


Abstract
The UNIfied database of TransMembrane Proteins (UniTmp) is a comprehensive and freely accessible resource of transmembrane protein structural information at different levels, from localization of protein segments, through the topology of the protein to the membrane-embedded 3D structure. We not only annotated tens of thousands of new structures and experiments, but we also developed a new system that can serve these resources in parallel. UniTmp is a unified platform that merges TOPDB (Topology Data Bank of Transmembrane Proteins), TOPDOM (database of conservatively located domains and motifs in proteins), PDBTM (Protein Data Bank of Transmembrane Proteins) and HTP (Human Transmembrane Proteome) databases and provides interoperability between the incorporated resources and an easy way to keep them regularly updated. The current update contains 9235 membrane-embedded structures, 9088 sequences with 536 035 topology-annotated segments and 8692 conservatively localized protein domains or motifs as well as 5466 annotated human transmembrane proteins. The UniTmp database can be accessed at https://www.unitmp.org.
Graphical Abstract
Graphical Abstract.
Introduction
Transmembrane proteins (TMP) play an important role in living cells, as they serve as a gatekeeper for cellular communication and transport of molecules across the membranes. They participate in cell signaling, maintaining cell structure and energy production. Despite their importance, their structure determination is rather laborious due to their hydrophobic nature, which needs to be retained in the lipid environment.
Numerous efforts were made to explore the uncharted space of membrane protein structures. By the end of the 90s, the majority of experimental information could be interpreted as topological data (e.g. the cellular compartment localization of a few residues, or sometimes the orientation of a longer connecting loop/tail region was defined (1,2) with a few revealed structures). In the 2000s structure determination yielded hundreds of important novel structures, meanwhile, structural genomic target selection projects aimed to pinpoint ‘important’ proteins that drove the field forward (3,4). Although cryo-electron microscopy boosted the number of solved TMP structures (5), they still lag far behind globular proteins in terms of structure determination. Not surprisingly considering the challenging experimental conditions, the next big step arrived with Artificial Intelligence (AI): AlphaFold2 (6) ‘solved’ the problem of predicting all structures, yet around one-third of the (predicted) human membrane proteome still has quality issues (7,8). The potential of AI is unquestionable, however, as in the case of classical topology prediction, integrating different types of information may significantly raise the accuracy of such methods. Thus, traditional resources providing structural and experimental information at many different levels may serve as a valuable addition when developing novel tools.
The UniTmp web resource provides an integrated platform for databases storing structural information at different levels: The Protein Data Bank of Transmembrane Proteins (PDBTM) (9,10) holding experimentally determined structures with the orientation of the lipid bilayer relative to the protein; the Topology Data Bank of Transmembrane Proteins (TOPDB) (11,12) containing all kinds of experimental topological data; TOPDOM (13,14) storing information about conserved protein domains and motifs located consistently on the same side of the membrane; The Human Transmembrane Proteome (HTP) (15) is an example of a significant step toward our complete understanding of TMP structures, achieved by storing all this information together. Depositing and connecting all these disparate pieces of information into a unified database helps researchers to find any kind of information at various structure levels of TMPs and paves the way for more reliable, AI-based structure predictions of them.
Materials and methods
Data resources
Four sources of data were utilized during the development: InterPro (16) (release: 5.62–94.0), UniProt (17) (release 2023_2), PDB (18) (until 25.08.2023) and Pubmed (until 01.08.2023).
Data processing
We used InterProScan (16) to search domains in CATH (19), NCBIfam (20), Panther (21), Pfam (22), Prints (23), ProSite (24), SMART (25) and SUPERFAMILY (26). We used TMDET (27) to reconstruct the most likely localization of the membrane bilayer using the original PDB structure. CCTOP (28) was used to predict signal peptides (notably we are using the latest SignalP6 (29) for this task), to discriminate TM and non-TM proteins and to predict TMP topology (Scampi-MSA (30) was replaced with Scampi2-MSA (31) for topology prediction and MemBrain (32) was removed). CCTOP automatically incorporates all experimental evidence when predicting topology for α-helical TMPs. In the case of β-barrel TMPs, we predicted topology using HMMTOP (33,34) with a slightly modified architecture (similarly as in TOPDB 1.0 and 2.0), using experimental evidence as constraints. We used BLAST (35) (e-value = 10–5, GOP = 11, GEP = 1) to search for homologous entries in the sequence pool. PDB (18) entries were assigned to UniProt (17) sequences using SIFTS (36) via PDBe updated mmCIF files. All temporary and final data are stored in a local MySQL database.
