

Summary
A solitary peripheral lung nodule was found in the left lung of a 52-year-old man. It was located in the lower lobe and measured 18.5 cm of major axis on chest computed tomography. A tru-cut core biopsy was obtained and a proliferation of bland, monomorphic, spindle cells in interlacing fascicles was observed. Accordingly, a surgical resection of the neoplasm was subsequently carried out. Macroscopically, the tumor appeared as a well-circumscribed nodule with a firm and whitish cut surface. Histologically, the neoplasm was predominantly composed of bland and monomorphic spindle cells, with a predominantly fascicular growth pattern, in which many tubular and cleft-like spaces of entrapped normal respiratory epithelium were involved. Myxoid change, stromal hyalinization and scattered bizarre mononucleated and multinucleated cells were also observed. Based on clinico-morphological, immunophenotypical and molecular features, we made a diagnosis of malignant transformation of pulmonary adenoleiomyomatous hamartoma into pulmonary leiomyosarcoma. As far as we know, this is the first described case of this exceptionally rare occurrence in an already rare neoplasm.
Key words: lung tumor, pulmonary hamartoma, pulmonary adenoleiomyomatous hamartoma, malignant transformation, primitive pulmonary leiomyosarcoma
Introduction
Lung neoplasms are the most commonly diagnosed tumors, with malignant epithelial neoplasms occurring significantly more frequently than benign tumors 1. Indeed, approximately only 8% of lung tumors are benign with the pulmonary hamartoma (PH) being the most common, and accounts for 75% to 77% of them 2,3. PH is defined by at least two mesenchymal components, mainly cartilaginous and fat tissue, with associated entrapped respiratory epithelium 4. PH usually represents an asymptomatic and incidental lesion during imaging studies worked out for thoracic disease-unrelated reasons, with radiographic feature being characterized by the histological components, frequently associated to the popcorn-like calcification 5. Based on mesenchymal elements, several histological types of PH have been described. In particular, cases with dominant smooth muscle tissue are defined pulmonary adenoleiomyomatous hamartoma (PAH) 4. Interestingly, malignant transformation has been rarely reported in PH and is still controversial 6-8. Herein we describe an unusual case of a PAH, developing a rare malignant transformation in a leiomyosarcoma; cliniopathological, immunohistochemical, and genetic features of the tumors are described in detail. We also carry out a thorough literature review on cases of PH with malignant transformation.
Case presentation
In our hospital, a 52-year-old man presented with non-specific respiratory symptoms including cough, progressive shortness of breath, dyspnea, and chest pain/pressure. His clinical history revealed several comorbidities, including high blood pressure, rheumatoid arthritis, class III obesity, and a history of bronchiolitis. Thus, an imaging study was performed and chest computed tomography (CT) revealed the presence of a bulky solitary lung nodule in the lower lobe of the left lung. The tumour measured 18.5 x 15.8 x 8.9 cm and showed inhomogeneous density. The lesion engulfed the left inferior pulmonary vein, compressed left atrium and displaced the bronchial tree (Fig. 1). In addition, systemic CT did not reveal any extrapulmonary tumoral localization. For histological diagnosis, tru-cut biopsy under CT guidance was carried out. The histological findings showed intersecting fascicles of bland, monotonous, spindle cells with indistinct borders, eosinophilic cytoplasm and hyperchromatic nuclei with tapered ends; no evidence of mitotic activity and tumor necrosis were detected (Fig. 2). Thus, we used a broad panel of immunohistochemical markers in order to define the immunophenotypical differentiation (Tab. I); only myogenic markers were