

Abstract
With the growing crisis of antimicrobial resistance, it is critical to continue seeking out sources of novel antibiotics. This need has led to renewed interest in natural product antimicrobials, specifically antimicrobial peptides. Non-lytic antimicrobial peptides are highly promising due to their unique mechanisms of action. One such peptide is apidaecin (Api), which inhibits translation termination through stabilization of the quaternary complex of ribosome-apidaecin-tRNA-release factor. Synthetic derivatives of apidaecin have been developed, but structure-guided modifications have yet to be considered. In this work we have focused on modifying key residues in the Api sequence that are responsible for the interactions that stabilize the quaternary complex. We present one of the first examples of a highly-modified Api peptide that maintains its antimicrobial activity and interaction with the translation complex. These findings establish a starting point for further structure-guided optimization of Api peptides.
Graphical Abstract
INTRODUCTION
The World Health Organization estimates that more than 700,000 people die each year from antibiotic-resistant infections1–3. Infections from bacteria resistant to drugs of last resort are increasingly common, so developing antimicrobials with novel structures and mechanisms of action has never been more important4–10. As a result, natural products have gained renewed interest as a source of antiobiotic leads11–13. Among the most recent classes of antibacterial natural products, antimicrobial peptides—including vancomycin, daptomycin, gramicidin D, oritavancin, dalbavancin, and telavancin—have been advanced as clinically useful therapeutics14–17.
Antimicrobial peptides can be categorized as either lytic or non-lytic18. Lytic antimicrobial peptides are bactericidal due to their ability to disrupt the bacterial membrane causing cell lysis19. Although effective, the non-specific activity of lytic peptides can lead to unwanted toxicity, making them suboptimal therapeutic agents20–22. Non-lytic peptides stop bacterial growth by passing through the bacterial cytoplasmic membrane and acting upon intracellular targets. In particular, many proline-rich antimicrobial peptides (PrAMPs) act upon the ribosome23–25. Among ribosome-targeting PrAMPs, a special place belongs to apidaecin, which is produced by honeybees (Apis mellifera) as a part of their innate immune response to bacterial infections26–28.
Similar to other PrAMPs, apidaecin exhibits activity against a range of Gram-negative bacteria, including Escherichia coli; however, in contrast to the majority of studied PrAMPs, apidaecin does not interfere with translation initiation or elongation, but instead arrests the ribosomes at stop codons of open reading frames (ORFs) (Figure 1), causing widespread disruption of translation termination29,30. When the translating ribosome reaches a stop codon, one of the class 1 release factors (RF1 or RF2) associates with the ribosome and facilitates the hydrolysis of peptidyl-tRNA, liberating the completed, synthesized protein. After dissociation of the new protein, apidaecin penetrates the peptide exit tunnel with its C-terminus oriented towards the catalytic peptidyl transferase center (PTC). Apidaecin forms interactions with ribosomal RNA and ribosomal proteins in the exit tunnel and, most critically, establishes specific contacts with the RF and the 2’−3’ diol of the 3’ terminal nucleotide of deacylated tRNA. The resulting apidaecin-ribosome complex remains stalled at the stop codon with a sequestered RF. Because ribosomes far outnumber the RF molecules in many bacteria, the apidaecin-mediated RF sequestration leads to translation termination impairment on other ribosomes, ultimately causing growth arrest.
Figure 1. Structure of the 70S ribosome in complex with apidaecin-137.
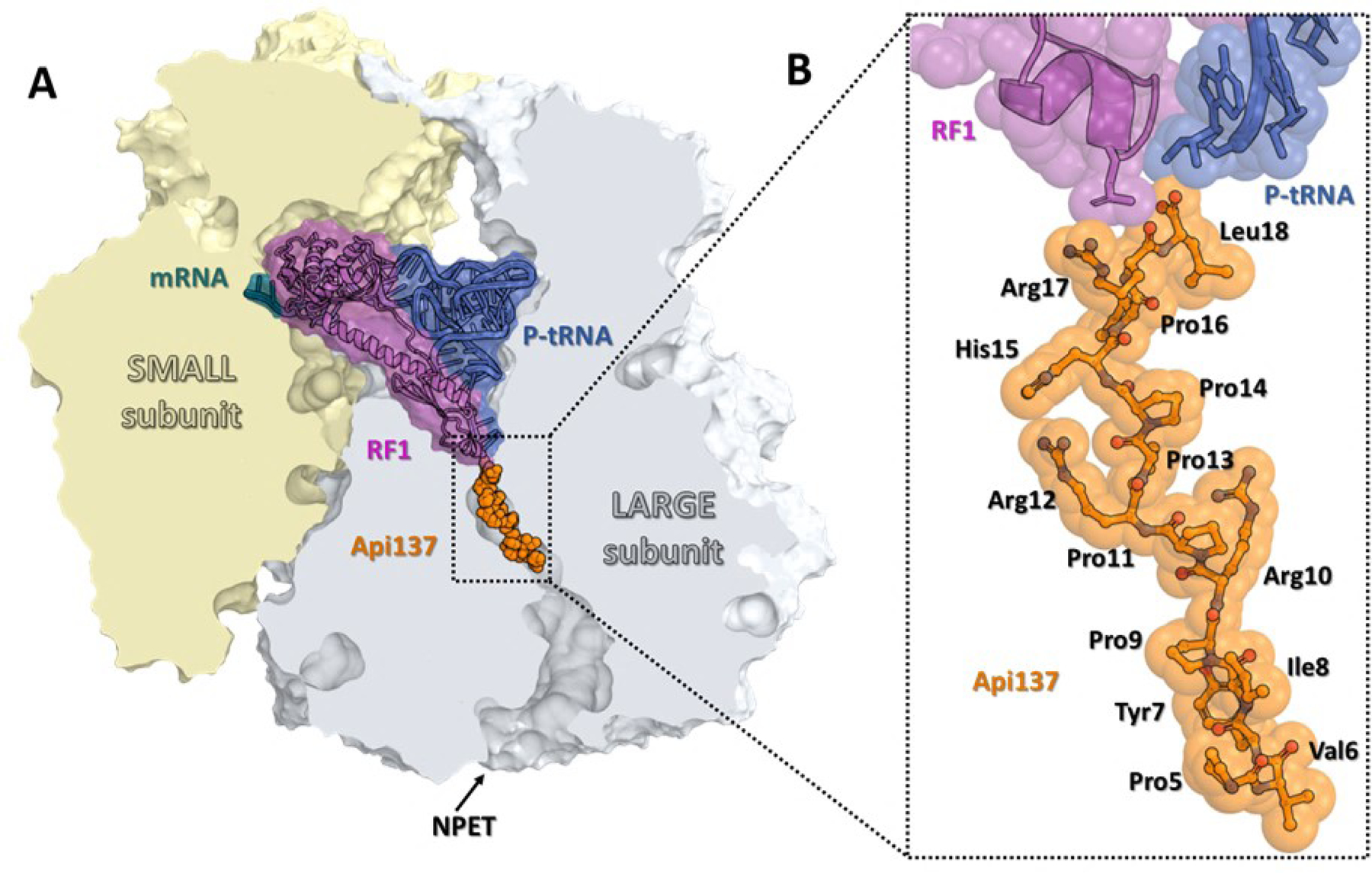
(A) Overview of the Api-binding site (orange) in the E. coli 70S ribosome carrying mRNA (teal), release factor 1 (RF1, magenta), and deacylated tRNA (navy) in the A and P sites, respectively, viewed as a cross-cut section through the nascent peptide exit tunnel (NPET). The small subunit is shown in light yellow; the large subunit is light gray. (B) Close-up view of Api137 bound in the exit tunnel of the 70S ribosome (PDB entry 5O2R29).
Apidaecin Ib, H-GNNRPVYIPQPRPPHPRL-OH, the active wildtype peptide produced by the honeybee, has been investigated as an antimicrobial against a variety of Gram-negative bacterial species31–39. A chemically modified derivative, Api-137 (gu-ONNRPVYIPRPRPPHPRL-OH, where gu = tetramethylguanidyl, O = l-ornithine), showed improved proteolytic stability in serum and was found to exhibit greater activity against E. coli, P. aeruginosa, and K. pneumonia31,40,41.
Our comprehensive genetic-based structure-activity relationship study (Figure 2) concluded that the pharmacophore of apidaecin is centered on the five C-terminal amino acids: P/z-H/z-P-R-X (where z = aromatic amino acid and X = any amino acid except A,S,G)34; however, structure-guided modification of synthetic apidaecins has not yet been investigated. Building on the results from the genetic-based structure-activity relationship study and those from Hoffmann and coworkers31–34,40,42–44, we have carried out structure-guided modifications of apidaecin to test analogs with novel modifications that could stabilize the quaternary complex of ribosome-apidaecin-tRNA-RF and/or gain proteolytic stability.
Figure 2.
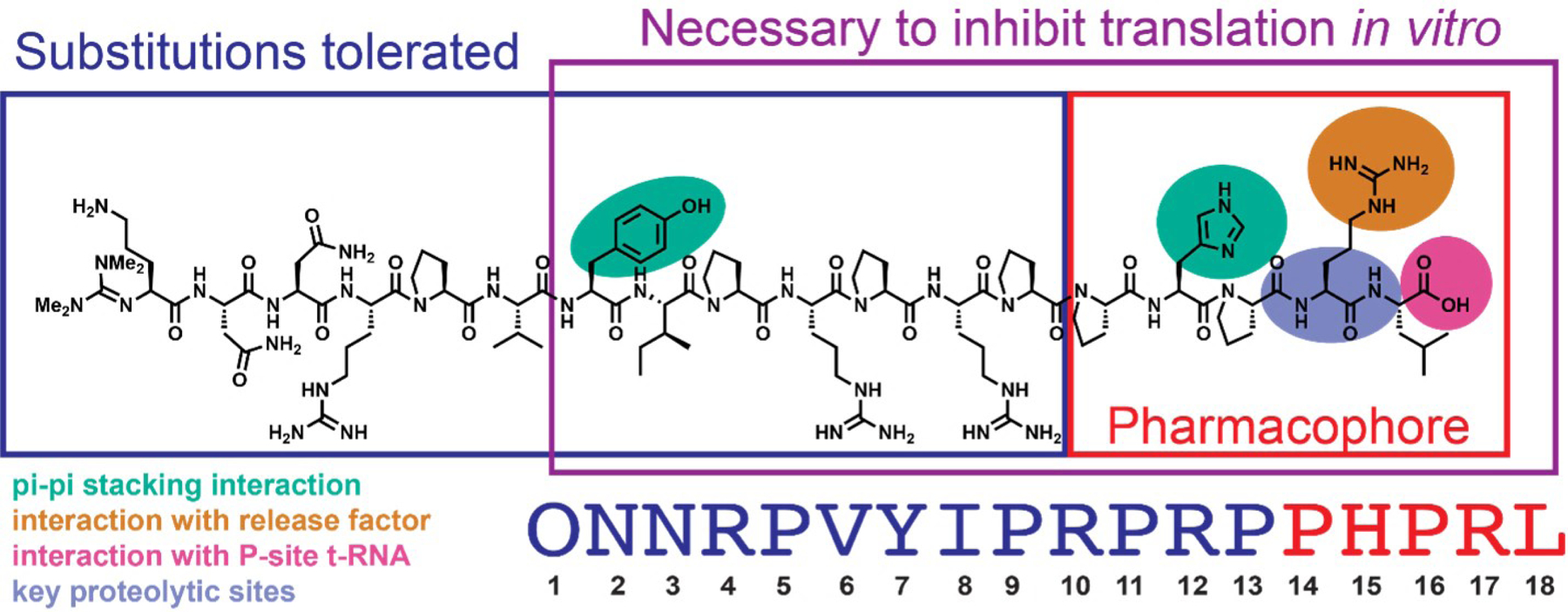
Key residues in the sequence of Api-137 as per Baliga et al. The pharmacophore residues are boxed in red. The residues necessary to arrest the ribosome at the stop codon in vitro are boxed in purple. The residues which tolerate substitutions while retaining the activity of apidaecin endogenously expressed in E. coli cells are shown in navy blue. The Api137 residues of interest in this work: residues with π-π stacking interactions with rRNA of the nascent peptide exit tunnel of the 70S ribosome (turquoise), residues with interactions with the release factor (orange), residues with interactions with the P-site tRNA (magenta), and residues which are sites of proteolysis in murine serum40 (violet).
