

Abstract
Introduction
Rothmund–Thomson syndrome (RTS) is a rare autosomal recessive disorder that has been reported in all ethnicities, with several identifiable pathogenic variants. There have been reported cases indicating that RTS may lead to low birth weight in fetuses, but specific data on the fetal period are lacking. Genetic testing for RTS II is currently carried out by identifying pathogenic variants in RECQL4.
Methods
In order to determine the cause, we performed whole‐genome sequencing (WGS) analysis on the patient and his parents. Variants detected by WGS were confirmed by Sanger sequencing and examined in family members.
Results
After analyzing the WGS data, we found a heterozygous nonsense mutation c.2752G>T (p.Glu918Ter) and a novel frameshift insertion mutation c.1547dupC (p.Leu517AlafsTer23) of RECQL4, which is a known pathogenic/disease‐causing variant of RTS. Further validation indicated these were compound heterozygous mutations from parents.
Conclusion
Our study expands the mutational spectrum of the RECQL4 gene and enriches the phenotype spectrum of Chinese RTS patients. Our information can assist the patient's parents in making informed decisions regarding their future pregnancies. This case offers a new perspective for clinicians to consider whether to perform prenatal diagnosis.
Keywords: pathogenic compound heterozygous mutations, prenatal diagnosis, RECQL4, Rothmund–Thomson syndrome
Rothmund–Thomson syndrome (RTS) is associated with intrauterine growth restriction during pregnancy. This case offers a new perspective for clinicians to consider whether to perform prenatal diagnosis and expands the mutational spectrum of the RECQL4 gene. Our information can assist the patient's parents in making informed decisions regarding their future pregnancies.
1. INTRODUCTION
Rothmund–Thomson syndrome (RTS; OMIM #268400) is a rare autosomal recessive disorder with a diagnostic hallmark of a facial rash in infancy, growth delay leading to short stature, and, in some cases, intrauterine growth restriction during pregnancy (although there are no specific data on the fetal period) (Larizza et al., 2013). Other clinical features include thinning hair, sparse or absent eyelashes and eyebrows, juvenile cataracts, and susceptibility to osteosarcoma (Kitao et al., 1999). The worldwide incidence of RTS II is less than 1/1,000,000 (Aymé, 2023), and more than 400 cases have been reported in the literature to date (Larizza et al., 2010; Larizza et al., 2013). The current method for genetic testing for RTS involves identifying biallelic pathogenic variants in ANAPC1 (RTS I) or RECQL4 (RTS II) through molecular genetic testing (Ajeawung et al., 2019; Wang et al., 2003). The RECQL4 gene is located in a small 6.5 kb genomic region on human chromosome 8q24.3 and belongs to the DNA helicase RecQ gene family. It spans 21 exons and contains many small introns of less than 100 bp (Kitao et al., 1999). Therefore, mutations in both introns and exons of RECQL4 can lead to Rothmund–Thomson syndrome (RTS) (Wang et al., 2002). RECQL4 mutations are typically predicted to result in premature stop codons, missense mutations, or frameshifts that are expected to truncate the protein before the helicase domain (encoded by exons 8‐14) (Siitonen et al., 2009) or disrupt the reading frame within (Siitonen et al., 2009). Over 100 clinically relevant mutations have been identified in the RECQL4 gene (Siitonen et al., 2009). The RECQL4 gene encodes a protein of 1208 amino acids in length, called the RECQL4 protein, and this protein contains a conserved core RecQ helicase domain (Kitao et al., 1998; Liu, 2010). The activity of this helicase is associated with various cellular processes, including the initiation of DNA replication, DNA damage repair, base and nucleotide excision repair, homologous recombination, chromosomal telomere maintenance, mitochondrial biogenesis, and genomic stability (Ghosh et al., 2012; Kim et al., 2021; Lu & Davis, 2021; Sangrithi et al., 2005; Xu & Liu, 2009).
2. MATERIALS AND METHODS
2.1. Ethics declaration
This study was approved and guided by the Shenzhen Maternity and Child Healthcare Hospital [project: SFYLS (2019)‐137] and was conducted in accordance with the criteria of the Declaration of Helsinki. Informed consent was obtained from the patient's parents.
2.2. Patient information
The patient of the study is a 3‐month‐old Chinese male infant with red rashes on his face, sparse hair, no eyelashes, and no eyebrows. We collected the umbilical cord blood and placenta of the patient and the peripheral blood of his parents from our cohort study on pregnancy. We also collected and sorted out the pregnancy examinations of the child's mother and follow‐up data of the patient from the Shenzhen Maternity and Child Healthcare Hospital.
2.3. Whole‐genome sequencing (WGS)
To determine the cause of these symptoms, we collected the patient's umbilical cord blood and placenta, along with peripheral blood from his parents in the prenatal cohort study of our research group. Whole‐genome sequencing (WGS) analysis was performed on all samples. The umbilical cord blood, placenta, and peripheral blood were stored at −20°C for a maximum of 3 days. Genomic DNA from the blood cells was extracted using the DNeasy Blood & Tissue Kit. The DNA concentration was measured using the Qubit Fluorometer 3.0, and WGS libraries were constructed using the BGISEQ‐500 Library Kit. Each sample's library was sequenced on the DNBSEQ‐T1 platform, generating at least 600 million reads in 2 × 100 paired‐end reads (~40× coverage from ~120 Gb raw data for each sample) (Kang et al., 2022).
