52. Moreover, mtDNA treatment promoted palmitate-induced activation of the AIM2 inflammasome and, subsequently, hepatocyte pyroptosis-exacerbated NAFLD52. In patients with type II diabetes, circulating cell-free mtDNAs induce AIM2 inflammasome-dependent caspase-1 activation and an increase in secreted IL-18 and IL-1β from macrophages53. Collectively, these studies suggest the close association of mtDNAs in human pathological conditions with inflammasome activation (Fig. 2).
Fig. 2. mtDNA-mediated immune signaling pathways.
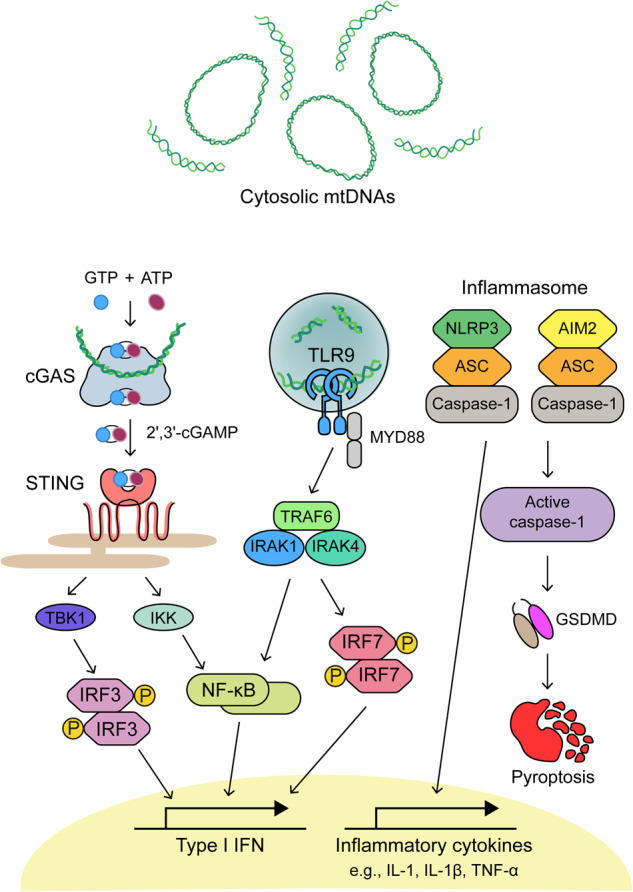
cGAS interacts with circular mtDNAs to form cGAMP, which then binds to STING on the ER. STING promotes the phosphorylation and dimerization of IRF3 through the actions of TBK1 and IKK to activate inflammatory gene expression. Upon mtDNA recognition, TLR9 in endosomal compartments activates NF-κB and IRF7 through the MyD88-dependent pathway and controls the expression of IFN-Is. Finally, mtDNAs activate cytosolic inflammasomes, leading to the maturation and secretion of inflammatory cytokines as well as the cleavage of caspase-1 to promote GSDMD-mediated pyroptosis.
Innate immune response to mt-dsRNAs
There is accumulating evidence that mtRNAs, in particular mt-dsRNAs, are important DAMPs that can activate the innate immune response and inflammatory pathways. Each cell contains thousands of copies of circular mtDNA genomes, which generate long dsRNAs via bidirectional transcription. The two complementary strands of the circular mitochondrial genome, the heavy (H) and the light (L) strands, are transcribed when mitochondrial transcription factor A (TFAM) recruits mitochondrial RNA polymerase (POLRMT) to promoters (denoted as HSP and LSP) via the POLRMT N-terminal extension54. The resulting two complementary strands of mtRNAs can bind to each other and form mt-dsRNAs that can be as long as the entire mitochondrial genome10.
mt-dsRNA recognition by dsRNA-sensing PRRs
Once released into the cytosol, mt-dsRNAs are detected by RLRs, such as MDA5 and RIG-I, which initiate the IFN-I response. The domain structures in MDA5 and RIG-I are similar, although MDA5 preferentially binds long dsRNAs, which activate it55. MDA5 interacts with dsRNAs through the C-terminal domain by binding to the RNA phosphate backbone and 2′-hydroxyl groups, resulting in sequence-independent RNA recognition55. The extended duplex structure of mt-dsRNAs serves as a platform for MDA5 oligomerization, which is required for MDA5 activation. As another member of the RLR family, RIG-I interacts with dsRNAs derived from mitochondria as well. However, RIG-I recognizes short blunt-ended dsRNAs bearing 5′-triphosphate or can also bind 5′-diphosphate dsRNAs, but with lower affinity than dsRNAs with 5′-triphosphate55. Transcription of short mtDNAs due to DNA double-stranded break results in the transcription run-off and generation of short dsRNAs carrying a 5′-end triphosphate, ideal substrates for RIG-I55. Thus, the mitochondrial genome can generate both long and short mt-dsRNAs that can activate MDA5 and RIG-I, respectively. Once activated, the two proteins transmit signals through MAVS to initiate a downstream innate immune response55.
PKR is another key cytosolic dsRNA sensor that interacts with mt-dsRNAs. PKR binding to dsRNAs is mediated through two dsRNA-binding domains, which results in the dimerization and the subsequent autophosphorylation of key residues, including Thr446 and Thr45156. Activated PKR binds to a conserved site in the alpha subunit of eukaryotic initiation factor 2 (eIF2α) and induces eIF2α phosphorylation at Ser51, blocking translation at the initiation step56. Moreover, PKR activation triggers the expression of inflammatory cytokine and IFN-I mediated by the IKK/NF-κB and TAK1/AP-1 pathways. Recently, many studies have revealed the roles of mt-dsRNAs as key activators of PKR in numerous human pathologies. During osteoarthritis development, mitochondrial dysfunction results in the cytosolic accumulation of mt-dsRNAs, which activate PKR and initiate apoptosis in chondrocytes26. Notably, blocking mtRNA transcription using a small chemical inhibitor of POLRMT, 2-C′-methyladenosine (2-CM), decreased the level of mtRNAs and attenuated PKR phosphorylation under osteoarthritis-mimicking stress conditions26. Similarly, PKR activation mediated by mt-dsRNAs has been reported in the context of autoimmune Sjögren’s disease (SjD)57. Specifically, polyinosinic-polycytidylic acid (poly I:C), a synthetic analog of viral dsRNAs, was used to stimulate IL-7 production and early inflammatory responses in salivary gland acinar cells to establish a model of SjD-like sialadenitis58. Interestingly, poly I:C stimulation led to the elevation of total and cytosolic mt-dsRNA levels, subsequent PKR phosphorylation, and ISG induction, although the molecular mechanism that drives the elevation of mt-dsRNAs remains to be investigated57. In this context, cytosolic mt-dsRNAs acted as positive feedback factors to augment the antiviral signaling initiated by poly I:C. Finally, the release of mt-dsRNAs and concomitant upregulation of innate immune signaling were observed in neurodegenerative disorder Huntington’s disease (HD) patients, where the mt-dsRNAs directly bound, activated PKR, and induced inflammatory response as well as programmed cell death in neurons59.
