

Abstract
Purpose:
FGFR inhibitors are effective in FGFR2-altered cholangiocarcinoma, leading to approval of reversible FGFR inhibitors, pemigatinib and infigratinib, and an irreversible inhibitor, futibatinib. However, acquired resistance develops, limiting clinical benefit. Some mechanisms of resistance have been reported, including secondary FGFR2 kinase domain mutations. Here, we sought to establish the landscape of acquired resistance to FGFR inhibition and to validate findings in model systems.
Experimental Design:
We examined the spectrum of acquired resistance mechanisms detected in circulating tumor DNA or tumor tissue upon disease progression following FGFR inhibitor therapy in 82 FGFR2-altered cholangiocarcinoma patients from 12 published reports. Functional studies of candidate resistance alterations were performed.
Results:
Overall, 49 of 82 patients (60%) had one or more detectable secondary FGFR2 kinase domain mutations upon acquired resistance. N550 molecular brake and V565 gatekeeper mutations were most common, representing 63% and 47% of all FGFR2 kinase domain mutations, respectively. Functional studies showed different inhibitors displayed unique activity profiles against FGFR2 mutations. Interestingly, disruption of the cysteine residue covalently bound by futibatinib (FGFR2 C492) was rare, observed in 1 of 42 patients treated with this drug. FGFR2 C492 mutations were insensitive to inhibition by futibatinib but showed reduced signaling activity, potentially explaining their low frequency.
Conclusions:
These data support secondary FGFR2 kinase domain mutations as the primary mode of acquired resistance to FGFR inhibitors, most commonly N550 and V565 mutations. Thus, development of combination strategies and next-generation FGFR inhibitors targeting the full spectrum of FGFR2 resistance mutations will be critical.
Translational Relevance.
This study defines the clinical landscape of potential resistance mechanisms to FGFR inhibitors in FGFR2-dependent cholangiocarcinoma. Our observations support that secondary FGFR2 resistance mutations may represent the most common mechanism of clinical acquired resistance, with N550 molecular brake and V565 gatekeeper alterations being the most frequent and critical. Disruption of C492, which is involved in the covalent binding reaction with futibatinib, was rare, potentially due to the negative impact of these mutations on FGFR2 signaling, supporting the promise of covalent FGFR inhibitors. These findings provide a guide for therapeutic development of FGFR inhibitors, emphasizing the importance of developing agents targeting the full spectrum of FGFR2 resistance mutations and combination strategies for overcoming signaling bypass processes.
Introduction
FGFR signaling is an essential oncogenic pathway in a subset of intrahepatic cholangiocarcinoma (ICC). Approximately 10%–15% ICCs show genetic alterations of FGFR2, most commonly involving in-frame fusions of FGFR2 exons 1–17 with a diversity of partners (1–8). These fusions, as well as other truncating events that are sometimes observed, contain the entire FGFR2 extracellular and kinase domains but lack a C-terminal negative regulatory domain encoded by the final exon 18, leading to constitutive kinase activity (9). Additional subsets of ICCs harbor activating point mutations or in-frame deletions in the FGFR2 extracellular domain (1). The development of selective FGFR kinase inhibitors has represented an important milestone for the treatment of patients with FGFR2-positive ICC. The reversible ATP-competitive FGFR1–3 inhibitors, pemigatinib (INCB-054828) and infigratinib (BGJ398), were approved for patients with previously treated, advanced ICC with FGFR2 rearrangements (10, 11). Moreover, clinical activity has been observed in phase I and phase II studies with multiple other selective ATP-competitive FGFR inhibitors, including zoligratinib (Debio1347), erdafitinib (JNJ-42756493), and rogaratinib (BAY 1163877; refs. 12–16).
While reversible ATP-competitive FGFR inhibitors provide benefit to most patients with FGFR2 fusion–positive ICC, with approximately 80% showing stable disease or partial response, progression is typically observed within less than 1 year. Initial analysis of serial tumor biopsies, circulating tumor DNA (ctDNA), and rapid autopsy specimens from ICC patients with acquired resistance to FGFR inhibitors (infigratinib, pemigatinib, and zoligratinib) revealed the emergence of secondary mutations in the FGFR2 kinase domain that block inhibitor binding as a common mechanism of resistance (1, 17–20). In several cases, these mutations were polyclonal, with testing of cell-free DNA or biopsy of individual lesions from the same patient revealing different FGFR2 kinase domain mutations (18, 19, 21, 22).
Irreversible kinase inhibitors that form covalent bonds with cysteines in the ATP-binding pocket can address some limitations of reversible ATP-competitive kinase inhibitors (23, 24). Futibatinib (TAS-120) is an irreversible, selective FGFR1–4 inhibitor, which binds the conserved P-loop cysteine residue (C492) in the FGFR2 kinase domain (25–27). Importantly, futibatinib has been shown to overcome acquired resistance in some patients with ICC who previously benefitted from reversible FGFR inhibitors (28). In these cases, ctDNA analysis revealed that clinical benefit correlated with decrease of specific FGFR2 kinase domain resistance mutations in the ctDNA. Unfortunately, resistance to futibatinib eventually developed both when this agent was provided as the first FGFR inhibitor or following progression on a reversible FGFR inhibitor.
Studies in preclinical models have provided further insights into FGFR inhibitor response and resistance in ICC. In FGFR2-altered ICC cell lines and patient-derived xenografts, FGFR inhibition leads to growth arrest and suppression of MEK/ERK signaling, underscoring a bona fide oncogene addiction phenotype and the importance of the downstream MEK/ERK pathway (28). Moreover, testing some of the FGFR2 kinase domain mutations reported to date reveal distinct activity profiles of different FGFR inhibitors. Thus, the clinical and preclinical findings support the strategic sequencing of FGFR inhibitors to overcome resistance and extend clinical benefit. In this regard, to effectively guide therapeutic development in FGFR2-positive ICC, it is important to more fully define the spectrum of potential resistance mechanisms observed in patients and to comprehensively determine the sensitivity of different FGFR2 mutations to FGFR inhibitors in model systems.
Here, we present an analysis of 82 FGFR2-altered cholangiocarcinoma patients from 12 published studies, in which postprogression ctDNA or tumor tissue was assessed after acquired resistance to FGFR inhibitors. These findings taken together with mechanistic experiments in preclinical models provide important insight into the current unmet therapeutic needs for this patient population.