expressed; in detail, the neoplastic population showed a strong and diffuse desmin (primary antibody, clone DE-R-11, Ventana), smooth muscle actin (SMA) (primary antibody, clone 1A4, Ventana) and h-caldesmon (primary antibody, clone E89, Ventana) expression. (Fig. 2). As a result, tru-cut biopsy findings supported the diagnosis of mesenchymal tumor with leiomyomatous differentiation. The patient underwent non-radical surgical removal of the tumor about 1.5 months later. Macroscopically, the tumour appeared as a well-circumscribed nodule of 10.5 x 7 x 5 cm, with a firm and whitish cut surface. Histologically, the neoplasm was predominantly composed of bland and monomorphic spindle cells, with a predominantly fascicular growth pattern, in which many tubular and cleft-like spaces of entrapped normal respiratory epithelium were involved (Fig. 3). Myxoid change, stromal hyalinization and very rare areas of mature adipose tissue were also observed (Fig. 3). However, we observed peculiar histological features such as a zonal pattern, composed of hypercellular and hypocellular areas and the widespread evidence of scattered bizarre mononucleated and multinucleated cells. These cells were characterized by bizarre and irregular shaped nuclei, with both coarse and finely dispersed chromatin with evident nucleoli (Fig. 4). Tumor necrosis was absent and low mitotic index (4 mitosis/10 HPF) was observed. However, when we evaluated the hypercellular areas, we observed peculiar cell crowding with neoplastic cells, which were different from those previously described, characterized by marked nuclear atypia, with nucleomegaly and nuclear hyperchromasia, and significant cellular pleomorphism (Fig. 4). The mitotic hot spots were observed in hypercellular areas, where rare atypical mitoses could also be observed (Fig. 4). These morphological features suggested a malignant biological behavior. The immunohistochemical study confirmed the myogenic differentiation with a strong and widespread expression of desmin (primary antibody, DE-R-11, Ventana), SMA (primary antibody, clone 1A4, Ventana) and h-caldesmon (primary antibody, clone E89, Ventana) both in hypercellular and hypocellular areas, weaker and more localized CD34 (primary antibody, clone QBEnd/10, Ventana) expression and a relatively low proliferation index (Ki-67; primary antibody, clone 30-9, Ventana) of about 10% (Fig. 5) (Tab. I). Moreover, we looked for the HMGA2 overexpression by immunohistochemistry, as a surrogate of the typical translocation t(3;12) (q27-q28;q14-q15) identifiable in most pulmonary hamartomas. This rearrangement results in HMGA2-LPP fusion gene, responsible for HMGA2 protein overexpression, a occurs in our case (No. 41878, Abcam) (Fig. 5). Thus, regarding for the overall clinical, morphological and immunophenotypical features, our definitive diagnosis was malignant transformation of pulmonary adenoleiomyomatous hamartoma in primitive pulmonary leiomyosarcoma. About one year after diagnosis, the tumor progressed, giving rise to bone metastases, despite chemotherapy, until the patient’s death after approximately 12 months.
Figure 1.
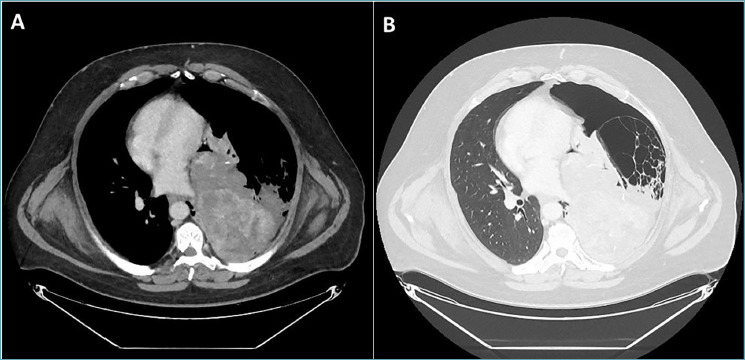
Chest computed tomography (CT) scan. (A, B) Bulky solitary mass with inhomogeneous density in the lower lobe of the left lung. The lesion engulfed the left inferior pulmonary vein, compressed the left atrium and displaced the bronchial tree.
Figure 2.
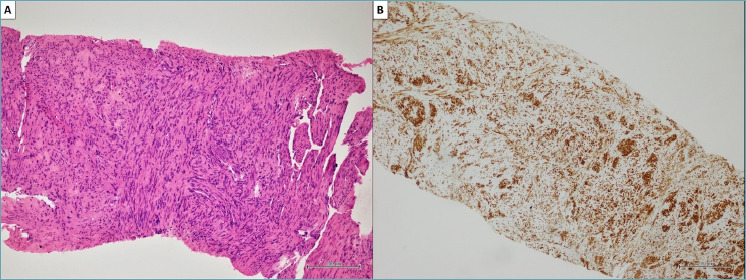
CT-guided tru-cut core biopsy. (A) Histopathological finding of intersecting fascicles of bland, monotonous, spindle cells with indistinct cellular borders, eosinophilic cytoplasm and hyperchromatic nuclei, with tapered ends. Lack of mitotic figures and foci of tumor necrosis (hematoxylin and eosin, original magnifications x200). (B) Immunohistochemistry showed a strong and diffuse expression of desmin by tumor cells (clone DE-R-11, Ventana) (original magnifications x200).
