

Abstract
Complex biological processes in cells are embedded in the interactome, representing the complete set of protein–protein interactions. Mapping and analyzing the protein structures are essential to fully comprehending these processes’ molecular details. Therefore, knowing the structural coverage of the interactome is important to show the current limitations. Structural modeling of protein–protein interactions requires accurate protein structures. In this study, we mapped all experimental structures to the reference human proteome. Later, we found the enrichment in structural coverage when complementary methods such as homology modeling and deep learning (AlphaFold) were included. We then collected the interactions from the literature and databases to form the reference human interactome, resulting in 117 897 non-redundant interactions. When we analyzed the structural coverage of the interactome, we found that the number of experimentally determined protein complex structures is scarce, corresponding to 3.95% of all binary interactions. We also analyzed known and modeled structures to potentially construct the structural interactome with a docking method. Our analysis showed that 12.97% of the interactions from HuRI and 73.62% and 32.94% from the filtered versions of STRING and HIPPIE could potentially be modeled with high structural coverage or accuracy, respectively. Overall, this paper provides an overview of the current state of structural coverage of the human proteome and interactome.
Keywords: human proteome, human interactome, protein complexes, structural coverage, PDB, homology modeling databases, AlphaFold2
INTRODUCTION
Protein interactions (PPIs) are key players in many cellular processes [1, 2]. Constructing a complete and accurate interactome (i.e. the network of protein–protein interactions) is crucial to understand better the fundamental working principles of cells, functions and disease mechanisms and eventually to identify key proteins or pathways for developing new treatment strategies. Several experimental and computational studies aimed to determine interactomes and were released either as resources or software [3–19]. These resources are curated and integrated to eliminate experimental artifacts and false-positive interactions, yielding up to millions of PPIs. Still, the number of studies incorporating the ever-increasing three-dimensional (3D) protein structures to the interactome has been limited, posing an ongoing challenge. Structurally characterized interactomes are essential to find detailed proteome-level functional annotations [20]. Utilizing these interactomes may assist in elucidating interactions that happen simultaneously and that are mutually exclusive [21]. The presence of mutations in protein–protein interfaces [22] and their impact as gain- or loss-of-function can also be revealed with structural information [23]. Drug design and repurposing similarly require structural characterization [24]. Overall, structural annotation of the PPI networks is necessary for molecular-level comprehension of the human interactome.
Understanding the atomic-level interactions between two proteins and the specific amino acids involved is essential for comprehending PPIs at the molecular level. In our previous studies, we developed the PRISM algorithm that uses known protein interfaces to accurately predict structural complexes of protein interactions [25, 26] and a prediction model for hotspots at protein interfaces, which are potential drug targets [27–29]. Other computational methods that are present, but not limited to, are Interactome3D [4] and Interactome INSIDER [3]. Predictions from these studies were further used to elaborate on the impact of mutations [30], structural modeling of signaling pathways [31–35] and analysis of post-translational modifications. The predictive performance of these computational methods highly depends on the completeness of the proteome and the availability of protein structures.
Human proteome is now 93.2% complete [36], and structural data are dramatically boosted with the accumulated experimental data (Protein Data Bank (PDB) [37]), homology modeling (ModBase [38], SWISS-MODEL [39]) and models from deep-learning methods such as AlphaFold (AF) [40]. AF is also adapted for modeling protein complex structures (i.e. AF2Complex) [41]. Other methods, such as AF2 followed by FoldDock, report promising results. However, these methods may have varying prediction performances on different organisms and be affected by the size of the protein complexes and post-translational modifications [42, 43]. Despite these advances, AF’s contribution to modeling the human structural interactome is still relatively limited, estimated to be less than 5% [44]. Intrinsically disordered proteins (IDPs) or regions (IDRs) are conformationally heterogeneous and do not have a fold under physiological conditions [45]. Their contribution to several biological processes and pathologies [46] makes them important targets that require rigorous structural elaboration. Approximately 22% of the human proteome is likely disordered [47]. These regions often pose challenges in structural modeling, making it difficult to obtain high-quality models [48, 49]. A significant portion of AF predicted very low– and low-confidence regions are found to have overlap with predicted IDRs [50].
In this study, we assess the structural coverage of human proteome and interactome by considering available known and predicted protein structures. We first estimated the experimental structural coverage of the reference human proteome. Next, we showed the improvement of structural proteome coverage when complementary methods like homology modeling (SWISS-MODEL, ModBase) and deep learning (AF; v2.0) were utilized (Figure 1A). We further assessed the structural coverage of the reconstructed interactomes (obtained by combining STRING, HuRI and HIPPIE and filtered to produce a comprehensive list of protein–protein interactions). Proteome analysis followed by interactome analysis allowed us to identify the portion of the human interactome that can be predicted using structure-based techniques. This assessment involves determining the proportion of the interactome for which there are complete structural models for both interactors (Figure 1B). This work assesses all existing 3D structural data mapped to human interactome with stringent filtering. These statistics reflect how close we are to reconstructing the complete structural interactome through experimental and computational methods.
Figure 1.
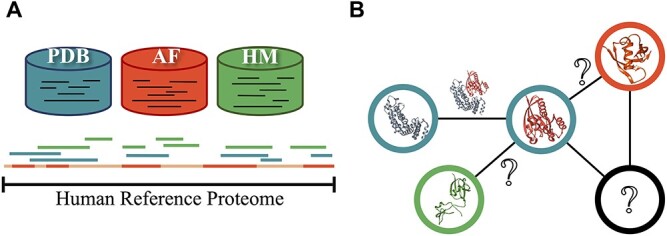
Concept figure. Each database is represented with the same coloring code in both sections. (A) for reference human proteome. PDB: Protein Data Bank, HM: homology modeling, AF: AlphaFold. Human reference proteome is shown by a long continuous line. Homology models and PDB structures might have overlapping regions that are represented by discrete lines. While AF provides a model for all of the proteome, we focus on regions that are modeled with high accuracy. (B) Sample network representation of the reference interactome. Protein structures are represented by nodes and interactions by edges. Question marks within the nodes show monomers that do not have any known 3D structure in any of the databases. Question marks on the edges show unknown 3D structures of the interactions (complex) between structurally known monomers.
RESULTS
Predictive methods improve structural coverage of human proteome
The human reference proteome has 18 401 proteins, of which 7085 are fully or partially covered in PDB (see Methods for coverage calculations). Our results are based on PDB structures containing at least 30 consecutive residues in the corresponding proteins with missing coordinates discarded for each PDB file, and sequence identity with a PDB chain is 100%. At residue resolution, the human reference proteome has 10 789 741 residues, of which 2 125 738 residues have coordinates in PDB that correspond to 19.70% of the proteome (Table 1) consistent with previous studies [47]. Sequence coverage categories at different coverage intervals and corresponding percentages of available structures in human reference proteome are given in Table 2. Calculations showed that 1663 proteins, which correspond to ~9.93% of the reference proteome, are almost fully (at least 90%) structurally covered by PDB. This result indicates that only a small subset of the experimental structures is available for applications that require detailed information, such as molecular simulations, structure-based drug design and structural interactome construction. Additional computational methods, including homology-based and AI-based structural modeling, are required to expand the set of structurally known proteins.
