

Abstract
Background
High-grade gliomas (HGG) in young children pose a challenge due to favorable but unpredictable outcomes. While retrospective studies broadened our understanding of tumor biology, prospective data is lacking.
Methods
A cohort of children with histologically diagnosed HGG from the SJYC07 trial was augmented with nonprotocol patients with HGG treated at St. Jude Children’s Research Hospital from November 2007 to December 2020. DNA methylome profiling and whole genome, whole exome, and RNA sequencing were performed. These data were integrated with histopathology to yield an integrated diagnosis. Clinical characteristics and preoperative imaging were analyzed.
Results
Fifty-six children (0.0–4.4 years) were identified. Integrated analysis split the cohort into four categories: infant-type hemispheric glioma (IHG), HGG, low-grade glioma (LGG), and other-central nervous system (CNS) tumors. IHG was the most prevalent (n = 22), occurred in the youngest patients (median age = 0.4 years), and commonly harbored receptor tyrosine kinase gene fusions (7 ALK, 2 ROS1, 3 NTRK1/2/3, 4 MET). The 5-year event-free (EFS) and overall survival (OS) for IHG was 53.13% (95%CI: 35.52–79.47) and 90.91% (95%CI: 79.66–100.00) vs. 0.0% and 16.67% (95%CI: 2.78–99.74%) for HGG (p = 0.0043, p = 0.00013). EFS and OS were not different between IHG and LGG (p = 0.95, p = 0.43). Imaging review showed IHGs are associated with circumscribed margins (p = 0.0047), hemispheric location (p = 0.0010), and intratumoral hemorrhage (p = 0.0149).
Conclusions
HGG in young children is heterogeneous and best defined by integrating histopathological and molecular features. Patients with IHG have relatively good outcomes, yet they endure significant deficits, making them good candidates for therapy de-escalation and trials of molecular targeted therapy.
Keywords: high-grade glioma, infant-type hemispheric glioma, outcomes, prospective, young children
Key Points.
HGG in young children is histologically, molecularly, and clinically diverse.
Detailed histologic and molecular analysis are needed for a more accurate diagnosis.
Among the diagnoses, infant-type hemispheric gliomas have excellent outcomes.
Despite excellent survival, there is a critical need to reduce morbidity.
Blinded imaging review shows different diagnoses have distinguishable features.
Importance of the Study.
Integrated histological, molecular, and clinical analyses show that HGGs in young children are heterogeneous, with vastly divergent outcomes. Tumors in our cohort could be divided into four major groups: (1) infant-type hemispheric glioma (IHG), which demonstrated excellent outcomes; (2) pediatric-type diffuse HGG, which had poor prognosis; (3) low-grade glioma (LGG) which displayed high proliferative indices yet demonstrated excellent outcomes; (4) other tumors that blur the lines between astrocytic, neuronal, and embryonal tumors, such as CNS tumor with BCOR internal tandem duplication or CIC-rearranged sarcoma; these had poor outcomes similar to HGGs. We conclude that integrated analysis improves risk stratification and better informs treatment planning and prognostication. Although survival is high in patients with IHG, therapy de-escalations and targeted therapy should be explored to reduce morbidity and improve quality of life.
High-grade glioma (HGG) in very young children is diagnostically and clinically challenging. While these tumors have favorable outcomes compared to older children and adolescents with HGGs,1–8 predicting an individual’s outcome has remained elusive. Indeed, up to 50% of young children with HGG progress on therapy and often die of the disease.9 Recent studies demonstrated that many HGGs in young children belong to a unique tumor type named infant-type hemispheric glioma (IHG).2,3,10,11 These retrospective studies showed that patients with IHG have a high overall survival explaining the often-observed better prognosis in the young.10 Still, prospective data on how IHG performs on therapy in young children is lacking.
To address this, we report the outcomes of young children (0–5 years old) with histopathologic diagnosis of HGG treated on the prospective multi-institutional phase II risk-adapted SJYC07 trial (NCT00602667). We supplemented this cohort with similar patients treated on nonprotocol treatment plans (NPTP). Genome-wide DNA methylome profiling and whole genome (WGS), whole exome (WES), and RNA (RNA-seq) sequencing were performed on available tumor tissue. The data was integrated with histopathology to yield an integrated diagnosis. Event-free (EFS) and overall survival (OS) were analyzed by the integrated diagnosis and molecular alterations.