Table I.
Complete immunohistochemical study performed on the core biopsy and surgical samples.
| IHC panel | Core biopsy | Surgical sample |
|---|---|---|
| Desmin | + | + |
| SMA | + | + |
| h-Caldesmon | + | + |
| S100 | - | - |
| CK-PAN (AE1/AE3) | - | - |
| TTF1 | - | - |
| Napsin A | - | - |
| CK7 | - | - |
| CK5/6 | - | - |
| WT1 | - | - |
| CD34 | NP | +/- |
| HMGA2 | NP | + |
| Vimentin | NP | + |
| Calretinin | - | - |
| p40 | - | - |
| p63 | - | - |
| HMB45 | - | NP |
| Melan-A | - | NP |
| CD30 | - | NP |
| STAT6 | - | NP |
| PLAP | - | NP |
| AFP | - | NP |
| HepPar1 | - | NP |
| BerEP4 | NP | - |
| CD99 | NP | - |
| Bcl2 | NP | - |
| Myogenin | NP | - |
| Ki-67 | NP | + in about 10% of tumor cells |
| IHC: immunohistochemistry; NP: not performed. | ||
Figure 3.
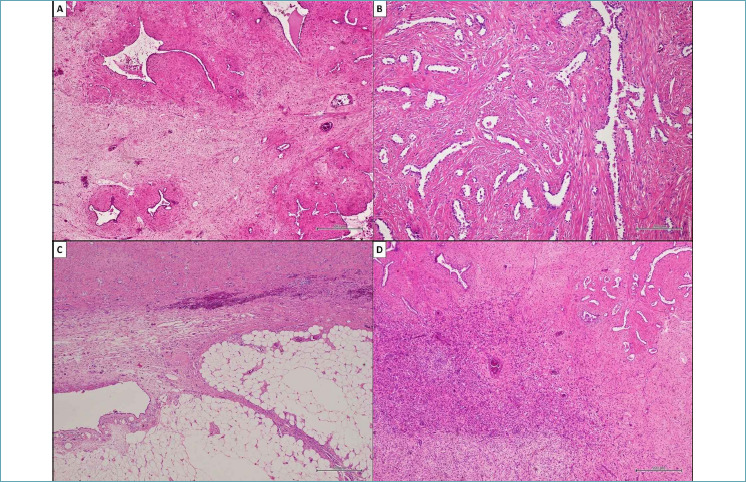
Histological findings observed in the surgical specimen. (A) The tumor consisting of bland and monomorphic spindle cells. Neoplastic population showed a predominant fascicular organization and a typical zonal pattern, with hypercellular and hypocellular areas in which many tubular and cleft-like spaces of entrapped normal respiratory epithelium were involved. (B) Detail of tubular and cleft-like spaces of entrapped respiratory epithelium. (C) Scattered foci of adipose tissue were also observed between neoplastic cells. (D) Detail of hypercellular area composed of relatively bland and monomorphic spindle cells. (A, C, D) Hematoxylin and eosin, original magnifications x500. (B) Hematoxylin and eosin, original magnifications x200.
Figure 4.
