

Abstract
Background
Cornelia de Lange syndrome (CdLS) is mainly characterized by specific facial features, growth retardation, and bone deformities. Seven genes reportedly cause CdLS. Recent research has reported that loss‐of‐function variants affecting MAU2, which encodes a regulator of the cohesin complex, can cause CdLS. Thus far, only one MAU2‐CdLS case has been reported worldwide.
Methods
We detected a novel variant in MAU2 gene, NM_015329, c.526C>T (p.Arg176Trp) in a Chinese patient with CdLS, constructed a plasmid for in vitro transcriptional and protein level analysis, and analyzed the interaction between the MAU2/NIPBL complex using molecular dynamics (MD).
Results
The results showed that the level of the exogenous MAU2 mutant protein was significantly reduced compared with that of the exogenous wild‐type protein. However, MD analysis predicted an increased binding free energy between the MAU2 and NIPBL proteins that may impact the structural stability of the complex.
Conclusion
We investigated a MAU2‐CdLS case in a Chinese family, which strengthens the association between MAU2 variants and CdLS phenotypes. We therefore propose that MAU2 be included in the CdLS gene screening list.
Keywords: CdLS, cohesin, Cornelia de Lange syndrome, genetic testing, MAU2
This is the first one to report a CdLS patient from China. In this study, a de novo heterozygous missense mutation of MAU2 gene was identified. In vitro functional studies showed that the mutation may affect the protein expression, which supported the evidence of biological harmfulness caused by the mutation.

1. INTRODUCTION
Cornelia de Lange syndrome (CdLS) is a genetic disease with neurodevelopmental disorders as its main clinical feature. It was first reported by Brachman in 1916 and further elucidated by Cornelia de Lange in 1933; it is therefore also referred to as Brachman de Lange syndrome (Boyle et al., 2015; Deschamps, 2022). The primary clinical manifestations of CdLS are intellectual disability, typical facial features, intrauterine and postnatal growth retardation, and multiple organ malformations. According to statistical and epidemiological evidence, one in every 10,000 to 30,000 people worldwide is affected by this disease (Bottai et al., 2019). Diagnosing children of different ages with CdLS is challenging due to the broad spectrum of symptoms (Allanson et al., 1997; Kline et al., 2007). Thus, careful attention must be paid to the early identification, diagnosis, follow‐up, and management of children with CdLS to improve their survival rate and quality of life.
The diagnosis of CdLS mainly relies on typical clinical features and molecular diagnostic techniques. The seven disease‐associated genes of CdLS (NIPBL, SMC1A, SMC3, RAD21, HDAC8, BRD4, and ANKRD11) are mostly the subunits or regulatory factors of a cohesin complex (NIPBL, SMC1A, SMC3, RAD21, and HDAC8). Therefore, CdLS is also considered to be a cohesinopathy disease (Kline et al., 2018). CdLS1, caused by NIPBL defects, is the most common CdLS subtype. While few subtypes have been reported, some patients with the typical CdLS phenotype have not been accurately diagnosed at the molecular level (Kline et al., 2018; Rohatgi et al., 2010). Parenti et al. reported a MAU2‐CdLS case and confirmed that the reported MAU2 variant can reduce the interaction NIPBL‐MAU2, suggesting that the reduced function of the NIPBL/MAU2 complex may be a potential mechanism of CdLS pathophysiology (Parenti et al., 2020).
Thus far, only one case of CdLS caused by a MAU2 variant has been reported worldwide (Parenti et al., 2020). Herein, we report a patient with CdLS from China, with clinical manifestations of atypical facial features, growth retardation, cardiac malformation, and kidney disease. In this patient, the trio‐whole exome sequencing (WES) test indicated that MAU2 was mutated. Our study enriched the variant and phenotype data of MAU2 and added a new reference basis for clarifying the correlation between MAU2 and CdLS. Hence, we suggest that MAU2 be included in the routine gene‐screening for CdLS diagnosis.
2. MATERIALS AND METHODS
2.1. Ethics approval
Informed consent was obtained from the patient's family. This study was approved by the Institutional Review Board of the Hospital (EYLL‐2020‐010).