RESULTS AND DISCUSSION
To test the key moieties involved in functionally important apidaecin interactions with the ribosome, RF, and tRNA, our synthetic effort was targeted against three segments of apidaecin (Figure 2).
Exploration of the interaction of Api with RF
The C-terminus has been identified as the pharmacophore region primarily responsible for apidaecin’s antimicrobial activity34,45,46 (Figure 2). To further understand the moieties important for this activity, we designed and synthesized derivatives of apidaecin with a modified penultimate Arg17 residue, whose sidechain guanidinium is involved in an H-bond network that includes Gln235 of RF and C2452 of the 23S rRNA29. We tested importance of these interactions by replacing Arg17 of Api137 with homoarginine (hArg) and citrulline (Cit) which differ from arginine in sidechain length (hArg) and in functional group (urea instead of guanidinium moiety in Cit; Figure 3), modifications that disrupt the H-bond network. The extra methylene of hArg derivative (1) is expected to distort the positioning of the guanidinium, whereas in the Cit derivative (2), one of the nitrogens of the guanidinium is replaced with an oxygen, thereby altering the H-bonding capacity. The hArg (1) and Cit (2) derivatives had minimum inhibitory concentrations (MICs) of 2.5 μM and 20 μM, respectively, as compared to 0.16 μM for Api-137 (Table 1), vividly underscoring the importance of H-bonding interactions revealed by the structural studies. The more significant loss of activity of the Cit derivative could be due to at least two differences between these groups: urea is neutrally charged, while guanidine is positively charged at physiological pH, and urea has two fewer H-bond donors than guanidinium. There are no apparent salt-bridge interactions between Api-137 and the ribosome or RF, so it is most likely that the lack of two H-bond donors is responsible for the loss in activity.
Figure 3.
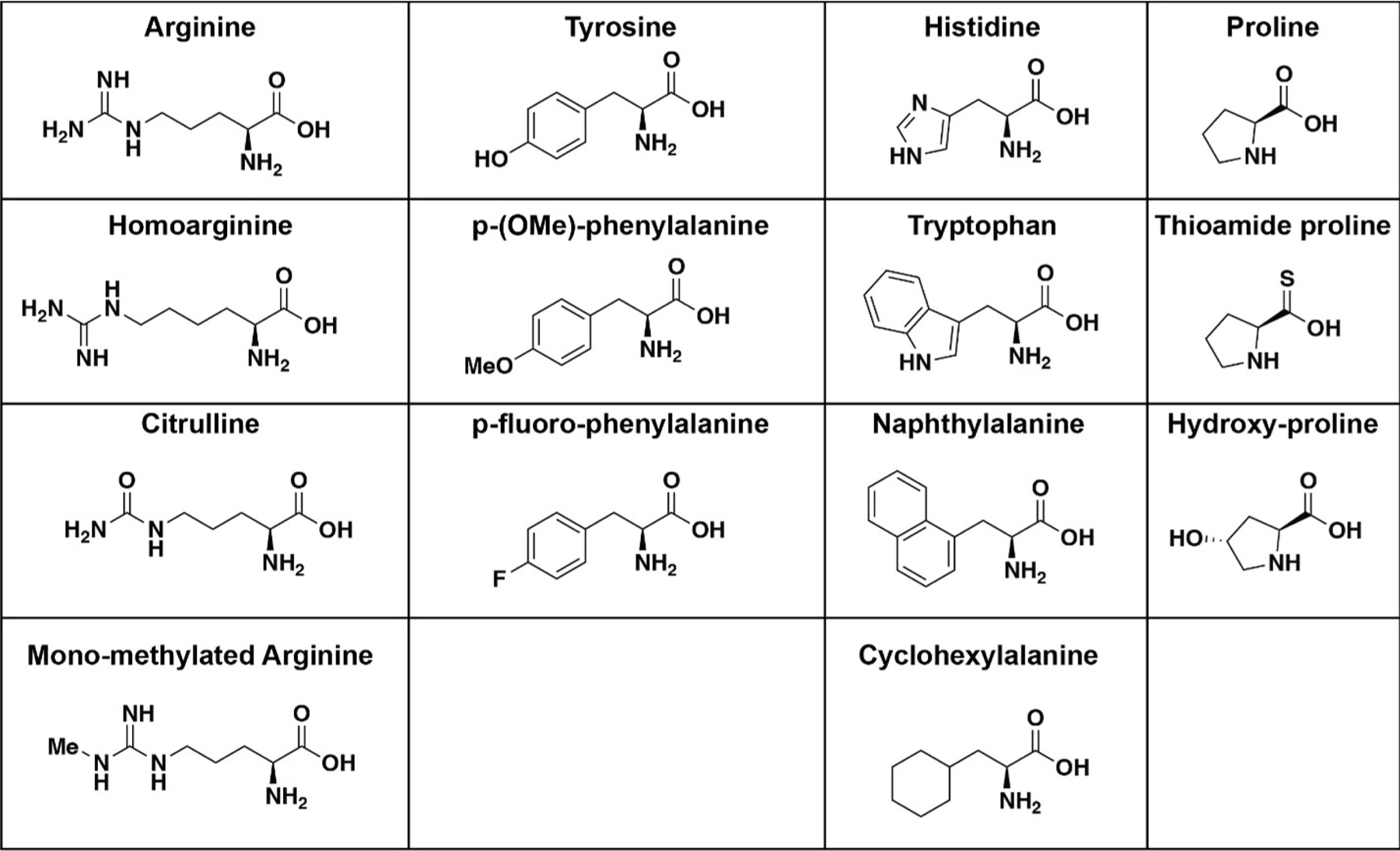
The structures of natural and unnatural amino acids incorporated into the Api-137 peptide. From left to right, arginine, homoarginine, citrulline, and mono-methylated arginine, tyrosine, p-(OMe)-phenylalanine, p-fluoro phenylalanine, histidine, tryptophan, naphthylalanine, and cyclohexylalanine, l-proline, l-thioamide proline, and l-trans-hydroxyproline.
Table 1.
The sequences and minimum inhibitory concentrations and fold changes for Api-137, the C-terminal leucine carboxylic acid modified derivatives (* = decarboxy; ** = primary alcohol), and the Arg17 modifications.
| Peptide | Sequence | Unnatural Amino Acid | MIC (μM) | Fold change |
|---|---|---|---|---|
| Api-137 | gu-ONNRPVYIPRPRPPHPRL-OH | none | 0.16–0.3 | 1 |
| 1 | gu-ONNRPVYIPRPRPPHPrL-OH | r = homoarginine | 2.5 | 16 |
| 2 | gu-ONNRPVYIPRPRPPHPrL-OH | r = citrulline | 20 | 128 |
| 3 | gu-ONNRPVYIPRPRPPHPRL-OH | R = mono-methyl arginine | 0.3 | 1 |
| 4 | gu-ONNRPVYIPRPRPPHPRl-* | l = decarboxy leucine | >40 | >800 |
| 5 | gu-ONNRPVYIPRPRPPHPRl-** | l = l-leucinol | 5 | 32 |
| 6 | gu-ONNRPVYIPRPRPPHPRl-** | l = d-leucinol | 10–20 | 62–128 |
| 7 | gu-ONNRPVYIPRPRPPHPRf-** | f = l-phenylalaninol | 5 | 32 |
| 8 | gu-ONNRPVYIPRPRPPHPRf-** | f = d-phenylalaninol | 40 | 800 |
To investigate whether other substitutions on the guanidinium would be allowed, we synthesized a derivative in which the sidechain of Arg17 was mono-methylated (3, Table 1). This derivative essentially retained the activity of Api-137 (MIC = 0.3 μM). A previous study has shown that methylation of arginine does not substantially change its pKa47, so it is unlikely that the charge state of mono-methylated 3 is different from Api-137. A cleft between the nucleobases of residues A2451 and C2452 of the 23S rRNA could accommodate a methyl group at Arg17, but because the activity does not substantially decrease, the methyl group may not make substantial van der Waals interactions with the complex. Regardless, to our knowledge, this result is the first example of successful replacement of Arg17 in the parent Api-137.
Exploration of the interaction of Api with P-site tRNA
The cryo-EM structure of the ribosome-bound Api-137 complex showed that the carboxylic oxygens of the C-terminal Leu18 are 3.1 and 3.4 Å away from the 2’ and 3’ hydroxyls, respectively, of the ribose of the 3’ terminal adenosine (A76) of the P-site tRNA29 (Figure 4B). To test the importance of this interaction for the inhibitory activity of Api, we synthesized several derivatives with modified Leu18 residues. We first synthesized a decarboxy-leucine Api peptide (Compound 4 in Table 1) that lacks the carboxylic acid altogether. The (decarboxy)Leu18 Api derivative was completely inactive (MIC > 40 μM), pointing to the critical role of the carboxy terminus of Api for antimicrobial activity. We then introduced more nuanced modifications at the C-terminus of Api. Specifically, we synthesized two derivatives in which the C-terminal carboxyl of Leu18 was replaced with an alcohol moiety, generating Api variants carrying l- or d-leucinol at the C-terminus. In addition, because C-terminal Leu can be replaced with Phe without loss of activity34, we also generated two peptides in which Leu18 was replaced with l- or d-phenylalaninol (Compounds 5–8 in Table 1). All of these derivatives retained antimicrobial activity, even though it was diminished compared to that of Api-137. The MIC for both the l-leucinol and l-phenylalaninol derivatives was 5 μM, while the MICs for the d-leucinol and d-phenylalaninol derivatives were 10–20 µM and 40 μM, respectively (Table 1). The dependence of the activity on stereochemistry of the C-terminal alcohol group reinforces the notion that Api’s action relies on the precise interaction of carboxylate oxygen atoms with the diol of the 3’ ribose of the deacylated tRNA.
Figure 4. Interactions of Api-137 with the residues of the 23S rRNA.