In the endosomal pathway, TLR3 recognizes mt-dsRNAs present in the extracellular space as both free-form and in exosomes. Studies showed that extracellular mt-dsRNAs undergo endocytosis and are transported into the endosomal lumen where they function as ligands of TLR326,27. Once TLR3 senses dsRNAs, the toll-IL-1 receptor (TIR) domain undergoes a conformational change and its tyrosine residues are phosphorylated, resulting in the formation of dually phosphorylated TLR360. Phosphorylated TLR3 then induces the oligomerization of TIR-domain-containing adapter-inducing IFN-β (TRIF), which subsequently activates IRF3 and NF-κB to produce proinflammatory cytokines and IFN-Is60. In an alcoholic liver disease (ALD) mouse model, extracellular mt-dsRNAs in exosomes were implicated in the increased expression of IL-1β and interleukins, especially IL-17A, in KCs27. In this context, the alcohol-associated stress in hepatocytes led to accumulated mt-dsRNAs in the cytosol and in the extracellular space due to a significant decrease in the expression of PNPT1. Another study showed that the overexpression of heat shock protein 60 (HSP60) alleviated HFD-elicited mtDNA and mt-dsRNA release into the extracellular space, thereby preventing the recognition of mt-NAs by TLR3 and MDA5 in the liver61. Moreover, mitochondrial dysfunction during osteoarthritis development resulted in the release of exosomal and cell-free mt-dsRNAs, which activated TLR3 and induced the expression of downstream factors, such as IRF3, TRAF3, and LY6E mRNAs in neighboring chondrocytes26. The implication of these cell line data was further supported by an analysis of synovial fluids from osteoarthritis patients, which showed elevated levels of mt-dsRNAs compared to those of mRNAs or noncoding RNAs (ncRNAs)26. Overall, the activation of PRRs by mt-dsRNAs may drive the development of several human diseases, including ALD27, HD59, and osteoarthritis26.
Autoantibodies associated with mtRNAs
Interestingly, mtRNAs are also recognized by autoantibodies characteristically generated in the context of autoimmune disorders. In SLE patients, both immunoglobulin G (IgG) and IgM subclass autoantibodies targeting mtRNAs have been detected62. These findings suggest that mtRNAs may be mitochondrial neoantigens and support the idea that mitochondria are important sources of circulating autoantigens in patients with SLE62. In addition, mt-dsRNAs are closely associated with the downstream response to an increase in the number of autoantibodies in SjD patients57. Under physiological conditions, acetylcholine (Ach), the endogenous ligand for muscarinic acetylcholine receptor subtype 3 (M3R), suppresses ISG induction partially by preventing the cytosolic release of mt-NAs63. However, SjD patients show an elevated level of anti-M3R autoantibodies, which counter the protective effect of Ach, aggravating the antiviral responses57. Collectively, studies show that mt-dsRNAs are potent cytosolic and extracellular activators of dsRNA sensors and are associated with a range of human diseases that are accompanied by inflammation and cell death (Fig. 3).
Fig. 3. mt-dsRNA-mediated immune signaling pathways.

Cytosolic mt-dsRNAs can activate PKR, MDA5, and RIG-I to initiate apoptotic programs as well as IFN-I and inflammatory responses. In addition, extracellular mt-dsRNAs are recognized by TLR3, resulting in the production of IFN-Is and proinflammatory cytokines.
Regulation of mt-NA-mediated immune activation
The efflux of mt-NAs and their immunostimulatory interactions with molecular sensors provoke critical activation of innate immune signaling pathways. To counter the potentially harmful effects, including apoptosis, caused by mitochondrial failure, cells have adapted countermeasures to prevent and resolve stress arising from the aberrant accumulation of mt-NAs. mtDNA transcription and replication give rise to mitochondrial R-loop and G-quadruplexes (G4) structures, which show immunostimulatory effects. R-loops are nucleic acid structures composed of an RNA–DNA hybrid and a displaced ssDNA. Silva et al. reported that the persistent and inadvertent formation of R-loops blocked replication and transcription machinery activity, leading to genome instability and, ultimately, the induction of human diseases such as neurodegenerative disorders and cancer64. Moreover, the mtEXO, formed by human suppressor of var1,3-like 1 (hSUV3) and PNPT1, is a guardian of mitochondrial genome integrity that counteracts the excessive accumulation of mitochondrial DNA–RNA hybrids64. In this context, an mtEXO binds and processes new transcripts to deter DNA–RNA hybridization and thus preserves individual DNA molecules. Alternatively, an mtEXO may play a more direct role where hSUV3 helicase unwinds the hybridized RNA moiety, which allows its immediate degradation by PNPT164. When DNA–RNA hybrids are not degraded, long stretches of cytosolic DNA–RNA hybrids are bound to cGAS, which triggers cGAS-STING-dependent expression of antiviral genes65.
In addition to R-loops, some mt-NA sequences rich in guanines adopt a non-canonical G4 conformation66. The guanine-rich heavy strand is a potential hotspot for G4 formation, which disrupts mitochondrial replication and transcription machinery actions66. Specifically, LSP transcription can be arrested by a hybrid DNA–RNA G4 structure formed at conserved block sequences66. Moreover, G4s are linked to origins of the mitochondrial genome instability and mtDNA deletion associated with human diseases, such as renal cell carcinomas67. Therefore, mitochondrial G4 structures can potentially be utilized as predictive markers for cancer associated with abnormal mtDNA metabolism67. Considering their high copy number and gene density, mtDNAs have the potential to amplify the effects of G4 dysregulation, resulting in compromised mitochondrial respiratory function and cell viability66. Hence, it is critical to identify and predict potential mitochondrial G4s and the proteins with which they interact to prevent defects in the mitochondrial genome.
The abundance of mt-dsRNAs is also regulated to prevent ectopic activation of immune responses. One of the regulatory proteins involved is RNA exonuclease 2 (REXO2), a 3′-to-5′ exonuclease that degrades small RNAs generated during mtRNA processing in the mitochondrial matrix68. REXO2 prevents the aberrant accumulation of short mt-dsRNAs that can induce an IFN response or the formation of R-loops that interfere with mtDNA maintenance68. Mitochondrial poly(A) polymerase (MTPAP) adds short poly(A) tails to the 3′ ends of most H- and L-strand RNAs to stabilize them69. Similarly, leucine-rich pentatricopeptide repeat motif-containing protein (LRPPRC) interacts with steroid receptor RNA activator-stem loop-interacting RNA-binding protein (SLIRP) via a PPR–RNA recognition motif interface70 to form a dimer that protects mtRNAs from mtEXO by providing a physical barrier71. The loss of LRPPRC also results in transcript-specific alterations in the steady-state levels of mtRNAs and their poly(A) tail length72, thereby suggesting that the LRPPRC/SLIRP complex acts as an RNA molecular chaperon to promote RNA polyadenylation and thus protects RNA from degradation72.