Materials and Methods
Patients
This study involved previously published studies of 82 patients with metastatic or unresectable intrahepatic cholangiocarcinoma with an FGFR2 fusion, mutation, or amplification who (i) received treatment with a selective FGFR inhibitor (infigratinib, pemigatinib, zoligratinib, and/or futibatinib), and (ii) underwent sequencing of post-progression ctDNA or tumor biopsy at the time of acquired resistance (1, 17–22, 28–32). All studies were performed under Institutional Review Board (IRB)-approved protocols with written informed consent from patients and in accordance with the Declaration of Helsinki.
Cell culture
CCLP-1 cells, previously characterized as FGFR dependent (28), were a kind gift from Dr. P.J. Bosma (Academic Medical Center, Amsterdam, the Netherlands). ICC13–7 is a cell line derived from a patient with ICC harboring FGFR2-OPTN fusion treated with futibatinib. Cells were passaged by trypsinization and adapted to uncoated tissue-culture plates in RPMI (CCLP-1, ICC13–7) or DMEM (HEK293T, NIH-3T3) supplemented with 10% FBS and 1% penicillin/streptomycin prior to functional studies. They were routinely checked to be Mycoplasma free. Cell lines were authenticated by short tandem repeat (STR) DNA profiling by the cell line bank from which they were obtained.
Generation of engineered cell lines expressing the wild-type or mutated FGFR2-PHGDH fusion
A FGFR2-PHGDH (FP) fusion construct, containing exons 1–17 of FGFR2-IIIb isoform fused to PHGDH (NM_006623.3) exons 6–12, was amplified from reverse transcribed cDNAs from an ICC patient sample and inserted into the pMSCV vector using the NEBuilder HiFi DNA Assembly (New England Biolabs). All FGFR2 mutations were introduced into the pMSCV vector using the same kit. Targeted Sanger sequencing was done to confirm the mutation generated. Retrovirus and lentivirus were generated by transfecting the indicated constructs and packaging plasmids into HEK293T cells. NRAS Q61K (Plasmid #120575) and PIK3CA E545K (Plasmid #12525) constructs were purchased from Addgene. After collection of virus, transfected HEK293T cells were tested by immunoblot to confirm protein expression from each construct. Retroviral infections of CCLP-1 cells, ICC13–7 cells, HEK 293T cells, and NIH-3T3 cells were performed in the presence of polybrene. Infected cells were selected in blasticidin (20–25 μg/mL) for one week or puromycin (4 μg/mL) for 2 days.
Cell viability assay
Cells were seeded at a density of 3,000 cells per well on 96-well plates and incubated overnight. Compounds were added the next day over a 9-point concentration range and then incubated for 3 days. The viability of cells was then measured by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. IC50 values were determined by GraphPad Prism using a 3-parameter dose–response model. Drugs were purchased from the following resources: futibatinib (S8848), infigratinib (S2183), pemigatinib (S0088), erdafitinib (S8401), zoligratinib (S7665), derazantinib (S8609). Rogaratinib was provided by Bayer.
Crystal violet staining
Cells were seeded in an equal number per well on 6-well plates and allowed to attach overnight. Fresh media with or without drug was replaced the next day and refreshed every 2–3 days until the end of the experiment. Cells were fixed in 4% paraformaldehyde solution for 10 minutes and subsequently stained with 0.5% crystal violet solution for 30 minutes. Plates were washed three times with H2O and dried overnight.
Immunoblot analysis
Snap-frozen tumor tissues were homogenized using a Precellys 24 homogenizer (Bertin Instruments) at 6,500 rpm. Tumor and cell protein lysates were prepared in Lysis Buffer [10% glycerol, 3% SDS, 12.5% solution buffer (0.5 mol/L Tris base, 0.4% SDS, adjust pH to 6.8 using HCl)] containing Pierce Protease Inhibitor (A32965) and Calbiochem phosphatase inhibitor cocktail set I and II. Protein concentration was determined by Pierce BCA Protein Assay. Twenty-microgram protein was used to perform analysis by SDS-PAGE, electro-transfer, and immunoblotting with specific antibodies. The following primary antibodies were used: from Cell Signaling Technology (all at 1:1,000 dilution), phospho-FGFR2 Y653/654 (3471, RRID:AB_331072), phospho-FRS2 Y196 (3864S, RRID:AB_2106222), SHP2 (3397S, RRID:AB_2174959), phospho-MEK1/2 S217/221 (9154S, RRID:AB_2138017), MEK1/2 (4694S, RRID:AB_10695868), phospho-ERK1/2 T202/Y204 (4370S, RRID:AB_2315112), ERK1/2 (4695S, RRID:AB_390779), RAS (3965, RRID:AB_2180216), PI3 Kinase p110α (4249, RRID:AB_2165248); from Abcam, FGFR2 (ab10648, RRID:AB_297369), phospho-SHP2 Y542 (ab62322, RRID:AB_945452) 1:1,000, FRS2 (ab183492) 1:1,000; from Sigma (1:5,000 dilution), β-actin (A5316, RRID:AB_476743).
In vivo studies
All mice were housed in a specific pathogen-free environment at the MGH and treated in strict accordance with protocols 2005N000148 and 2019N000116 approved by the Subcommittee on Research Animal Care at MGH. Six- to 10-week-old NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, 00557, The Jackson Laboratory) were used in the studies below. CCLP-1 cells engineered with FGFR2-PHGDH wild-type fusion or FGFR2-PHGDH-N550K mutant were injected subcutaneously with 1.5 × 106 cells. When tumors reached approximately 200 mm3, mice were randomized and started on treatment with vehicle (0.5 w/v% hydroxypropyl methylcellulose), or futibatinib 6 mg/kg daily for 10 days by oral gavage. Tumor volumes were monitored by digital calipers. Tumor samples (snap frozen in liquid nitrogen) were collected 4 hours after the last dose and apportioned for biochemical analysis. For NIH-3T3 cells in vivo fitness assay, 10% of each mutant cell line and 70% of FGFR2–PHGDH WT cells were pooled together at a total of 1 × 106 cells and injected subcutaneously. Tumors were allowed to grow for 16 days when euthanasia was required and then collected for ddPCR analysis.
Structural modeling
The diagram of clinically observed FGFR2 kinase domain mutations was generated in PyMol using the crystal structure of the FGFR2 kinase domain (PDB code: 2PVF).
Molecular dynamics simulations
Molecular dynamics simulations and trajectory analysis were carried out using Desmond in Schrodinger suite (2020-2 release). The explicit water model (TIP3P) was used in all simulations and default equilibration protocol was applied prior to production runs of 500 ns (33). The crystal structure of FGFR2 (PDB code: 2PVF) was used to construct the simulation systems. Missing loops were modeled in and energy optimized using PRIME in the Schrodinger suite. The 5′-adenylyl methylenediphosphonate (AMP-PCP) in the crystal structure was replaced by ATP in the simulations and the substrate was removed. FGFR2 protein was kept in the phosphorylated form. The simulation temperature was 300 K with periodic boundary condition. Orthorhombic simulation boxes were constructed with 12A margin. Default values were used for other parameters.