2.2. WES and sequential analysis
To clarify the patient's diagnosis, genomic DNA was extracted from the peripheral blood of the patient and his parents. A NovaSeq 6000 Sequencing platform (Illumina, San Diego, USA) was used to perform WES, and an IDT XGen Exome Research Panel v1.0 (Integrated DNA Technologies, Coralville, USA) was used to capture libraries. Raw data were processed by fastp (v0.20.1, https://github.com/OpenGene/fastp) for adapter removal and low‐quality reads filtering. Clean reads were defined as those remaining after adapter trimming and quality trimming bases below Q20 from 3′ ends using TrimGalore (v0.6.5, https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). The clean paired‐end reads were aligned to the Ensemble GRCh37/hg19 reference genome using BWA (v0.5.9, http://bio‐bwa.sourceforge.net/). Alignments were sorted, and duplicates were marked using Picard (v2.18.29, https://broadinstitute.github.io/picard/). Base quality score recalibration was performed with GATK (v4.1.7, http://bio‐bwa.sourceforge.net/) using dbSNP build 150 and Mills +1000G gold standard indels. Variants were called using GATK HaplotypeCaller (v4.1.7, http://bio‐bwa.sourceforge.net/) and filtered using GATK Variant Quality Score Recalibration (VQSR) at a 99% sensitivity tranche threshold. Variants were annotated using ANNOVAR (v2021Jul10, https://annovar.openbioinformatics.org) (McKenna et al., 2010). Through basic filtering, the following variants were filtered out: (1) wild‐type in the proband, (2) intronic variants deeper than 30 bp, (3) low‐quality SNPs (variant frequency <0.2 or sequencing depth <4X or quality value <35), (4) low‐frequency simple tandem repeat Indels (SSR >7 and AF <0.3), and (5) indels larger than 50 bp. Computational prediction tools were used to assess possible pathogenicity of missense variants, including SIFT (v5.2.2, www.sift.bii.a‐star.edu.sg), Polyphen‐2 (v2.2.2, www.genetics.bwh.harvard.edu/ph2), Provean (v1.1.5, http://provean.jcvi.org/index.php) and MutationTaster (NCBI 37/Ensembl 69, www.mutationtaster.org). To predict the functional change of variants on the splicing sites, MaxEntScan, dbscSNV, and GTAG software packages were used instead of product structure prediction software. The single nucleotide polymorphism database dbSNP (https://www.ncbi.nlm.nih.gov/snp/), the Genome Aggregation Database gnomAD (https://gnomad.broadinstitute.org/), the 1000 Genomes Project (http://www.1000genomes.org/home), the Exome Aggregation Consortium ExAC (http://exac.broadinstitute.org), the Exome Sequencing Project ESP (http://evs.gs.washington.edu/EVS/), and other frequency databases as well as Online Mendelian Inheritance in Man OMIM (https://www.omim.org/), the Human Gene Mutation Database HGMD (http://www.hgmd.cf.ac.uk/ac/index.php), ClinVar (Clinical relevant Sequence Variants, http://www.ncbi.nlm.nih.gov/clinvar/). Variant prioritization focused on rare variants with moderate or high predicted impacts (e.g. frameshift, nonsense, splicing, missense). The remaining variants were restricted to a panel of genes related to the patient's phenotype through Human Phenotype Ontology (HPO) terms. Trio analysis evaluated de novo, compound het, and recessive models. Variants were checked for previous reports in databases, such as ClinVar, HGMD, OMIM, and scientific literature. Top candidate variants were reviewed manually using IGV visualization, inheritance patterns, gene function, and patient phenotype. WES revealed potential pathogenic variants informed by the patient's phenotype and family history. All variants were evaluated according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015). Sanger sequencing was performed to validate the variation identified in the patient, his parents, and two younger brothers. For details of WES quality control and variant filtering, refer to Supplementary Materials 1 and 2, respectively.
2.3. Plasmid construction and real‐time fluorescent quantitative polymerase chain reaction (qPCR)
We constructed MAU2 wild‐type (MAU2‐WT) and mutant (MAU2‐MUT) plasmids (backbone: pcDNA3.1‐FLAG‐N, Beyotime Inc., Nanjing, China) and amplified the target fragment of MAU2‐WT with Phanta® Max Super‐Fidelity DNA Polymerase (#P505, Vazyme, Nanjing, China) high fidelity polymerase. Furthermore, the ClonExpress® II One Step Cloning Kit (#C112, Vazyme) was used to construct the MAU2‐WT plasmid, and the primer sequences, F: 5′‐aaggatgaggatgacaccaccttATGGCGGCTCAGGCG‐3′, and R: 5′‐agtggatccgagctcggtaccTCAGAGGCTGGCCAGG‐3′, were amplified. Subsequently, the MAU2‐MUT plasmid was constructed by point variant using Mut Express® MultiS Fast Mutagenesis Kit V2 (#C215, Vazyme), and the primer sequences, F: 5′‐tGGGTGGGGGATCGAATACACACGGCGCT‐3′, and R: 5′‐TCAGATCCCACCACCACCACCACGCGTACTCGGCCCCTAC‐3′, were amplified. The constructed plasmids were transfected into human 293T cells (cell density of 60%) using 5 μL of Lip2000 (Thermo Fisher Scientific Inc., Waltham, MA, USA). After 48 h of transfection, the RNA was extracted with Trizol lysate (Thermo Fisher Scientific Inc.). Finally, TIANScript II M‐MLV reverse transcriptase (#ER107, Tiangen Inc., China) and FastFire qPCR PreMix (Probe‐#FP208, Tiangen Inc.) were used for qPCR analysis, and the MAU2 primer sequences, F: 5′‐GAGAAGGCGAGAAGAGTACACG‐3′, R: 5′‐CAAGGCACATATGATG‐3′, and the GAPDH primer sequences, F: 5′‐TGTGGGCATCAATGGATTTGG‐3′, and R: 5′‐ACACCATGTATTCGGTCAAT‐3′, were amplified.
2.4. Protein level analysis
After the constructed MAU2‐WT and MAU2‐MUT plasmids (2 μg) were transfected into human 293T cells, protein (80 μg) was extracted with protein lysate (Beyotime Inc.) and analyzed by western blotting (WB). The experiment was repeated three times. The antibody information is as follows: (a) the primary antibodies were mouse anti‐human FLAG (cat#8146, CST Inc., USA) and mouse anti‐human β‐Actin (cat#3700, CST Inc.), at a dilution ratio of 1:1000; and (b) the secondary antibodies were anti‐mouse IgG and HRP‐linked antibody (cat#7076, CST Inc.), at a dilution ratio of 1:5000. Image J v1.46 software (NIH, Bethesda, MD, USA) was used to analyze the western blot bands.