Another important layer of posttranscriptional regulation of mtRNAs is provided by mitochondrial RNA granules (MRGs)73. MRGs are fluid-like condensates involved in RNA processing and degradation mediated by the mtEXO74. Specifically, hSUV3 unwinds RNA substrates, and the phosphate-dependent 3′-to-5′ exonuclease PNPT1 preferentially degrades the mtRNA strand transcribed from the L-strand11. Indeed, the downregulation of PNPT1 and/or hSUV3 results in the stabilization of L-strand mtRNAs and accumulation of mt-dsRNAs11,74. Recently, increasing attention has been focused on the protective role of PNPT1 in the mt-dsRNA-induced IFN response, emphasizing its importance in human health. Pathogenic mutations in both PNPT1 and hSUV3 genes led to the mt-dsRNA accumulation, which has been implicated in neurodegenerative syndromes, including Aicardi–Goutières syndrome75,76. Furthermore, an isoform of an mtRNA-binding protein, G-rich sequence factor 1 (GRSF1), regulates nascent RNA processing in MRGs77. Foci in the mitochondrial matrix contain GRSF1, nascent mtRNA, and RNase P, which is an enzyme responsible for classical and tRNA-less RNA precursor processing78. Defects in GRSF1-mediated mtRNA processing have been associated with several mitochondrial disorders in hepatocytes and OXPHOS dysfunction in skeletal muscles and bone marrows77.
mt-NAs as potential therapeutic targets
Due to their strong immunogenicity, mt-NAs are emerging as potential therapeutic targets to alleviate inflammatory responses. For example, inhibiting VDAC1 oligomerization by VBIT-4 attenuated immune activation and lupus-like symptoms by preventing the cytosolic release of mtDNAs17. In addition, factors mediating the downstream response to cytosolic mt-NAs can be targeted. One study showed that doxorubicin, a DNA-damaging chemotherapeutic agent, induced mtDNA instability and Z-DNA accumulation in cardiomyocytes, which triggered a robust cardiac IFN-I response that was dependent on ZBP1, STING, and IFNAR34. By targeting ZBP1, the cardiotoxicity induced by doxorubicin was reduced and heart function was improved, indicating that ZBP1 is a potential therapeutic target to prevent chemotherapy-related heart failure34.
Leveraging mitophagy to remove damaged mitochondria and their immunogenic debris is an alternative strategy for modulating mt-NA-mediated immune activation. The inhibition of mitophagy by downregulating PINK1 triggered the accumulation of mtDNAs in the cytosol, exacerbating RA-related symptoms41. By exploiting this effect, Xie et al. proposed targeting PINK1 as a strategy to enhance therapeutic responses to chemoimmunotherapy79. Similarly, autophagy can remove cytosolic mt-dsRNAs to prevent triggering a downstream immune response. Indeed, autophagy inducers, such as torin-1 and metformin, reduced the cytosolic level of mt-dsRNAs that had accumulated in response to mitochondrial stress in chondrocytes26. The decrease in mt-dsRNA levels subsequently attenuated the IFN-I response and rescued cell death. More importantly, this rescuing effect of autophagy inducers was diminished in mtRNA-deficient cells, suggesting that the effects were mediated partly by alterations to mtRNA levels26.
The expression of mtRNAs can be directly modulated. A recent study revealed a significant decrease in mt-dsRNA production and subsequent immunosuppression in breast cancer cells under hypoxic conditions80. This outcome suggests a novel mechanism of immunosuppression that can be utilized by hypoxia-inducing therapies80. In cell models of osteoarthritis and SjD, the attenuation of IFN signatures has been observed when the expression of mtRNAs was suppressed by treating the cells with 2-CM, an inhibitor of POLRMT26,57. In addition to 2-CM, a first-in-class specific inhibitor of mitochondrial transcription (IMT1B) has been developed, by Bonekamp et al.81, and it may be an alternative drug used for downregulating mt-dsRNA expression. In fact, IMT1B showed potent antitumor properties, and IMT1B treatment led to progressive loss of mtDNA levels and increased cell death82. Targeting POLRMT via IMT1B also suppressed endometrial carcinoma growth by impairing mitochondrial function, leading to mtDNA transcription inhibition, decreased mitochondrial membrane potential, ROS production, oxidative stress, and ATP loss83. Overall, modulating the expression of mtRNAs is a promising therapeutic strategy for not only attenuating symptoms and treating inflammatory diseases but also for enhancing the efficacy of chemoimmunotherapy.
Extracellular vesicles (EVs) enriched with mitochondrial contents (mitoEVs) are emerging as therapeutic materials. These vesicles deliver mitochondrial components that trigger the release mitochondrial DAMPs and alter cell signaling pathways in recipient cells under pathological conditions, such as inflammation, cancers, and lung diseases84. For instance, acute myeloid leukemia cells showed enriched levels of mitochondria in microvesicles (MVs) during differentiation, and inhibiting MV formation prevented myeloid differentiation85. In addition, mouse melanoma cells released mtDNA-rich EVs to induce cytokine production in macrophages, which suppressed the immune responses in the tumor microenvironment triggered by cytotoxic T cells86. On the other hand, mitoEVs from healthy cells are promising therapeutics for lung injuries because of their metabolic and/or immune regulatory effects84. For example, mitoEVs derived from the mesenchymal stem cells delivered healthy mitochondria to improve the oxygen consumption rate as well as the mtDNA and ATP levels in alveolar macrophages87. Considering that EV-mediated delivery of mt-NAs can be achieved in cells under either a physiological or pathological state, mitoEVs have promising diagnostic and therapeutic potential.
Physiological roles of mt-NAs
Due to their strong association with pathologies, it is unclear why mitochondria carry unmethylated circular DNAs and generate long dsRNAs that are detrimental to cells when released to the cytosol, as described above. Hence, discerning the potential physiological roles of mt-NAs remains as a challenge in the field. Recent studies have revealed strategies that cells employ to evade mt-NA-mediated immune activation and utilize cytosolic mt-NAs as beneficial signaling molecules. In particular, an increasing number of studies have reported on the role of mt-NAs during adipogenesis. Herein, using adipogenesis and beige adipocytes as case studies, we describe how mt-NAs can be used as upstream cues to trigger non-IFN responses.