2.4. Variant analysis
After the adapter trimming and filtering of the reads, Burrows–Wheeler Alignment Tool (BWA) was used to align the clean reads to the hg19 human genome reference. The Genome Analysis Toolkit (GATK) was utilized for the detection of single‐nucleotide variants (SNVs) and insertion–deletions (Indels) (Ren et al., 2018). Copy number variants (CNVs) were detected using PSCC software (Li et al., 2014). Public population databases, including the Single Nucleotide Polymorphism Database (dbSNP) (Sherry et al., 2001), 1000 Genomes (phase III) (Fairley et al., 2019), NHLBI Exome Sequencing Project (ESP) (Exome Variant Server, 2022), and the Genome Aggregation Database (gnomAD) (Karczewski et al., 2020), were used for reference. All variants in the coding region were subjected to in silico prediction tools for mutation pathogenicity, such as Scale‐Invariant Feature Transform (SIFT) (Kumar et al., 2009), Polymorphism Phenotyping v2 (PolyPhen2) (Adzhubei et al., 2010), likelihood‐ratio test (LRT), and MutationTaster (Schwarz et al., 2014).
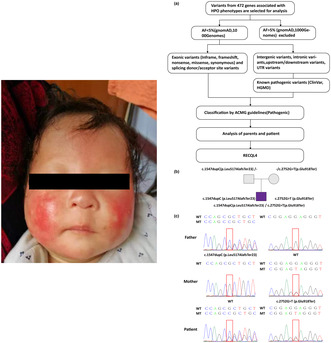
We screened thirteen terms related to the patient's phenotypes in Human Phenotype Ontology (HPO) (Köhler et al., 2021): Absent eyebrow (HP:0002223), Absent eyelashes (HP:0000561), Aplasia/Hypoplasia of the eyebrow (HP:0100840), Poikiloderma (HP:0001029), Erythema (HP:0001029), Facial erythema (HP:0001041), Growth delay (HP:0001510), Short stature (HP:0004322), Small for gestational age (HP:0001518), Sparse eyebrow (HP:0045075), Sparse eyelashes (HP:0000653), Sparse hair (HP:0008070), and Sparse or absent eyelashes (HP:0200102). According to the guidelines of the American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015), we interpreted all genes associated with these phenotypes, aiming to identify potentially pathogenic variants (shown in Figure 2a). In this study, we employed classification criteria for determining pathogenicity, with each pathogenic criterion assigned a weight as follows: very strong (PVS1), strong (PS1‐4), moderate (PM1‐6), or supporting (PP1‐5).
FIGURE 2.
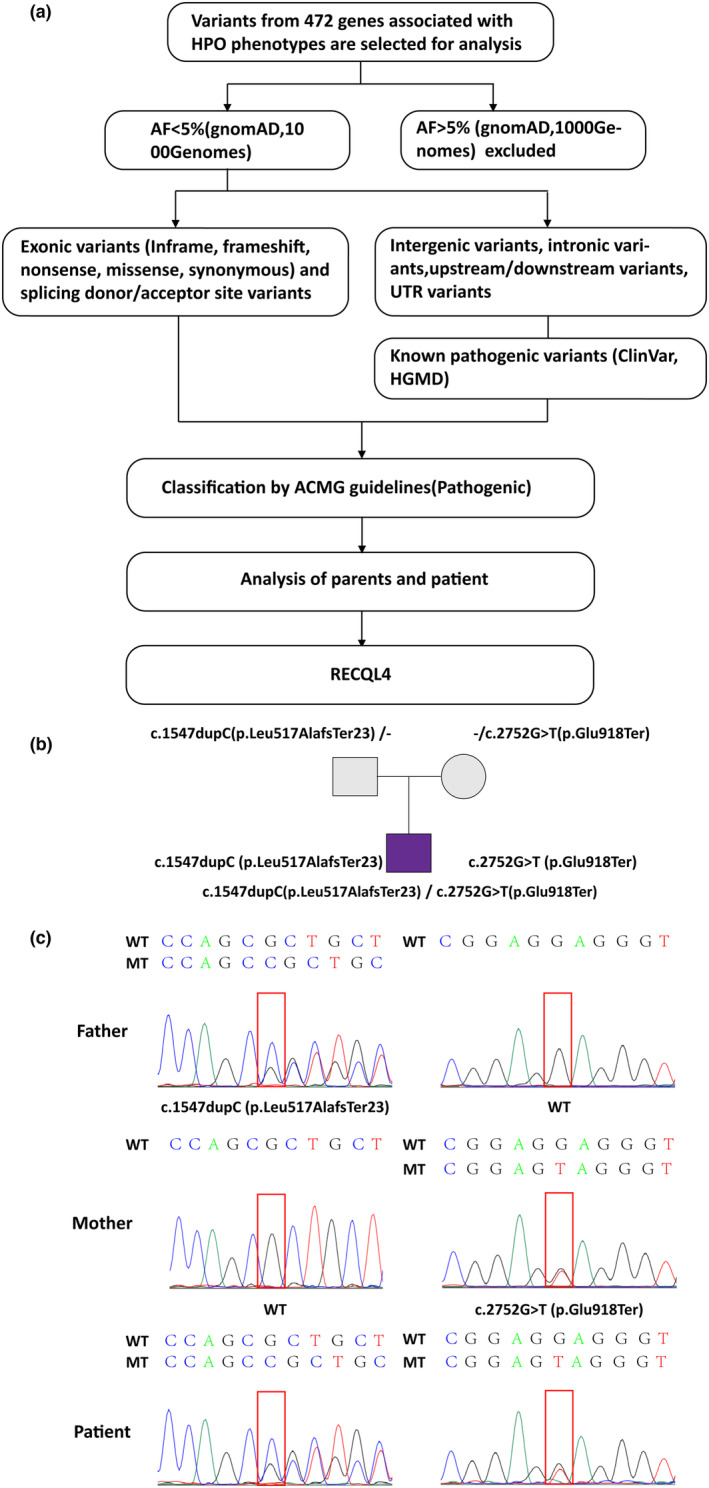