Table 1.
Residue-based coverage percentages of whole reference proteome by structure databases
| PDB | SWISS-MODEL (NA in PDB) | SWISS-MODEL | ModBase (NA in PDB) | ModBase | AFa | |
|---|---|---|---|---|---|---|
| % coverageb | 19.70 | 10.09 | 24.99 | 11.95 | 22.18 | 58.26 |
NA in PDB: not available in PDB.
aFor AF, structural data are considered available when a residue is predicted with ≥70% pLDDT score.
bThe number of amino acids where structural data are available is divided by the total number of residues in the reference proteome. Overlapping regions are counted only once, and missing residues are not included in the calculation.
Table 2.
Protein-based coverage/accuracy percentages of reviewed proteins in reference proteome by structure databases
| Protein residue coveragea (%) | PDB (%) | SWISS-MODEL (%) | ModBase (%) | AFb (%) |
|---|---|---|---|---|
| ≥90 | 9.93 | 13.59 | 17.12 | 17.04 |
| ≥70 | 19.25 | 25.99 | 26.07 | 52.52 |
| ≥50 | 24.85 | 32.53 | 31.10 | 74.66 |
| <50 | 13.66 | 10.33 | 15.73 | 23.61 |
aFor each protein, available structural data are combined. Coverage percentage is calculated by dividing the number of proteins that are modeled above an arbitrary threshold to the total number of proteins in the reference proteome, which is 18 401. Overlapping regions are counted only once, and missing residues are not included in the calculation.
bFor AF, accuracy was calculated instead of coverage.
For the proteins in the human reference proteome, we obtained high-quality models with 30 or more residues from the SWISS-MODEL and ModBase databases, resulting in 7886 proteins via SWISS-MODEL and 8618 proteins via ModBase. A total of 11 140 unique proteins were modeled, with 5364 proteins having predicted models in both SWISS-MODEL and ModBase. SWISS-MODEL and ModBase provided 24.99% and 22.18% residue-based coverage of the human proteome, respectively, both higher than the reported 19.70% coverage of PDB. The residue-based proteome coverage of the combination of these databases corresponds to 33.02% of the human reference proteome. These results show that homology modeling can cover approximately one-third of the human proteome without any contribution from PDB. To assess the contribution of homology models to reference proteome coverage, we excluded residues already covered by existing PDB structures and only used residues covered by homology modeling databases. We found 4904 proteins for SWISS-MODEL and 7300 proteins for ModBase. A total of 9096 unique proteins were modeled, with 3108 proteins having predicted models in both SWISS-MODEL and ModBase. Residue-based coverage of the proteome unavailable in PDB is 10.09% by SWISS-MODEL and 11.95% by ModBase. Residue-based coverage of their combination shows that homology models contribute to PDB structures by increasing human reference proteome coverage by 16.47%.
Next, we utilized the AF database produced by an AI-based method (AF v2.0). The accuracy of the predicted models is provided for each residue with a pLDDT score representing the per-residue estimate of its confidence. Residue positions with pLDDT ≥70% were considered high quality [40]. Residue-based proteome coverage by AF showed that 58.26% of residues have pLDDT ≥70%. For structural coverage, we labeled a protein as ‘accurately predicted’ if 85% of its residues were predicted with ≥70% pLDDT score. This constraint resulted in the loss of ~75% of the predicted structures in reference human proteome. Only 4930 (26.79%) of the AF predictions satisfy this condition. Among these, 2425 proteins already have at least a PDB structure, and the remaining 2505 predictions do not have any known structures deposited in any database before. As previously stated, 7085 proteins are fully or partially covered in PDB. All PDB data and accurately predicted AF models combined represent 9590 proteins (52.11%) for the human proteome.
We further assessed if AF’s prediction accuracy varies depending on the IDRs and the predicted protein class. Out of 1066 proteins that have IDRs according to the DisProt database [51], we found that AF predicts 797 proteins with low accuracy (LA; disorder percentage: 16.83%) and 256 proteins with high accuracy (HA; disorder percentage: 6.95%). Then, we selected three structural protein classes: mainly alpha, mainly beta and mixed alpha-beta proteins. We found that mainly alpha proteins have the lowest PDB coverage, considering the total protein count within the classes (Table 3). Next, we selected five functional protein classes: enzymes, immunoglobulins, membrane proteins, transcription factors (TFs) and transporter proteins. We discovered that TFs have the lowest count in terms of having a PDB structure and the lowest coverage. Likewise, AF predicts almost all the proteins, 1451 out of 1459, within the TF class, yet only 19 passed our HA thresholds. It is observed that 649 of the proteins from the TF class are zinc finger proteins. We searched for 20 consecutive residues with pLDDT<50 for TFs in order to mimic a disordered region scenario for an AF prediction. Of the 1279 proteins that meet these criteria that we have identified, 628 are zinc finger proteins. Similarly, AF predicts all 74 proteins belonging to immunoglobulins. However, it only predicts 17 of them with HA, showing that the prediction accuracy is positively correlated with structures deposited in the PDB and significantly varies between the classes (Table 4). In summary, despite the high number of predicted proteins by AF, the HA predictions are one-quarter of the total protein counts within the class for almost all protein classes.
Table 3.
Coverage of structural protein classes according to PDB and AF
| Structural class | Total protein count | Proteins [have PDB] (average PDB coverage, %) | Protein count [all predictions w/ AF] (average pLDDT score) | Protein count [high acc. predictions w/ AF] (average pLDDT score) |
|---|---|---|---|---|
| Mainly alpha | 6406 | 2830 (58.5) | 6294 (76.5) | 1735 (89.3) |
| Mainly beta | 4711 | 2395 (56.1) | 4577 (77.9) | 1220 (89.7) |
| Mixed alpha–beta | 6696 | 3268 (65.3) | 6585 (78.7) | 2367 (91.0) |
Proteins in reference proteome are classified according to the data provided by CATH-Gene3D. Proteins [have PDB] (average PDB coverage, %): The number of UniProt entries that belong to each class and have at least a PDB structure and average PDB coverage of the class calculated by taking the averages of UniProt entry’s corresponding PDB coverages (see Methods for coverage calculations). Protein count [all predictions w/ AF] (average pLDDT score, 0–100): The number of UniProt entries that belong to each class and have a predicted 3D structure produced by AF. In an AF prediction file, pLDDT scores of each residue were averaged and referred to as prediction accuracy of this corresponding UniProt entry. Protein count [high acc. predictions w/ AF] (average pLDDT score, 0–100): the number of UniProt entries that belong to each class and have a predicted 3D structure that 85% of the residues predicted with ≥70% pLDDT score. Only the UniProt entries within a class predicted with HA are considered and calculated by taking the averages of the UniProt entry’s corresponding AF prediction accuracies.
Table 4.