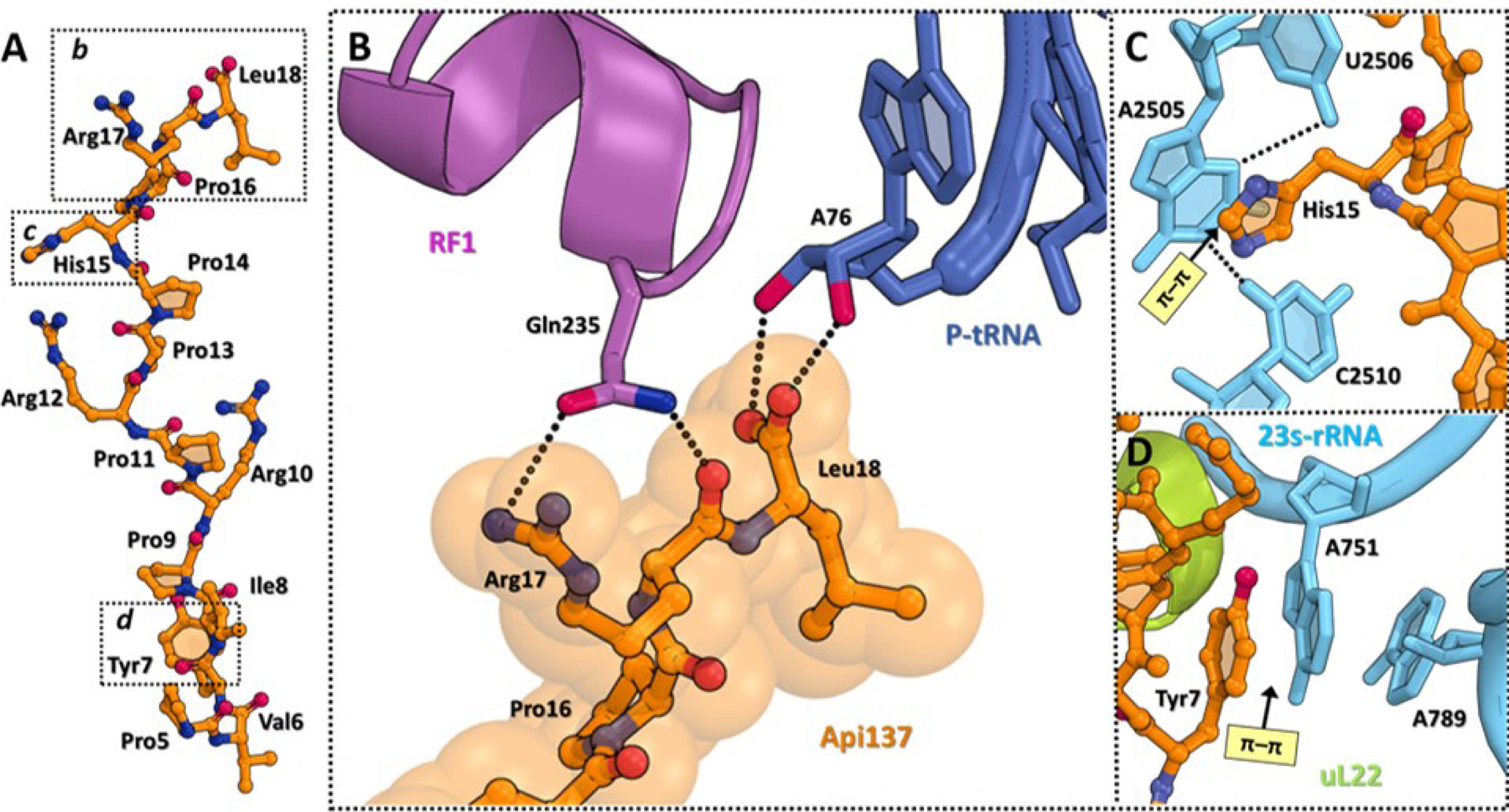
(A) Structure of ribosome-bound Api137 (orange). (B) Close-up view of the interactions of the C-terminal Arg17 and Leu18 residues of Api137 with the GGQ motif of RF1 (magenta) and A76 of the deacylated P-site tRNA (navy). H-bonds are shown with dotted lines. (C, D) π-π stacking interactions (light yellow) of Api residues (orange) with the nucleotides of the 23S rRNA (cyan). Ribosomal protein L22 is shown in light green. Figure rendered from PDB 5O2R.
Exploration of the interactions of Api with the elements of the ribosomal nascent peptide exit tunnel
To probe several of the specific interactions of Api with the ribosomal exit tunnel observed in cryo-EM reconstructions of the ribosome-Api-137 complex, we synthesized several derivatives with modifications at specific amino acid residues farther away from the C-terminus. Two apidaecin residues (Tyr7 and His15) closely approach the nucleobases of 23S rRNA of the walls of the exit tunnel29. Tyr7 makes a π-π sandwich stacking interaction with A751 of the rRNA (Figure 4D), whereas His15 forms π-π stacking interactions with the base of G2505 (Figure 4C). To explore the contributions of the Tyr7-A751 interaction, we synthesized Api derivatives with either a para-methoxyphenylalanine (p-OMe-Phe), para-fluorophenylalanine (p-F-Phe) or cyclohexylalanine (Cha; Figure 3; Compounds 9–11). If this interaction plays a role in Api binding, we would expect that the p-F-Phe could increase activity due to the presence of the electron-withdrawing group, which could strengthen π-π stacking. Conversely, the presence of an electron-donating group (p-OMe-Phe) or a lack of aromaticity (Cha) of the other two derivatives would be expected to decrease activity by weakening the π-π stacking interaction. In reasonable agreement with our expectations, MIC assay results indicated that the p-F-Phe peptide retains activity with an MIC of 0.35 μM, while the cyclohexylalanine and p-OMe-Phe peptides show decreased MICs (0.75 and 1.5 µM, respectively) compared to Api-137 (0.35 μM; Table 2). These data suggest that the π-π stacking interaction of the Tyr7 residue34 modestly contributes to the overall antimicrobial activity of the peptide.
Table 2.
The sequences and minimum inhibitory concentrations and fold changes for Api-137, Tyr7-modified peptides, and His15-modified peptides.
| Peptide | Sequence | Unnatural Amino Acid | MIC (μM) | Fold change |
|---|---|---|---|---|
| Api-137 | gu-ONNRPVYIPRPRPPHPRL-OH | none | 0.16–0.35 | 1 |
| 9 | gu-ONNRPVyIPRPRPPHPRL-OH | y = p-OMe-phenylalanine | 1.5 | 4 |
| 10 | gu-ONNRPVyIPRPRPPHPRL-OH | y = p-F-phenylalanine | 0.35 | 1 |
| 11 | gu-ONNRPVyIPRPRPPHPRL-OH | y = cyclohexylalanine | 0.75 | 2 |
| 12 | gu-ONNRPVYIPRPRPPhPRL-OH | h = naphthylalanine | 10 | 63 |
| 13 | gu-ONNRPVYIPRPRPPhPRL-OH | h = cyclohexylalanine | 5 | 32 |
| 14 | gu-ONNRPVYIPRPRPPWPRL-OH | W = tryptophan | 1.2–2.5 | 8–16 |
To explore the importance of Api H15 – 23S rRNA G2505 interactions, we replaced H15 with either naphthylalanine, cyclohexylalanine, or tryptophan (Figure 3; Compounds 12–14). All of these substitutions significantly decreased the activity of Api-137 (8–64 fold change in MIC relative to Api-137; Table 2). The naphthylalanine modification led to the largest decrease in activity, while the tryptophan modification led to the smallest activity decrease. Although generally useful, these results did not clarify whether His15-G2505 π-π stacking interactions contribute to the activity of Api; it may be that the pocket in which the histidine residue needs to fit may not accommodate a larger sidechain, which could explain the increase in MIC for the tryptophan and naphthylalanine derivatives.
Modifications of proline residues
The high proline content of the ribosome-targeting PrAMPs suggest their importance for peptide activity either because proline residues directly participate in interaction with the target, as they provide free Api with the conformation or rigidity that facilitates its intracellular stability or migration through the exit tunnel towards its binding site near the peptidyl transferase center, or because they facilitate the peptide uptake.
Peptides with high proline content may adopt an idiosyncratic conformation described as a polyproline II helix48. This secondary structure is characterized by three residues per turn and backbone dihedral angles of Φ ~ −75° and Ψ ~ +145°. To gain insights into the secondary structure of Api-137, we carried out a Ramachandran plot analysis of the well-resolved segment (Pro5-Leu18) of the ribosome-bound Api137 (PDB 5O2R; Figure 5). The data indicate that, in its ribosome-bound form, Api-137 has a general conformation resembling a polyproline type II helix. Specifically, the dihedral angles of pre-proline and trans-proline residues are within the range of values typical for the polyproline type II helix48. In addition to the structure of Api, previous studies have suggested that the prolines in the apidaecin sequence are partially responsible for the antimicrobial activity of these peptides40,49; therefore, we wanted to explore additional modifications of proline residues to determine their impact on activity.
Figure 5.