(a) The filtering and analysis process of variants from 472 genes associated with 13 phenotypes in HPO. (b) Two compound heterozygous mutations were confirmed. The patient inherited the missense and the nonsense mutations from both parents. (c) Sanger sequencing results of the compound heterozygous variants of RECQL4.
2.5. Sanger sequencing
Sanger sequencing was conducted for validating the variants identified in WGS data. Primers for amplification were designed based on the RECQL4 (GenBank NM_004260.3) sequence. The primer sequences were RECQL4_MIS_F: 5′‐GTTCAGACGGCAATGGGTAT‐3′; RECQL4_MIS_R: 5′‐GCTGTGAAGAGGCTGGTACA‐3′; RECQL4_INS_F: 5′‐GTTAGGGGACAAGCAGCAGT‐3′; and RECQL4_INS_R: 5′‐CAGTCATGCGGATCCTGTC‐3′. The corresponding gDNA fragments from the patient and his parents were amplified, and the PCR products were sequenced using Sanger sequencing.
3. RESULTS
3.1. Clinical characteristics
Prenatal ultrasound showed that the fetus was consistently about a week smaller (Leung et al., 2008) than expected and had decreased amniotic fluid in the later stages (shown in Table 1). After birth, the patient continued to experience growth retardation and was small in height and weight. At 2 months of age, he developed red rashes on his face which spread to his entire body. He had sparse hair, no eyelashes, and no eyebrows (shown in Figure 1).
TABLE 1.
Prenatal ultrasound.
| Measurement gestational weeks | BPD (cm) | AC (cm) | FL (cm) | HC (cm) | DVP (cm) | AFI (cm) | Fetal growth |
|---|---|---|---|---|---|---|---|
| 23 weeks + 2 days | 5.04 (5.1–6.3) | 17.86 (16.4–20.4) | 3.78 (3.5–4.3) | 20.40 (19.1–22.6) | 3.0 (3–7) | 8.9 (8–25) | 22 weeks + 1 day |
| 27 weeks + 6 days | 6.60 (6.4–7.7) | 22.30 (21.0–25.6) | 5.10 (4.5–5.4) | 24.80 (23.9–27.6) | 3.9 (3–7) | 11.3 (8–25) | 26 weeks + 5 days |
| 31 weeks + 6 days | 7.70 (7.4–8.8) | 26.50 (24.6–29.9) | 5.90 (5.3–6.3) | 28.10 (24.6–29.9) | 5.4 (3–7) | – | 30 weeks + 4 days |
| 36 weeks + 3 days | 8.70 (8.3–9.6) | 31.60 (28.3–34.3) | 6.62 (6.1–7.1) | 31.24 (30.0–34.0) | 3.7 (3–7) | 9.4 (8–25) | – |
| 39 weeks + 2 days | 8.86 (8.6–10.0) | 30.88 (30.4–36.8) | 7.19 (6.6–7.6) | 31.46 (30.9–35.0) | 1.6 (3–7) | 4.6 (8–25) | – |
Note: Reference ranges are shown in parentheses (−2 SD/+ 2 SD) (Schwarz et al., 2014).
Abbreviations: AC, abdominal circumference; AFI, amniotic fluid index; BPD, biparietal diameter; DVP, deepest vertical pocket; FL, femur length; HC, head circumference.
FIGURE 1.