Results
Data collection and curation
Data structure
For each protein, we store amino acid sequences, UniProt Accession, UniProt ID, solved PDB structure and domain/motif information from InterPro. Using the sequence pool (TMP proteins from SwissProt, human reference proteome and individual proteins with experimental data) we created a network of homologous proteins using BLAST. Therefore, when searching for entries, homologous proteins are also automatically listed. Although experimental information is transferred between entries via CCTOP, this way the source of information is better accessible.
PDBTM data curation
PDBTM has been updated weekly since its first release in 2004. During the weekly update, the TMDET algorithm is applied to each newly released PDB entry, and proteins identified as transmembrane by the TMDET are investigated and manually curated if needed. For integrating PDBTM into UniTmp, we scanned all PDB entries again by also applying homologous sequence information. We used SIFTS to assign the UniProt entry and the full protein sequence to PDB structures. We used TMDET on the PDB structures and CCTOP on the full protein sequences to automatically select candidate α-helical and β-barrel TMPs, which were then manually processed and corrected if needed. This way, several new PDB entries have been identified as transmembrane that were missed earlier, and several false positive hits were deleted from the database. In the current release, membrane-embedded structures of viral proteins are also included in the database that were formerly omitted, while PDB entries containing in silico predicted model structures have been removed. We remediate hundreds of entries that contain invalid region assignments, like re-entrant loops instead of transmembrane helices or invalid order of regions (e.g. the directly adjacent extra- and intracellular segments without an intervening transmembrane region, transmembrane regions connecting segments from the same side etc). Altogether 459, 145 and 856 entries were added, deleted and remediated, respectively, those modifications yielded 406 newly annotated TMPs.
Gathering and curating TOPDB data
Transmembrane protein structures in the PDBTM database provide only relative topological information, and it cannot be determined which one of the two non-membrane embedded parts of the protein (called side1 and side2) is situated inside and which is outside of the cell/organelle. Thus, we needed to add this information to all PDBTM structures by curation. Wherever possible, we manually assigned side definitions to PDB entries, using the original research article as the source for defining them. If a homologous entry has already been assigned, we transferred that annotation. Notably, we used a simplified partition that reflects the biochemical environment, and most cellular compartments are converted to a simplified binary definition: inside/outside. The only exception to this classification scheme was the bacterial and archaeal periplasmic space, which is located between the inner and outer membranes and cannot be easily classified using these terms. More information about side definitions has been made available at the TOPDB web resource, in the documents section. Notably, we added side definitions not only to the PDB entries containing transmembrane segments but also to entries that are soluble fragments of otherwise transmembrane proteins.
Another major source of topological information comes from the literature, including experiments performed on individual proteins as well as high-throughput experiments. We scanned PubMed and Google Scholar for results indicating protein topology, prioritizing articles published after the last major update of TOPDB (in 2016). Despite the limited number of new low-throughput studies, several new experimental methods have been invented since our major database release, necessitating the update of the methods section as well. These methods include novel split-protein reporters (37), new fusion protein based assays (38) and electron microscopy based techniques (39). Now we also regard experimentally validated eukaryotic linear motif based interactions with a known cytosolic, luminal, or extracellular partner as proof of topology. The latter information was inferred from the ELM database after manual curation of entries regarding transmembrane and topology status (40). We also imported low-throughput post-translational modification related data, whenever the partner and its location were identified (e.g. intracellularly localized or inward-facing enzyme dependent protein modification e.g. phosphorylation or lipidation, such as N-myristoylation (41)).
High-throughput mass spectrometry data regarding post-translational modifications expanded substantially in the past years, including topologically relevant modifications, such as the novel bacterial N-glycosylation (42). We also integrated an extensive amount of high-throughput eukaryotic N-glycosylation data from dedicated glycosylation databases, such as GlyGen (43) and GlyConnect (44) as well as further site annotations from UniProtKB based on experimental evidence. In all the cases before inclusion, it was also checked whether the sequence motifs around the collected sites met the criteria of the consensus sequence of N-type glycosylation (‘sequon’). Last but not least, we also included results from the numerous high-throughput surface labeling experiments carried out in our research group, yielding topologically reliable data (45–47).