2.5. Statistic analysis
GraphPad Prism8 (GraphPad Software Incorporation, San Diego, CA, USA) was used to conduct the statistical analyses and draw charts. Statistical significance is represented as follows: *p < 0.05; **p < 0.01; ***p < 0.001. Each experiment was repeated thrice.
2.6. Molecular dynamics (MD) prediction of the interaction between MAU2 and NIPBL
MAU2 interacts with NIPBL to form a complex to perform its biological functions (Parenti et al., 2020). Alphafold2 (https://alphafold.ebi.ac.uk/entry/) was used to build a 3D structure, and the “protein–protein interactions” function of the Rosetta software was used to obtain the best binding mode of the two proteins. The mutant complex structure was built on the basis of the wild‐type. MD simulation was completed using the Gromacs 5.14 software; the molecular force field was Gromos53a6, and the simple point charge water model was used for a total simulation time of 50 ns. The Particle Mesh Ewald method was used to calculate the electrostatic interactions, and the leapfrog algorithm was used to calculate the motion of each atom. The steepest descent energy method was used to minimize the energy of 400 steps, and the 50 ps position constraint simulation was carried out for each simulation system. The PyMOL v2.5, VMD, and Origin v8.5 software packages were used to generate the graphs.
3. RESULTS
3.1. Case presentation
The patient in this study was a 11‐year‐old boy hospitalized with symptoms of dizziness, weakness for four days, and abnormal renal function. He had non‐consanguineous Chinese parents and was born full term to a G1P1 mother. His birth weight was unknown, and there was no history of asphyxia at birth. Routine physical examination after birth showed that the left kidney was absent, but no further examination was performed. Atrial septal defect repair was performed at the age of one year. At the age of five years, he was treated with growth hormone for five years owing to his short stature. At 10 years of age, the patient was treated for attention deficit hyperactivity disorder with oral atomoxetine hydrochloride for more than one year.
The patient's physical examination showed that he had atypical facial features, including a hairy forehead and eyebrows with curved arches (Figure 1a). At present, the patient's height is 148 cm [−0.5 standard deviation (SD)], his body mass is 35.5 kg (−0.5 SD), head circumference is 55 cm (−0.5 SD), and his fingers and toes are short (Figure 1b). The child can communicate freely with others, but his self‐control abilities are poor. The evaluation result of the fifth edition of the Wechsler Intelligence Scale for Children is 71 points, which indicates mild intellectual disability.
FIGURE 1.
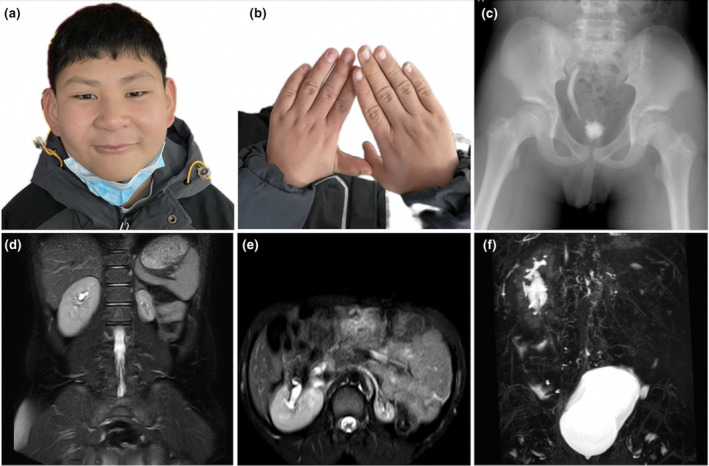
Clinical information of the patient with CdLS. (a) The child has facial features including a hairy forehead and face, thick eyebrows with curved arches, long philtrum, thin upper lip, and high jaw arch; (b) The fingers are short, and the fifth finger is bent; (c) Excretory cystourethrography showed right ureteral reflux, and less contrast medium remained in the bladder; (d–f) Abdominal magnetic resonance imaging showed a right duplicate kidney, two sets of renal pelvises converged at the renal hilum, and a slightly dilated proximal ureter. The left kidney was small with a thin wall. CdLS, Cornelia de Lange syndrome.
Color Doppler echocardiography revealed changes in the atrial‐septal defect post‐operation. No residual shunt or other structural or functional abnormalities of the heart were found. Excretory cystourethrography showed right ureteral reflux (I–II) and a less contrast agent that remained in the bladder (Figure 1c). Abdominal MRI showed a right duplicate kidney, two sets of renal pelvises converged at the renal hilum, a slightly dilated corresponding proximal ureter, full bladder, and a small left kidney with a thin wall (Figure 1d–f). His father had myocarditis, whereas his mother and two younger brothers were in good health. Table 1 depicts the patient's detailed clinical data.
TABLE 1.