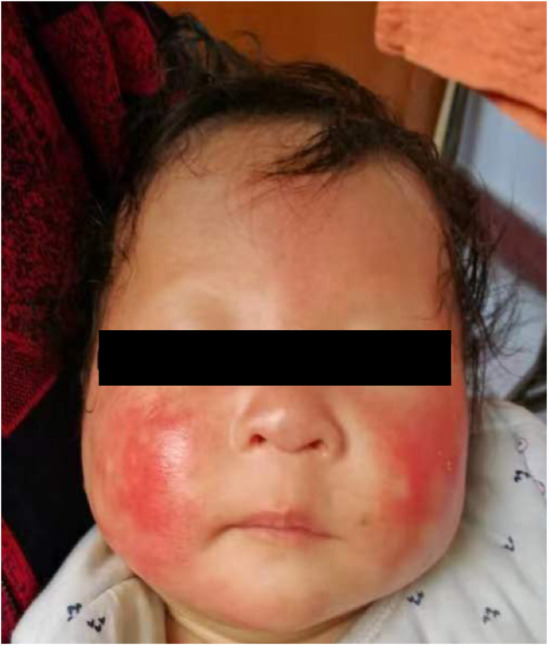
Clinical features of the patient: red rashes on the face, sparse hair, and absence of eyebrows.
3.2. Molecular genetic analysis
To determine the cause, we obtained all CNVs, SNVs, and Indels. The guidelines of ACMG were followed to identify pathogenic variants. Upon interpretation, none of the identified CNVs were classified as pathogenic or possibly pathogenic. However, two variants in RECQL4 (GenBank NM_004260.3) associated with RTS were identified, including a frameshift variant c.1547dupC, p.Leu517AlafsTer23 (exon 9), and a nonsense variant c.2752G>T, p.Glu918Ter (exon 16). The two compound heterozygous mutations were confirmed by Sanger sequencing, and it was also demonstrated that the patient inherited the frameshift and the nonsense mutations from both parents (shown in Figure 2b,c). Based on the ACMG guidelines, the two variants were categorized as pathogenic mutations. Due to the following evidence, the frameshift variant was classified as a pathogenic variation: (1) PVS1: null variant (frameshift) affecting gene RECQL4; (2) in ESP, 1000 Genomes Project, or gnomAD, the mutation is either absent from controls or has an extremely low frequency if recessive; (3) PM3: for autosomal recessive genes, a pathogenic variant detected in trans in the patient; and (4) PP3: the computational analysis concluded that the variant is pathogenic, as GERP predicted one pathogenic effect and no benign effects were detected. Due to the following evidence, the nonsense variant was classified as a pathogenic variation: (1) PVS1: null variant (nonsense) affecting gene RECQL4; (2) PM2: variant not found in gnomAD exomes and gnomAD genomes; (3) PM3: for autosomal recessive genes, a pathogenic variant detected in trans in the patient; (4) PP3: the computational assessment determined that the variant was pathogenic based on two pathogenic predictions from Genomic Evolutionary Rate Profiling (GERP) (Davydov et al., 2010) and Functional Analysis through Hidden Markov Models—Multiple Kernel Learning (FATHMM‐MKL) (Shihab et al., 2013), with no benign predictions; and (5) PP5: ClinVar (Landrum et al., 2020) classifies the variant as pathogenic with one star (above a minimum of 1 star), but there is no evidence from functional or in vivo studies to support PS3 (shown in Table 2).
TABLE 2.
Two variants were identified in RECQL4 (GenBank NM_004260.3) associated with RTS.
| Gene | Mode of inheritance | Phenotype | Transcript | Variants | Amino acid change | Function | Type | ACMG evidences | Pathogenicity |
|---|---|---|---|---|---|---|---|---|---|
| RECQL4 | AR |
Baller–Gerold syndrome (MIM#:218600) RAPADILINO syndrome (MIM#:266280) Rothmund–Thomson syndrome, type 2 (MIM#:268400) |
NM_004260.3 | c.1547dupC | p.Leu517AlafsTer23 | Frameshift | Het | PVS1 + PM2 + PM3 + PP3 | Pathogenic |
| RECQL4 | AR | NM_004260.3 | c.2752G>T | p.Glu918Ter | Nonsense | Het | PVS1 + PM2 + PM3 + PP3 + PP5 | Pathogenic |
Note: ACMG evidences (Adzhubei et al., 2010): The American College of Medical Genetics and Genomics (ACMG) has established classification criteria for determining pathogenicity, with each criterion assigned a weight as follows: very strong (PVS1), strong (PS1‐4), moderate (PM1‐6), or supporting (PP1‐5).
Abbreviations: AR, autosomal recessive; Het, heterozygous.
4. DISCUSSION/CONCLUSION
In this study, we carried out WGS of a trio with a RTS proband manifesting red rashes on face, sparse hair, no eyelashes, and no eyebrows and identified compound heterozygous mutations in the RECQL4 gene, which expands the mutation spectrum of RECQL4. We collected and analyzed follow‐up data from the patient and conducted pregnancy examinations of the child's mother. We found that the fetal growth of the patient was consistently small for approximately 1 week, enriching the phenotype spectrum of Chinese RTS patients.