Defining domain localizations for the TOPDOM database
We used CCTOP to predict the topology of α-helical TMPs in UniProtKB and subcellular localisations to extract the localization of non-TMPs, at first without incorporating any experimental information. From CCTOP, only predictions above 85% reliability were accepted. Domains from InterPro were assigned if they occurred at least 10 times, and in 99% they appeared on the same side of the membrane (inside/outside). At the second iteration, CCTOP was used again, however experimental information and domain information from the first iteration were also incorporated into the final prediction.
Combining all experimental and bioinformatic evidence for the HTP database
We used the CCTOP algorithm's TMP filtering ability on the human reference proteome to select α-helical TMPs. Using the network of homologous proteins, all experimental information from PDBTM, TOPDB and domain/motif information from TOPDOM is also incorporated.
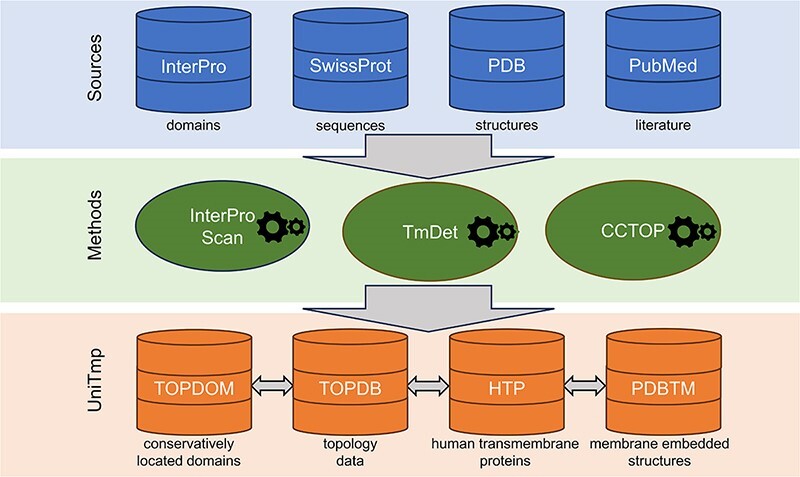
A schematic graph of data processing procedures is shown on Figure 1.
Figure 1.

Generation of the UniTmp resource. In UniTmp, we collect structural information at different levels: structures, domain localizations and topology data. UniTmp provides a shared, unified platform between TOPDOM, TOPDB, PDBTM and HTP databases. For more details see text.
Updating web backend and frontend
Data processing, SQL and backend
We used PHP 8.2 and Laravel 10.0 for reading and manipulating data and stored all data in a local MySQL server. BLAST searches and CCTOP predictions were made on our HPC.
Frontend development
While we aimed to keep the original look and feel of each database so accustomed users could quickly find everything, the engine was completely overhauled on each side. The original home pages of the TOPDB and the TOPDOM databases had been written in PHP earlier, but without using any framework, while the HTP and PDBTM sites were written in C++ using the WT toolkit. Now all four home pages have been rewritten in PHP 8.2 using Laravel 10.0 framework with integrated Eloquent SQL services and Blade template system. For visualizing 3D structures with the determined membrane orientation, we use a locally modified version of Mol* (48) (the modified software is available on our git server, https://git.enzim.ttk.hu/web/TmMolStar), while topology data are shown by using an in-house developed React based software, called JsvLib. Public API endpoints are also provided for all the four databases, for details see the Document and/or Usage menu item in the selected database.
Future plans
Data update schedules
We aim to update all four databases regularly. PDBTM has been updated every week after the PDB update, and we will keep on updating it as before. Adding side definitions to PDBTM entries, as well as updating alignments and the network of protein relatives is planned after the release of the new UniProt version (i.e. quarterly). Thus, TOPDB is going to be updated four times a year. TOPDOM and HTP updates will follow the TOPDB update.
Improving source resources
Our next goal is to update the TMDET algorithm to make it more robust, i.e. to be able to identify incorrect structures, non-biological oligomer forms and new features such as embedding proteins in curved membranes or bacterial protein complexes in double (inner and outer) membranes. We also plan to change the input processing so that not only ‘ent’ formatted files but CIF format will be also handled. We also plan to improve the CCTOP algorithm to be more accurate in discriminating between transmembrane and non-transmembrane proteins and to incorporate newly developed topology prediction methods such as TMBED (49) or DeepTMHMM (50).
Integrating other resources
We also plan to integrate more databases and resources into the common UniTmp platform, such as the TmAlphaFold database (8) and MemDis (51) prediction algorithm.