Adipose tissue plays a crucial role in metabolic regulation and homeostasis by controlling energy storage and expenditure. White adipose tissue (WAT) and brown adipose tissue are the two main types of fat tissue, with the former functioning as a reservoir of energy in a single large lipid droplets and the latter dissipating energy as heat through nonshivering thermogenesis mediated with highly dense mitochondria88. Additionally, a thermogenic population of adipocytes, known as beige adipose tissue, is also found interspersed among subcutaneous WATs (scWATs). During adipose tissue remodeling of browning, scWAT acquires brown fat-like features, such as an elevated level of the thermogenic mitochondrial protein uncoupling protein 1 (UCP1) and an increased number of mitochondria89. Even in adipocytes, dysregulation or considerable abundance of mitochondria is associated with the release of mt-NAs into the cytosol13. Impaired mitochondrial respiration and enhanced ROS production in adipose tissue have ben related to the cytosolic release of mtDNAs, which increase the possibility of STING-dependent inflammation and insulin resistance under conditions of obesity90. Indeed, the injection of mtDNAs or mtRNAs into the cytosol of adipocytes triggered an increase in Ifn-β expression in adult mice13. In turn, IFN immune responses damaged mitochondrial capacity for fat oxidation and thermogenesis, leading to inflammation and insulin resistance13.
During beige adipocyte differentiation, a developmental program that mitigates the potential risk of mtRNA efflux is activated because of an increase in the number of mitochondria13. Interestingly, despite carrying more mitochondria, adipocytes of young mice do not show Ifn responses after stimulation by mtRNAs due to the suppression of Irf7 production by vitamin D receptor (Vdr) signaling13. In adult mouse inguinal adipocytes, the Irf7-associated gene network, including ISGs such as Aim2, Ddx41, Zbp1, and Ifi205g genes, was highly expressed, promoting the initiation of the Ifn response to the increase in cytosolic mt-NA levels13. Consistent with these results observed in mice, the results observed in the adipocytes of lean children (1-4 years) revealed reduced expression of IFN-β in response to cytosolic mtRNAs compared to that of adipocytes of adolescents (16-17 years)13. In beige adipocytes of young mice, mtRNAs contributed to mitogenesis and mitochondrial thermogenesis under specific conditions but not to an Ifn response13. Cytosolic mtRNAs upregulated the expression of Ucp1, a key player in thermogenic mitochondrial function, and other beige adipocyte marker genes, namely, Ppargc1a, Cidea, and Dio213. Therefore, they increased the thermogenic activity of mitochondria. The absence of dsRNA sensors impeded the induction of thermogenic gene expression and ultimately resulted in the loss of beige adipocytes in young mice, indicating that the effects of cytosolic mtRNAs were facilitated through the dsRNA structure13. This study suggests the involvement of the Il-6 gene in the mtRNA-mediated control of adipocyte remodeling, as this gene enhances thermogenic adipocyte development13. The dsRNA sensors activated by mtRNAs increase Il-6 expression, and the inhibition of Il-6 signaling compromises the effect of mtRNAs on the beige adipocyte gene expression13. The molecular mechanisms underlying mtRNA-triggered signaling, originating in mitochondria and controlling transcription in the nucleus, remain to be characterized.
The impact of mtDNAs on thermogenic gene expression in adipocytes was revealed by Bai et al., who examined the effect of fat-specific knockout of the disulfide bond A oxidoreductase-like gene (DsbA-L), which encodes a chaperone-like mitochondrial protein91. DsbA-L knockout promoted the cytosolic release of mtDNAs, triggering the activation of the cGAS-STING pathway. As a result, the expression of Ucp1, Ppargc1a, Cebpb, and Prdm16, genes associated with the thermogenic function of beige and brown adipocytes, was suppressed, leading to decreased thermogenesis and increased adiposity91. The cGAS-STING pathway mediated the effects of DsbA-L knockout on thermogenic gene expression and metabolic phenotype acquisition by inhibiting protein kinase A (PKA) signaling through the downregulation of phosphodiesterase (PDE)-dependent adenosine 3′,5′-cyclic monophosphate (cAMP)91. Additionally, Aim2, a cytosolic inflammasome, has been implicated in adipogenesis through the Ifn-inducible gene Ifi202b92. These findings suggest that mtDNA modulates adipogenic and thermogenic programs in conjunction with immune response regulation (Fig. 4).
Fig. 4. mt-NAs-mediated regulation of thermogenic adipocytes.

A Mitochondria and small lipid vacuoles accumulate as adipogenic precursors differentiate into mature beige adipocytes. Mature beige adipocytes dissipate heat through nonshivering thermogenesis, which increases energy expenditure. B mtRNA-mediated stimulation of thermogenesis in young adipocytes with a suppressed Ifn response. Cytosolic mtRNAs recognized by dsRNA sensors activate thermogenic adipocyte differentiation through Il-6-Jak/Stat signaling in young mice and humans. In older or obese mice/humans, cytosolic mt-NAs induce an Ifn response and abrogate the expression of genes related to thermogenesis.
A high mtDNA copy number is associated with the potential physiological functions of mt-NAs. A decrease in the mtDNA copy number in the peripheral blood has been associated with chronic kidney diseases, suggesting the involvement of mtDNAs in renal function93. In addition, multiple mtDNA deletion mutations accumulate with age in tissues with high respiratory rates, such as the brain. A study by Nicholas et al. reported that the decreased expression of transcription factor B2 (TFB2M) led to increased risk of diabetes by disrupting the transcription of mtDNAs and reducing the mtDNA content94. Although the exact mechanism and the key factors regulating mt-NA levels in specific cellular and physiological contexts remain to be investigated, recent findings have implicated mt-NAs in many processes related to cell physiology.
Conclusion
This review summarizes the critical role of mt-NAs as endogenous ligands that activate various innate immune signaling pathways. In addition, it describes the potential physiological function of mt-NAs, especially during beige adipocyte differentiation. When released to the cytosol via MOMP, mtDNAs initiate cell signaling pathways such as the cGAS-STING and TLR9 pathways and induce inflammasome assembly, leading to the activation of transcription factors such as IRF3 and NF-κB and the production of IFN-Is and inflammatory cytokines. Furthermore, mt-dsRNAs, generated via the bidirectional transcription of the circular mitochondrial genome, are recognized by dsRNA sensors, such as MDA5, RIG-I, PKR, and TLR3, which induce the expression of proinflammatory cytokines, including IFN-Is, and mediate cell death via global suppression of translation. The recognition of mt-NAs by PRRs and the human diseases associated with mt-NAs are summarized in Table 1, along with information about the potential therapeutic benefits of targeting mt-NAs and their downstream signaling in Table 2.