The c.1547dupC (p.Leu517AlafsTer23) variant we discovered is a newly identified truncating mutation. The c.2752G>T (p.Glu918Ter) variant was previously reported as a nonsense mutation (Clinvar database: VCV000597188.3). Neither of these two mutations was found in the population, based on 1000 Genomes, ExAC, and East Asian population databases, or our local database. The c.2752G>A (p.Glu918Lys) and c.1547C>T (p.Ala516Val) variants were also identified in the Clinvar database (Clinvar database: VCV002387422.1 and VCV002189648.1) at the same site as our newly identified mutation. However, neither of these two variants had significant clinical significance or relevant case reports, indicating the variants we found were extremely rare in the general population. The RECQL4 gene comprises 21 exons (Ohlenschläger et al., 2012; Wang et al., 2003). The c.1547dupC (p.Leu517AlafsTer23) mutation occurred in the helicase domain (exons 9‐13) (Mann et al., 2005), while the other identified mutation, c.2752G>T (p.Glu918Ter), occurred downstream of the helicase domain. The c.2752G>T (p.Glu918Ter) mutation is a nonsense mutation that results in the substitution of an amino acid with a stop codon, leading to the termination of protein translation. The c.1547dupC (p.Leu517AlafsTer23) mutation is an insertion mutation that leads to reading frameshift and subsequent termination of protein translation. These mutations often lead to the destruction of the stability of mature mRNA through nonsense‐mediated decay (Larizza et al., 2010). We predict that the new mutation, c.1547dupC, would destroy the helicase domain, resulting in the deletion, truncation, or damage of the helicase activity of the RECQL4 protein.
The 3‐month‐old patient had red rashes on his face, sparse hair, no eyelashes, and no eyebrows, which is consistent with the main clinical diagnostic hallmark of RTS (Larizza et al., 2010; Wang et al., 2001). RTS patients are proportionately small in weight and height (Gui et al., 2018; Yadav et al., 2019; Zhang et al., 2021). Low birth weight, slow weight gain, and linear growth deficiency are present in at least two‐thirds of RTS patients (Wang et al., 2001). The patient in this study not only experienced growth retardation and small in height and weight after birth but also showed consistently about a week smaller as early as 23 gestational weeks estimated by prenatal ultrasound tests. RTS‐related bone abnormalities have been recapitulated in mouse models. Recql4 knockout mice died between embryonic days 3.5–6.5, showing that the DNA helicase functions fundamentally in embryonic development (Ichikawa et al., 2002). The homozygous Recql4 exon 13‐deleted mice exhibited severe growth retardation as early as embryonic day 14.5, showing short stature and low body weight (Hoki et al., 2003). Recql4 conditional knockout mice targeting the skeletal lineage had limb defects, showing shorter forelimbs and hindlimbs (Lu et al., 2015). An increased p53 response was seen in affected tissues, indicating Recql4 is critical for skeletal development by modulating p53 activity in vivo. Ng et al. (2015) also found that Recql4 bone cells specifically removed mice also had shortened bones. Castillo‐Tandazo et al. (2021) investigated mouse models with combination of a Recql4 truncating mutation and a conditional null allele and found that different mutations led to distinct phenotypes. Osx‐Cre Recql4 fl/R347X mice had lower body weight and shorter tibial length compared with Osx‐Cre Recql4 fl/+ mice, while Osx‐Cre Recql4 fl/G522Efs mice were not different (Yadav et al., 2019). Collectively, Recql4 functions in skeletogenesis as early as embryonic development, and different mutations have different effects. We can infer that the compound heterozygous mutations in RECQL4 may be involved in the occurrence of the slow fetal growth of the proband. Knockin mouse models could help to elucidate the molecular pathogenesis.
This patient exhibited slow fetal growth for approximately 1 week during the fetal stage, but he did not meet the diagnostic criteria for fetal growth restriction (FGR), manifesting as fetal ultrasound estimated weight or abdominal circumference below the 10th percentile for the corresponding gestational age (Fetal Medicine Subgroup, Chinese Society of Perinatal Medicine, Chinese Medical Association; Maternal‐Fetal Medicine Committee, Chinese Society of Obstetrics and Gynecology, Chinese Medical Association, et al., 2022). His phenotype could be overlooked by clinicians. Therefore, this case also makes some suggestions for clinicians that pregnant women whose fetal growth and development consistently exhibit being about 1 week behind and show no other significant abnormalities during pregnancy might consider whether prenatal diagnosis should be advised in addition to the necessary nutritional intervention. The use of high‐throughput genome sequencing to identify pathogenic variants is a valuable tool for early disease diagnosis and has significant clinical implications for genetic counseling and fertility guidance. Additionally, carrier testing for at‐risk relatives and preimplantation genetic diagnosis can assist the patient's parents in making informed decisions regarding their future fertility.
AUTHOR CONTRIBUTIONS
Clinical information collection: Juan Zeng and Sujun Zhu; sample collection and processing: Zhongzhen Liu, Jinghua Sun, Juan Zeng, and Sujun Zhu; data analysis: Qing Zhou, Wen‐Jing Wang, Jinghua Sun, and Jiayi Li; writing—original draft: Jiayi Li, Rui Liang, Yuwei Liu, and Lin Wang; and writing—review and editing: Juan Zeng, Wen‐Jing Wang, and Sujun Zhu. All authors have read and agreed to the published version of the manuscript.