The new UniTmp Database statistics
The complete UniTmp database holds 774 508 unique amino acid sequences from which 92 337 belong to transmembrane proteins. Regarding TMP sequences, experimental information is available for 9898 and 11 159 TMP sequences from UniProtKB and PDB, respectively. The number of TMPs in the PDBTM database has grown from 1700 to 9235 structures (8608 α-helical and 627 β-barrel proteins) since its last published release (10). The TOPDB database now contains 9088 entries, including 8783 α-helical and 305 β-barrel proteins) and 536035 topology data regions that more than doubles the number of entries and contains six times the topology data points since its last release. The number of conservatively localized domains also increased in the TOPDOM database from 5236 to 8692 domains (from 3699 to 7065 for inside localization and from 1537 to 1627 for outside localization). The HTP database now contains 5466 proteins, which covers 26.8% of the human proteome. By counting all experimental and bioinformatics evidence in HTP this means 704576 constraints helping the prediction derived from 3190 experimental and 558 domain-based sources. For 2906 (53.2%) proteins, there is at least one structural, other experimental, or domain-based evidence available.
Discussion
UniTmp is a novel resource that merges various databases developed for transmembrane proteins. UniTmp includes PDBTM, TOPDB, TOPDOM and the HTP databases and provides interoperability between them. These databases have existed for over 10 years and they provide structural information at different levels. They were used for a diverse range of research tasks. Manual annotation of experimental data is a unique and valuable addition, and our resources supplied training and testing benchmark data for state-of-the-art deep learning prediction algorithms, such as the SignalP series (29,52) or contact predictions (53). In contrast to most topology prediction algorithms, CCTOP incorporates experimental and domain information from homologous proteins, enabling these resources to serve as a solid base to perform surveys to analyze the impact of mutations and diseases (54–56). Information about the localization of domains and motifs can be utilized to construct filters when developing pipelines for Short Linear Motif analysis (57). Topology information can be also extremely useful to design wet-lab (58) or computational (59) experiments, to develop novel therapeutics acting on membrane proteins, or to rigorously benchmark high-throughput experiment design (45).
The continuous update and the reliable data in these databases allow the integration of their content into the largest resources of this field. Membrane proteins from the PDBTM database are shown on RCSB web pages since 2021 (60) as well as data are available on the PDBe-KB public FTP area in JSON format (https://ftp.ebi.ac.uk/pub/databases/pdbe-kb/annotations/PDBTM/) and they are also integrated into the PDBe graph database (https://www.ebi.ac.uk/pdbe/pdbe-kb/graph).
Comparing the contents of the PDBTM and the OPM (61) database, the other main source in the field of transmembrane PDB structure annotation, we found that among the bi- and polytopic membrane proteins that have defined TM regions and are at least 20 residues long there are 8687 common proteins, while PDBTM contains 796 TMPs that we could not find in OPM and 499 TMPs in OPM that are not in PDBTM. Note that OPM is for all proteins that interact with the membrane in some way, whereas PDBTM is for transmembrane proteins only. Therefore most of the proteins in the latter cluster are i, incorrectly annotated monotopic membrane proteins; ii, short peptides in the micelle that are intentionally omitted from PDBTM because they aren’t TMPs; iii, flagellar or pilus proteins that are also omitted from PDBTM because the membrane definitions of each chain in the homooligomeric structures are different; iv, proteins without an .ent file in the PDB database.
The most important feature of the UniTmp database is to collect and unify information from different sources to help better understand the structure and topology of proteins. For example, AT8A1_HUMAN is an ATPase mediating the translocation of phospholipids, playing an essential role in membrane trafficking and signaling pathways (62). Cryo-electron microscopy revealed the structure of six different type IV P-type adenosine triphosphatases (62), which are stored in the PDBTM database (Figure 2A). There are other experiments performed on this protein, that indirectly help to define the topology: for example, using fluorescent confocal microscopy and mutation analysis it was shown that the protein has an adaptin binding, dileucine-type endocytosis and sorting signal at position from 1105 to 1110 (63). Adaptin-binding dileucine motifs occur exclusively on the cytosolic side of membrane proteins, near to N- or C-termini of the protein (40). This information is stored in the TOPDB database, in addition to topology information derived from the paper descript the cryo-electron microscopy structure determination (Figure 2B). Furthermore, according to the CATH database (19) P-type ATPases possess a conserved domain, P-type ATPase cytoplasmic domain N (in this case from 479 to 655 residues), which we also store in the TOPDOM database as a domain always conservatively located on the same, cytoplasmic side of the membrane (Figure 2C). Transferring all this information helps to predict the most accurate topology for this protein, which is in turn stored in the HTP database (Figure 2D).