Coverage of functional protein classes according to PDB and AF
| Functional class | Total protein count | Proteins [have PDB] (average PDB coverage, %) | Protein count [all predictions w/ AF] (average pLDDT score) | Protein count [high acc. predictions w/ AF] (average pLDDT score) |
|---|---|---|---|---|
| Enzymes | 3618 | 2114 (72.2) | 3574 (83.5) | 1831 (91.6) |
| Immunoglobulins | 74 | 25 (85.6) | 74 (89.6) | 17 (90.5) |
| Membrane proteins | 5013 | 1500 (60.5) | 4939 (78.1) | 1391(88.8) |
| Transcription factors | 1459 | 372 (30.7) | 1451 (63.2) | 19 (86.9) |
| Transporter proteins | 1843 | 721 (67.8) | 1823 (78.6) | 517 (88.3) |
| Others | 8416 | 7085 (62.4) | 8304 (74.3) | 1929 (89.9) |
Proteins in reference proteome are classified according to the data provided by the HPA. Proteins [have PDB] (average PDB coverage, %): the number of UniProt entries that belong to each class and have at least a PDB structure and average PDB coverage of the class calculated by taking the averages of UniProt entry’s corresponding PDB coverages (see Methods for coverage calculations). Protein count [all predictions w/ AF] (average pLDDT score, 0–100): the number of UniProt entries that belong to each class and have a predicted 3D structure produced by AF. In an AF prediction file, pLDDT scores of each residue were averaged and referred to as prediction accuracy of this corresponding UniProt entry. Protein count [high acc. predictions w/ AF] (average pLDDT score, 0–100): the number of UniProt entries that belong to each class and have a predicted 3D structure that 85% of the residues predicted with ≥70% pLDDT score. Only the UniProt entries within a class predicted with HA are considered and calculated by taking the averages of the UniProt entry’s corresponding AF prediction accuracies.
Our results prove that combining PDB structures with homology and AF models increases the structural coverage (Table 2). In Figure 2A, we show the number of proteins in the human reference proteome shared by the structure databases when all accurate structures are considered. ModBase contributes the most by providing structures exclusively for 1599 proteins. Also, in Figure 2B, we show how the partial and complete coverage changes. PDB covers 1828 proteins (9.93%) when only highly covered (90%) structures are considered. The addition of high-coverage (≥90%) homology models and accurately predicted (85% of its residues were predicted with ≥70% pLDDT score) AF models raise this percentage to 33.16%.
Figure 2.

Structural coverage of proteins in the human reference proteome by databases. (A) The number of proteins modeled by PDB, SWISS-MODEL, ModBase, AF and their intersections are visualized. AF models with 85% of their residues predicted with ≥70% pLDDT score are used. (B) The number of proteins with no structure, partial structure, and complete structure. AF models with 85% (AF-85), 70% (AF-70) and 50% (AF-50) of their residues predicted with ≥70% pLDDT score are used for this demonstration. Partial structure denotes structure coverage <80% for PDB and homology models. For AF, it denotes an average accuracy of <80%. Similarly, complete structure means ≥80% coverage or average accuracy. Although it’s not visible, AF-85 has 13 models with partial structures.
A reference human interactome can be constructed by integrating multiple resources
Estimating the exact size of the human interactome still remains challenging [15]. Available interactions in databases, obtained via multiple techniques, are the best resource for reconstructing a complete human interactome. Here, we analyzed eight major human interaction databases, HuRI, STRING, BioPlex, BioGRID, HIPPIE, IID, APID and PICKLE, to evaluate the current status of the interactome coverage with available structural data and decided on which database to use toward the construction of a comprehensive structural human reference interactome. We investigated the number of proteins and interactions available after mapping the interactions to the human reference proteome and removing redundant interactions with the same UniProt identifier. Then, we quantified the number of proteins and interactions in each database with a structure and/or a model available in our structural data sources. These statistics are summarized in Table 5. HuRI is one of the most comprehensive experimental data providing direct physical interactions for 48 763 PPIs. The filtered STRING database, hereinafter referred to as STRINGF, resulted in 57 192 physical interactions with high confidence scores (>0.7), considering only experimental and database channels. In addition, there are 53 136 interactions in the BioPlex database. However, these are not restricted to binary physical interactions because the affinity purification–mass spectrometry (AP-MS) technique finds all physical/non-physical interactions in a complex [52]. Lastly, filtering the HIPPIE dataset, hereafter referred to as HIPPIEF, to binary high-confidence interactions resulted in 22 280 PPIs.
Table 5.
Statistics of the number of proteins and protein–protein interactions in interactome databases that are mapped to human reference proteome
| Database | Total number of | Number of proteins with available structures | Number of interactions with structures available for both interacting proteins | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Interactions | Proteins | PDB | SWISS-MODEL | ModBase | AFa | PDB | SWISS-MODEL | ModBase | AFa | |
| HuRI | 48 763 | 7889 | 3317 | 3414 | 3729 | 2026 | 7046 | 7014 | 8938 | 2737 |
| BioPlex v3.0 | 53 136 | 8806 | 3888 | 4242 | 4615 | 2819 | 14 036 | 13 945 | 15 162 | 5802 |
| STRING v11.5 | 57 192 | 7327 | 4518 | 4214 | 4095 | 2085 | 38 005 | 24 829 | 21 442 | 9941 |
| HIPPIE v2.3 | 22 280 | 7640 | 4164 | 3971 | 4231 | 1910 | 10 193 | 7795 | 8412 | 1586 |
| APID v2021_03 | 125 722 | 14 854 | 6508 | 6774 | 7236 | 3897 | 39 922 | 32 453 | 36 390 | 8394 |
| PICKLE v3.3 | 211 943 | 15 922 | 6852 | 4349 | 7719 | 4191 | 88 408 | 68 328 | 71 761 | 14 794 |
| BioGRID v4.4.216 | 719 566 | 17 100 | 6914 | 7499 | 8174 | 4587 | 292 081 | 233 361 | 239 226 | 70 455 |
| IID v2021_05 | 542 157 | 17 331 | 7015 | 7636 | 8250 | 4600 | 235 632 | 181 337 | 185 030 | 53 251 |
Additional filtering was applied to STRING and HIPPIE datasets.
aAF, 85% of the residues predicted with ≥70% pLDDT score.
Assessment of human interactomes from HuRI, HIPPIEF and STRINGF: mapping experimental and predicted 3D structures
Figure 3 shows the total number of interactions and the interactions with structures/models listed in Table 5. We chose two databases, HuRI and BioPlex, which output the results of major experimental studies, as well as the well-known databases STRING, which enables filtering the physical interactions by a confidence score, and the HIPPIE, which can be filtered with respect to experiment type and confidence score. We demonstrate that there is little overlap in terms of interactions among these four major databases (Figure 3A). Table 5 shows that the HuRI, HIPPIEF and STRINGF interaction networks contain 7889, 7640 and 7327 proteins, respectively. While most of the proteins are present in all three databases, a considerable number of proteins are exclusively present in one (Figure 4). We also found the number of interactions where both partners had PDB structures. The results show that STRINGF has the highest PDB coverage among interactome databases, while HuRI and HIPPIEF have poor PDB coverage (Figure 3B). The striking disparity might be due to ribosomal and mitochondrial proteins being significantly less represented in HuRI (protein count: 125; interaction count: 1348) and HIPPIEF (protein count: 186; interaction count: 2175) compared to STRINGF (protein count: 1801; interaction count: 16 453), taking part in so many interactions. Given that the Y2H technique detects the bulk of HIPPIEF interactions and all HuRI data, the missing proteins may be due to Y2H's technical limitations.