Materials and Methods
Study Cohort
Patients with histologically diagnosed HGG enrolled on the multicenter St Jude Young Child 07 trial (SJYC07) or treated on NPTP at St Jude Children’s Research Hospital (SJCRH) were included. SJYC07 was a multi-institutional phase II, risk-adapted study (NCT00602667) for children younger than three years old with newly diagnosed brain tumors between November 9, 2007, and April 19, 2017. The NPTP cohort was made up of patients diagnosed with HGG and treated at SJCRH between June 1, 2007, and December 31, 2020, who were not enrolled in SJYC07 due to age (> 3 and ≤ 5 years), ineligibility, or treatment after the protocol closed to accrual.
Treatment Strategies
The study design and approach of SJYC07 have been previously published.12,13 Briefly, for HGG, patients with localized disease (M0) were enrolled on the low-risk arm, and patients with metastatic (M+) or with extensive infiltrative disease not amenable to surgical resection (R+) were enrolled on the high-risk arm. Four cycles of induction chemotherapy (high-dose methotrexate/cisplatin/cyclophosphamide/vincristine; vinblastine added for high-risk disease) were given, followed by consolidation with two cycles of carboplatin, etoposide, and cyclophosphamide (SJYC07-low-risk), or two cycles of cyclophosphamide and topotecan (SJYC07-high-risk), and metronomic maintenance therapy. Patients in the NPTP cohort were risk stratified into SJYC07-like therapies (low-like and high-like), or if treatment differed, they were categorized as “Others.” These included focal radiation with/without chemotherapy, surgery only, and molecularly targeted therapies (Figure 1A).
Figure 1.
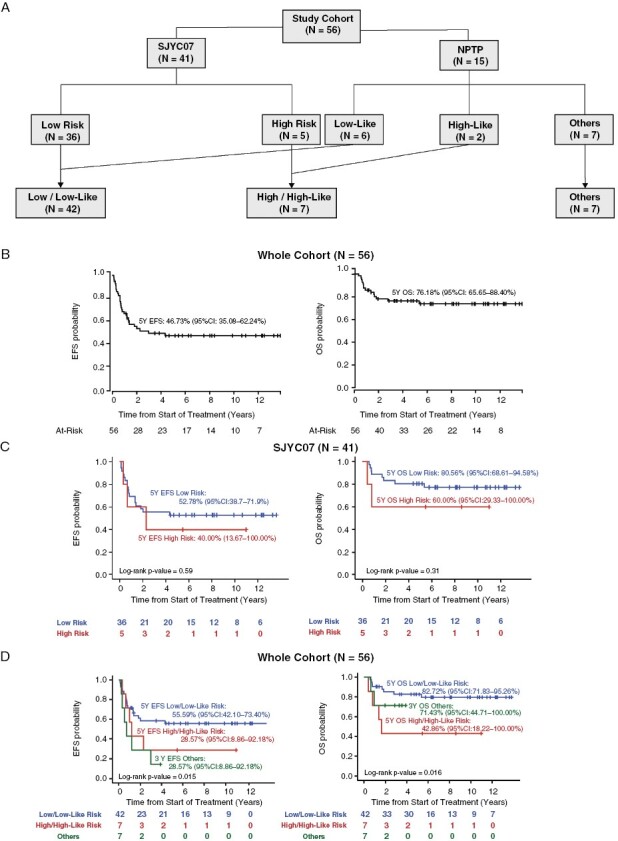
Cohort composition and key clinical outcome. (A) The cohort was assembled from a multicenter study, SJYC07 and an NPTP cohort. Patients were included based on histology at diagnosis or treatment initiation. Treatment was divided into low and high-risk categories for SJYC07 and low-like, high-like, and other categories for NPTP patients. (B) EFS and OS of the whole study cohort (N = 56). (C) EFS and OS based on treatment categories for SJYC07 patients (N = 41). (D) EFS and OS based on treatment categories for the whole cohort (N = 56).
Histopathology, Genome-wide DNA Methylation Profiling, and Genomic and Transcriptomic Analysis
All tumor samples were histopathologically centrally reviewed by neuropathologists specialized in pediatric central nervous system tumors (BAO, JC, and DWE) to confirm the diagnosis of HGG prior to trial enrollment and treatment.
Genomic DNA extracted from tumor samples was used for genome-wide methylation profiling and copy number variation (CNV) analysis by the Illumina Infinium Methylation EPIC platform, as previously described.14–16 For comparison, publicly available well-characterized reference methylation profiles of brain tumors were obtained from published brain tumor datasets.10,17 Tumors were also classified using the Molecular Neuropathology (MNP) brain tumor classifier (www.molecularneuropathology.org) version 12.5. A matching score higher than 0.9 was set for a given tumor type. WGS/WES was conducted on 45 samples, and transcriptomic analysis (RNA sequencing, RNA-seq) on 44. Details are available in the Supplemental Methods.