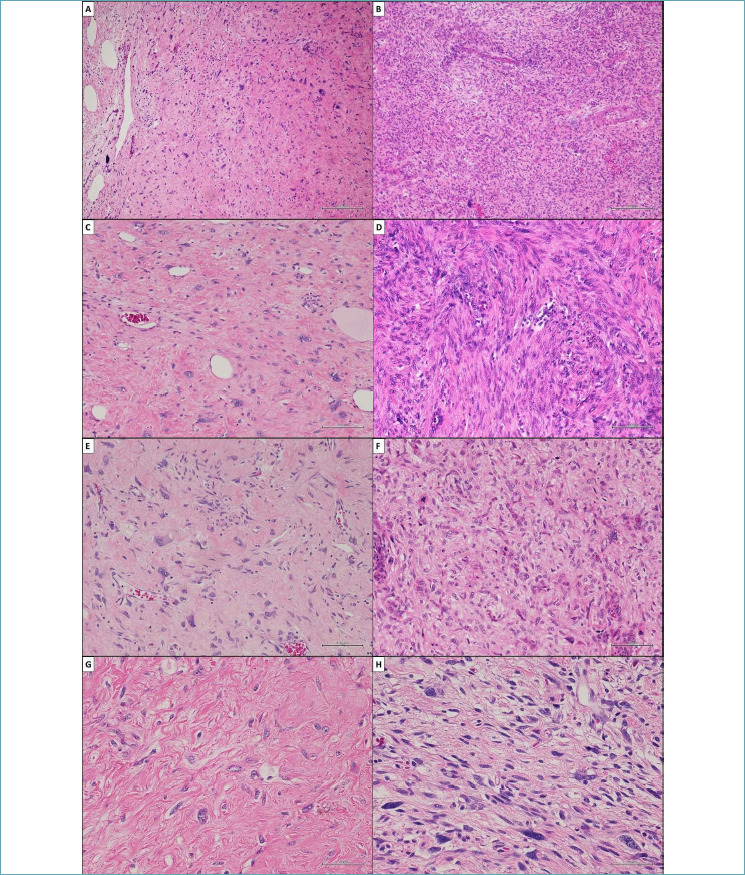
Comparison between histological features observed in hypocellular and hypercellular areas of the surgical specimen. (A, C, E, G) These cells in hypocellular areas were characterized by bizarre and irregular shaped nuclei, with both coarse and finely dispersed chromatin with evident nucleoli. Tumor necrosis was absent and low mitotic index (4 mitosis/10 HPF) was observed. (B, D, F, H) Conversely, a prominent cell crowding was observed in hypercellular areas; neoplastic cells were different from those previously described and they were characterized by marked nuclear atypia, with nucleomegaly and nuclear hyperchromasia, and significant cellular pleomorphism. The mitotic hot spots and scattered atypical mitotsis were observed. (A, B) Hematoxylin and eosin, original magnifications x200. (C, D, E, F) Hematoxylin and eosin, original magnifications x100. (G, H) Hematoxylin and eosin, original magnifications x50.
Figure 5.
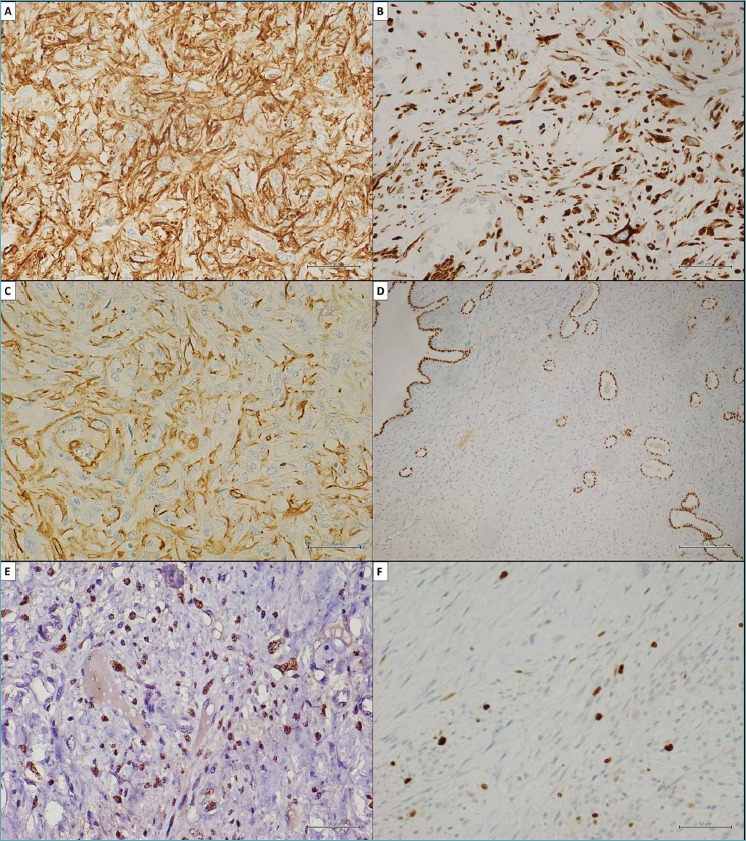
Immunohistochemical study confirmed the myogenic differentiation of the neoplasm. (A) Strong and diffuse cytoplasmic stain for h-caldesmon (clone E89, Ventana). (B) Strong and diffuse cytoplasmic stain for desmin (clone DE-R-11, Ventana). (C) Strong and diffuse membranous and cytoplasmic stain for smooth muscle actin (clone 1A4, Ventana). (D) Evidence of tubular and cleft-like spaces of entrapped respiratory epithelium by immunohistochemical staining for TTF1 (clone SP141, Ventana). (E) Immunohistochemical evidence of hmga2 nuclear overexpression in tumor cells (No. 41878, Abcam). (F) Low proliferation index (Ki-67) (clone 30-9, Ventana) in about 10% of the study population. (A, B, C, E, F) Original magnifications x50. (D) Original magnifications x200.