Figure 3.

Overview of PPIs found in HuRI, BioPlex, HIPPIEF, and STRINGF databases. (A) Interactions are filtered according to the reference proteome. (B) Interactions with a PDB structure for both interacting proteins filtered to the reference proteome. (C) Interactions with a high-quality homology model for both interacting proteins filtered to the reference proteome. (D) Interactions with an AF model that have 85% of their residues predicted with ≥70% pLDDT score for both interacting proteins filtered to the reference proteome. HM: homology modeling, AF: AlphaFold.
Figure 4.
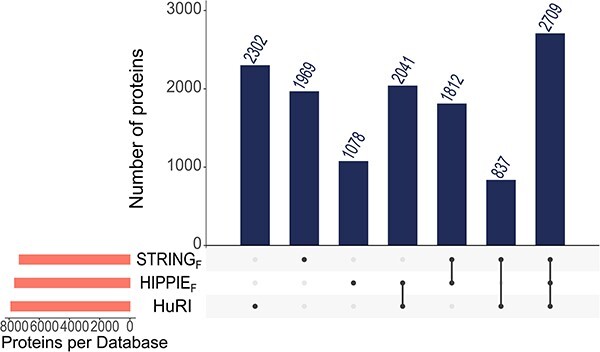
Total number of proteins found in STRINGF, HIPPIEF and HuRI databases.
Figure 3C and D show the number of interactions with homology and AF models for both interacting proteins, respectively. Homology models have better interaction coverage compared to AF. STRINGF has the most coverage in both scenarios, while HIPPIEF has the least. In general, the overlap between databases is minimal, reinforcing the notion that integrating these databases is critical since relying on a single database may result in incomplete interactomes.
Since many human proteins are multi-domain, and interactions may occur between single-domain and multi-domain proteins, we analyzed the differences across various interactome databases. The domain count distribution of interacting proteins, as depicted in Table 6, reveals an interesting trend. Most interactions occur between single-domain proteins, as evidenced by the significantly high number of interactions in the HuRI interactome, where 19 210 out of 41 705 interactions fall into this category. This observation suggests that single-domain–single-domain protein pairs predominantly mediating interactions in the HuRI dataset may be attributed to potential limitations in correctly expressing lengthy multi-domain proteins in yeast [53], which could result in an underrepresentation of interactions involving these proteins in the dataset.
Table 6.
Classification and observance percentages of domain types of the interacting protein pairs found in reference interactomes
| Single–single | Multi–multi | Single–multi | ||
|---|---|---|---|---|
| Reference interactomes |
HuRI (41 705) |
19 210 | 5403 | 17 092 |
| STRING (54 631) |
19 754 | 12 541 | 22 336 | |
| HIPPIE (20 601) |
5445 | 6248 | 8908 |
Additional filtering was applied to STRING and HIPPIE datasets.
These results have led us to select the HuRI, STRINGF and HIPPIEF databases as our reference interactome sources. When these three interactomes are combined, 12 748 non-redundant proteins participate in 117 897 interactions. Mapping these interactions to experimental 596 919 binary protein–protein complexes (https://github.com/ku-cosbi/interactome-structural-coverage/blob/main/data/PDB_interface_data.tsv) from PDB (as of December 2022) showed that very few experimentally resolved protein interactions are available and they account for only 3.95% of all non-redundant binary interactions. Next, we concentrated on the interactions and displayed the structural coverage of protein–protein interactions in interactome databases (Figure 5). As can be seen from the percentages, the STRINGF database outnumbers the rest (exp-exp, exp-model and model-model in the inset plot). Experimental and modeled structures of both interactors in HuRI are available for only ~40% of the whole interactome. In comparison, this figure rises to ~85% in STRINGF. Here, we did not distinguish between high- or low-coverage/accuracy structures.
Figure 5.
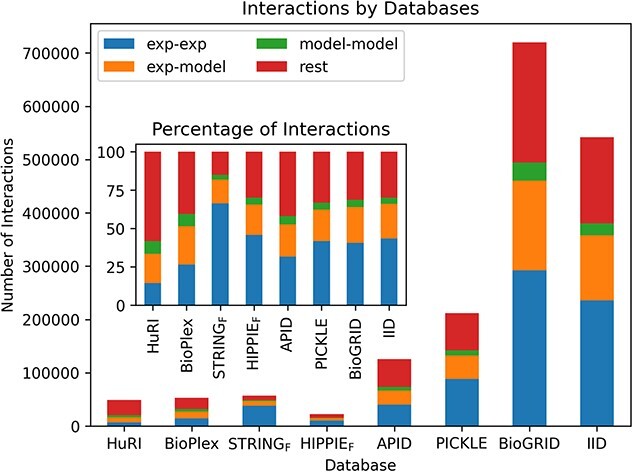
Structural coverage of protein–protein interactions in interactome databases. Exp-exp indicates interactions where both interacting partners have experimental structures from PDB . Exp-model represents interactions where only one interacting partner has an experimental structure from PDB, and the other partner has a model from ModBase, SWISS-MODEL or AF. Model-model indicates that both interacting partners do not have experimental structures but have a model. Lastly, rest is for the remaining interactions that have no structural data available. The inset plot shows the same concept in terms of percentages. AF models that have 85% of their residues predicted with ≥70% pLDDT score are considered.
Accurate 3D modeling of the interactions in HuRI, STRINGF and HIPPIEF interactomes requires structurally complete monomer protein structures. In other words, given that two proteins interact, structural knowledge for this protein–protein interaction can be obtained on the availability of the structures of both proteins. To assess the completeness of each interacting pair, proteins in HuRI, STRINGF and HIPPIEF interactomes were labeled as high coverage (HC) or low coverage (LC) along with the name of the data sources: PDB, SWISS-MODEL or ModBase. On the other hand, AF usually predicts the whole protein structure, yet the prediction quality of each structure, even each residue in a structure, is different. Therefore, rather than using HC and LC metrics, we preferred HA and LA metrics for the AF structures. Figure 6 shows a snapshot of the available structures in different databases. As stated in previous sections, we denoted 50% or higher coverage as HC; the rest as LC; 85% of the structure covered with 70% or higher accuracy for AF, is designated as HA, with the remaining predictions falling into the LA group. Numerous combinations of databases and coverages are available for an interaction because the 3D structures of each protein can be found across multiple databases with various coverages or accuracies. This is why the sum of the numbers in Figure 6 is greater than the total number of interactions in the reference interactome. AF's LA 3D structures dominate the HuRI, STRINGF, and HIPPIEF interactomes. PDB and homology modeling databases each add roughly equal numbers of structures to model a small number of interactions in HuRI and HIPPIEF. On the other hand, the PDB and homology modeling databases contain many 3D structures with HC that can be used to model the interactions in STRING. Only 12.97% of the HuRI and 32.94% of the HIPPIEF interactomes contained protein partners with HA or HC structures, while this percentage increased to 73.62% for the STRINGF interactome. Out of 117 897 interactions in total, 47 431 (40.23%) interactions have both protein partners with HA or HC structures.