Figure 2.

Same protein from different points of view: The human phospholipid-transporting ATPase IA protein. (A) 3D Structure and membrane localization from the PDBTM database. (B) Topology-related experimental evidence in the TOPDB database. (C) Conservatively localized protein domain information in the TOPDOM database. (D) All information combined and extended by topology prediction methods in the HTP database. For more details, see text.
Acknowledgements
Authors’ contributions: L.D.: Conceptualization, Methodology, Validation, Writing—original draft. C.G.: Web server programming, Testing, Visualization. S.T.: Annotation. T.L.: Annotation. A.Z.: Annotation. L.S.: Validation, Testing. K.K.: Annotation. G.E.T.: Conceptualization, Methodology, Validation, Web server programming, Testing, Annotation, Writing—draft, review & editing.
Contributor Information
László Dobson, Protein Bioinformatics Research Group, Institute of Enzymology, Research Centre for Natural Sciences, Budapest, Magyar Tudósok körútja 2, H-1117, Hungary; Department of Bioinformatics, Semmelweis University, Budapest, Tűzoltó u. 7, H-1094, Hungary.
Csongor Gerdán, Protein Bioinformatics Research Group, Institute of Enzymology, Research Centre for Natural Sciences, Budapest, Magyar Tudósok körútja 2, H-1117, Hungary.
Simon Tusnády, Department of Bioinformatics, Semmelweis University, Budapest, Tűzoltó u. 7, H-1094, Hungary.
Levente Szekeres, Protein Bioinformatics Research Group, Institute of Enzymology, Research Centre for Natural Sciences, Budapest, Magyar Tudósok körútja 2, H-1117, Hungary.
Katalin Kuffa, Protein Bioinformatics Research Group, Institute of Enzymology, Research Centre for Natural Sciences, Budapest, Magyar Tudósok körútja 2, H-1117, Hungary; Doctoral School of Biology, Institute of Biology, ELTE Eötvös Loránd University, Budapest, Pázmány P. stny. 1/C, H-1117, Hungary.
Tamás Langó, Protein Bioinformatics Research Group, Institute of Enzymology, Research Centre for Natural Sciences, Budapest, Magyar Tudósok körútja 2, H-1117, Hungary.
András Zeke, Protein Bioinformatics Research Group, Institute of Enzymology, Research Centre for Natural Sciences, Budapest, Magyar Tudósok körútja 2, H-1117, Hungary.
Gábor E Tusnády, Protein Bioinformatics Research Group, Institute of Enzymology, Research Centre for Natural Sciences, Budapest, Magyar Tudósok körútja 2, H-1117, Hungary; Department of Bioinformatics, Semmelweis University, Budapest, Tűzoltó u. 7, H-1094, Hungary.
Data availability
All data can be freely downloaded from the public webpage of the UniTmp database which is accessible at https://www.unitmp.org.
Funding
Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund [K132522]; K.K. was supported by the KDP-2021 Program of the Ministry of Innovation and Technology from the source of the National Research, Development and Innovation Fund. Funding for open access charge: Semmelweis University,Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund.
Conflict of interest statement. None declared.