Clinical data of our patient and another reported case of MAU2‐CdLS.
| Patient information | Our patient | Parenti et al. (2020) |
|---|---|---|
| Sex | M | M |
| Variant type | Missense | In‐frame deletion |
| Nucleic acid alteration | c.526C>T | c.927_947del |
| Amino acid variation | p.Arg176Trp | p.Gln310_Ala316del |
| RefSeq | NM_015329 | NM_015329 |
| Inheritance | De novo | De novo |
| CdLS type | Atypical | Classic |
| Basic feature a | ||
| Continuous eyebrows and/or thick eyebrows | − | + |
| Short nose, concave nasal ridge and/or nostril anteversion | − | + |
| Long‐featureless philtrum | + | + |
| Thin upper lip and/or downward corner of mouth | + | + |
| Oligodactyly and/or congenital anencephaly | − | − |
| Congenital diaphragmatic hernia | − | − |
| Suggestive features a | ||
| General retardation and/or intellectual disability | + | + |
| Intrauterine growth retardation (<x − 2 SD) | NA | + |
| Postnatal growth retardation (<x − 2 SD) | + | + |
| Microcephaly | − | + |
| Small hands and/or short feet | + | + |
| The fifth finger is stunted or undeveloped | + | + |
| Hirsutism | + | + |
| Other facial features | ||
| High arched palate | + | + |
| Cleft palate | − | − |
| Teeth (small, widely spaced) | + | − |
| Micrognathia | − | − |
| Other organs | ||
| Cardiac | ASD | − |
| Genitourinary | Right duplex kidney and left renal dysplasia | − |
| Gastroenterology | − | Gastroesophageal reflux disease |
| Feeding problems in infancy | − | + |
| Hearing loss | Hyperacusis | − |
| Ophthalmic problems | Ptosis | Hypertelorism |
| Myopia | + | + |
| Behavior | Temper tantrums | NA |
Abbreviations: ASD, atrial septal defect; CdLS, Cornelia de Lange syndrome; F, female; M, male; NA, not available; SD, standard deviation.
The classification of basic and suggestive features is based on the 2018 CdLS international consensus released by (Kline et al., 2018).
3.2. CdLS international consensus for clinical diagnosis
In 2018, Kline et al. published the first international consensus on CdLS, which divided the CdLS phenotype into basic and suggestive features to evaluate the diagnostic strategy (6). According to the consistent diagnostic criteria, the patient showed two basic features (long philtrum, thin lips) and two suggestive features (short fingers, 5th finger clinodactyly, hirsutism and growth retardation), with a comprehensive score of eight points. Therefore, the diagnosis needed to be further combined with the molecular diagnosis results.
3.3. Identification of the MAU2 variant causing CdLS
We identified a heterozygous missense variant, NM_015329, c.526C>T (p.Arg176Trp), in exon 5 of the patient's MAU2 gene; his parents are wild type, which indicates that it may be a de novo variant. This variant was not observed in population databases including dbSNP, 1000 Genomes, gnomAD, ExAC, ESP, or our internal database of exomes. No observations were found in disease databases OMIM, ClinVar, or HGMD. The prediction of the function of this missense variant with online software [SIFT (0.023, damaging), PolyPhen2_HDIV (0.999, probably damaging), PolyPhen2_HVAR (0.94, probably damaging), MutationTaster (0.999624, disease‐causing), and PROVEAN (−2.7, deleterious)] revealed that it is damaging or probably damaging. Only two variants of MAU2 were included in the ClinVar database, neither of which were pathogenic nor likely pathogenic. The NM_015329, c.526C>T (p.Arg176Trp) in our study was classified as likely pathogenic based on ACMG criteria: PS2—De novo occurrence with confirmed maternity and paternity; PM2—Absent from general population databases; PP2—Mutations in genes with a missense Z score ≥3.09 in the gnomAD database; PP3—Multiple lines of computational evidence support a deleterious effect. Sanger sequencing confirmed that the patient's MAU2 gene was mutated, while his parents and two brothers were both wild‐type (Figure 2a).
FIGURE 2.
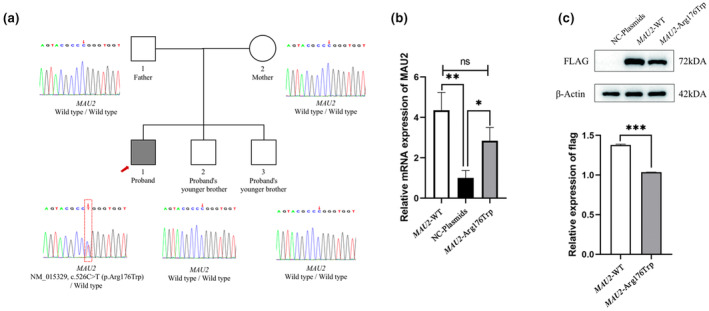
Mutation information of the patient. (a) The results of trio‐WES revealed a heterozygous missense mutation, NM_015329, c.526C>T (p.Arg176Trp), in the MAU2 gene of the patient. His parents and two younger brothers were wild‐type. Sanger sequencing confirmed the existence of the mutation; (b) qPCR results showed that the mRNA expression in the MAU2‐WT and MAU2‐MUT groups was significantly higher than that in the NC‐plasmids group, but there was no significant difference between the MAU2‐WT and MAU2‐MUT groups, indicating that the mutation may not affect the transcription process of MAU2; (c) Western blot analysis showed that the MAU2 protein expression of the MAU2‐MUT group (p.Arg176Trp) was significantly lower (22%) than that of the MAU2‐WT group. *p < 0.05; **p < 0.01; ***p < 0.001.