Discussion
PAH is a rare histological subtype of pulmonary hamartoma with very few described cases in literature 9-14. Like other types of PH, PAH is usually asymptomatic and it radiologically appears as a solitary, well-circumscribed, peripheral pulmonary coin nodule 15. The histological features defining this tumor are a predominant smooth muscle cell component with numerous epithelial inclusions, as tubular and cleft-like spaces of entrapped normal respiratory epithelium, and no cellular atypia, high mitotic index and atypical mitoses 12. Furthermore, also PAH frequently harbors t(3;12) (q27-q28;q14-q15) translocation, which has an excellent diagnostic sensitivity but very poor specificity in PH, leading to HMGA2 overexpression 16,17. Furthermore, when treated surgically, PAH has a great prognosis and a benign biological behavior 4. The relationship between PH and lung cancer has previously been questioned, and malignant transformation in PH, especially in PAH, is extremely uncommon and disputed (Tab. II) 18. In 1989 Basile et al. 8 reported a case of sarcomatous transformation in PH, including both cartilagenous or connective differentiation, occurring shortly after resection of a PH. Afterwards, Williams et al. 19 and Schenkel et al. 6 described two cases of malignant transformation of a chondroid PH into a pulmonary chondrosarcoma and a PH into a spindle cell malignancy with chondromatous differentiation, respectively; similarly, Rossi et al. 7 reported a case of a lipomatous PH with features of lipoma-like well-differentiated liposarcoma. Thus, the data about malignant transformation observed in PH are very limited and the chondrosarcoma is more frequent, because the predominant component of most PH was hyaline cartilage. Our case consisted of a voluminous lung neoplasia histologically showing a prevalent smooth muscle cell component as PAH. PAH is typically characterized by bland and monomorphic cells with no nuclear atypia. Differently, diffuse cells with bizarre nuclei, with irregular nuclear shape and frequent multinucleation, were widely represented in our case and showed a myogenic differentiation. However, this morphological feature was not convincing for malignancy diagnosis and we considered them as a leiomyoma with bizarre nuclei 20. Differently, the scattered hypercellular areas with cell crowding, marked cell pleomorphism and severe nuclear atypia, with prominent nucleomegaly and nuclear hyperchromasia, associated with a mitotic index of 4/10 HPF with scattered atypical mitoses oriented our diagnosis of leiomyosarcomatous transformation of PAH. Unlike many other lung neoplasms 21,22, there is lack of data about the molecular profile of these tumors. We performed a next generation sequencing (NGS) was performed on formalin-fixed paraffin-embedded (FFPE) tissue samples of PAH and pulmonary leiomyosarcoma. The DNA was extracted from 10-µm FFPE sections through the MGF03-Genomic DNA FFPE One-Step Kit, according to the manufacturer’s protocol (MagCore Diatech). DNA quality was established in using the FFPE QC Kit according to the manufacturer’s protocol (Illumina, San Diego, USA). The libraries were prepared with TruSigth Oncology 500 kit, the assay detects small nucleotide variants (SNVs), indels, splice variants, and immunotherapy biomarkers in 523 cancer-relevant genes. Sequencing was performed on an Illumina NovaSeq 6000 (San Diego, USA) platform. The resulting number of variants identified in both samples are reported in the Table III. The number of variants identified is comparable between the sets, but despite being the same absolute number, the impact of high impact variants in different sets is different. We performed comparations between the sets of variants and detected that there is a 10-fold increase in high impact variants between PAH set and pulmonary leiomyosarcoma (4 variants vs 42 variants, respectively).
Table II.