Figure 6.

3D Structure modeling assessment of ‘Protein 1-Protein 2’ pairs in reference interactomes in (A) HuRI, (B) STRINGF and (C) HIPPIEF. x–y axes labels show the 3D coverage or accuracy label of the databases. LC: low coverage, HC: high coverage, HA: high accuracy, LA: low accuracy, MB: ModBase, SM: SWISS-MODEL, AF: AlphaFold. The number in each cell indicates how many ‘Protein 1–Protein 2’ pairs of the reference interactome can be potentially constructed by using mentioned sources with given labeled coverage or accuracy.
Some of the proteins are highly studied; these usually correspond to disease-related proteins. Investigation of important proteins such as disease-related, COSMIC CGC [54], and drug targets in interactome databases shows that except for HuRI, interactomes contain ≥50% of COSMIC CGC and disease-related genes (Figure 7). Even though the number of interactions in the HIPPIEF dataset is less than HuRI, it is enriched in important genes. The coverage of drug targets, however, is often less extensive across most interactomes. When our selected reference interactomes are combined, drug target coverage increases by up to 69% while others surpass 75%.
Figure 7.
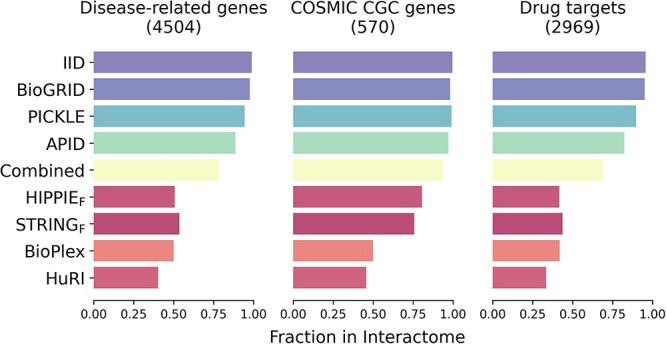
Investigation of important genes in interactome databases. Fraction of disease-related, COSMIC CGC and drug-targeting genes are investigated for all interactome databases. The term ‘combined’ represents merged interactomes of HIPPIEF, STRINGF and HuRI.
DISCUSSION
We comprehensively analyzed the current structural knowledge of the human proteome and interactomes (HuRI, STRINGF and HIPPIEF) by integrating experimental protein structures from PDB and predicted structures from ModBase, SWISS-MODEL, and AF. Our results showed that combining several resources improved the structural coverage of the human proteome and interactomes. Predicted protein structures bring an orthogonal layer of information toward having a more comprehensive structural proteome with varying degrees of accuracy. The availability of an experimental model significantly impacts the computational accuracy of the structure prediction. Moreover, we implicitly touched on the IDPs/IDRs in our analysis where we diligently separated out low-quality models. This quality control process has helped ensure the reliability of the structural data we have used for our analyses. Besides having a low number of available experimental structures from PDB, the functional class coverage for TFs was also impacted by the presence of IDRs [55] (Table 4). It is also mentioned that TFs with zinc finger domains are not accurately predicted by AF. Given that experimental structure determination is more difficult for the immunoglobulin class of proteins due to their long loop regions [56], our data demonstrate that the prediction ability is indeed restricted for these proteins. Proteome-level analysis shows we are still halfway to a structurally complete human proteome.
Integrating the structural data mapped to reference interactome databases at the interactome level reveals that STRINGF has the highest PDB coverage while HuRI has the lowest. This suggests that STRING may be a valuable resource for researchers seeking information about interactions involving experimental structures. While the number of proteins with PDB structures is similar between interactome databases, the number of interactions differs significantly, which might be attributed to the limited sensitivity of Y2H. Therefore, additional computational prediction methods, such as template-based or template-free docking and deep-learning-based methods, complement experimental methods as they capture interactions that Y2H or other experimental techniques may miss.
We also highlight that homology models show better interaction coverage than AF’s accurately predicted models. This implies that homology models may continue to be a useful tool for predicting PPIs when experimental data are lacking and may complement the capabilities of AF. Furthermore, we emphasize that there is minimal overlap between interactome databases. Consequently, depending solely on a single database may result in incomplete or biased results, as each database may have its strengths and limitations regarding coverage. Thus, integrating data from multiple databases can provide a more comprehensive and reliable picture of PPIs.
It has also been shown that protein pairs with HA or HC structures constitute a small portion of interactions in HuRI, a larger portion in HIPPIEF and the majority of interactions in STRINGF. HuRI is reported as a less-biased interactome containing genes that belonged to uncharted regions previously [15]. Such a characteristic might be the reason behind its lower coverage of interacting proteins than other interactomes. Considering this, bias toward well-studied proteins may impact the structural coverage of the interactome because less-studied proteins may have fewer interactions reported in the interactome. Despite the increased coverage provided by computational methods, the number of HA/HC interactions remains low compared to all existing interactions. We should note that there are valuable thermodynamic and kinetic data accessible for the stability of human proteins and binding affinity of protein–protein interactions in databases such as ProThermDB, SKEMPI and PROXiMATE [57–59]; however, these data are not exhaustive to cover the entire proteome or interactome. This altogether highlights the need for continued efforts in both experimental and computational methods to improve our understanding of protein–protein interactions and the structural coverage of the human interactome.
METHODS
Reference proteome analysis
A recent version of the UP000005640 was obtained from the UniProtKB proteome database (UniProt release 2022_04). This text-based data contain information for 20 360 reviewed human proteins. First, protein names, including keywords such as ‘putative’ or ‘uncharacterized’, are eliminated from the initial proteome dataset. Second, only the proteins with proven existence belonging to experimental evidence at the protein level and experimental evidence at transcript level classes is kept in the dataset. Last, proteins with less than 30 amino acids are removed from the dataset. After the three-step filtering, the final reference proteome dataset contained 18 401 proteins. Domain information was incorporated into this dataset from the PFAM database cross-referenced in UniProt. We considered a protein ‘multi-domain’ if multiple PFAM IDs are assigned to its UniProt ID.
PDB coverage
We retrieved all PDB structures cross-referenced in UniProt to obtain the structural proteome and its coverage (as of 1 June 2022). We parsed all mm/CIF files by using Biopython’s Bio.PDB package [60] to find the protein chains that match UniProt identifiers in the reference proteome. In total, we parsed 128 696 protein chains for 7376 unique human proteins. Calculations run on Koc University High-Performance Computing (KUACC HPC) cluster. In the end, we have an exact begin–end residue range for each PDB chain corresponding to the original UniProt sequence and missing residues that fall into that region. The corresponding PDB file is not considered for further analysis if the resulting region does not contain at least 30 consecutive amino acids. To eliminate redundancies, we removed duplications representing identical residues and excluded missing coordinates only once if there were any repeats in another coordinate file. For all PDB coverage calculations, Equation 1 is used.
 |
(1) |
AF coverage
The DeepMind team released structures predicted by AF v2.0 [61] in October 2022. Predictions for UP000005640 proteome downloaded from the AF Protein Structure Database (https://ftp.ebi.ac.uk/pub/databases/alphafold). There were 23 391 predictions deposited for the reference proteome. After eliminating the predictions previously labeled as unreviewed by UniProt, we ended up with 20 315 reviewed/canonical protein predictions. AF predicts the proteins as fractions whose amino acid count exceeds 2700. Those predictions for 207 unique protein entries are also eliminated. Confidence in the AF predictions is measured by pLDDT, a per-residue accuracy estimate on a scale from 0 to 100. A pLDDT score ≥70% is declared as an indicator of a good backbone prediction [40]. Therefore, in our analysis, we applied a structural constraint. We only assume ‘accurately predicted’ if a protein’s 85% of the residues predicted with ≥70% pLDDT score.