Statistical analysis
Statistics were performed and graphs were generated in GraphPad Prism 9 (RRID:SCR_002798). All experimental data were analyzed by Student t test (two tailed). P < 0.05 was considered to be significant.
Data availability
This work includes a meta-analysis of published data from 12 studies (1, 17–22, 28–32). The data generated in this study are available in the article and its Supplementary Data. Raw data are available upon reasonable request from the corresponding authors.
Results
Spectrum of secondary FGFR2 clinical acquired resistance mutations
We assessed a cohort of 82 patients with FGFR2-altered advanced ICC from 12 published studies who underwent next-generation sequencing analysis of postprogression ctDNA and/or tumor biopsy upon acquired resistance to selective FGFR inhibitors (1, 17–22, 28–32). Of the combined 82 patients, 10 were treated with pemigatinib, 9 with infigratinib, 1 with zoligratinib, 17 with an unspecified reversible inhibitor, 36 with futibatinib, and 3 with an unknown FGFR inhibitor. Six patients were treated with both a reversible and irreversible inhibitor. The cohort consisted of 77 patients with FGFR2 fusions or rearrangements downstream of exon 17, 2 with extracellular domain in-frame deletions, 2 with FGFR2 amplification, and 1 with a point mutation (Supplementary Table S1). Demographics data were available for a subset of patients, which were majority female, consistent with known enrichment of FGFR2 alterations in this group (ref. 34; Supplementary Table S2).
Upon acquired resistance to FGFR inhibitors, 60% (49/82) of patients developed secondary FGFR2 kinase domain mutations, detected in postprogression ctDNA or tumor biopsy (Fig. 1A). Among patients with acquired FGFR2 mutations, the most commonly observed mutations affected the molecular brake residue N550, occurring in 63% (31/49) of patients (38%; 31/82 of the total cohort), and the gatekeeper residue V565, occurring in 47% (23/49) of patients (28%; 23/82 of the total cohort). The gatekeeper controls accessibility of ATP-competitive inhibitors to the hydrophobic pocket, whereas the molecular brake residues form a hydrogen bond network that constrains kinase activation (35).
Figure 1.

Landscape of acquired alterations on FGFR inhibitors. A, Frequency of FGFR2 kinase domain mutations on FGFR inhibitors. Ribbon diagram (B) and schematic (C) showing location and overall frequency of the secondary FGFR2 kinase domain mutations. (B, Created with PyMOL. C, Created with BioRender.com.)
In total, 128 distinct FGFR2 mutations affecting the kinase domain were detected, resulting in amino acid changes at 20 different residues (Figs. 1B and C). Three different amino acid changes were identified at the gatekeeper residue (V565F, V565I, V565L) and four at the molecular brake residue N550 (N550D, N550H, N550K, N550T).
Notably, an acquired mutation at the site of covalent binding of futibatinib, C492F, was observed at the time of resistance in 1 of 42 patients who received futibatinib at any time (Fig. 1C). This mutation was not observed in patients who received reversible inhibitors. Analogous mutations, which remove a critical cysteine residue in the tyrosine kinase domain, are observed as mechanisms of resistance to other irreversible receptor tyrosine kinase inhibitors, such as osimertinib and ibrutinib (36, 37).
Other candidate resistance mechanisms
While FGFR2 secondary mutations appeared to be the dominant mechanism of acquired resistance, some patients developed “bypass” mutations in signaling pathways downstream of FGFR, including the RAS and PI3K pathways. Of note, not all patients were evaluated for bypass pathway alterations in every study so overall frequencies of these alterations cannot be calculated. Although PI3K pathway alterations, including PIK3CA and PTEN mutations, emerged in some patients progressing on FGFR inhibitors, PIK3CA mutations were also detected in a subset of patients prior to FGFR inhibitor treatment, in agreement with reports of the co-occurrence of FGFR2 alterations and PIK3CA mutations in 15%–20% of baseline ICC (7, 11, 21). Thus, it is not clear whether PIK3CA mutations represent additional drivers of acquired resistance in some cases, or whether they represent pre-existing subclonal alterations that are present in certain clones that emerge due to a different resistance mechanism. Consistent with this latter hypothesis, expression of mutant PIK3CA did not lead to FGFR inhibitor resistance in vitro, as compared with an NRAS mutation, which was observed as an emergent bypass mutation in 3 patients (1, 30, 31). In particular, PIK3CA E545K did not affect futibatinib sensitivity of the FGFR-dependent ICC cell line, CCLP-1, engineered with FGFR2-PHGDH fusion (CCLP-1 FP; ref. 28), whereas NRAS Q61K caused resistance in the CCLP-1 FP model and in the patient-derived ICC13–7 model (Supplementary Fig. S1).
Distinct activity profiles of different FGFR inhibitors against the spectrum of FGFR2 kinase domain mutations
Given that diverse FGFR2 kinase mutations represented the most commonly identified mechanisms of acquired resistance, we evaluated the functional consequences of many of the mutations identified in the context of treatment with a range of clinical FGFR inhibitors (Fig. 2A). The FGFR-dependent ICC cell line, CCLP-1, was engineered to express the FGFR2-PHGDH fusion containing the different kinase domain mutations and the activity (IC50) of each FGFR TKI was determined for these alleles (represented as fold change relative to the fusion containing a wild-type kinase domain from an individual batch experiment and as absolute IC50 in Fig. 2A). These clinical inhibitors include ATP-competitive compounds with a substituted phenyl moiety that occupies the back hydrophobic pocket and contacts with the V565 gatekeeper residue of FGFR2. This moiety consists of a dimethoxyphenyl ring (infigratinib, pemigatinib, erdafitinib) or a methoxy benzothiophene group (rogaratinib). Futibatinib also has a dimethoxyphenyl group in the hydrophobic pocket interacting with V565, and an acrylamide moiety that forms a covalent bond to C492 in FGFR2 in addition to dual hinge hydrogen bonds. Although zoligratinib and derazantinib have distinct structural features, molecular models indicate that they also contain an analogous moiety occupying the same back pocket (i.e., the benzyl imidazole in zoligratinib and fluorophenyl group in derazantinib). Notably, each of the candidate resistance mutations identified is located in proximity to the ATP binding site with the exception of K715R and Q747 L (Fig. 1B).
Figure 2.