3.4. Mutated MAU2 transcription and protein level
After the three groups—no carrier control (NC‐plasmids), MAU2‐MUT, and MAU2‐WT—were transfected into human 293T cells, the mRNA level of MAU2 was analyzed using qPCR. The results showed that the mRNA level of the MAU2‐MUT and MAU2‐WT groups was significantly higher than that of the NC‐plasmids group, but there was no significant difference between the MAU2‐MUT and MAU2‐WT groups (p = 0.076), suggesting that the variant did not affect its mRNA level (Figure 2b). However, WB results showed that the protein level of the MAU2‐MUT group was 22% lower than that of the MAU2‐WT group (p < 0.001; Figure 2c). The in vitro experimental results showed that the level of the exogenous MAU2 mutant protein was significantly reduced compared with that of the exogenous wild‐type protein. The uncut WB gel film is included in Supplementary Material 3.
3.5. Mutated MAU2 may have a weakened interaction with NIPBL
The root mean square deviation (RMSD) was used to monitor the average deviation between the protein conformation and original structure (Supplementary Material 4). The wild‐type and mutant complexes reached equilibrium at the same time, and the corresponding average RMSD values were approximately 2.45 and 2.47 Å, respectively (Figure 3a). In the wild‐type system, a hydrogen bond was formed between MAU2 Arg176 and Glu173 and between Arg184 and Glu181. In the NIPBL/MAU2 complex, a hydrogen bond was formed between NIPBL Cys2392 and MAU2 Glu173 and between NIPBL Thr1943 and MAU2 Arg176. In the mutant system, MAU2 Arg176Trp broke the hydrogen bond with Glu173, while Arg184 formed a hydrogen bond with Glu173. In the NIPBL/MAU2 complex, NIPBL Cys2392 formed a hydrogen bond with mutant MAU2 Glu173, and mutant MAU2 Trp176 formed a hydrophobic interaction with NIPBL, which may weaken the interactions between proteins in the mutant system (Figure 3b). The results of the binding free energy of protein interaction showed that the total binding free energy of the wild‐type (−223.97 kJ/mol) was lower than that of the mutant system (−161.35 kJ/mol), with the main difference observed in the van der Waals forces (Table 2).
FIGURE 3.
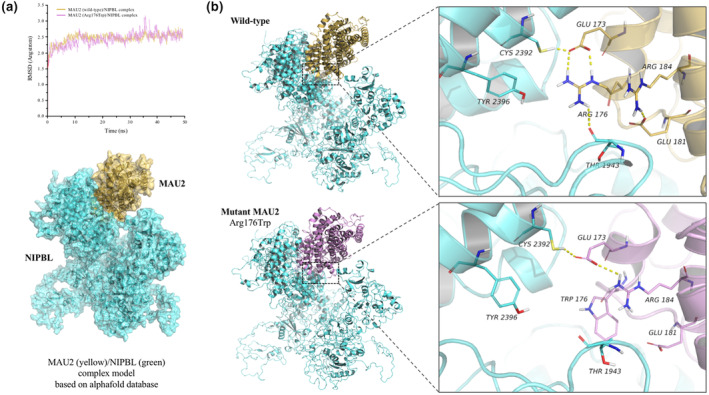
Structural prediction and analysis of the mutant protein. (a) The root mean square evolution was used to monitor the balance of wild‐type and mutant MAU2/NIPBL complexes, and the wild‐type complex structure diagram was formed; (b) A hydrogen bond was formed between the wild‐type MAU2 Arg176 and Glu173, and between the surrounding residues Arg184 and Glu181. In the wild‐type NIPBL/MAU2 complex, a hydrogen bond was formed between NIPBL Cys2392 and MAU2 Glu173, and between NIPBL Thr1943 and MAU2 Arg176. However, mutant MAU2 p.Arg176Trp broke the hydrogen bond with Glu173, while Arg184 formed a hydrogen bond with Glu173. In the mutant NIPBL/MAU2 complex, NIPBL Cys2392 formed a hydrogen bond with mutant MAU2 Glu173, and mutant MAU2 Trp176 formed a hydrophobic interaction with NIPBL, which may weaken the stability of the mutant NIPBL/MAU2 complex. CdLS, Cornelia de Lange syndrome; MAU2‐MUT, MAU2‐mutation plasmids; MAU2‐WT, MAU2‐wild type plasmids; NC‐plasmids, no carrier control plasmids; qPCR, quantitative polymerase chain reaction.
TABLE 2.
Energy analysis of MAU2/NIPBL complex interaction.
| Contribution term | MAU2 (wild‐type)/NIPBL complex energy value (kJ/mol) | MAU2 (mutant‐type)/NIPBL complex energy value (kJ/mol) |
|---|---|---|
| △Evdw | −275.91 ± 1.56 | −201.22 ± 1.21 |
| △Eele | −25.33 ± 0.64 | −19.09 ± 0.59 |
| △Gpolar | 102.35 ± 1.01 | 85.25 ± 1.25 |
| △Gnonpolar | −25.08 ± 0.17 | −26.29 ± 0.22 |
| △Gbind | −223.97 ± 1.52 | −161.35 ± 1.33 |
Abbreviations: Eele, Electrostatic interaction; Evdw, van der Waals force; Gbind, Binding free energy; Gnonpolar, Nonpolar solvation energy; Gpolar, Polar solvation energy.