Pulmonary hamartoma with sarcomatous transformation, review of the literature.
| Authors | Age (year) | Size (cm) | PH histological subtype | Sarcomatous transformation type | Follow-up |
|---|---|---|---|---|---|
| Basile et al. 8 | 62 | 12 (diameter) | PH | Pleomorphic sarcoma | ***MRD |
| Williams et al. 19 | 74 | 6.5x5.5 | Chondroid PH | Pulmonary chondrosarcoma | NA* |
| Schenkel et al. 6 | 67 | 9.6x7.9 | Chondroid PH | Pulmonary chondrosarcoma | ***MRD |
| Rossi et al. 7 | 60 | 5 (diameter) | Lipomatous PH | Lipoma-like well-differentiated liposarcoma | DFS** at 7 years |
| *NA: not available; **DFS: disease free survival; ***MRD: malignancy related death. | |||||
Table III.
Comparison between the number of genetic variants identified in both PAH and pulmonary leiomyosarcoma samples by next generation sequencing.
| Sample | Number of variants |
|---|---|
| PAH | 1025 |
| Pulmonary leiomyosarcoma | 953 |
Conclusions
Our case represents the first case of malignant transformation of a PAH into pulmonary leiomyosarcoma, as far as we know. To date, the pathogenetic mechanisms underlying these malignant changes are still unknown. However, all the evidence, including the molecular signatures of these neoplasms, suggests that malignant histologic transformation in PH, although exceptionally rare, can occur. These findings are very interesting and could influence the management of PH, especially the diagnostic-therapeutic algorithm, given the risk of malignant transformation.
CONFLICTS OF INTEREST
Authors declare no conflict of interest.
FUNDING
We have not disclosure of funding for this work.
AUTHORS’ CONTRIBUTIONS
All listed authors contributed to the production of this manuscript and are listed in the appropriate order.
Figures and tables
References
- 1.Lucà S, Zannini G, Morgillo F, et al. The prognostic value of histopathology in invasive lung adenocarcinoma: a comparative review of the main proposed grading systems. Expert Rev Anticancer Ther 2023;23:265-277. https://doi.org/10.1080/14737140.2023.2179990 10.1080/14737140.2023.2179990 [DOI] [PubMed] [Google Scholar]
- 2.Li B, Xin Z, Xue W, et al. Lung hamartoma resembling lung cancer: a report of three cases. J Int Med Res 2022;50:3000605221132979. https://doi.org/10.1177/03000605221132979 10.1177/03000605221132979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ettinger DS. Ten years of progress in non-small cell lung cancer. J Natl Compr Canc Netw 2012;10(3):292-5. https://doi.org/10.6004/jnccn.2012.0029 10.6004/jnccn.2012.0029 [DOI] [PubMed] [Google Scholar]
- 4.WHO Classification of Tumours Editorial Board. WHO Classification of Tumours - Thoracic Tumours. 5th ed. Lyon: IARC, 2021. [Google Scholar]
- 5.Guo W, Zhao YP, Jiang YG, et al. Surgical treatment and outcome of pulmonary hamartoma: a retrospective study of 20-year experience. J Exp Clin Cancer Res 2008;27:8. https://doi.org/10.1186/1756-9966-27-8 10.1186/1756-9966-27-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schenkel R, Altfillisch C, Chung J, et al. Malignant Degeneration of Biopsy-Proven Hamartoma to Chondrosarcoma. Cureus 2020;12:e12150. https://doi.org/10.7759/cureus.12150 10.7759/cureus.12150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rossi G, Cavazza A, Valli R, et al. Atypical lipomatous tumour (lipoma-like well-differentiated liposarcoma) arising in a pulmonary hamartoma and clinically presenting with pneumothorax. Lung Cancer 2003;39:103-106. https://doi.org/10.1016/s0169-5002(02)00393-8 10.1016/s0169-5002(02)00393-8 [DOI] [PubMed] [Google Scholar]