FGFR inhibitors show differential activity profiles against the spectrum of clinically observed FGFR2 kinase domain mutations. A, Activity profiles of the indicated FGFR kinase inhibitors against FGFR-dependent CCLP-1 cells expressing the FGFR2-PHGDH fusion (FP) with a WT kinase domain or with different FGFR2 kinase domain mutations. The table reports IC50 values and fold changes. Mutants were tested in groups. FC denotes fold-change of IC50 for FGFR2 mutant normalized to the IC50 for the WT kinase within an individual group. Each value was representative of the mean of two biological replicates. B, Immunoblot analysis of signaling proteins in CCLP-1 cells engineered to express the indicated FGFR2-PHGDH fusion alleles. Cells were treated with vehicle, 50 nmol/L futibatinib, or 100 nmol/L pemigatinib for 4 hours. (A, Created with ChemDraw.)
The IC50 for the compounds against cells harboring the wild-type (WT) fusion ranged from <10 nmol/L (futibatinib, infigratinib, pemigatinib, erdafitinib) to approximately 200 nmol/L (derazantinib) with rogaratinib (approx. 50 nmol/L) and zoligratinib (approx. 100 nmol/L) having intermediate potency (Fig. 2A). Other than the L551F, L552F, and K715R variants, each mutation resulted in reduced activity (
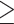 3-fold IC50 shift) for at least one of the inhibitors. Consistent with their related structural features and interaction with the gatekeeper residue, infigratinib, pemigatinib, erdafitinib, and rogaratinib have broadly similar activity profiles, and suffer pronounced resistance (>100-fold increase in IC50) to mutations of the gatekeeper residue (V565F and V565L; Fig. 2A). These larger amino acids create steric clashes with the phenyl moiety of inhibitors in the back pocket. The N550K mutation that disturbs the autoinhibitory molecular brake was also highly resistant to these agents (>100-fold IC50 increase for all except erdafitinib). Interestingly, among the reversible inhibitors, only erdafitinib makes a single-hinge hydrogen bond in the FGFR2 binding pocket. This might be the reason that it is less sensitive to N550K, which may disturb the hydrogen bond and salt bridge network of the hinge residue E566 to release the molecular brake triad. Zoligratinib and derazantinib showed distinct profiles, although their in vitro potency against WT FGFR2 is significantly weaker than the other compounds tested. Zoligratinib largely retained activity against V565F, presumably due to the lack of bulky substitution on the phenyl group like the methoxy group in other inhibitors, as reported previously, but was strongly compromised against V565L (45-fold). Derazantinib was only modestly affected (<6-fold) by each of the mutations tested, but the lack of potency and high baseline IC50 of this molecule may limit the likelihood of observing large shifts in FGFR2-specific IC50 in this viability assay.
3-fold IC50 shift) for at least one of the inhibitors. Consistent with their related structural features and interaction with the gatekeeper residue, infigratinib, pemigatinib, erdafitinib, and rogaratinib have broadly similar activity profiles, and suffer pronounced resistance (>100-fold increase in IC50) to mutations of the gatekeeper residue (V565F and V565L; Fig. 2A). These larger amino acids create steric clashes with the phenyl moiety of inhibitors in the back pocket. The N550K mutation that disturbs the autoinhibitory molecular brake was also highly resistant to these agents (>100-fold IC50 increase for all except erdafitinib). Interestingly, among the reversible inhibitors, only erdafitinib makes a single-hinge hydrogen bond in the FGFR2 binding pocket. This might be the reason that it is less sensitive to N550K, which may disturb the hydrogen bond and salt bridge network of the hinge residue E566 to release the molecular brake triad. Zoligratinib and derazantinib showed distinct profiles, although their in vitro potency against WT FGFR2 is significantly weaker than the other compounds tested. Zoligratinib largely retained activity against V565F, presumably due to the lack of bulky substitution on the phenyl group like the methoxy group in other inhibitors, as reported previously, but was strongly compromised against V565L (45-fold). Derazantinib was only modestly affected (<6-fold) by each of the mutations tested, but the lack of potency and high baseline IC50 of this molecule may limit the likelihood of observing large shifts in FGFR2-specific IC50 in this viability assay.
Finally, futibatinib remained at least partially active against most of these mutations, other than the gatekeeper mutation, V565F, and C492F (IC50 increases of 138-fold and 604-fold, respectively), the latter, consistent with the covalent binding of futibatinib to this residue. All other mutations caused <10-fold increase in IC50, with the exception of N550K (12-fold IC50 increase) and E566A (10-fold IC50 increase), which were among the most common emergent mutations observed in futibatinib-treated patients (28). FGFR signaling analysis corroborated the distinct activity profiles of FGFR inhibitors against key mutations, demonstrating that futibatinib activity was modestly decreased by V565L and V565I, but markedly decreased by V565F and C492F, whereas pemigatinib was compromised against all gatekeeper mutations, while being effective against the FGFR2 C492F allele (Fig. 2B). Thus, futibatinib shows favorable activity against many mutations that cause pronounced resistance to reversible FGFR inhibitors.
The performance profiles of the FGFR inhibitors were largely in line with clinically observed resistance data. For example, V565F was one of the most commonly observed acquired mutations in patients treated with reversible inhibitors, including pemigatinib and infigratinib, and futibatinib and conferred a very high degree of resistance to these compounds in vitro. L618F was found to emerge in a patient progressing on zoligratinib and resulted in in vitro resistance to this drug, whereas this allele did not emerge in patients treated with futibatinib, matching the in vitro response data (Fig. 2A). The N550K and C492F mutations were notable exceptions to these correlations between in vitro resistance and acquired mutations in vivo. First, N550K emerged as a common resistance mechanism to futibatinib, despite driving only a relatively modest approximately 12-fold loss of potency for futibatinib in vitro. Second, while the C492F mutation led to complete insensitivity to futibatinib, it was only seen in 1 of 42 patients progressing on futibatinib (Fig. 1C). This rarity contrasts with clinical resistance to the irreversible EGFR inhibitor, osimertinib, in lung cancer and irreversible BTK inhibitors (e.g., ibrutinib) in lymphomas, which are frequently associated with acquired mutations of the residue for covalent inhibitor binding (EGFR-C797S and BTK-C481S or C481R; refs. 36, 37).