Homology modeling coverage
We have utilized two widely known homology modeling databases: SWISS-MODEL and ModBase. We downloaded homology models for reference proteome based on the UniProtKB release 2022_04 from the SWISS-MODEL Repository. Then, we filtered the models based on their QMeanDisCo Global score, a composite scoring function that estimates model quality [62]. We selected models with a QMeanDisCo Global score greater than 0.7 as they are considered confident models by the providers. For ModBase, we downloaded the file named modbase_models_all-latest.xz with a last modified date of 28 February 2014 from the downloads section. We eliminated models that are not present in the filtered human reference proteome and have less than 30 amino acids for both databases. We selected models with a ModPipe Quality Score (MPQS) ≥1.1 if available, or if MPQS is not available, sequence identity ≥30% and a model score (GA341) ≥0.7 were selected as good-quality models. MPQS is a composite quality score that includes e-value, z-Dope, GA341, coverage and sequence identity to the template [63]. Also, we selected the model with a higher-quality score for both databases if there were models with precisely the same amino acid start and end positions. We have reported residue-based and protein-based coverage results for these models. For residue-based coverage calculations, a simplified version of Equation (1) is used where the missing residues are not considered because of the nature of homology models. To understand the contribution of homology modeling to experimentally determined protein structures, we further eliminated the models that cover the identical residues with available PDB structures. We have utilized the former case where we do not eliminate models concerning PDB data for the analysis of interactome coverage.
Coverage of IDRs, structural and functional protein classes
In this study, we used IDR data from DisProt [51], structural protein classes from CATH-Gene3D [64, 65] and functional classification data of the proteins from the Human Protein Atlas (HPA) [66]. We examined the accuracy of IDRs predicted with AF and computed the disorder percentage of predicted proteins by subtracting the end and start positions of the residues and dividing by the total length of the protein. Three main structural protein classes: mainly alpha, mainly beta and mixed alpha–beta together with five main functional protein classes: membrane proteins, TFs, immunoglobulins and transporter proteins, are investigated in terms of their structural coverage either experimentally (PDB) or computationally (AF). From these datasets, we only include the proteins that are part of our reference proteome. As AF produces whole-protein predictions, its coverage is evaluated in terms of prediction accuracy rather than the count of the protein residues predicted. In this manner, average counts of the proteins in any class are calculated by dividing the number of proteins with a 3D structure by the protein count within the class.
Analysis of interactome databases
We reviewed interactome databases currently available online, including HuRI [15], STRING [9], BioPlex [5], BioGRID [14], HIPPIE [10], Interactome INSIDER [3], Interactome3D [4] hu.MAP [16], IID [7], APID [6] and PICKLE [12]. An overview of these databases can be found in Supplementary Table 1 available online at http://bib.oxfordjournals.org/.. We downloaded the protein–protein interactions from HuRI, STRING, BioPlex, BioGRID, HIPPIE, IID, APID, and PICKLE databases to investigate the number of PPIs available (as of 5 December 2022). Three interactomes were investigated in detail. We downloaded the HuRI.tsv file of the 2020 publication from http://www.interactome-atlas.org/download. A STRING physical interaction dataset named ‘9606.protein.physical.links.detailed.v11.5.txt’ was downloaded from the Downloads tab at https://string-db.org, and rescoring has been performed using the Python script from https://stringdb-static.org/download/combine_subscores.py, which STRING creators provide. We selected models with a combined score of experimental and database channels greater than 0.7. For HIPPIE, we downloaded the current release (v2.3) from http://cbdm-01.zdv.uni-mainz.de/~mschaefer/hippie/download.php. We filtered this dataset and have only the binary PPI detection methods that are two-hybrid, atomic force microscopy and fluorescent resonance energy transfer [1]. Then, we set the quality threshold at 0.73 (high quality) and removed interactions smaller than this cutoff value. We converted any different protein identifiers to UniProt identifiers for standardization and removed redundant protein–protein interactions. We filtered the datasets for the proteins present in our reference proteome. Then, we found the number of unique proteins and unique interactions in each interactome database; the number of unique proteins with at least one available structure in PDB; and the number of unique proteins with at least one model in SWISS-MODEL, ModBase and AF databases. Furthermore, we found a number of unique interactions in which both interacting pairs had a structure and/or a model.
Gene Ontology: cellular component analysis and investigation of important genes in interactome databases
For cellular component analysis, we focused only on the proteins available in STRING to reveal the reason behind the difference in the interaction counts between interactome databases. We collected the PFAM IDs of these proteins and found UniProt entries in the reference proteome containing at least one PFAM domain. A common pattern in terms of frequent GO cellular component annotations [67], ribosome (GO:0005739) and/or mitochondrion (GO:0005840) is detected for this protein subset. Then, we checked the existence of such proteins with these annotations in STRINGF, HuRI and HIPPIEF. Later, we investigated the coverage of disease-related, COSMIC CGC and drug-targeting genes in interactome databases. For disease-related genes, we downloaded variant summary data from ClinVar [68] (last modified date: 21 January 2023) and filtered them to take genes in assembly GRCh38 and have ‘pathogenic’ clinical significance. For COSMIC CGC genes, we downloaded cancer gene census data from COSMIC (version 97) and restricted them to Tier 1 genes [54]. Lastly, for drug targets, we utilized the complete target data from IUPHAR/BPS Guide to Pharmacology (2022.4 version) [69].We filtered all datasets for the proteins present in our reference proteome. Then, we calculated the fraction of these important genes in interactome databases to find their coverage.
Key Points
We employed a large scale computational analysis of protein structures (known or predicted) in human proteome and interactomes to understand how close we are to potentially construct the complete structural interactome with experimental or computational methods.
We showed that 33.16% of the human proteome can be represented with high accuracy and high coverage protein structures obtained with experimental (PDB) or predicted (homology modeling and AlphaFold) structures.
In turn, although AlphaFold significantly enriches the structural human proteome (up to 52.11%), we are still halfway in obtaining the high accuracy complete structures.
The structural representation of interactomes is even more limited than the proteome. However, there is a huge opportunity for 3D modeling techniques to predict the rest where we showed that 40.23% of the interactome is predictable.
Supplementary Material
Kayra Kosoglu is a third-year PhD student in the Computational Sciences and Engineering Department at Koc University, Istanbul. She has been working on protein-protein interactions.