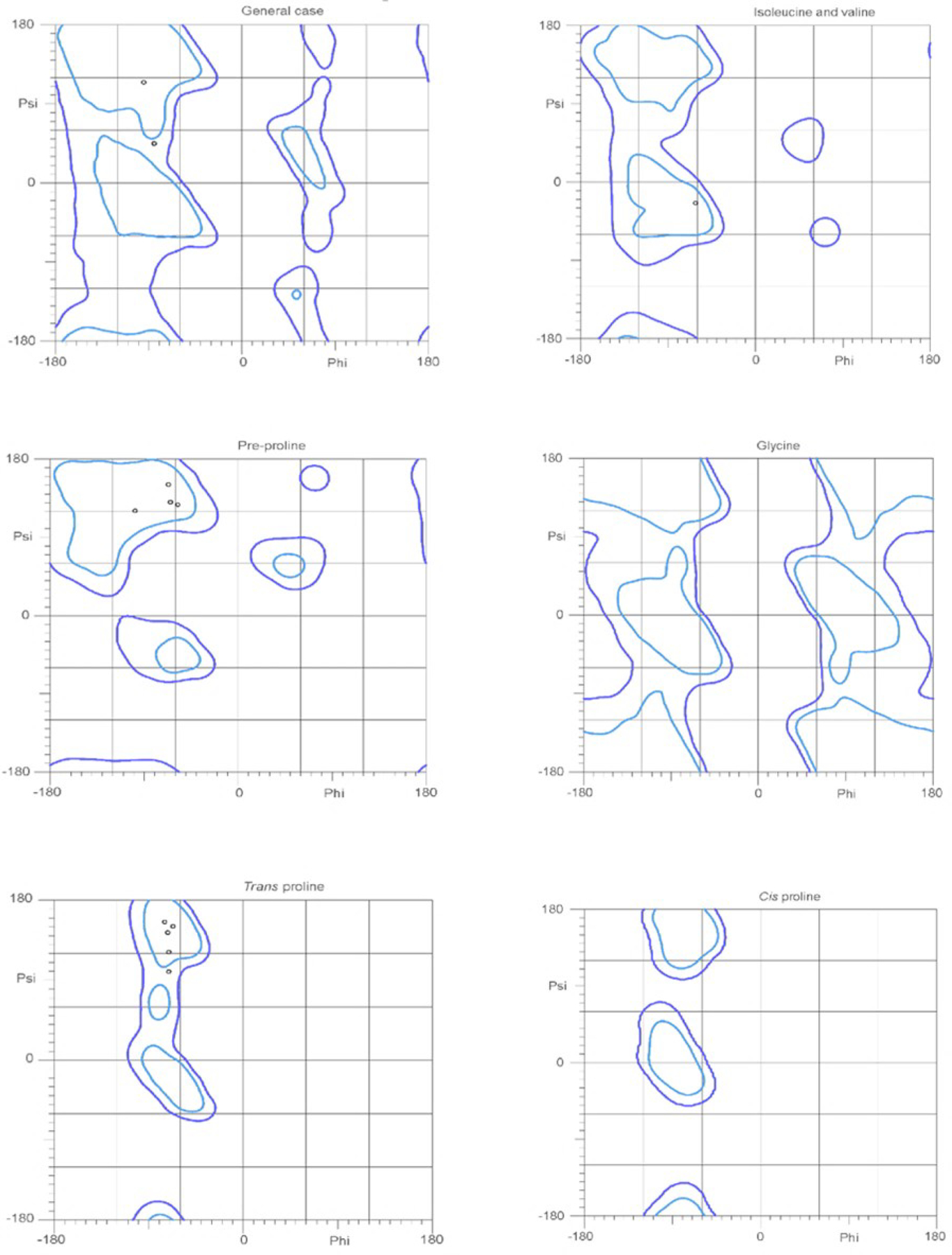
The Ramachandran plot analysis generated for Api-137(Pro5-Leu18) indicating a polyproline type II helix secondary structure. Residues that make up a polyproline type II helix are typically located at −75°, +150°. In the general case, pre-proline and trans proline plots of all of the residues are located near −75°, +150°. The only outlier is the isoleucine and valine plots which have the residue near −60°, +20°. The black circles are the individual Api peptide residues from PDB 5O2R. The blue shapes indicate the possibility of these residues being present in these areas. As the individual resides (black circles) are present in the areas associated with polyproline type II helix secondary structures, these data support a polyproline type II helix structure for Api-137.
Thioamide substitutions may impact activity, proteolytic stability, and secondary structure of biologically active peptides with polyproline II helical secondary structures50,51. We explored whether the incorporation of thioamide prolines could affect the activity of apidaecin (Figure 3)52–54. None of the five synthesized derivatives (Compounds 15–19 in Table 3) maintained the same activity as Api-137. Replacement of Pro11, Pro13, and Pro16 with thioamide prolines led to a four-fold increase in MIC, replacement of Pro9 increased MIC 15-fold, and replacement of Pro14 led to a 125-fold increase in MIC (Table 3). Thioamide and peptide bonds are isosteric, but the lengths of the C=S and C=O bonds are different (1.66 Å and 1.22 Å, respectively)55. Although the structure of the ribosome-Api complex does not reveal any direct interactions of Pro14 carbonyl with the elements of the exit tunnel, the thioamide substitution may alter the structure or positioning of the peptide in the tunnel. Furthermore, the propensity of thioamides for forming hydrogen bonds can vary greatly depending on whether they act as hydrogen bond donor or acceptor55. Therefore, these data indicate that thioamide proline substitutions are not optimal for developing a more active and more proteolytically stable Api-137 derivative.
Table 3.
The sequences and minimum inhibitory concentrations and fold change for Api-137, thioamide proline (15–19) and hydroxy-proline (20–26) modified peptides.
| Peptide | Sequence | MIC (μM) | Fold change |
|---|---|---|---|
| Api-137 | gu-ONNRPVYIPRPRPPHPRL-OH | 0.08–0.3 | 1 |
| 15 | gu-ONNRPVYIPRPRPPHPRL-OH | 0.62 | 4 |
| 16 | gu-ONNRPVYIPRPRPPHPRL-OH | 20 | 125 |
| 17 | gu-ONNRPVYIPRPRPPHPRL-OH | 0.62 | 4 |
| 18 | gu-ONNRPVYIPRPRPPHPRL-OH | 0.62 | 4 |
| 19 | gu-ONNRPVYIPRPRPPHPRL-OH | 2.5 | 15 |
| 20 | gu-ONNRPVYIPRPRPPHPRL-OH | 10 | 63 |
| 21 | gu-ONNRPVYIPRPRPPHPRL-OH | 0.3–0.6 | 2–4 |
| 22 | gu-ONNRPVYIPRPRPPHPRL-OH | 0.6 | 2–4 |
| 23 | gu-ONNRPVYIPRPRPPHPRL-OH | 0.16–0.3 | 2 |
| 24 | gu-ONNRPVYIPRPRPPHPRL-OH | 1.2 | 4–8 |
| 25 | gu-ONNRPVYIPRPRPPHPRL-OH | 0.08–0.16 | 1 |
| 26 | gu-ONNRPVYIPRPRPPHPRL-OH | 0.08–0.16 | 1 |
Prompted by the importance of hydroxyprolines in the polyproline II helical structure of collagen, we incorporated hydroxylated prolines in Api-137. Hydroxyproline differs from l-proline by the presence of a hydroxy group on the γ-carbon. We synthesized seven derivatives of Api137 in which either all or individual proline residues were replaced with hydroxyprolines (Compounds 20–26 in Table 3). This modification had been previously incorporated into the Api88 sequence as single modifications40, but the combination had never been tested. Our current results match the previously published data, since the l-trans-hydroxyproline substitutions, especially near the N-terminus of Api, are well tolerated throughout the sequence (Table 3). l-3-cis-hydroxyproline substitutions were previously shown to match the trends of the l-4-trans-hydroxyproline substututions40. Since none of the modifications led to a more highly active derivative, we have not considered incorporating the l-3-cis-hydroxyproline substitutions. Even simultaneous replacement of all the prolines in the Api-137 sequence with l-trans-hydroxyproline leads to a marginally active molecule, with an MIC of 10 μM (Table 3). The proline residues play an important role in the antimicrobial activity of Api peptides; however, certain modifications are tolerated, indicating the possibility for developing Api derivatives with modified structures and activity. These data confirm that incorporation of highly modified proline residues may allow for the development of more active and more stable Api peptides. Furthermore, the tolerated proline modifications were mostly centered around the N-terminus of the peptide, which support previous data on the pharmacophore of Api34.
C-terminal backbone methylation
As an unmodified peptide, apidaecin can be cleaved by cellular and serum proteases. Incorporation of non-proteinogenic amino acids in the Api structure could increase its activity by improving its proteolytic stability56–58. Apidaecin’s C-terminus, which is critical for activity, is the segment particularly vulnerable to the preferential proteolytic cleavage in blood, serum, and plasma40 (Figure 2). Backbone modifications, including methylation of backbone amides, could influence the activity and/or proteolytic stability of Api59; therefore, we wanted to test whether methylation of the backbone amide group would affect the antibacterial properties of Api.
We synthesized two derivatives in which the backbone amides of Leu18 or Arg17 were methylated. The MIC of (N-Me)Leu18 Api (27, Table 4) was 0.3 μM, which matched the activity of Api-137, whereas the (N-Me)Arg17 derivative (28) displayed an MIC of 20 μM, which is 67-fold higher than that of Api-137. Analysis of an available cryo-EM structure of Api-137 bound to the ribosome shows that there is space available near the backbone amide of Leu18 that could be occupied by the methyl group without altering the orientation of the C-terminus of Api, which is critical for interaction with the tRNA (Figure 4B). This amide does not appear to interact with the ribosome, tRNA or RF, which explains the comparable activity of the (N-Me)Leu18 derivative to the parental peptide. In contrast, the backbone amide of Arg17 H-bonds with N1 of A2062 residue of the 23S rRNA (Figure 4B). The (N-Me)Arg17 modification likely disrupts this interaction, underscoring its importance for the activity of Api. This interaction may aid in orienting the C-terminus for optimal interaction with the tRNA and the RF. In a previously published study, derivatives of Api88 (which differs from Api-137 only by the presence of a C-terminal amide instead of a carboxylic acid) with Arg17 replaced by (N-Me)Arg or Leu18 substituted with (N-Me)Leu40 were inactive; however, our results indicate that the (N-Me)Leu18 Api-137 maintains antimicrobial activity. It is unclear why there would be a difference in activity, but it may be that the bioactive conformations of Api88 and Api-137 are different at the C-termini and that N-methylation disfavors the bioactive conformation of Api88 but not of Api-137.
Table 4.
The sequences and minimum inhibitory concentrations and fold change for Api-137, N-methyl leucine derivative (27), N-methyl arginine (28), and combination peptide 29 derivatives.
| Peptide | Sequence | MIC (μM) | Fold change |
|---|---|---|---|
| Api-137 | gu-ONNRPVYIPRPRPPHPRL-OH | 0.3–0.35 | 1 |
| 27 | gu-ONNRPVYIPRPRPPHPRl-OH | 0.3 | 1 |
| 28 | gu-ONNRPVYIPRPRPPHPrL-OH | 20 | 67 |
| 29 | gu-ONNRPVYIPRPRPPHPRl-OH | 0.35 | 1 |
Testing simultaneous modifications in a combination peptide
We wondered whether Api-137 would tolerate several modifications simultaneously. From all the modifications that were previously tested, we selected four that positively (or minimally) affected the activity and synthesized a peptide with an l-trans-hydroxyproline instead of Pro5, p-F-Phe in place of Tyr7, Arg(Me) in the Arg17 position, and N-Me-Leu in the Leu18 position (Compound 29, Table 4, Figure 6). Although the modifications to Arg17 and Leu18 were selected within the pharmacophore, Pro5 and Tyr7 are outside of the pharmacophore region. These modifications were selected to determine whether changes outside of the pharmacophore region could also have a synergistic impact on the antimicrobial activity of the peptide that had changes to the pharmacophore region. Despite the presence of multiple alterations in the chemical makeup of Api, the combination peptide exhibited antibacterial activity on par with Api-137 (MIC 0.35 μM; Table 4). This result indicates that multiple modifications can be incorporated into the Api-137 sequence without causing a decrease in the activity of the molecule. To our knowledge, this is the first example of an Api-137 derivative that has multiple modifications at the C-terminus that are tolerated and do not lead to decreases in activity. Future incorporation of multiple or non-proteinogenic amino acids into the sequence of Api-137 could help further improve the proteolytic stability or other pharmacological properties of ribosome-targeting antibacterial peptides.
Figure 6.