FUNDING INFORMATION
This work was supported by the Science Technology and Innovation Commission of Shenzhen Municipality under Grant No. JCYJ20170412152854656.
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest to declare.
ETHICS STATEMENT
This study was conducted according to the criteria of the Declaration of Helsinki and was approved and guided by the Shenzhen Maternity and Child Healthcare Hospital [project: SFYLS (2019) NO137]. Written informed consent was obtained from the patient's parents for the publication of this paper.
ACKNOWLEDGMENTS
We would like to express our gratitude to the BGI research group and the doctors at Shenzhen Maternal and Child Health Hospital for their valuable contributions and insightful discussions. We are also deeply appreciative of the patient and his parents for their support in participating in this study. Furthermore, we extend our thanks to China National GeneBank (CNGB) for their support throughout the study.
Zeng, J. , Li, J. , Liu, Y. , Liang, R. , Wang, L. , Zhou, Q. , Sun, J. , Liu, Z. , Wang, W.‐J. , & Zhu, S. (2024). A Chinese patient with Rothmund–Thomson syndrome. Molecular Genetics & Genomic Medicine, 12, e2347. 10.1002/mgg3.2347
Juan Zeng and Jiayi Li should be regarded as joint first authors.
Contributor Information
Wen‐Jing Wang, Email: wangwenjing@genomics.cn.
Sujun Zhu, Email: zhusj1223@126.com.
DATA AVAILABILITY STATEMENT
This study's supporting data can be obtained by contacting the corresponding author. Please note that due to privacy and ethical restrictions, the data are not publicly available.
REFERENCES
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , Kondrashov, A. S. , & Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajeawung, N. F. , Nguyen, T. T. M. , Lu, L. , Kucharski, T. J. , Rousseau, J. , Molidperee, S. , Atienza, J. , Gamache, I. , Jin, W. , Plon, S. E. , Lee, B. H. , Teodoro, J. G. , Wang, L. L. , & Campeau, P. M. (2019). Mutations in ANAPC1, encoding a scaffold subunit of the anaphase‐promoting complex, cause Rothmund‐Thomson syndrome type 1. American Journal of Human Genetics, 105(3), 625–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymé, S . Schmidtke [Internet] The portal for rare diseases and orphan drugs. 2023. [cited 2023 Jan 12]. https://www.orpha.net/consor/cgi‐bin/index.php.
- Castillo‐Tandazo, W. , Frazier, A. E. , Sims, N. A. , Smeets, M. F. , & Walkley, C. R. (2021). Rothmund‐Thomson syndrome‐like RECQL4 truncating mutations cause a haploinsufficient low‐bone‐mass phenotype in mice. Molecular and Cell Biology, 41(3), e0059020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov, E. V. , Goode, D. L. , Sirota, M. , Cooper, G. M. , Sidow, A. , & Batzoglou, S. (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Computational Biology, 6(12), e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exome Variant Server . NHLBI GO Exome Sequencing Project (ESP). 2022. [cited 2022 Oct 31]. http://evs.gs.washington.edu/EVS/
- Fairley, S. , Lowy‐Gallego, E. , Perry, E. , & Flicek, P. (2019). The international genome sample resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Research, 48(D1), D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetal Medicine Subgroup, Chinese Society of Perinatal Medicine, Chinese Medical Association; Maternal‐Fetal Medicine Committee, Chinese Society of Obstetrics and Gynecology, Chinese Medical Association , Sun, L. , Hu, Y. , & Qi, H. (2022). A summary of Chinese expert consensus on fetal growth restriction (an update on the 2019 version). Maternal‐Fetal Medicine, 4(3), 162–168. [Google Scholar]
- Ghosh, A. K. , Rossi, M. L. , Singh, D. K. , Dunn, C. , Ramamoorthy, M. , Croteau, D. L. , Liu, Y. , & Bohr, V. A. (2012). RECQL4, the protein mutated in Rothmund‐Thomson syndrome, functions in telomere maintenance. The Journal of Biological Chemistry, 287(1), 196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui, B. , Song, Y. , Hu, X. , Li, H. , Qin, Z. , Su, J. , Li, C. , Fan, X. , Li, M. , Luo, J. , Feng, Y. , Song, L. , Chen, S. , Gong, C. , & Shen, Y. (2018). Novel pathogenic RECQL4 variants in Chinese patients with Rothmund‐Thomson syndrome. Gene, 654, 110–115. [DOI] [PubMed] [Google Scholar]
- Hoki, Y. , Araki, R. , Fujimori, A. , Ohhata, T. , Koseki, H. , Fukumura, R. , Nakamura, M. , Takahashi, H. , Noda, Y. , Kito, S. , & Abe, M. (2003). Growth retardation and skin abnormalities of the Recql4 ‐deficient mouse. Human Molecular Genetics, 12(18), 2293–2299. [DOI] [PubMed] [Google Scholar]