We explored both of these observations experimentally. Each of the pan-FGFR inhibitors has dose-limiting toxicities such that the clinically achievable dose results in intratumor compound concentrations that are only moderately above the level needed for effective target inhibition (38). In this regard, while the 12-fold increase in in vitro IC50 of futibatinib against the N550K variant compared with WT FGFR2 was modest compared with the >100-fold increase seen with V565F and C492F mutants, it exceeded the shift seen for all other variants analyzed. Accordingly, treatment of either ICC13–7 cells or CCLP-1 cells expressing the FGFR2-PHGDH (FP) fusion harboring the N550K mutation with a clinically relevant concentration of 10 nmol/L futibatinib (26) failed to suppress downstream signaling (ICC13–7) or produced a partial effect (CCLP-1), while signaling by FP fusion with a wild-type or L618V mutant kinase domain was effectively inhibited (Fig. 3A). Pemigatinib at the clinically equivalent dose (20 nmol/L) was inactive against both of these mutations (39). We tested the efficacy of futibatinib in vivo against CCLP1-FP-N550K and CCLP-1-FP-WT xenografts. Once tumors reached approximately 200 mm3, mice were treated with the clinically relevant dose of 6 mg/kg futibatinib or with vehicle (Fig. 3B shows schematic of study). While CCLP-1-FP-WT xenografts were highly sensitive to futibatinib treatment, CCLP1-FP-N550K tumors showed minimal decreases in tumor growth (Fig. 3C). Correspondingly, signaling analysis demonstrated that futibatinib was largely ineffective against the N550K mutation, with limited effects on reduction of phosphorylation of FRS2 and of downstream MEK/ERK signaling (Fig. 3D). Thus, the shift in IC50 of futibatinib toward the N550K mutation is sufficient to drive resistance to exposures that are clinically achievable in vivo, consistent with the emergence of these mutation in patients progressing on futibatinib, similar to data presented in our prior studies (28).
Figure 3.

The recurrent molecular brake mutation, N550K, drives resistance to clinically achievable pan-FGFR TKI dose levels. A, Immunoblot analysis of signaling proteins in CCLP-1 or ICC13–7 cells engineered to express the FGFR2–PHGDH fusion protein with wild-type kinase domain or with the N550K or L618V mutations. Cells were treated with vehicle, 10 nmol/L futibatinib, or 20 nmol/L pemigatinib for 4 hours. B–D,In vivo assessment of futibatinib efficacy against the FGFR2 molecular brake mutation, N550K. B, Schematic diagram of experiment design. Mice harboring FGFR-dependent CCLP-1 xenografts expressing the indicated FGFR2-PHGDH fusion (FP) with a WT kinase domain or with the N550K mutation were treated with vehicle (n = 4) or futibatinib 6 mg/kg (n = 4) daily for 10 days. Treatment was started once tumors reached a volume approximately 200 mm3. C, Relative fold change of tumor volume (left) or tumor weight (right) compared with vehicle treatment at the end point. **, P < 0.01. Data are shown as mean ± SD. D, Immunoblot analysis of tumor lysates. Tumors were harvested 4 hours after the last dose of treatment. (B, Created with BioRender.com.)
Effects of resistance mutations on FGFR2 activity may contribute to differences in observed frequencies upon acquired resistance
The effectiveness of a mutation in driving clinical resistance is a function both of its insensitivity to pharmacologic inhibition and its relative fitness in activating downstream oncogenic signaling. Indeed, most of the recurrent mutations associated with FGFR inhibitor resistance both block inhibitor binding and increase kinase activity (18, 28, 40). To gain insight into the rarity of C492 mutations in the context of progression on futibatinib (relative to mutations disrupting the cysteine required for activity of other covalent kinase inhibitors, such as osimertinib and ibrutinib), we tested the signaling activity of the FGFR2-PHGDH fusion harboring each of the amino acid alterations generated by the single-nucleotide changes at the codon encoding C492 (Fig. 4A). Viral vectors expressing these alleles were introduced into immortalized human embryonic kidney cells (HEK293 cells) grown in low serum conditions (1% FBS) and FGFR2 activity was determined by immunoblot compared with empty vector (EV) control (Fig. 4B). As a reference, we also generated lines expressing the gatekeeper mutations, V565L and V565F, which stimulate FGFR2 signaling modestly by stabilizing the active form of the kinase. Cells expressing FGFR2-PHGDH with a wild-type kinase domain showed autophosphorylation of the fusion (FGFR2 Y653/654) and increased phosphorylation of downstream effectors (FRS2, SHP2, MEK1/2, and ERK1/2), consistent with constitutive kinase activity, and the V565L and V565F mutations resulted in a further increase in activity (Fig. 4B and C). In comparison, despite being expressed at higher protein levels, each of the C492 mutants had significantly impaired signaling relative to the wild-type fusion. Whereas autophosphorylation was only moderately attenuated (ratio of pFGFR2 Y653/Y654 to total FGFR2), there was marked reduction in phosphorylation of the FRS2, which binds to the FGFR2 juxta membrane region and serves as a scaffold mediating downstream MEK/ERK activation. Accordingly, there was pronounced decrease in ERK phosphorylation, ranging from approximately 35% (C492F) to >95% (C492R).
Figure 4.

Mutation of FGFR2 C492 compromises FGFR2 signaling. A–C, Signaling analysis of FGFR2 fusion alleles harboring mutations of C492. A, Schematic view of signaling studies. 293T cells were engineered by retroviral transduction to stably express empty vector (EV) or the FGFR2–PHGDH fusion harboring a WT kinase domain, each mutation generated by single nucleotide changes at FGFR2 codon 492, or mutations at the gatekeeper (V565) residue. Immunoblot analysis of signaling proteins (B) and quantification (C). Quantifications were generated from two independent experiments. Data are shown as mean ± SD. Statistical comparison of WT vs. C492 variants by one-way ANOVA (C). D and E, Clonal fitness of cysteine mutations. D, Schematic view of in vivo fitness assay. NIH-3T3 cells engineered with the indicated mutants were pooled and injected subcutaneously into NSG mice. After 16 days, tumors were harvested and processed for ddPCR analysis. E, Clonal abundance of indicated mutants in vivo. Each point represents 3 replicates. Data are shown as mean ± SD. Statistical analysis of input versus tumor by unpaired t test. F–G, Molecular dynamics simulations of select FGFR2 C492 mutations. F, Fluctuations of the G-loop measured by root mean square deviation (RMSD). G, Representative structures from the simulation trajectory. (A and D, Created with BioRender.com. F, Created with R. G, Created with PyMOL.)
To extend these findings, we tested the clonal fitness associated with C492 mutations by examining their transforming potential in NIH3T3 fibroblasts. Cell pools were generated and injected into immunodeficient (NSG) mice (Fig. 4D). Clonal representation of the parental population and within the fully formed tumors was compared, revealing that clones expressing FGFR2 N550K readily outcompeted FGFR2 C492F- and C492R-mutant clones in the absence of drug selection (Fig. 4E). These data reinforce the conclusion that this series of C492 mutations is inefficient in driving clonal outgrowth, despite evading inhibition by futibatinib, consistent with their hypomorphic signaling activity.