Imaging Review
All available preoperative contrast- and noncontrast-enhanced cross-sectional imaging exams (MRI and CT scans) were reviewed by a pediatric neuroradiologist (SNP) blinded to the diagnosis. The following characteristics were recorded: tumor location, margins, cellularity (diffusion restriction), T1 and T2 signal, calcification, enhancement, hemorrhage, cystic changes, degrees of associated edema, leptomeningeal metastases, and complications, including hydrocephalus, midline shift, and transtentorial herniation.
Neurocognitive Outcome Analysis
Patients enrolled in SJYC07 were offered serial neurocognitive testing at baseline (postsurgery or early in adjuvant therapy from enrollment up to 60 days), six months after baseline, end of treatment, and yearly following, as previously described.18 Random coefficients models with patient-specific intercepts and slopes were used to investigate changes in intellectual outcome scores over time.
Statistical Analysis
Fisher’s exact tests and Wilcoxon rank sum tests were used to examine associations between variables. Event-free survival (EFS) was defined as the time interval from the date of treatment initiation of the primary tumor (depending on the risk classification of the patient the treatment may have included surgery, chemotherapy, focal radiation, molecular-directed therapy, or a combination of several of these modalities as described in the previous section) to the earliest date of disease progression, second malignancy, or death due to any cause for patients who experienced events or to the date of last follow-up for patients without events. Progression was defined as an increase of >25% of tumor size (any measurable lesion) by MRI imaging, appearance of a new radiographically demonstratable lesion, or the conversion of CSF cytology to positive in two consecutive cytologic evaluations. Overall survival (OS) was defined as the time interval from the date of treatment initiation (as defined above) to the date of death from any cause or to the date of the last follow-up for survivors. Survival outcomes were estimated with the survival package (version 3.3.1) in R (Version 4.2.1) (https://cran.r-project.org/web/packages/survival/index.html) using the Kaplan–Meier method with 95% confidence intervals (CIs). Differences in survival distributions were examined by log-rank test. A significance threshold of 0.05 was used throughout this manuscript without adjusting for multiplicity.
Data Portal
We extended the capabilities of Protein Paint to interactively visualize the cohort metadata, clinical features, and oncoplots using the D3.js library for all the frontend charting and interactive features on the browser.19 The data portal allows users to perform customization on the dataset, and then survival analysis, metadata, and molecular information for the customized data. The users can select these datasets either based on terms in the patient metadata or based on gene alterations or based on lasso function in the methylation scatter plot. The portal also allows users to create groups within the data to perform comparative analysis.
Ethics Statement
SJYC07 was approved by the institutional review board (IRB # Pro00000332) with written informed consent obtained from patients and families. This trial is registered at clinicaltrials.gov, number NCT00602667 and was permanently closed to accrual in April 2017. The review of nonprotocol patients was approved by the IRB with waiver for consent (IRB# 21-0805).
Results
Demographics and Clinical Characteristics
Fifty-six patients with histological diagnoses of HGG were included (Table 1). The median age at diagnosis was 1.22 years (0.00–4.40 years). Thirty-two (57%) were male, and 24 (43%) were female. Forty-one were treated on the SJYC07 clinical trial and 15 on an NPTP. All SJYC07 patients and 8 NPTP patients (49/56, 88%) were younger than three years old; 7 NPTP patients (7/56, 12%) were between 3 and 5 years old at the time of diagnosis. Seven patients had M + disease at diagnosis: 6 were treated in the high/high-like risk category, and 1 in the others category with molecular targeted therapy. One patient with M0, R + disease was treated in the high-risk like category (Figure 1A).
Table 1.