- Ichikawa, K. , Noda, T. , & Furuichi, Y. (2002). Preparation of the gene targeted knockout mice for human premature aging diseases, Werner syndrome, and Rothmund‐Thomson syndrome caused by the mutation of DNA helicases. Nippon Yakurigaku Zasshi, 119, 219–226. [DOI] [PubMed] [Google Scholar]
- Kang, J. , Zhou, Q. , Chen, N. , Liu, Z. , Zhang, Y. , Sun, J. , Ma, C. , Chen, F. , Ma, Y. , Wang, L. , Zhu, L. , & Wang, W. (2022). Clinical and genetic characteristics of a cohort with distal vaginal atresia. International Journal of Molecular Sciences, 23(21), 12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alföldi, J. , Wang, Q. , Collins, R. L. , Laricchia, K. M. , Ganna, A. , Birnbaum, D. P. , Gauthier, L. D. , Brand, H. , Solomonson, M. , Watts, N. A. , Rhodes, D. , Singer‐Berk, M. , England, E. M. , Seaby, E. G. , Kosmicki, J. A. , … MacArthur, D. G. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H. , Choi, H. , Im, J. S. , Park, S. Y. , Shin, G. , Yoo, J. H. , Kim, G. , & Lee, J. K. (2021). Stable maintenance of the Mre11‐Rad50‐Nbs1 complex is sufficient to restore the DNA double‐strand break response in cells lacking RECQL4 helicase activity. The Journal of Biological Chemistry, 297(4), 101148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao, S. , Lindor, N. M. , Shiratori, M. , Furuichi, Y. , & Shimamoto, A. (1999). Rothmund‐Thomson syndrome responsible gene, RECQL4: Genomic structure and products. Genomics, 61(3), 268–276. [DOI] [PubMed] [Google Scholar]
- Kitao, S. , Ohsugi, I. , Ichikawa, K. , Goto, M. , Furuichi, Y. , & Shimamoto, A. (1998). Cloning of two new human helicase genes of the RecQ family: Biological significance of multiple species in higher eukaryotes. Genomics, 54(3), 443–452. [DOI] [PubMed] [Google Scholar]
- Köhler, S. , Gargano, M. , Matentzoglu, N. , Carmody, L. C. , Lewis‐Smith, D. , Vasilevsky, N. A. , Danis, D. , Balagura, G. , Baynam, G. , Brower, A. M. , Callahan, T. J. , Chute, C. G. , Est, J. L. , Galer, P. D. , Ganesan, S. , Griese, M. , Haimel, M. , Pazmandi, J. , Hanauer, M. , … Robinson, P. N. (2021). The human phenotype ontology in 2021. Nucleic Acids Research, 49(D1), D1207–D1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, P. , Henikoff, S. , & Ng, P. C. (2009). Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nature Protocols, 4(7), 1073–1081. [DOI] [PubMed] [Google Scholar]
- Landrum, M. J. , Chitipiralla, S. , Brown, G. R. , Chen, C. , Gu, B. , Hart, J. , Hoffman, D. , Jang, W. , Kaur, K. , Liu, C. , Lyoshin, V. , Maddipatla, Z. , Maiti, R. , Mitchell, J. , O'Leary, N. , Riley, G. R. , Shi, W. , Zhou, G. , Schneider, V. , … Kattman, B. L. (2020). ClinVar: Improvements to accessing data. Nucleic Acids Research, 48(D1), D835–D844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larizza, L. , Roversi, G. , & Verloes, A. (2013). Clinical utility gene card for: Rothmund‐Thomson syndrome. European Journal of Human Genetics, 21(7), 792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larizza, L. , Roversi, G. , & Volpi, L. (2010). Rothmund‐Thomson syndrome. Orphanet Journal of Rare Diseases, 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung, T. N. , Pang, M. W. , Daljit, S. S. , Leung, T. Y. , Poon, C. F. , Wong, S. M. , & Lau, T. K. (2008). Fetal biometry in ethnic Chinese: Biparietal diameter, head circumference, abdominal circumference and femur length. Ultrasound in Obstetrics & Gynecology, 31(3), 321–327. [DOI] [PubMed] [Google Scholar]
- Li, X. , Chen, S. , Xie, W. , Vogel, I. , Choy, K. W. , Chen, F. , Christensen, R. , Zhang, C. , Ge, H. , Jiang, H. , Yu, C. , Huang, F. , Wang, W. , Jiang, H. , & Zhang, X. (2014). PSCC: Sensitive and reliable population‐scale copy number variation detection method based on low coverage sequencing. PLoS One, 9(1), e85096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. (2010). Rothmund‐Thomson syndrome helicase, RECQ4: On the crossroad between DNA replication and repair. DNA Repair (Amst), 9(3), 325–330. [DOI] [PubMed] [Google Scholar]
- Lu, H. , & Davis, A. J. (2021). Human RecQ helicases in DNA double‐Strand break repair. Frontiers in Cell and Development Biology, 9, 640755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, L. , Harutyunyan, K. , Jin, W. , Wu, J. , Yang, T. , Chen, Y. , Joeng, K. S. , Bae, Y. , Tao, J. , Dawson, B. C. , Jiang, M. M. , Lee, B. , & Wang, L. L. (2015). RECQL4 regulates p53 function in vivo during skeletogenesis. Journal of Bone and Mineral Research, 30(6), 1077–1089. [DOI] [PubMed] [Google Scholar]