Table 1.
Summary of mt-NAs, their interacting receptors, and the associated human diseases.
| mt-NA | Interacting receptors | Associated diseases |
|---|---|---|
| mtDNA | cGAS |
SARS-CoV-236 Nonalcoholic steatohepatitis38 Acute kidney injury40 Rheumatoid arthritis41 Amyotrophic lateral sclerosis42 |
| TLR9 |
SARS-CoV-2 infection45 Systemic inflammatory response syndrome46 |
|
| NLRP3 |
Acute myocardial infarction50 Systemic lupus erythematosus50 Rheumatoid arthritis51 Type II diabetes51 Parkinson’s disease, Alzheimer’s disease, and multiple sclerosis51 |
|
| AIM2 |
Nonalcoholic fatty acid liver disease and hepatocyte pyroptosis52 Type II diabetes53 |
|
| mtRNA | RLRs | Aberrant immune responses11 |
| TLR3 |
Osteoarthritis26 |
|
| PKR |
Osteoarthritis26 Sjögren’s disease57 Huntington’s disease59 |
Table 2.
Summary of potential therapeutic effects expected by targeting mt-NAs.
| Associated diseases | mt-NA-mediated therapeutic effects | |
|---|---|---|
| Target | Potential therapeutic effects | |
| SARS-CoV-2 | STING36 | STING inhibition reduces SARS-CoV-2-induced inflammation in mice. |
| TLR945 | Targeting TLR9 rescues compromised endothelial cell function due to reduced expression of nitric oxide synthase. | |
| Non-alcoholic steatohepatitis | STING38 | STING functions as an mtDNA sensor in KCs of the liver under lipid overload to induce NF-κB-dependent inflammation in the context of NASH. |
| Acute kidney injury | cGAS-STING40 | Reduced activation of the cGAS-STING pathway prevents cytosolic mtDNA-mediated inflammation in cisplatin-induced AKI. |
| Rheumatoid arthritis | cGAS-STING41 | cGAS deficiency ameliorates arthritic symptoms and reduces ISG induction. |
| NLRP3 inflammasome51 | Targeting NLRP3 or downstream caspases suppresses IL-1 production in the RA context. | |
| Amyotrophic lateral sclerosis | cGAS-STING42 | cGAS and STING inhibitors prevent TDP-43-induced inflammation and improve the symptoms of neuronal decline in patients with diseases involving TDP-43 proteinopathy. |
| Liver disease | TLR327 | Targeting mt-dsRNA-mediated TLR3 activation prevents the recruitment of γδ T cells and expression of IL‐17A in the early stage of ALD. |
| HSP6061 | Overexpression of HSP60 significantly reduces weight gain and fat accumulation in adipose tissue as well as mt-dsRNA-mediated inflammation in chronic kHFD. | |
| Osteoarthritis | POLRMT26 | Reduced expression of mt-dsRNAs by 2-CM treatment rescues the activation of PKR and eIF2α and reduces ISG induction. |
| Sjögren’s disease | POLRMT57 | Downregulation of mtRNAs reduces the phosphorylation of PKR and attenuates ISG induction, which rescues the viability of salivary gland acinar cells. |
mt-dsRNAs, in particular, are emerging as underlying immune activators that drive the pathogenesis of a wide range of human diseases, which opens up new insights and leading to questions that need to be answered. A recent report by Kim et al. described three complementary approaches to analyze the expression of mt-dsRNAs in multiple cell types95. Technical advancements in mt-dsRNA analysis will allow researchers to carry out the additional mechanistic and biochemical studies required to elucidate the formation of mt-dsRNAs, the posttranscriptional regulation of mt-dsRNAs, and the precise molecular structures that are recognized by PRRs. In addition to that directed to mt-dsRNAs, research is needed on the mitochondrial genome, which encodes long ncRNAs, small ncRNAs, and even circular RNAs96. Notably, a conserved 7S ncRNA transcribed from LSP inhibits mitochondrial transcription by functioning as a scaffold for POLRMT dimerization97. Moreover, other mitochondrial ncRNAs contribute to the regulation of mitochondrial gene expression, metabolism, and homeostasis98. Yet, the biological function of these mitochondrial ncRNAs remains to be investigated.
Recent studies highlighted the function of mt-NAs beyond innate immunity. Interestingly, mt-NAs serve as key mediators of the signaling crosstalk between mitochondria and the nucleus. The cellular signaling initiated by chronic mtDNA stress conditions triggers the nuclear DNA repair system and protects the nuclear genome99. The efflux of mtRNAs also stimulates the transcription of nuclear-encoded genes related to mitobiogenesis and thermogenesis in early adipocyte development13. Moreover, mtRNA modifications can alter cellular metabolic pathways and cell homeostasis, which depends on OXPHOS stimulation and oxidative stress neutralization100. Indeed, dysregulation of effectors of RNA modifications leads to metabolic alterations that are similar to those related to obesity phenotypes100. Additionally, mitochondrial tRNA modification accelerates the metastasis of head and neck cancer by functions as a metabolic switch101. In summary, investigating the biogenesis of mt-NAs, the posttranscriptional modifications that regulate mt-NA functions, and their downstream effects on cell signaling will allow us to further elucidate the important roles of mt-NAs under both physiological and pathological conditions.
Acknowledgements
We thank the members of the Lee and Kim laboratories for their helpful discussions. This study was supported by the Ministry of Health & Welfare (grant number HI21C1501 to Y.K.) and the Basic Science Research Program of the National Research Foundation of Korea (NRF-RS-2023-00219563 to M.L.), funded by the Korean government’s Ministry of Science and ICT.
Author contributions
J.Y., S.K., M.L. and Y.K. organized, wrote, and revised the manuscript. J.Y., S.K. and M.L. prepared the figures. M.L. and Y.K., supervised manuscript preparation.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Jimin Yoon, Sujin Kim.
Contributor Information
Mihye Lee, Email: mihyelee@sch.ac.kr.
Yoosik Kim, Email: ysyoosik@kaist.ac.kr.