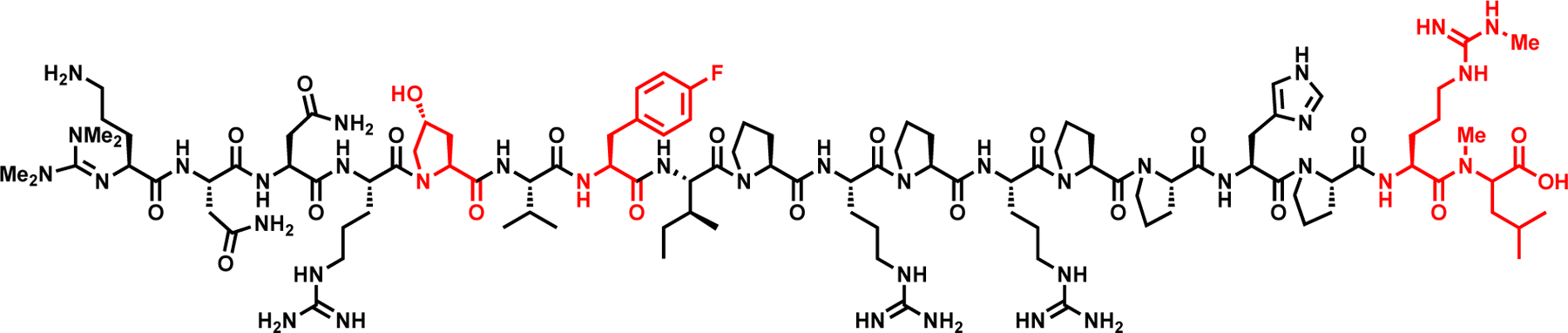
The structure of the Api-137 derivative with a combination of tolerated modifications (Compound 29). Labeled in red are l-trans-hydroxyproline is in the 5th position, l-p-fluorophenylalanine in the 7th position, l-arginine (Me) in the 17th position and l-N-methyl leucine in the 18th position.
An important factor that must be considered when developing peptidic antimicrobials is proteolytic stability. Human plasma was used to determine proteolytic stability of key compounds. Neither the multiply-substituted Api (Compound 29, Table 4) nor (N-Me)Leu18 Api (Compound 27, Table 4) showed significant change in proteolytic stability. Similar to the previously reported stability of Api-137 in mouse serum40, approximately 80% of either of these peptides remained intact after a 2 hour incubation in human serum (Figure 7A). While incorporation of unnatural amino acids will often induce higher proteolytic stability, in this case the proteolytic stability remained similar. This could indicate that further changes must be incorporated into the sequence to eliminate additional points of metabolism. In a similar fashion, synthetic antimicrobial peptide mimics have been developed that retain very high proteolytic stability; however, few compounds have non-lytic mechanisms of action60–64.
Figure 7.
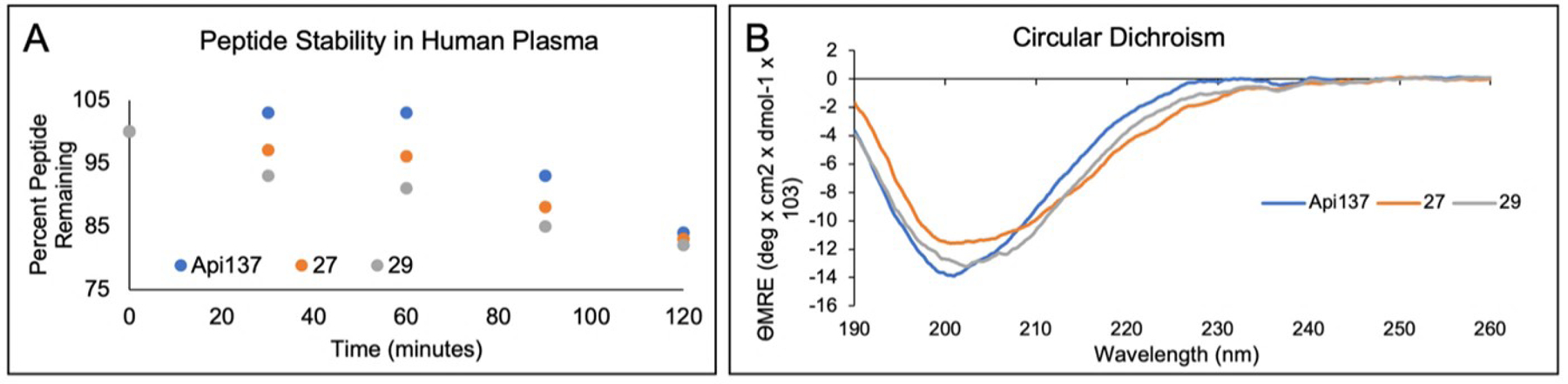
A) Plasma stability of Api-137, compound 27 and compound 29. The values represent the average of two runs. B) Circular dichroism of Api-137, compound 27 and compound 29.
The secondary structure of antimicrobial peptides often impacts its mechanism of action; specifically, alpha-helical AMPs will often become lytic. We collected circular dichroism spectra to determine the secondary structure of key compounds. Upon incorporation of modifications, compounds 27 and 29 maintained spectra similar to Api-137. Therefore, these modifications do not cause significant perturbations to the secondary structure. Furthermore, these spectra lack the key characteristics of alpha-helical structures; therefore, they are not as likely to have lytic properties. These data can be interpreted as supporting a polyproline-II-like helical structure for these compounds due to the presence of a negative band at around 200 nm48,65,66.
Confirming the mechanism of action of Api derivatives
Incorporation of modifications could impact the unique mechanism of action of Api peptides; therefore, it is critical to confirm the antimicrobial activity of the key compounds. We determined their MIC values using multiple strains of E. coli (Table 5). We used the standard BL21 as a comparison for the mutant strains. Compounds 3, 27, and 29 had antimicrobial activity similar to Api-137 against the BL21 strain, as expected based on our previous data above (Table 5). Api-137 requires the sbmA transporter for uptake into bacterial cells; therefore, we tested our compounds against a knockout strain to determine whether their uptake mechanism is the same. Compounds 3, 27, and 29, as well as Api-137, have no antimicrobial activity against the ΔsbmA mutant E. coli strain. This indicates that these compounds all require the transporter for their antimicrobial activity and do not have a lytic mechanism of action, as they are inactive without the transporter. Resistance mechanisms against Api-137 have been determined and include mutations in the release factor, specifically R262C and Q280L29. These mutations in the RF cause Api-137 to be inactive. Against the RF2 R262C and RF2 Q280L strains, Compounds 3 and 27 are also inactive, indicating that the activity of these compounds relies on an interaction with the RF. The same strains appear to retain some sensitivity to compound 29, which may be indicative of its overcoming release factor-based resistance; however, further investigation is required to fully confirm these results.
Table 5.
The minimum inhibitory concentration of Api-137, compound 3, 27 and 29 against the following E. coli strains: BL21, BL21 ΔsbmA, BL21 RF2(R262C) and BL21 RF2(Q280L) to confirm whether the compounds have the same mechanism of action as Api-137.
| Compound | BL21 | BL21 ΔsbmA | BL21 RF2(R262C) | BL21 RF2(Q280L) |
|---|---|---|---|---|
| Api 137 | 1.5 | >25 | >25 | >25 |
| 3 | 3.1 | >25 | >25 | 25 |
| 27 | 1.5 | >25 | >25 | 12.5 |
| 29 | 3.1 | >25 | 6.2 | 3.1 |
One of the unique effects of the mechanism of Api is its ability to induce stop codon readthrough. The ability of the synthetic peptides to induce stop codon readthrough activity was tested using the pRXG reporter plasmid that carries the rfp and gfp genes encoding red and green fluorescent proteins, respectively67. In the pRXG(UGA) plasmid, the in-frame fused rfp and gfp genes are separated by a UGA stop codon 68. Placing a drop of the PrAMP on surface of agar plate inoculated with E. coli carrying the pRXG(UGA) reporter generates a gradient of the peptide concentration. At the high PrAMP concentrations (near the site of application) cells are killed, but at subinhibitory concentrations, PrAMPs with the mechanisms of action like that of Api-137 generate a halo of GFP fluorescence due to induction of the stop codon readthrough. As can be seen in Fig. 8, all the three tested compounds (3, 27 and 29) induce appearance of a halo of GFP fluorescence revealing their ability to induce stop codon readthrough and confirming that their mode of action resembles that of Api-137.
Figure 8.
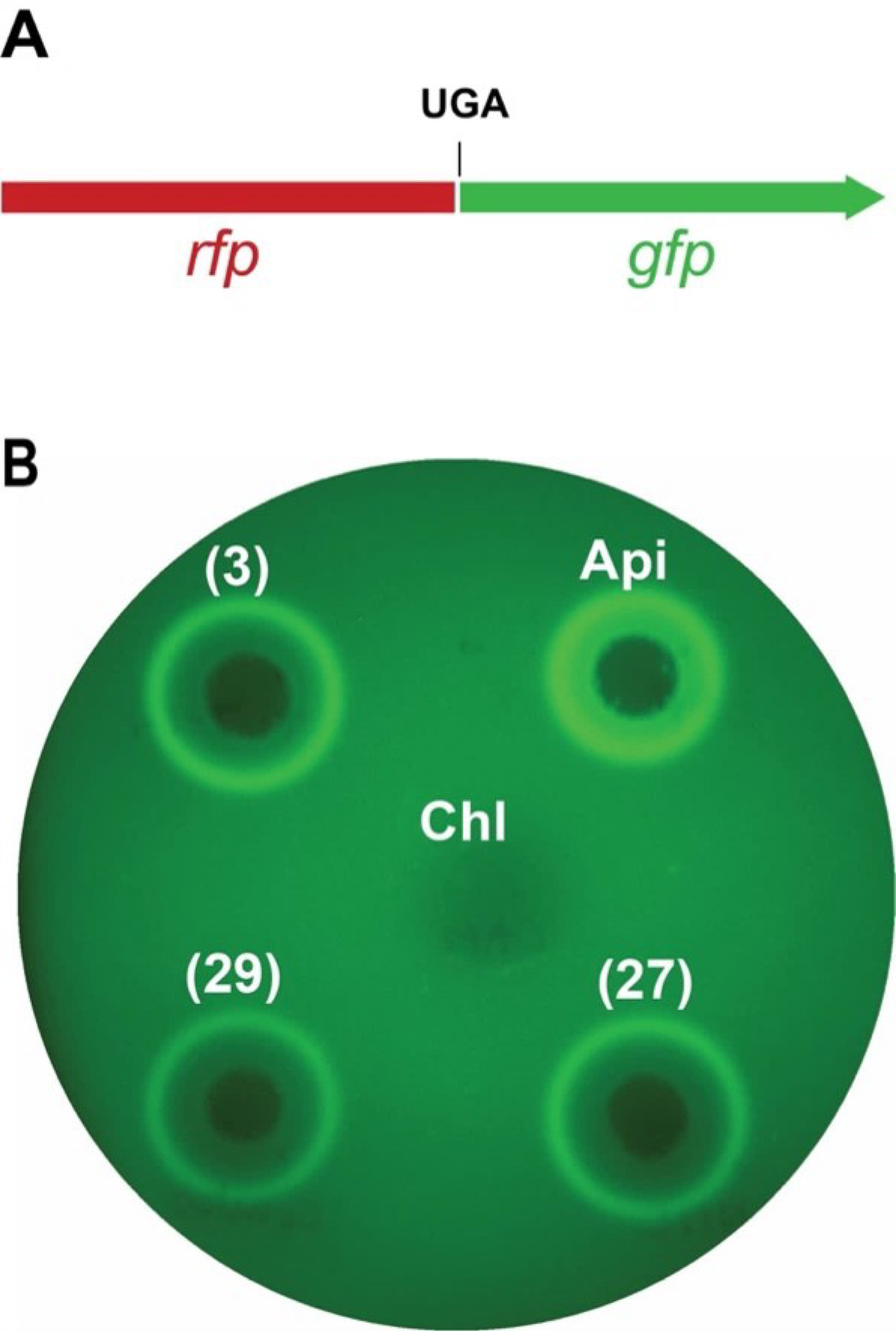
A) In the pRXG(UGA) reporter plasmid, the fused in-frame rfp-gfp genes are separated by a stop codon UGA. Stop codon readthrough is required for expressing the GFP activity. B) Drop diffusion test on agar plate inoculated with the E. coli cells carrying the pRXG(UGA) reporter plasmid. Two μL droplets of 0.2 mM solutions of compounds 3, 27 and 29 or of the positive control Api-137, and 1 μL drop of a 1 mg/mL solution of a negative control antibiotic chloramphenicol that does not induce stop codon readthrough, were applied to the plates. Shown is the false-colored image of the plate imaged in the Cy2 channel after overnight incubation at 37 °C.