References
- 1. Manoil C., Beckwith J.. TnphoA: a transposon probe for protein export signals. Proc. Natl. Acad. Sci. U.S.A. 1985; 82:8129–8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Broome-Smith J.K., Tadayyon M., Zhang Y.. Beta-lactamase as a probe of membrane protein assembly and protein export. Mol. Microbiol. 1990; 4:1637–1644. [DOI] [PubMed] [Google Scholar]
- 3. Punta M., Love J., Handelman S., Hunt J.F., Shapiro L., Hendrickson W.A., Rost B.. Structural genomics target selection for the New York consortium on membrane protein structure. J. Struct. Funct. Genomics. 2009; 10:255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Varga J., Dobson L., Reményi I., Tusnády G.E.. TSTMP: target selection for structural genomics of human transmembrane proteins. Nucleic Acids Res. 2017; 45:D325–D330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thonghin N., Kargas V., Clews J., Ford R.C.. Cryo-electron microscopy of membrane proteins. Methods. 2018; 147:176–186. [DOI] [PubMed] [Google Scholar]
- 6. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A.et al.. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jambrich M.A., Tusnady G.E., Dobson L.. How AlphaFold shaped the structural coverage of the human transmembrane proteome. 2023; bioRxiv doi:18 April 2023, preprint: not peer reviewed 10.1101/2023.04.18.537193. [DOI] [PMC free article] [PubMed]
- 8. Dobson L., Szekeres L.I., Gerdán C., Langó T., Zeke A., Tusnády G.E.. TmAlphaFold database: membrane localization and evaluation of AlphaFold2 predicted alpha-helical transmembrane protein structures. Nucleic Acids Res. 2023; 51:D517–D522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tusnády G.E., Dosztányi Z., Simon I.. PDB_TM: selection and membrane localization of transmembrane proteins in the protein data bank. Nucleic Acids Res. 2005; 33:D275–D278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kozma D., Simon I., Tusnády G.E.. PDBTM: protein Data Bank of transmembrane proteins after 8 years. Nucleic Acids Res. 2013; 41:D524–D529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tusnády G.E., Kalmár L., Simon I.. TOPDB: topology data bank of transmembrane proteins. Nucleic Acids Res. 2008; 36:D234–D239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dobson L., Langó T., Reményi I., Tusnády G.E.. Expediting topology data gathering for the TOPDB database. Nucleic Acids Res. 2015; 43:D283–D289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tusnády G.E., Kalmár L., Hegyi H., Tompa P., Simon I.. TOPDOM: database of domains and motifs with conservative location in transmembrane proteins. Bioinformatics. 2008; 24:1469–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Varga J., Dobson L., Tusnády G.E.. TOPDOM: database of conservatively located domains and motifs in proteins. Bioinformatics. 2016; 32:2725–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dobson L., Reményi I., Tusnády G.E.. The human transmembrane proteome. Biol. Direct. 2015; 10:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paysan-Lafosse T., Blum M., Chuguransky S., Grego T., Pinto B.L., Salazar G.A., Bileschi M.L., Bork P., Bridge A., Colwell L.et al.. InterPro in 2022. Nucleic Acids Res. 2023; 51:D418–D427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. UniProt Consortium UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023; 51:D523–D531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Burley S.K., Bhikadiya C., Bi C., Bittrich S., Chao H., Chen L., Craig P.A., Crichlow G.V., Dalenberg K., Duarte J.M.et al.. RCSB Protein Data Bank (RCSB.org): delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 2023; 51:D488–D508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sillitoe I., Bordin N., Dawson N., Waman V.P., Ashford P., Scholes H.M., Pang C.S.M., Woodridge L., Rauer C., Sen N.et al.. CATH: increased structural coverage of functional space. Nucleic Acids Res. 2021; 49:D266–D273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li W., O’Neill K.R., Haft D.H., DiCuccio M., Chetvernin V., Badretdin A., Coulouris G., Chitsaz F., Derbyshire M.K., Durkin A.S.et al.. RefSeq: expanding the Prokaryotic Genome Annotation Pipeline reach with protein family model curation. Nucleic Acids Res. 2021; 49:D1020–D1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thomas P.D., Ebert D., Muruganujan A., Mushayahama T., Albou L.-P., Mi H.. PANTHER: making genome-scale phylogenetics accessible to all. Protein Sci. 2022; 31:8–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G.A., Sonnhammer E.L.L., Tosatto S.C.E., Paladin L., Raj S., Richardson L.J.et al.. Pfam: the protein families database in 2021. Nucleic Acids Res. 2021; 49:D412–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Attwood T.K., Bradley P., Flower D.R., Gaulton A., Maudling N., Mitchell A.L., Moulton G., Nordle A., Paine K., Taylor P.et al.. PRINTS and its automatic supplement, prePRINTS. Nucleic Acids Res. 2003; 31:400–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sigrist C.J.A., de Castro E., Cerutti L., Cuche B.A., Hulo N., Bridge A., Bougueleret L., Xenarios I.. New and continuing developments at PROSITE. Nucleic Acids Res. 2013; 41:D344–D347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Letunic I., Khedkar S., Bork P.. SMART: recent updates, new developments and status in 2020. Nucleic Acids Res. 2021; 49:D458–D460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gough J., Karplus K., Hughey R., Chothia C.. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J. Mol. Biol. 2001; 313:903–919. [DOI] [PubMed] [Google Scholar]