Zeynep Aydin is a third-year PhD student in the Computational Sciences and Engineering Department at Koc University, Istanbul. She has been working on protein-protein interactions.
Nurcan Tuncbag is an associate professor in the Chemical and Biological Engineering Department and School of Medicine and she is the principal investigator of the Network Modeling Research Group at Koc University, Istanbul. Her research focuses on computational systems biology and network modeling.
Attila Gursoy is a professor in the Computer Engineering Department and the principal investigator of the Computational and Systems Biology Group at Koc University, Istanbul. His research focuses on the computational studies of protein interactions and parallel computing.
Ozlem Keskin is a professor in the Chemical and Biological Engineering Department and the principal investigator of the Computational and Systems Biology Group at Koc University, Istanbul. Her research focuses on the computational studies of protein interactions and dynamics.
Contributor Information
Kayra Kosoglu, Computational Sciences and Engineering, College of Engineering, Koc University, 34450 Istanbul, Turkey.
Zeynep Aydin, Computational Sciences and Engineering, College of Engineering, Koc University, 34450 Istanbul, Turkey.
Nurcan Tuncbag, School of Medicine, Koc University, 34450 Istanbul, Turkey; Department of Chemical and Biological Engineering, College of Engineering, Koc University, 34450 Istanbul, Turkey.
Attila Gursoy, Department of Computer Engineering, College of Engineering, Koc University, 34450 Istanbul, Turkey.
Ozlem Keskin, Department of Chemical and Biological Engineering, College of Engineering, Koc University, 34450 Istanbul, Turkey.
FUNDING
This work has been partially supported by the Scientific and Technological Research Council of Turkey (TUBITAK), grant number 120C120.
DATA AND SOFTWARE AVAILABILITY
All source code and datasets are freely available at https://github.com/ku-cosbi/interactome-structural-coverage.
References
- 1. Shoemaker BA, Panchenko AR. Deciphering protein-protein interactions. Part I. Experimental techniques and databases. PLoS Comput Biol 2007;3(3):e42. 10.1371/journal.pcbi.0030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Garland W, Benezra R, Chaudhary J. Chapter fifteen—targeting protein–protein interactions to treat cancer—recent progress and future directions. In: Desai MC (ed). Annual Reports in Medicinal Chemistry, Vol. 48., Massachusetts: Academic Press, 2013, 227–45. [Google Scholar]
- 3. Meyer MJ, Beltrán JF, Liang S, et al. . Interactome INSIDER: a structural interactome browser for genomic studies. Nat Methods 2018;15(2):107–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mosca R, Céol A, Aloy P. Interactome3D: adding structural details to protein networks. Nat Methods 2013;10(1):47–53. [DOI] [PubMed] [Google Scholar]
- 5. Huttlin EL, Ting L, Bruckner RJ, et al. . The BioPlex network: a systematic exploration of the human interactome. Cell 2015;162(2):425–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alonso-López D, Campos-Laborie FJ, Gutiérrez MA, et al. . APID database: redefining protein-protein interaction experimental evidences and binary interactomes. Database (Oxford) 2019;2019:baz005. 10.1093/database/baz005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kotlyar M, Pastrello C, Ahmed Z, et al. . IID 2021: towards context-specific protein interaction analyses by increased coverage, enhanced annotation and enrichment analysis. Nucleic Acids Res 2022;50(D1):D640–d647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou Y, Chen H, Li S, Chen M. mPPI: a database extension to visualize structural interactome in a one-to-many manner. Database 2021;2021:baab036. 10.1093/database/baab036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Szklarczyk D, Gable AL, Lyon D, et al. . STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47(D1):D607–d613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alanis-Lobato G, Andrade-Navarro MA, Schaefer MH. HIPPIE v2.0: enhancing meaningfulness and reliability of protein-protein interaction networks. Nucleic Acids Res 2017;45(D1):D408–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xenarios I, Rice DW, Salwinski L, et al. . DIP: the database of interacting proteins. Nucleic Acids Res 2000;28(1):289–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dimitrakopoulos GN, Klapa MI, Moschonas NK. PICKLE 3.0: enriching the human meta-database with the mouse protein interactome extended via mouse-human orthology. Bioinformatics (Oxford, England) 2020;37:145–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chatr-aryamontri A, Ceol A, Palazzi LM, et al. . MINT: the molecular INTeraction database. Nucleic Acids Res 2007;35:D572–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stark C, Breitkreutz BJ, Reguly T, et al. . BioGRID: a general repository for interaction datasets. Nucleic Acids Res 2006;34:D535–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luck K, Kim DK, Lambourne L, et al. . A reference map of the human binary protein interactome. Nature 2020;580(7803):402–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Drew K, Wallingford JB, Marcotte EM. Hu.MAP 2.0: integration of over 15,000 proteomic experiments builds a global compendium of human multiprotein assemblies. Mol Syst Biol 2021;17(5):e10016. 10.15252/msb.202010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dapeng X, et al. . 3D structural human interactome reveals proteome-wide perturbations by disease mutations. bioRxiv [Preprint]. 2023;2023.