CONCLUSIONS
We have carried out structure-guided modifications of antimicrobial peptide apidaecin to test if its derivatives would tolerate substitutions that alter the interaction of the peptide with ribosome—tRNA—RF and/or gain proteolytic stability. We have found several amino acid substitutions and modifications that preserve the antibacterial activity of this PrAMP. Modifications, such as specific methylations, can be tolerated at the C-terminus of the molecule; however, methylation at the backbone nitrogen of Arg17 is detrimental to the antibacterial activity of Api. Single amino acid modifications at Tyr7 are allowed, while His15 does not tolerate substitution to large aromatic side chains. Hydroxylation of prolines is tolerated throughout the sequence to a limited degree, while thioamide proline activity varies substantially. A combination of several well-tolerated substitutions retained the activity of Api-137. Furthermore, incorporation of these modifications in the pharmacophore region does not disrupt the unique mechanism of action of Apidaecins and may actually lead to overcoming a resistance mechanism, which needs further investigation. These results provide further insight into the acceptable modifications of apidaecin peptides and may guide further development of more active, proteolytically stable and mutant-resistant derivatives.
EXPERIMENTAL SECTION
Chemistry
Commercially available Fmoc-amino acids, 2-chlorotrityl chloride resin, and amino alcohols were purchased from Novabiochem, Sigma-Aldrich or Chem-Impex and used without further purification. 6-Chloro-benzotriazole-1-yloxy-tris-pyrrolidinophosphonium hexafluorophosphate (PyClocK), N,N’-diisopropylcarbodiimide (DIC), ethyl cyanohydroxyiminoacetate (Oxyma Pure), 3-[Bis(dimethylamino)methyliumyl]-3H-benzotriazol-1-oxide hexafluorophosphate (HBTU), 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridiniom 3-oxide hexafluorophosphate (HATU), 1H-1,2,3-benzotriazol-1-ol (HOBt hydrate), were purchased from Sigma-Aldrich or Chem-Impex.
N,N-dimethylformamide (DMF), dichloromethane (DCM), piperidine, trifluoroacetic acid (TFA), triisopropylsilane (TIPS), N,N-diisopropylethylamine (DIPEA), hexafluoroisopropanol (HFIP), and HPLC-grade acetonitrile were purchased from Sigma-Aldrich, Fisher, or Chem-Impex and used without further purification. All compounds are >95% by HPLC analysis with the exception of compound 16 and 19 which are >90%.
General manual Fmoc-based solid-phase peptide synthesis
Solid-phase peptide synthesis was carried out using standard Fmoc-based protocols at 60 or 100 μmol scale using DIC and Oxyma Pure or PyClocK as the activating agents.
Loading of 2-chlorotrityl chloride resin with Fmoc-Leu-OH, Fmoc N-Me-Leu-OH, Fmoc-Arg-OH
The appropriate amount of resin for 60 or 100 μmol scale was weighed out and swelled with dry CH2Cl2. 5 eq of Fmoc-protected amino acid was dissolved in dry CH2Cl2 and 10 eq of DIPEA were added to the resin. The loading was allowed to proceed for 2–18 hours. The resin loading was determined using standard protocols as follows: 5–10 mg of dried, loaded resin were weighed out. 1 mL of 20% piperidine in DMF was added to each sample. The deprotection reaction was allowed to proceed for 20 minutes. After completion of the deprotection reaction, 100 μL of the solution was added to 10 mL DMF. 2 mL of this solution was added to a cuvette and the absorbance of the solution at 289 nm and 301 nm was measured. Using the following equation, the resin loading was determined.
Where A = measured absorbance; ε = molar absorption coefficient (at 289.8nm = 6089 l mol−1cm−1; at 301.0nm = 8021 l mol−1cm−1), w = weight of sample resin in milligrams, d = path length (usually 1 centimeter).
Coupling after N-Me Leu or N-Me Arg
Following an N-Me amino acid, the coupling of the following amino acid required alternative coupling conditions. 2 eq Fmoc-protected amino acid were dissolved in DMF. 2 eq HATU, 0.1 M HOBt, and 4 eq DIPEA were added to the solution and was added to the resin. The coupling proceeded overnight.
General Fmoc deprotection procedure
Fmoc deprotection was carried out for 20–30 minutes using 20–25% piperidine in DMF with 0.1M HOBt at room temperature.
General procedure for amino acid coupling
Amino acids were coupled using 5 equivalents (eq) of Fmoc-protected amino acid, 5 eq of activating agent, 5 eq of Oxyma (if using DIC) and 10 eq of DIPEA (if using PyClocK) in DMF for 30–60 minutes. Double coupling was used if coupling was incomplete after first round.
General procedure for guanidinilation (introduction of N,N,N′,N′-tetramethylguanidine)
After the N-terminal Fmoc-protecting group was deprotected, 10 eq of TBTU or HBTU with 10 eq DIPEA were added to the resin for at 2–16 hours, preferably overnight. The resin was washed with DMF, CH2Cl2 twice, and dried down to prepare for global deprotection and cleavage, or selective removal from resin.
Coupling of amino alcohols and decarboxy leucine
Amino alcohols were coupled to a 17mer peptide with a free carboxylic acid at the C-terminus, guanidinilated N-terminus and protected sidechains. 2 eq amino alcohols were added to the peptide with 2 eq DIC, 2 eq Oxyma Pure in dry CH2Cl2 for 18–24 hours. The reaction was monitored via MALDI-TOF, and upon reaction completion, the solution was dried down. The residue was exposed to standard global deprotection and cleavage conditions and was purified to yield the target compounds.
Global deprotection and cleavage or selective cleavage from the resin
Global deprotection and cleavage from resin was completed using 95:2.5:2.5 TFA/TIPS/H2O for 2–4 hours. Selective cleavage of the peptide from 2-chlorotrityl resin was completed using 1:5 HFIP/DCM for 20 minutes, twice. The resin was filtered, the solution was collected and dried to yield crude sidechain-protected peptide. The peptide could be further purified through precipitation from cold diethyl ether. It was used for further chemistry without further purification.
Peptide purification and characterization
The crude peptides were purified by semipreparative HPLC to >95% purity (solvent system MeCN:H2O with 0.1% formic acid; 0–5 min, 15% MeCN; 5–19 min 15–20% MeCN; 19–20 min, 20% MeCN; 20–22 min, 20–15% MeCN. Column: Phenomenex Luna 5 μm C18(2), 100 Å, 250 ✕ 10 mm). Fractions containing pure peptide were lyophilized. The purity of the peptides was established using HPLC on a Shimadzu LC-20AB (Solvent system MeCN:H2O with 0.1% formic acid; 0–2 min, 4% MeCN; 2–12 min, 4–70% MeCN; 12–13 min, 70% MeCN, 13–14 min, 70–4% MeCN, 14–18 min, 4% MeCN. Column: Phenomenex Luna C8, 5 μm, 100 Å, 50 ✕ 4.6 mm). A Bruker MALDI-TOF spectrometer was used to verify the m/z of purified peptides.
Purification of the decarboxy leucine peptide did not follow the standard solvent system used for the remainder of peptides and specified above. This peptide was purified by semipreparative HPLC (solvent system MeCN:H2O with 0.1% formic acid; 0–5 min, 15% MeCN; 5–19 min 15–20% MeCN; 19–20 min, 20% MeCN; 20–22 min, 20–15% MeCN. Column: Phenomenex Luna 5 μm C18(2), 100 Å, 250 ✕ 10 mm).
Synthesis of thioamide prolines
Thioamide prolines were synthesized following a previously published protocols52–54
Antimicrobial Activity Assays
In vivo inhibition of growth of E. coli BL21:
The Minimum Inhibitory Concentrations (MIC’s) of designed Api-variant peptides were determined by microbroth dilution technique in 96-well plates. Specifically, exponentially growing E. coli BL21 cells were diluted to the final density OD600 = 0.002 in 0.3 Tryptic Soy Broth (corresponding to 1% (w:v))42 and 100 mL of the diluted culture were placed in the wells, and after the addition of the peptide, plates were incubated overnight at 37 °C. The minimal concentration of the peptide preventing appearance of the visible cell density was recoded as the MIC. The assay was run in duplicate.
The potency of the peptide was separately confirmed by determining the Zone of Inhibition. This was done by spotting 2 mL of 2 mM concentration of each peptide solution on a lawn of E. coli cells growing on 0.3 Tryptic Soy plates with 1.5% (w:v) Agar, incubating at 37 °C overnight, and measuring the diameter of the clearance zone seen around the site of application of the peptide.
In both the experiments, the antimicrobial effects of the variant peptides were compared to that of Api-137 to obtain a fold-change in efficacy.
Stop codon readthrough assay:
E. coli cells (strain BL21) transformed with the pRXG(UGA) plasmid were grown overnight in LB medium supplemented with 50 μg /mL of kanamycin. Cell cultures were diluted 1:10 into fresh LB/kanamycin medium and grown to the late exponential phase (O.D.600 ~ 1). Cells were pelleted, washed with M9 minimal medium supplemented with 2 mM MgSO4, 0.1 mM CaCl2, 10 μg/mL thiamine (supplemented M9 medium), and then resuspended in supplemented M9 medium to O.D. 600 ~ 1. 3 mL of cell suspension were poured over the surface of agar plate prepared with supplemented M9 medium containing 0.2 mM IPTG and 50 μg/mL kanamycin in the ⊘ 10 cm Petri dish. Plates were rapidly swirled to evenly distribute cell culture, and the remaining liquid was aspirated with a pipet from the corner of the tilted plate.
Plates were allowed to dry without the lid in the laminar flow hood. 2 μL drops of the 0.2 mM solutions of the tested PrAMPs were applied to the surface of the plate. The control antibiotic chloramphenicol (1 μL of 1 mg/mL) that does not induce readthrough, was used as a negative control. Plates were incubated for ~ 18 hr at 37 °C and imaged in ChemiDoc MP imaging system (BioRad) using Cy3 channel for RFP and Cy2 channel for GFP fluorescence.