- 27. Tusnády G.E., Dosztányi Z., Simon I.. TMDET: web server for detecting transmembrane regions of proteins by using their 3D coordinates. Bioinformatics. 2005; 21:1276–1277. [DOI] [PubMed] [Google Scholar]
- 28. Dobson L., Reményi I., Tusnády G.E.. CCTOP: a Consensus Constrained TOPology prediction web server. Nucleic Acids Res. 2015; 43:W408–W412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Teufel F., Almagro Armenteros J.J., Johansen A.R., Gíslason M.H., Pihl S.I., Tsirigos K.D., Winther O., Brunak S., von Heijne G., Nielsen H.. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022; 40:1023–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bernsel A., Viklund H., Falk J., Lindahl E., von Heijne G., Elofsson A.. Prediction of membrane-protein topology from first principles. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:7177–7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peters C., Tsirigos K.D., Shu N., Elofsson A.. Improved topology prediction using the terminal hydrophobic helices rule. Bioinformatics. 2016; 32:1158–1162. [DOI] [PubMed] [Google Scholar]
- 32. Shen H., Chou J.J.. MemBrain: improving the accuracy of predicting transmembrane helices. PLoS One. 2008; 3:e2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tusnády G.E., Simon I.. Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J. Mol. Biol. 1998; 283:489–506. [DOI] [PubMed] [Google Scholar]
- 34. Tusnády G.E., Simon I.. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001; 17:849–850. [DOI] [PubMed] [Google Scholar]
- 35. Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J.. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dana J.M., Gutmanas A., Tyagi N., Qi G., O’Donovan C., Martin M., Velankar S.. SIFTS: updated Structure Integration with Function, Taxonomy and Sequences resource allows 40-fold increase in coverage of structure-based annotations for proteins. Nucleic Acids Res. 2019; 47:D482–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hatlem D., Trunk T., Linke D., Leo J.C.. Catching a SPY: using the SpyCatcher-SpyTag and Related Systems for Labeling and Localizing Bacterial Proteins. Int. J. Mol. Sci. 2019; 20:2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rousset F., Zhang L., Lardy B., Morel F., Nguyen M.V.C.. Transmembrane Nox4 topology revealed by topological determination by Ubiquitin Fusion Assay, a novel method to uncover membrane protein topology. Biochem. Biophys. Res. Commun. 2020; 521:383–388. [DOI] [PubMed] [Google Scholar]
- 39. Mavylutov T., Chen X., Guo L., Yang J.. APEX2- tagging of Sigma 1-receptor indicates subcellular protein topology with cytosolic N-terminus and ER luminal C-terminus. Protein Cell. 2018; 9:733–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kumar M., Michael S., Alvarado-Valverde J., Mészáros B., Sámano-Sánchez H., Zeke A., Dobson L., Lazar T., Örd M., Nagpal A.et al.. The Eukaryotic Linear Motif resource: 2022 release. Nucleic Acids Res. 2022; 50:D497–D508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Utsumi T., Hosokawa T., Shichita M., Nishiue M., Iwamoto N., Harada H., Kiwado A., Yano M., Otsuka M., Moriya K.. ANKRD22 is an N-myristoylated hairpin-like monotopic membrane protein specifically localized to lipid droplets. Sci. Rep. 2021; 11:19233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cain J.A., Dale A.L., Cordwell S.J.. Exploiting Oligosaccharyltransferase-Positive and -Negative and a Multiprotease Digestion Strategy to Identify Novel Sites Modified by N-Linked Protein Glycosylation. J. Proteome Res. 2021; 20:4995–5009. [DOI] [PubMed] [Google Scholar]
- 43. York W.S., Mazumder R., Ranzinger R., Edwards N., Kahsay R., Aoki-Kinoshita K.F., Campbell M.P., Cummings R.D., Feizi T., Martin M.et al.. GlyGen: computational and Informatics Resources for Glycoscience. Glycobiology. 2020; 30:72–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alocci D., Mariethoz J., Gastaldello A., Gasteiger E., Karlsson N.G., Kolarich D., Packer N.H., Lisacek F.. GlyConnect: glycoproteomics Goes Visual, Interactive, and Analytical. J. Proteome Res. 2019; 18:664–677. [DOI] [PubMed] [Google Scholar]