C492 is located in the G-loop (GEGCFG) of FGFR2, which upon ATP binding undergoes a conformational change that positions ATP for efficient transfer of γ-phosphate to tyrosine residues on substrates (41). We employed molecular dynamics simulations to study the kinase domain structure and plasticity of the C492 mutants to gain insight into the underlying structural mechanism of their impaired signaling transduction. Four mutations were chosen (i.e., C492F, C492Y, C492R, and C492S) as representatives based on mutant residue size and polarity. We calculated the root mean square deviation (RMSD) of the G-loop as a measure of the fluctuations of this segment (based on 500 ns simulations in explicit water; Fig. 4F). The analysis shows that the wild-type G-loop is more stable than all C492 mutants, with C492R exhibiting the largest flexibility (RMSDavg = 2.1A, compared to RMSDavg = 1.1A for wild-type; see RMSD distribution in Fig. 4F, right). Figure 4G illustrates representative structures from the simulation trajectory, highlighting the increased swing of G-loop of the C492 mutants. These simulations suggest that increased flexibility induced by the mutations might impact dimer formation, ATP binding, and/or FRS2 binding to the dimer.
Discussion
Acquired resistance presents a challenge to prolonged clinical efficacy of FGFR-directed therapies in ICC. The overall response rate of approximately 25%–45% and median duration of response of 7–9 months seen with approved FGFR inhibitors (pemigatinib, infigratinib, futibatinib) falls short of responses seen with targeted therapies in other oncogene-driven cancers like non–small cell lung cancer, in part due to resistance and dose-limiting off-target toxicities (42–48). In this study, we present a systematic analysis of clinical acquired resistance to FGFR inhibitors in FGFR2-positive ICC across a set of reversible FGFR inhibitors and the irreversible inhibitor, futibatinib. Combined analysis of 82 FGFR-treated ICC patients shows that secondary mutations in the FGFR2 kinase domain are the predominant mechanism of acquired resistance, occurring in 60% of patients. Moreover, mutations in the N550 molecular brake residues and V565 gatekeeper are the most frequent such mutations, arising in 63% and 47% of patients, respectively. These data further support that FGFR2 secondary mutations represent the primary mode of acquired resistance to current clinical FGFR inhibitors and that developing novel inhibitors capable of surmounting these alterations represents a critical need.
Interestingly, however, while on-target FGFR2 mutations were the most frequently observed genomic resistance event, mutation of the cysteine residue (C492) that mediates covalent binding of futibatinib was rare, occurring in only 1/42 patients. This is in stark contrast to other covalent inhibitor resistance paradigms, where disruption of the cysteine required for covalency is a common event, such as mutation of C797 in osimertinib resistance and C481 in ibrutinib resistance (36, 37). Our mechanistic studies suggest that mutation of C492 in FGFR2 may result in hypomorphic activity, thus reducing its fitness and providing a potential explanation for its infrequency. This observation also has key implications for the development of covalent inhibitors in general, demonstrating that loss of the critical cysteine residue may not always provide a facile mechanism of acquired resistance.
Similar to the pattern seen with KRASG12C inhibitors and several other targeted therapies, emergence of polyclonal acquired resistance is common with FGFR inhibitors (21, 49–51). This heterogeneous and diverse landscape of mutations presents a challenge for current and future therapies. Specifically, the inability to overcome recurrent V565F and N550K mutations and resistance driven by bypass pathway activation is a universal weakness of all currently approved FGFR TKIs for FGFR2-altered ICC. In this regard, in addition to the mutations in NRAS reported here, our recent work in preclinical models has pointed to feedback activation of EGFR signaling as another bypass mechanism to pan-FGFR inhibition (6, 52). Thus, development of novel agents capable of overcoming common resistance mutations, combination therapies to constrain bypass signaling, and optimal sequencing of FGFR inhibitors may be key avenues to improve outcomes in this patient population. Moreover, serial ctDNA assays to monitor tumor evolution under selective therapeutic pressure can facilitate early detection of acquired resistance alterations, even prior to evidence of radiographic progression, thus providing insight into the etiology of eventual therapeutic failure.
The data demonstrate a diverse landscape of on-target mutations and alternative mechanisms conferring resistance to FGFR inhibitors in cholangiocarcinoma, which can guide optimal selection of approved therapies and clinical development of new drugs. There are several next-generation FGFR inhibitors currently in preclinical and clinical development, including FGFR1-sparing, FGFR2-specific inhibitors and pan-FGFR inhibitors potentially capable of broader coverage of resistance mutations to address these limitations. In particular, FGFR2-specific inhibitors are hypothesized to achieve higher exposures and higher FGFR2 target engagement by minimizing toxicity due to inhibition of other FGFR isoforms (20, 53). Because our data suggest that some frequently observed resistance alterations lead to only modest (e.g., 5- to 10-fold) shifts in IC50, simply increasing the clinically achievable drug concentration could help to prevent outgrowth of some resistance mutations, as we have discussed previously (28). Ultimately, however, given that 60% of patients were observed to develop secondary FGFR2 mutations in this study, and that gatekeeper and molecular brake alterations were the most common, engineering any new therapy to overcome these key resistance alterations would be important.
Supplementary Material
Alternative resistance pathways
Supplementary Table 1. Baseline clinical characteristics of patients with FGFR2-altered intrahepatic cholangiocarcinoma (N=82)
Supplementary Table S2. Representativeness of study.