Acknowledgments.
This work was supported by the National Institute of Allergy and Infectious Diseases (R56 AI162961 and R01 AI162961 to A.S.M., T.W.M., and Y.P.). The authors are grateful to Nora Vázquez-Laslop, University of Illinois Chicago, for helpful discussions. The authors dedicate this publication to the 60th anniversary of the MIKIW Meeting-in-Miniature.
Abbreviations Used.
- Api
apidaecin
- Cha
cyclohexylalanine
- Cit
citrulline
- DIC
N,N’-diisopropylcarbodiimide
- DIPEA
N,N-diisopropylethylamine
- hArg
homoarginine
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridiniom 3-oxide hexafluorophosphate
- HBTU
3-[Bis(dimethylamino)methyliumyl]-3H-benzotriazol-1-oxide hexafluorophosphate
- HFIP
hexafluoroisopropanol
- HOBt
1H-1,2,3-benzotriazol-1-ol
- NPET
nascent peptide exit tunnel
- ORF
Open reading frame
- PrAMP
proline-rich antimicrobial peptide
- PTC
peptidyl transferase center
- PyClocK
6-Chloro-benzotriazole-1-yloxy-tris-pyrrolidinophosphonium hexafluorophosphate
- RF
release factor
- TBTU
2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate
Footnotes
ANCILLARY INFORMATION
Supporting Information. MALDI-TOF spectra, and HPLC traces (PDF).
REFERENCES
- (1).Thiemann S; Smit N; Strowig T Antibiotic Resistance: Problems and New Opportunities; 2016; Vol. 398. [Google Scholar]
- (2).Pendleton JN; Gorman SP; Gilmore BF Clinical Relevance of the ESKAPE Pathogens. Expert Rev. Anti. Infect. Ther 2013, 11 (3), 297–308. 10.1586/eri.13.12. [DOI] [PubMed] [Google Scholar]
- (3).O’Neill JR on A. R. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. Arch. Pharm. Pract 2016. 10.4103/2045-080x.186181. [DOI] [Google Scholar]
- (4).Organization, W. H. World Health Organization Model List of Essential Medicines 2021, No. 22, 66. [Google Scholar]
- (5).Sabnis A; Hagart KLH; Klöckner A; Becce M; Evans LE; Furniss RCD; Mavridou DAI; Murphy R; Stevens MM; Davies JC; et al. Colistin Kills Bacteria by Targeting Lipopolysaccharide in the Cytoplasmic Membrane. Elife 2021, 10, 1–26. 10.7554/ELIFE.65836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Carlet J; Jarlier V; Harbarth S; Voss A; Goossens H; Pittet D Ready for a World without Antibiotics? The Pensières Antibiotic Resistance Call to Action. Antimicrob. Resist. Infect. Control 2012, 1, 1–13. 10.1186/2047-2994-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Carlet J; Collignon P; Goldmann D; Goossens H; Gyssens IC; Harbarth S; Jarlier V; Levy SB; N’Doye B; Pittet D; et al. Society’s Failure to Protect a Precious Resource: Antibiotics. Lancet 2011, 378 (9788), 369–371. 10.1016/S0140-6736(11)60401-7. [DOI] [PubMed] [Google Scholar]
- (8).AMR Indistry Alliance: 2020 Progress Report 2020, No. January, 1–128. [Google Scholar]
- (9).Miethke M; Pieroni M; Weber T; Brönstrup M; Hammann P; Halby L; Arimondo PB; Glaser P; Aigle B; Bode HB; et al. Towards the Sustainable Discovery and Development of New Antibiotics. Nat. Rev. Chem 2021, 5 (10), 726–749. 10.1038/s41570-021-00313-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Shore C; Trusts PC New Types of Antibiotics Are Key to Fighting Drug-Resistant Bacteria 2017. [Google Scholar]
- (11).Rossiter SE; Fletcher MH; Wuest WM Natural Products as Platforms to Overcome Antibiotic Resistance. Chem. Rev 2017, 117 (19), 12415–12474. 10.1021/acs.chemrev.7b00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Moloney MG Natural Products as a Source for Novel Antibiotics. Trends Pharmacol. Sci 2016, 37 (8), 689–701. 10.1016/j.tips.2016.05.001. [DOI] [PubMed] [Google Scholar]
- (13).Igarashi M New Natural Products to Meet the Antibiotic Crisis: A Personal Journey. J. Antibiot. (Tokyo) 2019, 72 (12), 890–898. 10.1038/s41429-019-0224-6. [DOI] [PubMed] [Google Scholar]
- (14).Czaplewski L; Bax R; Clokie M; Dawson M; Fairhead H; Fischetti VA; Foster S; Gilmore BF; Hancock REW; Harper D; et al. Alternatives to Antibiotics-a Pipeline Portfolio Review. Lancet Infect. Dis 2016, 16 (2), 239–251. 10.1016/S1473-3099(15)00466-1. [DOI] [PubMed] [Google Scholar]
- (15).Mahlapuu M; Håkansson J; Ringstad L; Björn C Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol 2016, 6 (December), 1–12. 10.3389/fcimb.2016.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dijksteel GS; Ulrich MMW; Middelkoop E; Boekema BK H. L. Review: Lessons Learned From Clinical Trials Using Antimicrobial Peptides (AMPs). Front. Microbiol 2021, 12 (February). 10.3389/fmicb.2021.616979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chen CH; Lu TK Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics 2020, 9 (1). 10.3390/antibiotics9010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Huan Y; Kong Q; Mou H; Yi H Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol 2020, 11 (October), 1–21. 10.3389/fmicb.2020.582779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Benfield AH; Henriques ST Mode-of-Action of Antimicrobial Peptides: Membrane Disruption vs. Intracellular Mechanisms. Front. Med. Technol 2020, 2 (December), 25–28. 10.3389/fmedt.2020.610997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bacalum M; Radu M Cationic Antimicrobial Peptides Cytotoxicity on Mammalian Cells: An Analysis Using Therapeutic Index Integrative Concept. Int. J. Pept. Res. Ther 2015, 21 (1), 47–55. 10.1007/s10989-014-9430-z. [DOI] [Google Scholar]
- (21).Laverty G Cationic Antimicrobial Peptide Cytotoxicity. SOJ Microbiol. Infect. Dis 2014, 2 (1). 10.15226/sojmid.2013.00112. [DOI] [Google Scholar]
- (22).Greco I; Molchanova N; Holmedal E; Jenssen H; Hummel BD; Watts JL; Håkansson J; Hansen PR; Svenson J Correlation between Hemolytic Activity, Cytotoxicity and Systemic in Vivo Toxicity of Synthetic Antimicrobial Peptides. Sci. Rep 2020, 10 (1), 1–13. 10.1038/s41598-020-69995-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Li W; Tailhades J; O’Brien-Simpson NM; Separovic F; Otvos L; Hossain MA; Wade JD Proline-Rich Antimicrobial Peptides: Potential Therapeutics against Antibiotic-Resistant Bacteria. Amino Acids 2014, 46 (10), 2287–2294. 10.1007/s00726-014-1820-1. [DOI] [PubMed] [Google Scholar]
- (24).Mishra AK; Choi J; Moon E; Baek KH Tryptophan-Rich and Proline-Rich Antimicrobial Peptides. Molecules 2018, 23 (4), 1–23. 10.3390/molecules23040815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Graf M; Mardirossian M; Nguyen F; Seefeldt AC; Guichard G; Scocchi M; Innis CA; Wilson DN Proline-Rich Antimicrobial Peptides Targeting Protein Synthesis. Nat. Prod. Rep 2017, 34 (7), 702–711. 10.1039/c7np00020k. [DOI] [PubMed] [Google Scholar]
- (26).Casteels-Josson K; Capaci T; Casteels P; Tempst P Apidaecin Multipeptide Precursor Structure: A Putative Mechanism for Amplification of the Insect Antibacterial Response. EMBO J 1993, 12 (4), 1569–1578. 10.1002/j.1460-2075.1993.tb05801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Casteels P; Tempst P Apidaecin-Type Peptide Antibiotics Function through a Nonporeforming Mechanism Involving Stereospecificity. Biochemical and Biophysical Research Communications 1994, pp 339–345. 10.1006/bbrc.1994.1234. [DOI] [PubMed] [Google Scholar]
- (28).Li WF; Ma GX; Zhou XX Apidaecin-Type Peptides: Biodiversity, Structure-Function Relationships and Mode of Action. Peptides 2006, 27 (9), 2350–2359. 10.1016/j.peptides.2006.03.016. [DOI] [PubMed] [Google Scholar]
- (29).Florin T; Maracci C; Graf M; Karki P; Klepacki D; Berninghausen O; Beckmann R; Vázquez-Laslop N; Wilson DN; Rodnina MV; et al. An Antimicrobial Peptide That Inhibits Translation by Trapping Release Factors on the Ribosome. Nat. Struct. Mol. Biol 2017, 24 (9), 752–757. 10.1038/nsmb.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Graf M; Huter P; Maracci C; Peterek M; Rodnina MV; Wilson DN Visualization of Translation Termination Intermediates Trapped by the Apidaecin 137 Peptide during RF3-Mediated Recycling of RF1. Nat. Commun 2018, 9 (1), 1–11. 10.1038/s41467-018-05465-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Bluhm MEC; Knappe D; Hoffmann R Structure-Activity Relationship Study Using Peptide Arrays to Optimize Api137 for an Increased Antimicrobial Activity against Pseudomonas Aeruginosa. Eur. J. Med. Chem 2015, 103, 574–582. 10.1016/j.ejmech.2015.09.022. [DOI] [PubMed] [Google Scholar]
- (32).Czihal P; Knappe D; Fritsche S; Zahn M; Berthold N; Piantavigna S; Müller U; Van Dorpe S; Herth N; Binas A; et al. Api88 Is a Novel Antibacterial Designer Peptide to Treat Systemic Infections with Multidrug-Resistant Gram-Negative Pathogens. ACS Chem. Biol 2012, 7 (7), 1281–1291. 10.1021/cb300063v. [DOI] [PubMed] [Google Scholar]
- (33).Bluhm MEC; Schneider VAF; Schäfer I; Piantavigna S; Goldbach T; Knappe D; Seibel P; Martin LL; Veldhuizen EJA; Hoffmann R N-Terminal Ile-Orn- and Trp-Orn-Motif Repeats Enhance Membrane Interaction and Increase the Antimicrobial Activity of Apidaecins against Pseudomonas Aeruginosa. Front. Cell Dev. Biol 2016, 4 (May). 10.3389/fcell.2016.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Baliga C; Brown TJ; Florin T; Colon S; Shah V; Skowron KJ; Kefi A; Szal T; Klepacki D; Moore TW; et al. Charting the Sequence-Activity Landscape of Peptide Inhibitors of Translation Termination. Proc. Natl. Acad. Sci. U. S. A 2021, 118 (10). 10.1073/pnas.2026465118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Dosselli R; Tampieri C; Ruiz-González R; De Munari S; Ragàs X; Sánchez-García D; Agut M; Nonell S; Reddi E; Gobbo M Synthesis, Characterization, and Photoinduced Antibacterial Activity of Porphyrin-Type Photosensitizers Conjugated to the Antimicrobial Peptide Apidaecin 1b. J. Med. Chem 2013, 56 (3), 1052–1063. 10.1021/jm301509n. [DOI] [PubMed] [Google Scholar]