References
- 1.Kolesnikov AA, Gerasimov ES. Diversity of mitochondrial genome organization. Biochemistry. 2012;77:1424–1435. doi: 10.1134/S0006297912130020. [DOI] [PubMed] [Google Scholar]
- 2.Bauer TM, Murphy E. Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ. Res. 2020;126:280–293. doi: 10.1161/CIRCRESAHA.119.316306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson AR, et al. Spontaneous DNA damage to the nuclear genome promotes senescence, redox imbalance and aging. Redox. Biol. 2018;17:259–273. doi: 10.1016/j.redox.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011;11:389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Rehwinkel J, Gack MU. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020;20:537–551. doi: 10.1038/s41577-020-0288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang ED, Wang CY. TRAF5 is a downstream target of MAVS in antiviral innate immune signaling. PLoS One. 2010;5:e9172. doi: 10.1371/journal.pone.0009172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beck MA, et al. DNA hypomethylation leads to cGAS-induced autoinflammation in the epidermis. EMBO J. 2021;40:e108234. doi: 10.15252/embj.2021108234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017;17:363–375. doi: 10.1038/nri.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young PG, Attardi G. Characterization of double-stranded RNA from HeLa cell mitochondria. Biochem. Biophys. Res. Commun. 1975;65:1201–1207. doi: 10.1016/s0006-291x(75)80357-3. [DOI] [PubMed] [Google Scholar]
- 11.Dhir A, et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature. 2018;560:238–242. doi: 10.1038/s41586-018-0363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim Y, et al. PKR senses nuclear and mitochondrial signals by interacting with endogenous double-stranded RNAs. Mol. Cell. 2018;71:1051–1063.e6. doi: 10.1016/j.molcel.2018.07.029. [DOI] [PubMed] [Google Scholar]
- 13.Hoang AC, et al. Mitochondrial RNA stimulates beige adipocyte development in young mice. Nat. Metab. 2022;4:1684–1696. doi: 10.1038/s42255-022-00683-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moehlman, A. T. & Youle, R. J. Mitochondrial quality control and restraining innate immunity. Annu. Rev. Cell Dev. Biol.36, 265–289. 10.1146/annurev-cellbio-021820-101354 (2020). [DOI] [PubMed]
- 15.Khalfi, P. et al. Antagonism of ALAS1 by the measles virus V protein contributes to degradation of the mitochondrial network and promotes interferon response. PLoS Pathog.19, e1011170 (2023). [DOI] [PMC free article] [PubMed]
- 16.Berry, N. et al. Herpes simplex virus type 1 infection disturbs the mitochondrial network, leading to type i interferon production through the RNA polymerase III/RIG-I pathway. mBio12, e0255721 (2021). [DOI] [PMC free article] [PubMed]
- 17.Kim J, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science. 2019;366:1531–1536. doi: 10.1126/science.aav4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pérez-Treviño P, Velásquez M, García N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. Biochim. Biophys. Acta. 2020;1866:165761. doi: 10.1016/j.bbadis.2020.165761. [DOI] [PubMed] [Google Scholar]
- 19.Suh DH, Kim MK, Kim HS, Chung HH, Song YS. Mitochondrial permeability transition pore as a selective target for anti-cancer therapy. Front. Oncol. 2013;3:40670. doi: 10.3389/fonc.2013.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Broekemeier KM, Dempsey ME, Pfeiffer DR. Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J. Biol. Chem. 1989;264:7826–7830. [PubMed] [Google Scholar]
- 21.Guarino, F. et al. NRF-1 and HIF-1α contribute to modulation of human VDAC1 gene promoter during starvation and hypoxia in HeLa cells. Biochim. Biophys. Acta Bioenerg.1861, 148289 (2020). [DOI] [PubMed]
- 22.Cosentino K, et al. The interplay between BAX and BAK tunes apoptotic pore growth to control mitochondrial-DNA-mediated inflammation. Mol. Cell. 2022;82:933–949.e9. doi: 10.1016/j.molcel.2022.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McArthur, K. et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science359, eaao6047 (2018). [DOI] [PubMed]
- 24.White MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159:1549. doi: 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sprenger HG, et al. Cellular pyrimidine imbalance triggers mitochondrial DNA–dependent innate immunity. Nat. Metab. 2021;3:636–650. doi: 10.1038/s42255-021-00385-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim, S. et al. Mitochondrial double-stranded RNAs govern the stress response in chondrocytes to promote osteoarthritis development. Cell Rep.40, 111178 (2022). [DOI] [PubMed]
- 27.Lee JH, et al. Mitochondrial double-stranded RNA in exosome promotes interleukin-17 production through toll-like receptor 3 in alcohol-associated liver injury. Hepatology. 2020;72:609–625. doi: 10.1002/hep.31041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoon J, et al. Resveratrol attenuates the mitochondrial RNA-mediated cellular response to immunogenic stress. Int. J. Mol. Sci. 2023;24:7403. doi: 10.3390/ijms24087403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jackisch L, et al. Tunicamycin-induced endoplasmic reticulum stress mediates mitochondrial dysfunction in human adipocytes. J. Clin. Endocrinol. Metab. 2020;105:2905–2918. doi: 10.1210/clinem/dgaa258. [DOI] [PubMed] [Google Scholar]
- 30.Vringer E, Tait SWG. Mitochondria and cell death-associated inflammation. Cell Death Differ. 2022;30:304–312. doi: 10.1038/s41418-022-01094-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X, Bai XC, Chen ZJ. Structures and mechanisms in the cGAS-STING innate immunity pathway. Immunity. 2020;53:43–53. doi: 10.1016/j.immuni.2020.05.013. [DOI] [PubMed] [Google Scholar]
- 32.Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 2020;21:501–521. doi: 10.1038/s41580-020-0244-x. [DOI] [PubMed] [Google Scholar]
- 33.Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS–STING pathway in health and disease. Nat. Rev. Genet. 2019;20:657–674. doi: 10.1038/s41576-019-0151-1. [DOI] [PubMed] [Google Scholar]
- 34.Lei, Y. et al. Cooperative sensing of mitochondrial DNA by ZBP1 and cGAS promotes cardiotoxicity. Cell186, 3013-3032.e22 (2023). [DOI] [PMC free article] [PubMed]
- 35.Yousefi S, et al. In vivo evidence for extracellular DNA trap formation. Cell Death Dis. 2020;11:1–15. doi: 10.1038/s41419-020-2497-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Domizio JD, et al. The cGAS–STING pathway drives type I IFN immunopathology in COVID-19. Nature. 2022;603:145–151. doi: 10.1038/s41586-022-04421-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen, R., Du, J., Zhu, H. & Ling, Q. The role of cGAS-STING signalling in liver diseases. JHEP Rep.3, 100324 (2021). [DOI] [PMC free article] [PubMed]