Acknowledgments
The authors would like to acknowledge the clinicians who cared for the patients for whom data was analyzed in this study. This work was supported by grants from the NIH (P50CA127003), TargetCancer Foundation, DOD (CA160216 and CA210849), and a V Foundation Translational Research Award. R.B. Corcoran was supported by NIH/NCI Moonshot DRSN U54CA224068. Q. Wu was supported by a Cholangiocarcinoma Foundation Marion U. Schwartz Memorial Research Fellowship. H. Ellis was supported by the Irving W. Janock Fellowship. Y. Zhen was supported by the Cholangiocarcinoma Foundation Mark R. Clements Memorial Research Fellowship.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
J. Che reports personal fees from Allorion Therapeutics, Matchpoint Therapeutics, and Soltego Therapeutics; other support from M3 Bioinformatics & Technology; and grants from Springworks Inc outside the submitted work. R.B. Corcoran reports personal fees and other support from Kinnate Biopharma, Alterome Therapeutics, Sidewinder Therapeutics, Cogent Biosciences, C4 Therapeutics, Nested Therapeutics, nRichDx, Remix Therapeutics, and Revolution Medicines; grants and personal fees from Pfizer; other support from Avidity Biosciences and Erasca; personal fees from AbbVie, Elicio, FOG Pharma, Guardant Health, Mirati Therapeutics, Natera, Qiagen, and Taiho; and grants from Lilly and Novartis outside the submitted work. N. Bardeesy reports grants from NIH, NCI, Department of Defense, V Foundation, Cholangiocarcinoma Foundation, and TargetCancer Foundation during the conduct of the study, as well as grants from Taio Pharmaceutical, Tyra Biosciences, and Kinnate Biopharma outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
Q. Wu: Conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. H. Ellis: Conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. G. Siravegna: Data curation, investigation. A.G. Michel: Investigation. B.L. Norden: Investigation. F. Fece de la Cruz: Investigation. E.R. Balasooriya: Investigation, writing–review and editing. Y. Zhen: Investigation, writing–review and editing. V.S. Silveira: Writing–review and editing. J. Che: Formal analysis, investigation, visualization, methodology, writing–original draft, writing–review and editing. R.B. Corcoran: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, methodology, writing–original draft, project administration, writing–review and editing. N. Bardeesy: Conceptualization, resources, formal analysis, supervision, funding acquisition, visualization, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Cleary JM, Raghavan S, Wu Q, Li YY, Spurr LF, Gupta HV, et al. FGFR2 extracellular domain in-frame deletions are therapeutically targetable genomic alterations that function as oncogenic drivers in cholangiocarcinoma. Cancer Discov 2021;11:2488–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jusakul A, Cutcutache I, Yong CH, Lim JQ, Huang MN, Padmanabhan N, et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov 2017;7:1116–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nakamura H, Arai Y, Totoki Y, Shirota T, Elzawahry A, Kato M, et al. Genomic spectra of biliary tract cancer. Nat Genet 2015;47:1003–10. [DOI] [PubMed] [Google Scholar]
- 4. Arai Y, Totoki Y, Hosoda F, Shirota T, Hama N, Nakamura H, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014;59:1427–34. [DOI] [PubMed] [Google Scholar]
- 5. Graham RP, Barr Fritcher EG, Pestova E, Schulz J, Sitailo LA, Vasmatzis G, et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol 2014;45:1630–8. [DOI] [PubMed] [Google Scholar]
- 6. Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov 2013;3:636–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lowery MA, Ptashkin R, Jordan E, Berger MF, Zehir A, Capanu M, et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: potential targets for intervention. Clin Cancer Res 2018;24:4154–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sia D, Losic B, Moeini A, Cabellos L, Hao K, Revill K, et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun 2015;6:6087. [DOI] [PubMed] [Google Scholar]
- 9. Zingg D, Bhin J, Yemelyanenko J, Kas SM, Rolfs F, Lutz C, et al. Truncated FGFR2 is a clinically actionable oncogene in multiple cancers. Nature 2022;608:609–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abou-Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al-Rajabi R, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol 2020;21:671–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Javle M, Roychowdhury S, Kelley RK, Sadeghi S, Macarulla T, Weiss KH, et al. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol Hepatol 2021;6:803–15. [DOI] [PubMed] [Google Scholar]
- 12. Cleary JMVM, Meric-Bernstam F, Hierro C, Heist RS, Ishii N, et al. Safety and efficacy of the selective FGFR inhibitor Debio 1347 in phase I study patients with FGFR genomically activated advanced biliary tract cancer (BTC). J Clin Oncol 2018;36:447. [Google Scholar]
- 13. Pant S, Schuler M, Iyer G, Witt O, Doi T, Qin S, et al. Erdafitinib in patients with advanced solid tumours with FGFR alterations (RAGNAR): an international, single-arm, phase 2 study. Lancet Oncol 2023;24:925–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schuler M, Cho BC, Sayehli CM, Navarro A, Soo RA, Richly H, et al. Rogaratinib in patients with advanced cancers selected by FGFR mRNA expression: a phase 1 dose-escalation and dose-expansion study. Lancet Oncol 2019;20:1454–66. [DOI] [PubMed] [Google Scholar]
- 15. Voss MH, Hierro C, Heist RS, Cleary JM, Meric-Bernstam F, Tabernero J, et al. A phase I, open-label, multicenter, dose-escalation study of the oral selective FGFR inhibitor Debio 1347 in patients with advanced solid tumors harboring FGFR gene alterations. Clin Cancer Res 2019;25:2699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bahleda R, Italiano A, Hierro C, Mita A, Cervantes A, Chan N, et al. Multicenter phase I study of erdafitinib (JNJ-42756493), oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced or refractory solid tumors. Clin Cancer Res 2019;25:4888–97. [DOI] [PubMed] [Google Scholar]
- 17. Goyal L, Baiev I, Zhang K, Dalgai S, Shroff RT, Kelley RK, et al. Acquired resistance to selective FGFR inhibitors in FGFR-altered cholangiocarcinoma. Eur J Cancer 2020;138:S21–2. [Google Scholar]
- 18. Silverman IM, Hollebecque A, Friboulet L, Owens S, Newton RC, Zhen H, et al. Clinicogenomic analysis of FGFR2-rearranged cholangiocarcinoma identifies correlates of response and mechanisms of resistance to pemigatinib. Cancer Discov 2021;11:326–39. [DOI] [PubMed] [Google Scholar]
- 19. Varghese AM, Patel J, Janjigian YY, Meng F, Selcuklu SD, Iyer G, et al. Noninvasive detection of polyclonal acquired resistance to FGFR inhibition in patients with cholangiocarcinoma harboring FGFR2 alterations. JCO Precis Oncol 2021;5:PO.20.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Subbiah V, Sahai V, Maglic D, Bruderek K, Toure BB, Zhao S, et al. RLY-4008, the first highly selective FGFR2 inhibitor with activity across FGFR2 alterations and resistance mutations. Cancer Discov 2023;13:2012–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goyal L, Saha SK, Liu LY, Siravegna G, Leshchiner I, Ahronian LG, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov 2017;7:252–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krook MA, Lenyo A, Wilberding M, Barker H, Dantuono M, Bailey KM, et al. Efficacy of FGFR inhibitors and combination therapies for acquired resistance in FGFR2-fusion cholangiocarcinoma. Mol Cancer Ther 2020;19:847–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov 2018;17:353–77. [DOI] [PubMed] [Google Scholar]