- Mann, M. B. , Hodges, C. A. , Barnes, E. , Vogel, H. , Hassold, T. J. , & Luo, G. (2005). Defective sister‐chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II Rothmund‐Thomson syndrome. Human Molecular Genetics, 14(6), 813–825. [DOI] [PubMed] [Google Scholar]
- Ng, A. J. M. , Walia, M. K. , Smeets, M. F. , Mutsaers, A. J. , Sims, N. A. , Purton, L. E. , Walsh, N. C. , Martin, T. J. , & Walkley, C. R. (2015). The DNA helicase Recql4 is required for normal osteoblast expansion and osteosarcoma formation. PLoS Genetics, 11(4), e1005160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlenschläger, O. , Kuhnert, A. , Schneider, A. , Haumann, S. , Bellstedt, P. , Keller, H. , Saluz, H. P. , Hortschansky, P. , Hänel, F. , Grosse, F. , Görlach, M. , & Pospiech, H. (2012). The N‐terminus of the human RECQL4 helicase is a homeodomain‐like DNA interaction motif. Nucleic Acids Research, 40(17), 8309–8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, S. , Bertels, K. , & Al‐Ars, Z. (2018). Efficient acceleration of the pair‐HMMs forward algorithm for GATK HaplotypeCaller on graphics processing units. Evolutionary Bioinformatics Online, 14, 1176934318760543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangrithi, M. N. , Bernal, J. A. , Madine, M. , Philpott, A. , Lee, J. , Dunphy, W. G. , & Venkitaraman, A. R. (2005). Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund‐Thomson syndrome. Cell, 121(6), 887–898. [DOI] [PubMed] [Google Scholar]
- Schwarz, J. M. , Cooper, D. N. , Schuelke, M. , & Seelow, D. (2014). MutationTaster2: Mutation prediction for the deep‐sequencing age. Nature Methods, 11(4), 361–362. [DOI] [PubMed] [Google Scholar]
- Sherry, S. T. , Ward, M. H. , Kholodov, M. , Baker, J. , Phan, L. , Smigielski, E. M. , & Sirotkin, K. (2001). dbSNP: The NCBI database of genetic variation. Nucleic Acids Research, 29(1), 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihab, H. A. , Gough, J. , Cooper, D. N. , Stenson, P. D. , Barker, G. L. , Edwards, K. J. , Day, I. N. M. , & Gaunt, T. R. (2013). Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Human Mutation, 34(1), 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siitonen, H. A. , Sotkasiira, J. , Biervliet, M. , Benmansour, A. , Capri, Y. , Cormier‐Daire, V. , Crandall, B. , Hannula‐Jouppi, K. , Hennekam, R. , Herzog, D. , Keymolen, K. , Lipsanen‐Nyman, M. , Miny, P. , Plon, S. E. , Riedl, S. , Sarkar, A. , Vargas, F. R. , Verloes, A. , Wang, L. L. , … Kestilä, M. (2009). The mutation spectrum in RECQL4 diseases. European Journal of Human Genetics, 17(2), 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. L. , Gannavarapu, A. , Kozinetz, C. A. , Levy, M. L. , Lewis, R. A. , Chintagumpala, M. M. , Ruiz‐Maldanado, R. , Contreras‐Ruiz, J. , Cunniff, C. , Erickson, R. P. , Lev, D. , Rogers, M. , Zackai, E. H. , & Plon, S. E. (2003). Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund‐Thomson syndrome. Journal of the National Cancer Institute, 95(9), 669–674. [DOI] [PubMed] [Google Scholar]
- Wang, L. L. , Levy, M. L. , Lewis, R. A. , Chintagumpala, M. M. , Lev, D. , Rogers, M. , & Plon, S. E. (2001). Clinical manifestations in a cohort of 41 Rothmund‐Thomson syndrome patients. American Journal of Medical Genetics, 102(1), 11–17. [DOI] [PubMed] [Google Scholar]
- Wang, L. L. , Worley, K. , Gannavarapu, A. , Chintagumpala, M. M. , Levy, M. L. , & Plon, S. E. (2002). Intron‐size constraint as a mutational mechanism in Rothmund‐Thomson syndrome. American Journal of Human Genetics, 71(1), 165–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X. , & Liu, Y. (2009). Dual DNA unwinding activities of the Rothmund‐Thomson syndrome protein, RECQ4. The EMBO Journal, 28(5), 568–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav, S. , Thakur, S. , Kohlhase, J. , Bhari, N. , Kabra, M. , & Gupta, N. (2019). Report of two novel mutations in Indian patients with Rothmund‐Thomson syndrome. Journal of Pediatric Genetics, 8(3), 163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Qin, W. , Wang, H. , Lin, Z. , Tang, Z. , & Xu, Z. (2021). Novel pathogenic variants in the RECQL4 gene causing Rothmund‐Thomson syndrome in three Chinese patients. The Journal of Dermatology, 48(10), 1511–1517. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study's supporting data can be obtained by contacting the corresponding author. Please note that due to privacy and ethical restrictions, the data are not publicly available.