4. DISCUSSION
Cohesin is a trimolecular ring‐shaped complex that encircles chromatin and performs a series of different functions throughout the cell cycle (Sedeño Cacciatore & Rowland, 2019). This complex consists of four core subunits: SMC1A, SMC3, RAD21, and STAG1/2. Besides these, many other proteins are involved in effective adhesion protein complex functions, such as cohesive protein loading complex NIPBL/MAU2 or cohesive protein releasing factor WAPAL (Nasmyth & Haering, 2009). Next‐generation sequencing technology has revealed gene variants related to the cohesin complex to be the main cause of CdLS, and these variants have been gradually identified in patients with CdLS (Kline et al., 2018; Selicorni et al., 2021). However, MAU2‐CdLS cases are rare. Until now, only one such case has been reported worldwide (Parenti et al., 2020). The patient we reported has an atypic CdLS appearance. According to the international consensus of CdLS, the patient was scored eight points and finally diagnosed by molecular detection. The child also had atypical growth retardation, cardiac malformation, and nephrosis with a low incidence rate. It is worth noting that patients with CdLS seeking medical treatment are usually diagnosed because of their specific facial features or abnormal nervous system development, whereas our patient was diagnosed with CdLS because of renal dysplasia. Therefore, in addition to the general process of international consensus, CdLS diagnosis should include detailed organ function screening.
In this study, we used trio‐WES technology to detect the variant in the child and his parents, but did not find any pathogenic variants in the seven known pathological genes of CdLS. In addition, we also screened related syndromes overlapping with CdLS phenotypes, such as KBG syndrome (ANKRD11 variant‐related), CHOP syndrome (AFF4 variant‐related), and Rubinstein‐Taybi syndrome (EP300 variant‐related), but no suspected pathogenic variants in these disease‐related genes were found (Bestetti et al., 2022; Lee et al., 2022; Raible et al., 2019; Selicorni et al., 2021). Finally, we found a de novo variant, c.526C>T (p.Arg176Trp), on exon 5 of MAU2, which was located in the unknown domain between TPR‐1 and TPR‐3 of the MAU2 protein (Hinshaw et al., 2015). Through structural model analysis, the variant site was found to interact with the NIPBL protein structure. The hydrophilic wild‐type Arg176 formed a hydrogen bond with Glu173. However, p.Arg176Trp destroyed the hydrogen bond structure, leading to the reduced local structural stability of MAU2. In vitro functional experiments also showed that the expression level of the mutant protein was significantly reduced. p.Arg176Trp also formed a hydrophobic interaction with NIPBL, which may affect the structural stability between the proteins. MD results also confirmed that the increased binding free energy may lead to decreased stability of the NIPBL/MAU2 complex.
MAU2 and NIPBL proteins can form a heterodimer complex called kollerin, which can load cohesin onto chromatin (Kline et al., 2018). In general, the structural stability of kollerin is crucial. An altered or missing structure of MAU2 or NIPBL may destroy the structural stability of kollerin, affecting cohesin loading and transcriptional regulation (Zuin et al., 2014). It is noteworthy that the pathogenesis of MAU2 may be more complex than expected. For example, the cases reported by Parenti et al. [MAU2 de novo variant, NM_015329, c.927_947del, p.(Gln310_Ala316del)] mainly showed classic CdLS symptoms, accompanied by severe mental, language, and motor retardations, and brain structural abnormalities (Parenti et al., 2020). However, the two CdLS patients reported by Braunholz et al. showed special conditions, with MAU2 variant in both unrelated families, c.9_23del (p.Gln4_Ala8del). The unaffected mother of one patient with classic CdLS symptoms also carried the variant. Another patient carried the variant and had an additional deletion of approximately 5 Mb on chromosome 2. However, functional experiments have shown that the variant does not significantly affect the interaction with NIPBL, so the researchers do not believe that the variant was a pathogenic factor for the two patients (Braunholz et al., 2012). In our study, the disease was caused by a missense de novo variant in MAU2. To find out whether this is mildly related to the phenotype in this case (for example, there is no synophrys, bushy eyebrows, or serious neurological developmental disorders), more cases need to be analyzed.
Usually, CdLS can be easily diagnosed during childhood owing to the typical facial features. For CdLS cases confirmed by the international consensus of disease diagnosis, pediatricians should comprehensively monitor and evaluate the possible manifestations of multi‐system or organ abnormalities, including congenital heart disease, and bone and kidney deformities. At present, there is no specific treatment plan for CdLS, and only symptomatic treatment is provided. However, it is imperative to determine the genotype to formulate personalized treatments and nursing plans. For example, growth retardation is a common phenotype of CdLS. NIPBL variants may lead to short stature due to low growth hormone levels; therefore, good height recovery can be achieved with growth hormone treatment (de Graaf et al., 2017). Other CdLS pathogenic genes may cause mild growth retardation due to gastrointestinal abnormalities or growth hormone disorders (Huisman et al., 2017). Our patient presented with a severely short stature. Although we were unable to obtain previous growth and developmental data, the continuous use of recombinant human growth hormone over many years helped him achieve the standard height‐for‐age, indicating that the MAU2 variant may show symptoms of low growth hormone levels similar to NIPBL‐CdLS. In addition, this child had nephrotic characteristics. Therefore, for children with CdLS, multidisciplinary joint consultation is required to formulate a comprehensive treatment and nursing plan.