- (36).Gobbo M; Benincasa M; Bertoloni G; Biondi B; Dosselli R; Papini E; Reddi E; Rocchi R; Tavano R; Gennaro R Substitution of the Arginine/Leucine Residues in Apidaecin Ib with Peptoid Residues: Effect on Antimicrobial Activity, Cellular Uptake, and Proteolytic Degradation. J. Med. Chem 2009, 52 (16), 5197–5206. 10.1021/jm900396a. [DOI] [PubMed] [Google Scholar]
- (37).Gobbo M; Biondi L; Filira F; Gennaro R; Benincasa M; Scolaro B; Rocchi R; Filira F; Biondi L; Rocchi R; et al. Antimicrobial Peptides: Synthesis and Antibacterial Activity of Linear and Cyclic Drosocin and Apidaecin 1b Analogues. J. Med. Chem 2002, 45 (20), 4494–4504. 10.1021/jm020861d. [DOI] [PubMed] [Google Scholar]
- (38).Dosselli R; Gobbo M; Bolognini E; Campestrini S; Reddi E Porphyrin-Apidaecin Conjugate as a New Broad Spectrum Antibacterial Agent. ACS Med. Chem. Lett 2010, 1 (1), 35–38. 10.1021/ml900021y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).ZHOU XU-XIA, a, b W.-F. Li. and Pan Y-J; Zhou XX; Li WF; Pan Y-J Functional and Structural Characterization of Apidaecin and Its N-Terminal and C-Terminal Fragments. J. Pept. Sci 2008, 14 (December 2007), 697–707. 10.1002/psc. [DOI] [PubMed] [Google Scholar]
- (40).Berthold N; Czihal P; Fritsche S; Sauer U; Schiffer G; Knappe D; Alber G; Hoffmann R Novel Apidaecin 1b Analogs with Superior Serum Stabilities for Treatment of Infections by Gram-Negative Pathogens. Antimicrob. Agents Chemother 2013, 57 (1), 402–409. 10.1128/AAC.01923-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Schmidt R; Knappe D; Ostorházi E; Wende E; Hoffmann R; Ostorházi E; Hoffmann R In Vivo Efficacy and Pharmacokinetics of Optimized Apidaecin Analogs. Front. Chem 2017, 5 (March), 1–13. 10.3389/fchem.2017.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Czihal P; Hoffmann R Mapping of Apidaecin Regions Relevant for Antimicrobial Activity and Bacterial Internalization. Int. J. Pept. Res. Ther 2009, 15 (2), 157–164. 10.1007/s10989-009-9178-z. [DOI] [Google Scholar]
- (43).Holfeld L; Hoffmann R; Knappe D Correlating Uptake and Activity of Proline-Rich Antimicrobial Peptides in Escherichia Coli. Anal. Bioanal. Chem 2017, 409 (23), 5581–5592. 10.1007/s00216-017-0496-2. [DOI] [PubMed] [Google Scholar]
- (44).Krizsan A; Volke D; Weinert S; Sträter N; Knappe D; Hoffmann R Insect-Derived Proline-Rich Antimicrobial Peptides Kill Bacteria by Inhibiting Bacterial Protein Translation at the 70 S Ribosome. Angew. Chemie - Int. Ed 2014, 53 (45), 12236–12239. 10.1002/anie.201407145. [DOI] [PubMed] [Google Scholar]
- (45).Castle M; Nazarian A; Yi SS; Tempst P Lethal Effects of Apidaecin on Escherichia Coli Involve Sequential Molecular Interactions with Diverse Targets. J. Biol. Chem 1999, 274 (46), 32555–32564. 10.1074/jbc.274.46.32555. [DOI] [PubMed] [Google Scholar]
- (46).Taguchi S; Ozaki A; Nakagawa K; Momose H Functional Mapping of Amino Acid Residues Responsible for the Antibacterial Action of Apidaecin. Appl. Environ. Microbiol 1996, 62 (12), 4652–4655. 10.1128/aem.62.12.4652-4655.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Evich M; Stroeva E; Zheng YG; Germann MW Effect of Methylation on the Side-Chain PKa Value of Arginine. Protein Sci 2016, 25 (2), 479–486. 10.1002/pro.2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Adzhubei AA; Sternberg MJE; Makarov AA Polyproline-II Helix in Proteins: Structure and Function. J. Mol. Biol 2013, 425 (12), 2100–2132. 10.1016/j.jmb.2013.03.018. [DOI] [PubMed] [Google Scholar]
- (49).Lai P-KK; Tresnak DT; Hackel BJ Identification and Elucidation of Proline-Rich Antimicrobial Peptides with Enhanced Potency and Delivery. Biotechnol. Bioeng 2019, 116 (10), 0–3. 10.1002/bit.27092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Walters CR; Szantai-Kis DM; Zhang Y; Reinert ZE; Horne WS; Chenoweth DM; Petersson EJ The Effects of Thioamide Backbone Substitution on Protein Stability: A Study in α-Helical, β-Sheet, and Polyproline II Helical Contexts. Chem. Sci 2017, 8 (4), 2868–2877. 10.1039/c6sc05580j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Chen X; Mietlicki-Baase EG; Barrett TM; McGrath LE; Koch-Laskowski K; Ferrie JJ; Hayes MR; Petersson EJ Thioamide Substitution Selectively Modulates Proteolysis and Receptor Activity of Therapeutic Peptide Hormones. J. Am. Chem. Soc 2017, 139 (46), 16688–16695. 10.1021/jacs.7b08417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Butler E; Florentino L; Cornut D; Gomez-Campillos G; Liu H; Regan AC; Thomas EJ Synthesis of Macrocyclic Precursors of the Vioprolides. Org. Biomol. Chem 2018, 16 (38), 6935–6960. 10.1039/C8OB01756E. [DOI] [PubMed] [Google Scholar]
- (53).Mukherjee S; Verma H; Chatterjee J Efficient Site-Specific Incorporation of Thioamides into Peptides on a Solid Support. Org. Lett 2015, 17 (12), 3150–3153. 10.1021/acs.orglett.5b01484. [DOI] [PubMed] [Google Scholar]
- (54).Liu H; Thomas EJ Synthesis of the (E)-Dehydrobutyrine-Thiazoline-Proline-Leucine Fragment of Vioprolides B and D. Tetrahedron Lett 2013, 54 (24), 3150–3153. 10.1016/j.tetlet.2013.04.017. [DOI] [Google Scholar]
- (55).Lampkin BJ; VanVeller B Hydrogen Bond and Geometry Effects of Thioamide Backbone Modifications. J. Org. Chem 2021, 86 (24), 18287–18291. 10.1021/acs.joc.1c02373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Huhmann S; Koksch B Fine-Tuning the Proteolytic Stability of Peptides with Fluorinated Amino Acids. European J. Org. Chem 2018, 2018 (27), 3667–3679. 10.1002/ejoc.201800803. [DOI] [Google Scholar]
- (57).Evans BJ; King AT; Katsifis A; Matesic L; Jamie JF Methods to Enhance the Metabolic Stability of Peptide-Based PET Radiopharmaceuticals. Molecules 2020, 25 (10). 10.3390/molecules25102314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Lu J; Xu H; Xia J; Ma J; Xu J; Li Y; Feng J D- and Unnatural Amino Acid Substituted Antimicrobial Peptides With Improved Proteolytic Resistance and Their Proteolytic Degradation Characteristics. Front. Microbiol 2020, 11 (November), 1–17. 10.3389/fmicb.2020.563030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Erak M; Bellmann-Sickert K; Els-Heindl S; Beck-Sickinger AG Peptide Chemistry Toolbox – Transforming Natural Peptides into Peptide Therapeutics. Bioorganic Med. Chem 2018, 26 (10), 2759–2765. 10.1016/j.bmc.2018.01.012. [DOI] [PubMed] [Google Scholar]
- (60).Gabriel GJ; Madkour AE; Dabkowski JM; Nelson CF; Nüsslein K; Tew GN Synthetic Mimic of Antimicrobial Peptide with Nonmembrane-Disrupting Antibacterial Properties. Biomacromolecules 2008, 9 (11), 2980–2983. 10.1021/bm800855t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Thaker HD; Som A; Ayaz F; Lui D; Pan W; Scott RW; Anguita J; Tew GN Synthetic Mimics of Antimicrobial Peptides with Immunomodulatory Responses. J. Am. Chem. Soc 2012, 134 (27), 11088–11091. 10.1021/ja303304j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Dey R; De K; Mukherjee R; Ghosh S; Haldar J Small Antibacterial Molecules Highly Active against Drug-Resistant: Staphylococcus Aureus. Medchemcomm 2019, 10 (11), 1907–1915. 10.1039/c9md00329k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Mai S; Mauger MT; Niu L. na; Barnes JB; Kao S; Bergeron BE; Ling J. qi; Tay FR Potential Applications of Antimicrobial Peptides and Their Mimics in Combating Caries and Pulpal Infections. Acta Biomater 2017, 49, 16–35. 10.1016/j.actbio.2016.11.026. [DOI] [PubMed] [Google Scholar]
- (64).Findlay B; Zhanel GG; Schweizer F Cationic Amphiphiles, a New Generation of Antimicrobials Inspired by the Natural Antimicrobial Peptide Scaffold. Antimicrob. Agents Chemother 2010, 54 (10), 4049–4058. 10.1128/AAC.00530-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Woody RW Circular Dichroism Spectrum of Peptides in the Poly(Pro)II Conformation. J. Am. Chem. Soc 2009, 131 (23), 8234–8245. 10.1021/ja901218m. [DOI] [PubMed] [Google Scholar]
- (66).Lopes JLS; Miles AJ; Whitmore L; Wallace BA Distinct Circular Dichroism Spectroscopic Signatures of Polyproline II and Unordered Secondary Structures: Applications in Secondary Structure Analyses. Protein Sci 2014, 23 (12), 1765–1772. 10.1002/pro.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Monk JW; Leonard SP; Brown CW; Hammerling MJ; Mortensen C; Gutierrez AE; Shin NY; Watkins E; Mishler DM; Barrick JE Rapid and Inexpensive Evaluation of Nonstandard Amino Acid Incorporation in Escherichia Coli. ACS Synth. Biol 2017, 6 (1), 45–54. 10.1021/acssynbio.6b00192. [DOI] [PubMed] [Google Scholar]
- (68).Mangano K; Klepacki D; Ohanmu I; Baliga C; Huang W; Brakel A; Krizsan A; Polikanov YS; Hoffmann R; Vázquez-laslop N; et al. Inhibition of Translation Termination by the Antimicrobial Peptide Drosocin. Nat. Chem. Bioology 2023. 10.1038/s41589-023-01300-x. [DOI] [PMC free article] [PubMed] [Google Scholar]