- 8.Basile A, Gregoris A, Antoci B, et al. Malignant change in a benign pulmonary hamartoma. Thorax 1989;44:232-233. https://doi.org/10.1136/thx.44.3.232 10.1136/thx.44.3.232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kara M, Onder S, Firat P, et al. A rare histological presentation of a common lung tumor: adenoleiomyomatous hamartoma. Acta Chir Belg 2012;112:74-76. https://doi.org/10.1080/00015458.2012.11680800 10.1080/00015458.2012.11680800 [DOI] [PubMed] [Google Scholar]
- 10.Itoh H, Yanagi M, Setoyama T, et al. Solitary fibroleiomyomatous hamartoma of the lung in a patient without a pre-existing smooth-muscle tumor. Pathol Int 2001;51:661-5. https://doi.org/10.1046/j.1440-1827.2001.01245.x 10.1046/j.1440-1827.2001.01245.x [DOI] [PubMed] [Google Scholar]
- 11.Takeshima Y, Furukawa K, Inai K. ‘Adenomyomatous’ hamartoma of the lung. Pathol Int 2000;50:984-986. https://doi.org/10.1046/j.1440-1827.2000.01136.x 10.1046/j.1440-1827.2000.01136.x [DOI] [PubMed] [Google Scholar]
- 12.Ichiki Y, Kawasaki J, Hamatsu T, et al. A rare pulmonary hamartoma: fibroleiomyomatous hamartoma. Surg Case Rep 2016;2:53. https://doi.org/10.1186/s40792-016-0184-z 10.1186/s40792-016-0184-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takeshima Y, Furukawa K, Inai K. ‘Adenomyomatous’ hamartoma of the lung. Pathology International 2000;50:984-986. https://doi.org/10.1046/j.1440-1827.2000.01136.x 10.1046/j.1440-1827.2000.01136.x [DOI] [PubMed] [Google Scholar]
- 14.Rossi G, Cavazza A, Comin C, et al. Mucinous Adenomyomatous Pulmonary Hamartoma: Clinicopathologic, Immunohistochemical, and Molecular Features of 6 Cases. Int J Surg Pathol 2021;29:273-280. https://doi.org/10.1177/1066896920945016 10.1177/1066896920945016 [DOI] [PubMed] [Google Scholar]
- 15.Hutter J, Reich-Weinberger S, Hutarew G, et al. Giant pulmonary hamartoma--a rare presentation of a common tumor. Ann Thorac Surg 2006;82:e5-7. https://doi.org/10.1016/j.athoracsur.2006.05.032 10.1016/j.athoracsur.2006.05.032 [DOI] [PubMed] [Google Scholar]
- 16.von Ahsen I, Rogalla P, Bullerdiek J. Expression patterns of the LPP-HMGA2 fusion transcript in pulmonary chondroid hamartomas with t(3;12)(q27 approximately 28;q14 approximately 15). Cancer Genet Cytogenet 2005;163:68-70. https://doi.org/10.1016/j.cancergencyto.2005.02.023 10.1016/j.cancergencyto.2005.02.023 [DOI] [PubMed] [Google Scholar]
- 17.Piton N, Angot É, Marguet F, et al. HMGA2 immunostaining is a straightforward technique which helps to distinguish pulmonary fat-forming lesions from normal adipose tissue in small biopsies: a retrospective observational study about a series of 13 lung biopsies. Diagn Pathol 2017;12:21. https://doi.org/10.1186/s13000-017-0603-x 10.1186/s13000-017-0603-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bateson EM. Histogenesis of intrapulmonary and endobronchial hamartomas and chondromas (cartilage-containing tumours): a hypothesis. J Pathol 1970;101:77-83. https://doi.org/10.1002/path.1711010202 10.1002/path.1711010202 [DOI] [PubMed] [Google Scholar]
- 19.Williams E, Pratt J. Malignant Degeneration of a Pulmonary Hamartoma Into a Pulmonary Chondrosarcoma. Chest 2013; Vol 144, No. 4_MeetingAbstracts [Google Scholar]
- 20.Guo E, Li C, Hu Y, et al. Leiomyoma with Bizarre Nuclei: A Current Update. Int J Womens Health 2022;14:1641-1656. https://doi.org/10.2147/IJWH.S388278 10.2147/IJWH.S388278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lucà S, Franco R, Napolitano A, et al. PATZ1 in Non-Small Cell Lung Cancer: A New Biomarker That Negatively Correlates with PD-L1 Expression and Suppresses the Malignant Phenotype. Cancers (Basel) 2023;15:2190. https://doi.org/10.3390/cancers15072190 10.3390/cancers15072190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang P, Cheng CL, Chang YH, et al. Molecular gene signature and prognosis of non-small cell lung cancer. Oncotarget 2016;7:51898-51907. https://doi.org/10.18632/oncotarget.10622 10.18632/oncotarget.10622 [DOI] [PMC free article] [PubMed] [Google Scholar]