5. CONCLUSION
A novel variant in MAU2 was found in a child with atypical CdLS appearance using gene detection. The variant may affect the structural stability of the MAU2/NIPBL complex. This study also confirmed that CdLS has the characteristics of genetic heterogeneity and expanded the variant‐phenotype data of MAU2. Further research on MAU2‐CdLS cases can improve the molecular diagnosis rate of CdLS as well as our understanding of the mechanisms underlying this disease.
AUTHOR CONTRIBUTIONS
Yin Peng and Fang Deng: Designed the study. Yin Peng and Ying Zhu: Wrote the manuscript. Yin Peng and Fang Deng: Revised the article. Yin Peng, Ying Zhu, and Lin Wu: Collected the data. All authors contributed to the article and approved the submitted version.
FUNDING INFORMATION
This research was sponsored by the Natural Science Foundation of Anhui Province (No. 2108085MH262).
CONFLICT OF INTEREST STATEMENT
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study was conducted according to the guidelines of the World Medical Association (Declaration of Helsinki) and approved by the Ethics Committee of the Anhui Provincial Children's Hospital.
PATIENT CONSENT FOR PUBLICATION
The patient's parents have signed an informed consent form.
Supporting information
Supplementary material 1.
Supplementary material 2.
Supplementary material 3.
Supplementary material 4.
ACKNOWLEDGMENTS
We express our gratitude to the patient and his parents for their participation in this study.
Peng, Y. , Zhu, Y. , Wu, L. , & Deng, F. (2024). Clinical study and genetic analysis of Cornelia de Lange syndrome caused by a novel MAU2 gene variant in a Chinese boy. Molecular Genetics & Genomic Medicine, 12, e2318. 10.1002/mgg3.2318
DATA AVAILABILITY STATEMENT
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Allanson, J. E. , Hennekam, R. C. , & Ireland, M. (1997). De Lange syndrome: Subjective and objective comparison of the classical and mild phenotypes. Journal of Medical Genetics, 34(8), 645–650. 10.1136/jmg.34.8.645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestetti, I. , Crippa, M. , Sironi, A. , Tumiatti, F. , Masciadri, M. , Smeland, M. F. , Naik, S. , Murch, O. , Bonati, M. T. , Spano, A. , Cattaneo, E. , Mariani, M. , Gotta, F. , Crosti, F. , Cavalli, P. , Pantaleoni, C. , Natacci, F. , Bedeschi, M. F. , Milani, D. , … Finelli, P. (2022). Expanding the molecular Spectrum of ANKRD11 gene defects in 33 patients with a clinical presentation of KBG syndrome. International Journal of Molecular Sciences, 23(11), 5912. 10.3390/ijms23115912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottai, D. , Spreafico, M. , Pistocchi, A. , Fazio, G. , Adami, R. , Grazioli, P. , Canu, A. , Bragato, C. , Rigamonti, S. , Parodi, C. , Cazzaniga, G. , Biondi, A. , Cotelli, F. , Selicorni, A. , & Massa, V. (2019). Modeling Cornelia de Lange syndrome in vitro and in vivo reveals a role for cohesin complex in neuronal survival and differentiation. Human Molecular Genetics, 28(1), 64–73. 10.1093/hmg/ddy329 [DOI] [PubMed] [Google Scholar]
- Boyle, M. I. , Jespersgaard, C. , Brøndum‐Nielsen, K. , Bisgaard, A. M. , & Tümer, Z. (2015). Cornelia de Lange syndrome. Clinical Genetics, 88(1), 1–12. 10.1111/cge.12499 [DOI] [PubMed] [Google Scholar]
- Braunholz, D. , Hullings, M. , Gil‐Rodríguez, M. C. , Fincher, C. T. , Mallozzi, M. B. , Loy, E. , Albrecht, M. , Kaur, M. , Limon, J. , Rampuria, A. , Clark, D. , Kline, A. , Dalski, A. , Eckhold, J. , Tzschach, A. , Hennekam, R. , Gillessen‐Kaesbach, G. , Wierzba, J. , Krantz, I. D. , … Kaiser, F. J. (2012). Isolated NIBPL missense mutations that cause Cornelia de Lange syndrome alter MAU2 interaction. European Journal of Human Genetics, 20(3), 271–276. 10.1038/ejhg.2011.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf, M. , Kant, S. G. , Wit, J. M. , Willem Redeker, E. J. , Eduard Santen, G. W. , Henriëtta Verkerk, A. J. M. , Uitterlinden, A. G. , Losekoot, M. , & Oostdijk, W. (2017). Successful growth hormone therapy in Cornelia de Lange syndrome. Journal of Clinical Research in Pediatric Endocrinology, 9(4), 366–370. 10.4274/jcrpe.4349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschamps, G. N. (2022). Cornelia de Lange Syndrome. Neonatal Network, 41(3), 145–149. 10.1891/NN-2021-0011 [DOI] [PubMed] [Google Scholar]