- 18. O'Reilly FJ, Graziadei A, Forbrig C, et al. . Protein complexes in cells by AI-assisted structural proteomics. Mol Syst Biol 2023;19(4):e11544. 10.15252/msb.202311544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang QC, Petrey D, Garzón JI, et al. . PrePPI: a structure-informed database of protein-protein interactions. Nucleic Acids Res 2013;41:D828–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aloy P, Russell RB. Structural systems biology: modelling protein interactions. Nat Rev Mol Cell Biol 2006;7(3):188–97. [DOI] [PubMed] [Google Scholar]
- 21. Tuncbag N, Kar G, Gursoy A, et al. . Towards inferring time dimensionality in protein–protein interaction networks by integrating structures: the p53 example. Mol Biosyst 2009;5(12):1770–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xiong D, Lee D, Li L, et al. . Implications of disease-related mutations at protein-protein interfaces. Curr Opin Struct Biol 2022;72:219–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fraser JS, Gross JD, Krogan NJ. From systems to structure: bridging networks and mechanism. Mol Cell 2013;49(2):222–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petrey D, Honig B. Structural bioinformatics of the interactome. Annu Rev Biophys 2014;43:193–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tuncbag N, Gursoy A, Nussinov R, Keskin O. Predicting protein-protein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using PRISM. Nat Protoc 2011;6(9):1341–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Keskin O, Tuncbag N, Gursoy A. Predicting protein-protein interactions from the molecular to the proteome level. Chem Rev 2016;116(8):4884–909. [DOI] [PubMed] [Google Scholar]
- 27. Cukuroglu E, Engin HB, Gursoy A, Keskin O. Hot spots in protein–protein interfaces: towards drug discovery. Prog Biophys Mol Biol 2014;116(2):165–73. [DOI] [PubMed] [Google Scholar]
- 28. Engin HB, Keskin O, Nussinov R, Gursoy A. A strategy based on protein–protein Interface motifs may help in identifying drug off-targets. J Chem Inf Model 2012;52(8):2273–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kar G, Kuzu G, Keskin O, Gursoy A. Protein-protein interfaces integrated into interaction networks: implications on drug design. Curr Pharm Des 2012;18(30):4697–705. [DOI] [PubMed] [Google Scholar]
- 30. Tuncbag N, Keskin O, Nussinov R, Gursoy A. Prediction of protein interactions by structural matching: prediction of PPI networks and the effects of mutations on PPIs that combines sequence and structural information. Methods Mol Biol 2017;1558:255–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guven-Maiorov E, Keskin O, Gursoy A, et al. . The architecture of the TIR domain signalosome in the toll-like receptor-4 signaling pathway. Sci Rep 2015;5:13128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Engin HB, Guney E, Keskin O, et al. . Integrating structure to protein-protein interaction networks that drive metastasis to brain and lung in breast cancer. PloS One 2013;8(11):e81035. 10.1371/journal.pone.0081035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Acuner-Ozbabacan ES, Engin BH, Guven-Maiorov E, et al. . The structural network of Interleukin-10 and its implications in inflammation and cancer. BMC Genomics 2014;15:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Acuner Ozbabacan SE, Gursoy A, Nussinov R, Keskin O. The structural pathway of interleukin 1 (IL-1) initiated signaling reveals mechanisms of oncogenic mutations and SNPs in inflammation and cancer. PLoS Comput Biol 2014;10(2):e1003470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guven-Maiorov E, Keskin O, Gursoy A, Nussinov R. A structural view of negative regulation of the toll-like receptor-mediated inflammatory pathway. Biophys J 2015;109(6):1214–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Omenn GS, Lane L, Overall CM, et al. . The 2022 report on the human proteome from the HUPO human proteome project. J Proteome Res 2023;22(4):1024–1042. 10.1021/acs.jproteome.2c00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berman H, Henrick K, Nakamura H. Announcing the worldwide protein data Bank. Nat Struct Biol 2003;10(12):980. [DOI] [PubMed] [Google Scholar]
- 38. Pieper U, Webb BM, Dong GQ, et al. . ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res 2014;42:D336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bienert S, Waterhouse A, de Beer TAP, et al. . The SWISS-MODEL repository-new features and functionality. Nucleic Acids Res 2017;45(D1):D313–d319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jumper J, Evans R, Pritzel A, et al. . Highly accurate protein structure prediction with AlphaFold. Nature 2021;596(7873):583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gao M, Nakajima An D, Parks JM, Skolnick J. AF2Complex predicts direct physical interactions in multimeric proteins with deep learning. Nat Commun 2022;13(1):1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bryant P, Pozzati G, Elofsson A. Improved prediction of protein-protein interactions using AlphaFold2. Nat Commun 2022;13(1):1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lamb J, Elofsson A. pyconsFold: a fast and easy tool for modeling and docking using distance predictions. Bioinformatics 2021;37(21):3959–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Burke DF, Bryant P, Barrio-Hernandez I, et al. . Towards a structurally resolved human protein interaction network. Nat Struct Mol Biol 2023;30(2):216–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tompa P, Fersht A. Structure and Function of Intrinsically Disordered Proteins, New York: Chapman and Hall/CRC, 2009.
- 46. Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol 2015;16(1):18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Porta-Pardo E, Ruiz-Serra V, Valentini S, Valencia A. The structural coverage of the human proteome before and after AlphaFold. PLoS Comput Biol 2022;18(1):e1009818. 10.1371/journal.pcbi.1009818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tunyasuvunakool K, Adler J, Wu Z, et al. . Highly accurate protein structure prediction for the human proteome. Nature 2021;596(7873):590–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Akdel M, Pires DEV, Pardo EP, et al. . A structural biology community assessment of AlphaFold2 applications. Nat Struct Mol Biol 2022;29(11):1056–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ruff KM, Pappu RV. AlphaFold and implications for intrinsically disordered proteins. J Mol Biol 2021;433(20):167208. [DOI] [PubMed] [Google Scholar]
- 51. Quaglia F, Mészáros B, Salladini E, et al. . DisProt in 2022: improved quality and accessibility of protein intrinsic disorder annotation. Nucleic Acids Res 2022;50(D1):D480–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim EDH, Sabharwal A, Vetta AR, Blanchette M. Predicting direct protein interactions from affinity purification mass spectrometry data. Algorithms for Molecular Biology 2010;5(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Galletta BJ, Rusan NM. A yeast two-hybrid approach for probing protein-protein interactions at the centrosome. Methods Cell Biol 2015;129:251–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tate JG, Bamford S, Jubb HC, et al. . COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res 2019;47(D1):D941–d947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Minezaki Y, Homma K, Kinjo AR, Nishikawa K. Human transcription factors contain a high fraction of intrinsically disordered regions essential for transcriptional regulation. J Mol Biol 2006;359(4):1137–49. [DOI] [PubMed] [Google Scholar]
- 56. Ruffolo JA, Chu LS, Mahajan SP, Gray JJ. Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies. Nat Commun 2023;14(1):2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nikam R, Kulandaisamy A, Harini K, et al. . ProThermDB: thermodynamic database for proteins and mutants revisited after 15 years. Nucleic Acids Res 2021;49(D1):D420–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jankauskaitė J, Jiménez-García B, Dapkūnas J, et al. . SKEMPI 2.0: an updated benchmark of changes in protein–protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics 2019;35(3):462–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jemimah S, Yugandhar K, Michael Gromiha M. PROXiMATE: a database of mutant protein–protein complex thermodynamics and kinetics. Bioinformatics 2017;33(17):2787–8. [DOI] [PubMed] [Google Scholar]
- 60. Cock PJA, Antao T, Chang JT, et al. . Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics 2009;25(11):1422–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Varadi M, Anyango S, Deshpande M, et al. . AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 2022;50(D1):D439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Studer G, Rempfer C, Waterhouse AM, et al. . QMEANDisCo—distance constraints applied on model quality estimation. Bioinformatics 2019;36(6):1765–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Eramian D, Eswar N, Shen MY, Sali A. How well can the accuracy of comparative protein structure models be predicted? Protein Sci 2008;17(11):1881–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sillitoe I, Bordin N, Dawson N, et al. . CATH: increased structural coverage of functional space. Nucleic Acids Res 2021;49(D1):D266–d273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lewis TE, Sillitoe I, Dawson N, et al. . Gene3D: extensive prediction of globular domains in proteins. Nucleic Acids Res 2018;46(D1):D1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Karlsson M, Zhang C, Méar L, et al. . A single-cell type transcriptomics map of human tissues. Sci Adv 2021;7(31). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Roncaglia P, Martone ME, Hill DP, et al. . The gene ontology (GO) cellular component ontology: integration with SAO (subcellular anatomy ontology) and other recent developments. J Biomed Semantics 2013;4(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Landrum MJ, Lee JM, Benson M, et al. . ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 2018;46(D1):D1062–d1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Harding SD, Armstrong JF, Faccenda E, et al. . The IUPHAR/BPS guide to PHARMACOLOGY in 2022: curating pharmacology for COVID-19, malaria and antibacterials. Nucleic Acids Res 2022;50(D1):D1282–d1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All source code and datasets are freely available at https://github.com/ku-cosbi/interactome-structural-coverage.