- 38.Yu Y, et al. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Invest. 2019;129:546–555. doi: 10.1172/JCI121842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen L, et al. Loss of Sam50 in hepatocytes induces cardiolipin-dependent mitochondrial membrane remodeling to trigger mtDNA release and liver injury. Hepatology. 2022;76:1389–1408. doi: 10.1002/hep.32471. [DOI] [PubMed] [Google Scholar]
- 40.Maekawa H, et al. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep. 2019;29:1261–1273.e6. doi: 10.1016/j.celrep.2019.09.050. [DOI] [PubMed] [Google Scholar]
- 41.Willemsen, J. et al. TNF leads to mtDNA release and cGAS/STING-dependent interferon responses that support inflammatory arthritis. Cell Rep.37, 109977 (2021). [DOI] [PubMed]
- 42.Yu CH, et al. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell. 2020;183:636. doi: 10.1016/j.cell.2020.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Latz E, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat. Immunol. 2004;5:190–198. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- 44.Heilig R, Lee J, Tait SWG. Mitochondrial DNA in cell death and inflammation. Biochem. Soc. Trans. 2023;51:457. doi: 10.1042/BST20221525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Costa, T. J. et al. Mitochondrial DNA and TLR9 activation contribute to SARS-CoV-2-induced endothelial cell damage. Vascul. Pharmacol.142, 106946 (2022). [DOI] [PMC free article] [PubMed]
- 46.Zhang JZ, Liu Z, Liu J, Ren JX, Sun TS. Mitochondrial DNA induces inflammation and increases TLR9/NF-κB expression in lung tissue. Int J. Mol. Med. 2014;33:817–824. doi: 10.3892/ijmm.2014.1650. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Sharma BR, Karki R, Kanneganti TD. Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. Eur. J. Immunol. 2019;49:1998. doi: 10.1002/eji.201848070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coll RC, Holley CL, Schroder K. Mitochondrial DNA synthesis fuels NLRP3 inflammasome. Cell Res. 2018;28:1046–1047. doi: 10.1038/s41422-018-0112-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang YY, Liu XL, Zhao R. Induction of pyroptosis and its implications in cancer management. Front. Oncol. 2019;9:971. doi: 10.3389/fonc.2019.00971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang, Z. et al. NLRP3 inflammasome and inflammatory diseases. Oxid. Med. Cell Longev.2020, (2020). [DOI] [PMC free article] [PubMed]
- 51.Fusco R, Siracusa R, Genovese T, Cuzzocrea S, Paola RD. Focus on the role of NLRP3 inflammasome in diseases. Int J. Mol. Sci. 2020;21:1–25. doi: 10.3390/ijms21124223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu L, et al. Mitochondrial DNA enables AIM2 inflammasome activation and hepatocyte pyroptosis in nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2021;320:G1034–G1044. doi: 10.1152/ajpgi.00431.2020. [DOI] [PubMed] [Google Scholar]
- 53.Bae, J. H. et al. Circulating cell-free mtDNA contributes to AIM2 inflammasome-mediated chronic inflammation in patients with type 2 diabetes. Cells8, 328 (2019). [DOI] [PMC free article] [PubMed]
- 54.D’Souza AR, Minczuk M. Mitochondrial transcription and translation: overview. Essays Biochem. 2018;62:309. doi: 10.1042/EBC20170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan, X., Sun, L., Chen, J. & Chen, Z. J. Detection of microbial infections through innate immune sensing of nucleic acids. Annu. Rev. Microbiol.72, 447–478 10.1146/annurev-micro-102215-095605 (2015). [DOI] [PubMed]
- 56.Dabo S, Meurs EF. dsRNA-dependent protein kinase PKR and its role in stress, signaling and HCV infection. Viruses. 2012;4:2598. doi: 10.3390/v4112598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoon J, et al. Mitochondrial double-stranded RNAs as a pivotal mediator in the pathogenesis of Sjӧgren’s syndrome. Mol. Ther. Nucleic Acids. 2022;30:257–269. doi: 10.1016/j.omtn.2022.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jin, J. O., Shinohara, Y. & Yu, Q. Innate immune signaling induces interleukin-7 production from salivary gland cells and accelerates the development of primary Sjögren’s syndrome in a mouse model. PLoS One8, e77605 (2013). [DOI] [PMC free article] [PubMed]
- 59.Lee H, et al. Cell type-specific transcriptomics reveals that mutant huntingtin leads to mitochondrial RNA release and neuronal innate immune activation. Neuron. 2020;107:891–908.e8. doi: 10.1016/j.neuron.2020.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu L, et al. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science. 2008;320:379–381. doi: 10.1126/science.1155406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang, Y. H. et al. Heat shock protein 60 restricts release of mitochondrial dsRNA to suppress hepatic inflammation and ameliorate non-alcoholic fatty liver disease in mice. Int. J. Mol. Sci.23, 577 (2022). [DOI] [PMC free article] [PubMed]
- 62.Becker Y, et al. Autoantibodies in systemic lupus erythematosus target mitochondrial RNA. Front. Immunol. 2019;10:1026. doi: 10.3389/fimmu.2019.01026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu B, et al. α7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol. Med. 2014;20:350–358. doi: 10.2119/molmed.2013.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Silva S, Camino LP, Aguilera A. Human mitochondrial degradosome prevents harmful mitochondrial R loops and mitochondrial genome instability. Proc. Natl Acad. Sci. USA. 2018;115:11024–11029. doi: 10.1073/pnas.1807258115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mankan AK, et al. Cytosolic RNA:DNA hybrids activate the cGAS–STING axis. EMBO J. 2014;33:2937–2946. doi: 10.15252/embj.201488726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Falabella M, Fernandez RJ, Johnson FB, Kaufman BA. Potential roles for G-quadruplexes in mitochondria. Curr. Med Chem. 2019;26:2918. doi: 10.2174/0929867325666180228165527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bharti SK, et al. DNA sequences proximal to human mitochondrial DNA deletion breakpoints prevalent in human disease form G-quadruplexes, a class of DNA structures inefficiently unwound by the mitochondrial replicative Twinkle helicase. J. Biol. Chem. 2014;289:29975–29993. doi: 10.1074/jbc.M114.567073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Szewczyk M, et al. Human REXO2 controls short mitochondrial RNAs generated by mtRNA processing and decay machinery to prevent accumulation of double-stranded RNA. Nucleic Acids Res. 2020;48:5572–5590. doi: 10.1093/nar/gkaa302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bratic, A. et al. Mitochondrial polyadenylation is a one-step process required for mRNA integrity and tRNA maturation. PLoS Genet. 12, e1006028 (2016). [DOI] [PMC free article] [PubMed]