- 45. Langó T., Róna G., Hunyadi-Gulyás É., Turiák L., Varga J., Dobson L., Várady G., Drahos L., Vértessy B.G., Medzihradszky K.F.et al.. Identification of Extracellular Segments by Mass Spectrometry Improves Topology Prediction of Transmembrane Proteins. Sci. Rep. 2017; 7:42610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Müller A., Langó T., Turiák L., Ács A., Várady G., Kucsma N., Drahos L., Tusnády G.E.. Covalently modified carboxyl side chains on cell surface leads to a novel method toward topology analysis of transmembrane proteins. Sci. Rep. 2019; 9:15729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Langó T., Kuffa K., Tóth G., Turiák L., Drahos L., Tusnády G.E.. Comprehensive discovery of the accessible primary amino group-containing segments from cell surface proteins by fine-tuning a high-throughput biotinylation method. Int. J. Mol. Sci. 2022; 24:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sehnal D., Bittrich S., Deshpande M., Svobodová R., Berka K., Bazgier V., Velankar S., Burley S.K., Koča J., Rose A.S.. Mol* Viewer: modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021; 49:W431–W437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bernhofer M., Rost B.. TMbed: transmembrane proteins predicted through language model embeddings. BMC Bioinf. 2022; 23:326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hallgren J., Tsirigos K.D., Pedersen M.D., Armenteros J.J.A., Marcatili P., Nielsen H., Krogh A., Winther O.. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. 2022; bioRxiv doi:10 April 2022, preprint: not peer reviewed 10.1101/2022.04.08.487609. [DOI]
- 51. Dobson L., Tusnády G.E.. MemDis: predicting disordered regions in transmembrane proteins. Int. J. Mol. Sci. 2021; 22:12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Almagro Armenteros J.J., Tsirigos K.D., Sønderby C.K., Petersen T.N., Winther O., Brunak S., von Heijne G., Nielsen H.. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019; 37:420–423. [DOI] [PubMed] [Google Scholar]
- 53. Lin P., Yan Y., Tao H., Huang S.-Y.. Deep transfer learning for inter-chain contact predictions of transmembrane protein complexes. Nat. Commun. 2023; 14:4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Molnár J., Szakács G., Tusnády G.E.. Characterization of disease-associated mutations in human transmembrane proteins. PLoS One. 2016; 11:e0151760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kulandaisamy A., Binny Priya S., Sakthivel R., Tarnovskaya S., Bizin I., Hönigschmid P., Frishman D., Gromiha M.M.. MutHTP: mutations in human transmembrane proteins. Bioinformatics. 2018; 34:2325–2326. [DOI] [PubMed] [Google Scholar]
- 56. Dobson L., Mészáros B., Tusnády G.E.. Structural principles governing disease-causing germline mutations. J. Mol. Biol. 2018; 430:4955–4970. [DOI] [PubMed] [Google Scholar]
- 57. Tusnády G.E., Zeke A., Kálmán Z.E., Fatoux M., Ricard-Blum S., Gibson T.J., Dobson L.. LeishMANIAdb: a comparative resource for Leishmania proteins. Database. 2023; baad074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mohamed S.A., Samir T.M., Helmy O.M., Elhosseiny N.M., Ali A.A., El-Kholy A.A., Attia A.S.. A novel surface-exposed polypeptide is successfully employed as a target for developing a prototype one-step immunochromatographic strip for specific and sensitive direct detection of causing neonatal sepsis. Biomolecules. 2020; 10:1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sanches R.C.O., Tiwari S., Ferreira L.C.G., Oliveira F.M., Lopes M.D., Passos M.J.F., Maia E.H.B., Taranto A.G., Kato R., Azevedo V.A.C.et al.. Immunoinformatics design of multi-epitope peptide-based vaccine against using transmembrane proteins as a target. Front. Immunol. 2021; 12:621706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bittrich S., Rose Y., Segura J., Lowe R., Westbrook J.D., Duarte J.M., Burley S.K.. RCSB Protein Data Bank: improved annotation, search and visualization of membrane protein structures archived in the PDB. Bioinformatics. 2022; 38:1452–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lomize M.A., Pogozheva I.D., Joo H., Mosberg H.I., Lomize A.L.. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012; 40:D370–D376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hiraizumi M., Yamashita K., Nishizawa T., Nureki O.. Cryo-EM structures capture the transport cycle of the P4-ATPase flippase. Science. 2019; 365:1149–1155. [DOI] [PubMed] [Google Scholar]
- 63. Kook S., Wang P., Meng S., Jetter C.S., Sucre J.M.S., Benjamin J.T., Gokey J.J., Hanby H.A., Jaume A., Goetzl L.et al.. AP-3-dependent targeting of flippase ATP8A1 to lamellar bodies suppresses activation of YAP in alveolar epithelial type 2 cells. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2025208118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data can be freely downloaded from the public webpage of the UniTmp database which is accessible at https://www.unitmp.org.