- Hinshaw, S. M. , Makrantoni, V. , Kerr, A. , Marston, A. L. , & Harrison, S. C. (2015). Structural evidence for Scc4‐dependent localization of cohesin loading. eLife, 4, e06057. 10.7554/eLife.06057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisman, S. , Mulder, P. A. , Redeker, E. , Bader, I. , Bisgaard, A. M. , Brooks, A. , Cereda, A. , Cinca, C. , Clark, D. , Cormier‐Daire, V. , Deardorff, M. A. , Diderich, K. , Elting, M. , van Essen, A. , FitzPatrick, D. , Gervasini, C. , Gillessen‐Kaesbach, G. , Girisha, K. M. , Hilhorst‐Hofstee, Y. , … Hennekam, R. C. (2017). Phenotypes and genotypes in individuals with SMC1A variants. American Journal of Medical Genetics. Part A, 173(8), 2108–2125. 10.1002/ajmg.a.38279 [DOI] [PubMed] [Google Scholar]
- Kline, A. D. , Grados, M. , Sponseller, P. , Levy, H. P. , Blagowidow, N. , Schoedel, C. , Rampolla, J. , Clemens, D. K. , Krantz, I. , Kimball, A. , Pichard, C. , & Tuchman, D. (2007). Natural history of aging in Cornelia de Lange syndrome. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 145C(3), 248–260. 10.1002/ajmg.c.30137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline, A. D. , Moss, J. F. , Selicorni, A. , Bisgaard, A. M. , Deardorff, M. A. , Gillett, P. M. , Ishman, S. L. , Kerr, L. M. , Levin, A. V. , Mulder, P. A. , Ramos, F. J. , Wierzba, J. , Ajmone, P. F. , Axtell, D. , Blagowidow, N. , Cereda, A. , Costantino, A. , Cormier‐Daire, V. , FitzPatrick, D. , … Hennekam, R. C. (2018). Diagnosis and management of Cornelia de Lange syndrome: First international consensus statement. Nature Reviews. Genetics, 19(10), 649–666. 10.1038/s41576-018-0031-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y. R. , Lin, Y. C. , Chang, Y. H. , Huang, H. Y. , Hong, Y. K. , Aala, W. J. F. , Tu, W. T. , Tsai, M. C. , Chou, Y. Y. , & Hsu, C. K. (2022). Genetic diagnosis of Rubinstein‐Taybi syndrome with multiplex ligation‐dependent probe amplification (MLPA) and whole‐exome sequencing (WES): Case series with a novel CREBBP variant. Frontiers in Genetics, 13, 848879. 10.3389/fgene.2022.848879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. , & DePristo, M. A. (2010). The genome analysis toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth, K. , & Haering, C. H. (2009). Cohesin: Its roles and mechanisms. Annual Review of Genetics, 43, 525–558. 10.1146/annurev-genet-102108-134233 [DOI] [PubMed] [Google Scholar]
- Parenti, I. , Diab, F. , Gil, S. R. , Mulugeta, E. , Casa, V. , Berutti, R. , Brouwer, R. W. W. , Dupé, V. , Eckhold, J. , Graf, E. , Puisac, B. , Ramos, F. , Schwarzmayr, T. , Gines, M. M. , van Staveren, T. , van IJcken, W. F. J. , Strom, T. M. , Pié, J. , Watrin, E. , … Wendt, K. S. (2020). MAU2 and NIPBL variants impair the heterodimerization of the Cohesin loader subunits and cause Cornelia de Lange syndrome. Cell Reports, 31(7), 107647. 10.1016/j.celrep.2020.107647 [DOI] [PubMed] [Google Scholar]
- Raible, S. E. , Mehta, D. , Bettale, C. , Fiordaliso, S. , Kaur, M. , Medne, L. , Rio, M. , Haan, E. , White, S. M. , Cusmano‐Ozog, K. , Nishi, E. , Guo, Y. , Wu, H. , Shi, X. , Zhao, Q. , Zhang, X. , Lei, Q. , Lu, A. , He, X. , … Izumi, K. (2019). Clinical and molecular spectrum of CHOPS syndrome. American Journal of Medical Genetics. Part A, 179(7), 1126–1138. 10.1002/ajmg.a.61174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi, S. , Clark, D. , Kline, A. D. , Jackson, L. G. , Pie, J. , Siu, V. , Ramos, F. J. , Krantz, I. D. , & Deardorff, M. A. (2010). Facial diagnosis of mild and variant CdLS: Insights from a dysmorphologist survey. American Journal of Medical Genetics. Part A, 152A(7), 1641–1653. 10.1002/ajmg.a.33441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedeño Cacciatore, Á. , & Rowland, B. D. (2019). Loop formation by SMC complexes: Turning heads, bending elbows, and fixed anchors. Current Opinion in Genetics & Development, 55, 11–18. 10.1016/j.gde.2019.04.010 [DOI] [PubMed] [Google Scholar]
- Selicorni, A. , Mariani, M. , Lettieri, A. , & Massa, V. (2021). Cornelia de Lange syndrome: From a disease to a broader spectrum. Genes, 12(7), 1075. 10.3390/genes12071075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuin, J. , Franke, V. , van Ijcken, W. F. , van der Sloot, A. , Krantz, I. D. , van der Reijden, M. I. , Nakato, R. , Lenhard, B. , & Wendt, K. S. (2014). A cohesin‐independent role for NIPBL at promoters provides insights in CdLS. PLoS Genetics, 10(2), e1004153. 10.1371/journal.pgen.1004153 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 1.
Supplementary material 2.
Supplementary material 3.
Supplementary material 4.
Data Availability Statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request.