- 70.Spåhr H, et al. SLIRP stabilizes LRPPRC via an RRM–PPR protein interface. Nucleic Acids Res. 2016;44:6868–6882. doi: 10.1093/nar/gkw575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pajak, A. et al. Defects of mitochondrial RNA turnover lead to the accumulation of double-stranded RNA in vivo. PLoS Genet.15, e1008240 (2019). [DOI] [PMC free article] [PubMed]
- 72.Honarmand S, Shoubridge EA. Poly (A) tail length of human mitochondrial mRNAs is tissue-specific and a mutation in LRPPRC results in transcript-specific patterns of deadenylation. Mol. Genet Metab. Rep. 2020;25:100687. doi: 10.1016/j.ymgmr.2020.100687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pearce SF, et al. Regulation of mammalian mitochondrial gene expression: recent advances. Trends Biochem. Sci. 2017;42:625–639. doi: 10.1016/j.tibs.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Borowski LS, Dziembowski A, Hejnowicz MS, Stepien PP, Szczesny RJ. Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res. 2013;41:1223–1240. doi: 10.1093/nar/gks1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bamborschke D, et al. PNPT1 mutations may cause Aicardi-Goutières-Syndrome. Brain Dev. 2021;43:320–324. doi: 10.1016/j.braindev.2020.10.005. [DOI] [PubMed] [Google Scholar]
- 76.van Esveld SL, et al. Mitochondrial RNA processing defect caused by a SUPV3L1 mutation in two siblings with a novel neurodegenerative syndrome. J. Inherit. Metab. Dis. 2022;45:292–307. doi: 10.1002/jimd.12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jourdain AA, et al. GRSF1 regulates RNA processing in mitochondrial RNA granules. Cell Metab. 2013;17:399. doi: 10.1016/j.cmet.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jourdain AA, Boehm E, Maundrell K, Martinou JC. Mitochondrial RNA granules: compartmentalizing mitochondrial gene expression. J. Cell Biol. 2016;212:611. doi: 10.1083/jcb.201507125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xie XQ, et al. Targeting ATAD3A-PINK1-mitophagy axis overcomes chemoimmunotherapy resistance by redirecting PD-L1 to mitochondria. Cell Res. 2023;33:215. doi: 10.1038/s41422-022-00766-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Arnaiz E, et al. Hypoxia regulates endogenous double-stranded RNA production via reduced mitochondrial DNA transcription. Front. Oncol. 2021;11:4970. doi: 10.3389/fonc.2021.779739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bonekamp NA, et al. Small-molecule inhibitors of human mitochondrial DNA transcription. Nature. 2020;588:712–716. doi: 10.1038/s41586-020-03048-z. [DOI] [PubMed] [Google Scholar]
- 82.Mennuni M, et al. Metabolic resistance to the inhibition of mitochondrial transcription revealed by CRISPR-Cas9 screen. EMBO Rep. 2022;23:e53054. doi: 10.15252/embr.202153054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li SP, Ou L, Zhang Y, Shen FR, Chen YG. A first-in-class POLRMT specific inhibitor IMT1 suppresses endometrial carcinoma cell growth. Cell Death Dis. 2023;14:1–11. doi: 10.1038/s41419-023-05682-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou X, et al. MitoEVs: a new player in multiple disease pathology and treatment. J. Extracell. Vesicles. 2023;12:12320. doi: 10.1002/jev2.12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhao, F. et al. Increased release of microvesicles containing mitochondria is associated with the myeloid differentiation of AML-M5 leukaemia cells. Exp. Cell Res.395, 112213 (2020). [DOI] [PubMed]
- 86.Cheng, A. N. et al. Mitochondrial Lon-induced mtDNA leakage contributes to PD-L1-mediated immunoescape via STING-IFN signaling and extracellular vesicles. J. Immunother. Cancer8, e001372 (2020). [DOI] [PMC free article] [PubMed]
- 87.Antunes, M. A. et al. Mesenchymal stromal cells from emphysematous donors and their extracellular vesicles are unable to reverse cardiorespiratory dysfunction in experimental severe emphysema. Front. Cell Dev. Biol.9, 661385 (2021). [DOI] [PMC free article] [PubMed]
- 88.Townsend K, Tseng Y-H. Brown adipose tissue: recent insights into development, metabolic function and therapeutic potential. Adipocyte. 2012;1:13. doi: 10.4161/adip.18951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ikeda K, Yamada T. UCP1 dependent and independent thermogenesis in brown and beige adipocytes. Front. Endocrinol. 2020;11:544307. doi: 10.3389/fendo.2020.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bai J, et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc. Natl Acad. Sci. USA. 2017;114:12196–12201. doi: 10.1073/pnas.1708744114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bai J, et al. Mitochondrial stress-activated cGAS-STING pathway inhibits thermogenic program and contributes to overnutrition-induced obesity in mice. Commun. Biol. 2020;3:1–14. doi: 10.1038/s42003-020-0986-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gong Z, et al. Deficiency in AIM2 induces inflammation and adipogenesis in white adipose tissue leading to obesity and insulin resistance. Diabetologia. 2019;62:2325–2339. doi: 10.1007/s00125-019-04983-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tin A, et al. Association between mitochondrial DNA copy number in peripheral blood and incident CKD in the atherosclerosis risk in communities study. J. Am. Soc. Nephrol. 2016;27:2467–2473. doi: 10.1681/ASN.2015060661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nicholas LM, et al. Mitochondrial transcription factor B2 is essential for mitochondrial and cellular function in pancreatic β-cells. Mol. Metab. 2017;6:651. doi: 10.1016/j.molmet.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim, S., Yoon, J., Lee, K. & Kim, Y. Analysis of mitochondrial double-stranded RNAs in human cells. STAR Protoc.4, 102007 (2023). [DOI] [PMC free article] [PubMed]
- 96.Ren B, Guan MX, Zhou T, Cai X, Shan G. Emerging functions of mitochondria-encoded noncoding RNAs. Trends Genet. 2023;39:125–139. doi: 10.1016/j.tig.2022.08.004. [DOI] [PubMed] [Google Scholar]
- 97.Zhu X, et al. Non-coding 7S RNA inhibits transcription via mitochondrial RNA polymerase dimerization. Cell. 2022;185:2309–2323.e24. doi: 10.1016/j.cell.2022.05.006. [DOI] [PubMed] [Google Scholar]
- 98.Sun W, et al. Mitochondrial non-coding RNAs are potential mediators of mitochondrial homeostasis. Biomolecules. 2022;12:1863. doi: 10.3390/biom12121863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wu Z, et al. Mitochondrial DNA stress signalling protects the nuclear genome. Nat. Metab. 2019;1:1209–1218. doi: 10.1038/s42255-019-0150-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Boughanem H, et al. The emergent role of mitochondrial RNA modifications in metabolic alterations. Wiley Interdiscip. Rev. RNA. 2023;14:e1753. doi: 10.1002/wrna.1753. [DOI] [PubMed] [Google Scholar]
- 101.Delaunay S, et al. Mitochondrial RNA modifications shape metabolic plasticity in metastasis. Nature. 2022;607:593–603. doi: 10.1038/s41586-022-04898-5. [DOI] [PMC free article] [PubMed] [Google Scholar]