- 24. Chaikuad A, Koch P, Laufer SA, Knapp S. The cysteinome of protein kinases as a target in drug development. Angew Chem Int Ed Engl 2018;57:4372–85. [DOI] [PubMed] [Google Scholar]
- 25. Sootome H, Fujita H, Ito K, Ochiiwa H, Fujioka Y, Ito K, et al. Futibatinib is a novel irreversible FGFR 1–4 inhibitor that shows selective antitumor activity against FGFR-deregulated tumors. Cancer Res 2020;80:4986–97. [DOI] [PubMed] [Google Scholar]
- 26. Bahleda R, Meric-Bernstam F, Goyal L, Tran B, He Y, Yamamiya I, et al. Phase I, first-in-human study of futibatinib, a highly selective, irreversible FGFR1–4 inhibitor in patients with advanced solid tumors. Ann Oncol 2020;31:1405–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goyal L, Meric-Bernstam F, Hollebecque A, Valle JW, Morizane C, Karasic TB, et al. Futibatinib for FGFR2-rearranged intrahepatic cholangiocarcinoma. N Engl J Med 2023;388:228–39. [DOI] [PubMed] [Google Scholar]
- 28. Goyal L, Shi L, Liu LY, Fece de la Cruz F, Lennerz JK, Raghavan S, et al. TAS-120 overcomes resistance to ATP-competitive FGFR inhibitors in patients with FGFR2 fusion-positive intrahepatic cholangiocarcinoma. Cancer Discov 2019;9:1064–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Krook MA, Bonneville R, Chen HZ, Reeser JW, Wing MR, Martin DM, et al. Tumor heterogeneity and acquired drug resistance in FGFR2-fusion-positive cholangiocarcinoma through rapid research autopsy. Cold Spring Harb Mol Case Stud 2019;5:a004002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lim M, Lynch PT, Bai X, Hsiehchen D. Oncogenic RAS drives resistance to pemigatinib in cholangiocarcinoma harboring a FGFR2 delins disrupting ligand binding. JCO Precis Oncol 2023;7:e2200340. [DOI] [PubMed] [Google Scholar]
- 31. DiPeri TPZM, Moss T, Kahle M, Rauli P, Lee SS, et al. Convergent MAPK pathway alterations mediate acquired resistance to FGFR inhibitors in cholangiocarcinoma with FGFR fusions/rearrangements. Cancer Res 2022;82:2618. [Google Scholar]
- 32. Kasi PM. Favorable outcomes in FGFR fusion-positive cholangiocarcinomas and evolution on treatment noted on circulating tumor DNA liquid biopsies. Case Rep Oncol 2020;13:941–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys 1983;79:926–35. [Google Scholar]
- 34. Sridharan V, Neyaz A, Chogule A, Baiev I, Reyes S, Barr Fritcher EG, et al. FGFR mRNA expression in cholangiocarcinoma and its correlation with FGFR2 fusion status and immune signatures. Clin Cancer Res 2022;28:5431–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen H, Ma J, Li W, Eliseenkova AV, Xu C, Neubert TA, et al. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell 2007;27:717–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N Engl J Med 2014;370:2286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med 2015;21:560–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krook MA, Reeser JW, Ernst G, Barker H, Wilberding M, Li G, et al. Fibroblast growth factor receptors in cancer: genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br J Cancer 2021;124:880–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Subbiah V, Iannotti NO, Gutierrez M, Smith DC, Feliz L, Lihou CF, et al. FIGHT-101, a first-in-human study of potent and selective FGFR 1–3 inhibitor pemigatinib in pan-cancer patients with FGF/FGFR alterations and advanced malignancies. Ann Oncol 2022;33:522–33. [DOI] [PubMed] [Google Scholar]
- 40. Byron SA, Chen H, Wortmann A, Loch D, Gartside MG, Dehkhoda F, et al. The N550K/H mutations in FGFR2 confer differential resistance to PD173074, dovitinib, and ponatinib ATP-competitive inhibitors. Neoplasia 2013;15:975–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marsiglia WM, Katigbak J, Zheng S, Mohammadi M, Zhang Y, Traaseth NJ. A conserved allosteric pathway in tyrosine kinase regulation. Structure 2019;27:1308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vogel A, Segatto O, Stenzinger A, Saborowski A. FGFR2 inhibition in cholangiocarcinoma. Annu Rev Med 2023;74:293–306. [DOI] [PubMed] [Google Scholar]
- 43. Shaw AT, Solomon BJ, Besse B, Bauer TM, Lin CC, Soo RA, et al. ALK resistance mutations and efficacy of lorlatinib in advanced anaplastic lymphoma kinase-positive non-small-cell lung cancer. J Clin Oncol 2019;37:1370–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer 2019;121:725–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lin JJ, Choudhury NJ, Yoda S, Zhu VW, Johnson TW, Sakhtemani R, et al. Spectrum of mechanisms of resistance to crizotinib and lorlatinib in ROS1 fusion-positive lung cancer. Clin Cancer Res 2021;27:2899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lin JJ, Liu SV, McCoach CE, Zhu VW, Tan AC, Yoda S, et al. Mechanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small-cell lung cancer. Ann Oncol 2020;31:1725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Russo M, Siravegna G, Blaszkowsky LS, Corti G, Crisafulli G, Ahronian LG, et al. Tumor heterogeneity and lesion-specific response to targeted therapy in colorectal cancer. Cancer Discov 2016;6:147–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cocco E, Lee JE, Kannan S, Schram AM, Won HH, Shifman S, et al. TRK xDFG mutations trigger a sensitivity switch from type I to II kinase inhibitors. Cancer Discov 2021;11:126–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, et al. Clinical acquired resistance to KRAS(G12C) inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov 2021;11:1913–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Parikh AR, Leshchiner I, Elagina L, Goyal L, Levovitz C, Siravegna G, et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat Med 2019;25:1415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired resistance to KRAS(G12C) inhibition in cancer. N Engl J Med 2021;384:2382–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wu Q, Zhen Y, Shi L, Vu P, Greninger P, Adil R, et al. EGFR inhibition potentiates FGFR inhibitor therapy and overcomes resistance in FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov 2022;12:1378–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hollebecque A, Borad M, Goyal L, Schram A, Park JO, Cassier PA, et al. Efficacy of RLY-4008, a highly selective FGFR2 inhibitor in patients (pts) with an FGFR2-fusion or rearrangement (f/r), FGFR inhibitor (FGFRi)-naïve cholangiocarcinoma (CCA): ReFocus trial. Ann Oncol 2022;33 (Suppl 7):S808–69. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Alternative resistance pathways
Supplementary Table 1. Baseline clinical characteristics of patients with FGFR2-altered intrahepatic cholangiocarcinoma (N=82)
Supplementary Table S2. Representativeness of study.
Data Availability Statement
This work includes a meta-analysis of published data from 12 studies (1, 17–22, 28–32). The data generated in this study are available in the article and its Supplementary Data. Raw data are available upon reasonable request from the corresponding authors.