Clinical Characteristic of the Study Cohort:
| SJYC07 | NPTP | Entire Cohort | |
|---|---|---|---|
| N = 41 | N = 15 | N = 56 | |
| Age on Study | |||
| < 3 years | 41 (100%) | 8 (53%) | 49 (88%) |
| 3–5 years | 0 (0%) | 7 (47%) | 7 (12%) |
| Gender | |||
| Male | 27 (67%) | 5 (33%) | 32 (57%) |
| Female | 14 (33%) | 10 (67%) | 24 (43%) |
| Histology at Diagnosis | |||
| HGG | 41 (100%) | 15 (100%) | 56 (100%) |
| Metastatic Status | |||
| Nonmetastatic (M0) | 36 (88%) | 13 (87%) | 49 (88%) |
| Metastatic (M+) | 5 (12%) | 2 (13%) | 7 (12%) |
| Tumor Location | |||
| Hemispheric | 29 (71%) | 12 (80%) | 41 (73%) |
| Midline | 8 (19%) | 3 (20%) | 11 (20%) |
| Cerebellum | 4 (10%) | 0 (0%) | 4 (7%) |
| Extent of Resection | |||
| GTR | 21 (51%) | 7 (47%) | 28 (50%) |
| NTR | 4 (10%) | 1 (6%) | 5 (9%) |
| STR | 8 (20%) | 3 (20%) | 11 (20%) |
| Biopsy | 8 (20%) | 4 (27%) | 12 (21%) |
| Treatment Risk group | |||
| SJYC07-Low Risk/NPTP-Low Like | 36 (88%) | 6 (40%) | 42 (75%) |
| SJYC07-High Risk/NPTP-High Like | 5 (12%) | 2 (13%) | 5 (9%) |
| NPTP-Others | 0 (0%) | 7 (47%) | 7 (12%) |
| Total | 41 (73%) | 15 (27%) | 56 (100%) |
Tumors were more commonly supratentorial (86%) than infratentorial (14%): 41 (73%) were in the cerebral hemispheres, 11 (20%) midline (6 thalamic, 3 brainstem, 1 third ventricle, 1 spinal cord), and 4 (7%) cerebellar. Twenty-eight patients (50%) had gross total resection (GTR) of the primary tumor (2 after 2 cycles of chemotherapy), and 5 (9%) had near total resection (NTR—residual tumor volume measure ≤ 1.5 cm2). Twelve patients (21%) had biopsied only, and 11 (20%) were sub-totally resected (STR- residual tumor measures > 1.5 cm2). None of the patients received craniospinal irradiation (CSI) as initial therapy, and no patients enrolled on the SJYC07 clinical trial received focal radiation as part of their initial therapy. Four NPTP patients received focal RT as part of their initial treatment plan. Twelve patients (21%) received focal RT, and one received CSI as treatment after disease progression. Neurocognitive data were available for 24 patients among the 41 patients treated on SJYC07.
Clinical Risk Categories and Outcomes
The 5-year EFS and OS of the entire cohort were 46.73% (95%CI 35.08–62.24%) and 76.18% (95%CI 65.65–88.40%), respectively (Figure 1B). For patients on SJYC07 protocol (N = 41), the 5-year EFS and OS were 51.06% (95% CI 37.99–69.05%) and 78.05% (95%CI 66.35–91.80%), and there was no significant difference in survival between the low-risk and high-risk strata (5-year EFS 52.78% 95%CI 38.75–71.89% vs. 40.00% 95%CI 13.67–100.00%, p = 0.59; 5-Year OS 80.56% 95%CI 68.61–94.58% vs. and 60.00% 95%CI 29.33–100.00%, p = 0.31) (Figure 1C). However, despite no significant difference in EFS and OS between the SJYC07 and NPTP cohorts (Supplemental Figure 1A), there was a difference in EFS and OS for low/low-like risk and high/high-like risk for the combined cohort (Figure 1D, p = 0.015 and p = 0.016, respectively). Metastatic status did not significantly influence EFS and OS (Supplemental Figure 1B). A greater extent of tumor resection was associated with a higher EFS (p = 0.03) but not OS (p = 0.088). Hemispheric tumor location and female gender were associated with better OS (p = 0.016 and p = 0.003, respectively) but not EFS (p = 0.19 and p = 0.23, respectively) (Supplemental Figure 1C-E).
Molecular Analysis and Integrated Diagnosis
Tumors in the study cohort demonstrated a broad range of histopathologic features, reflecting our experience with infant HGGs. All 56 tumors were originally classified using criteria defined in the WHO classification of CNS tumors applicable to when the patient presented.20 This identified 14/56 (25%) anaplastic astrocytomas (AA), 16/56 (29%) glioblastomas (GB), 12/56 (21%) HGGs, 1/56 (2%) anaplastic pilocytic astrocytoma (APA), 1/56 (2%) high-grade pilomyxoid glioma (PMG), and 12/56 (21%) high-grade neuroepithelial tumors (HGNET) (Supplemental Table 1). Of the 56, DNA methylation profiling was performed on 51 (91%); WES/WGS was performed on 45 (80%). and RNA sequencing was performed on 44 (78.5%). Five cases had insufficient sample for any molecular analysis and were marked “not-defined” (ND) (Supplemental Table 1).
Thirty-four (61%) of the 51 tumors with sufficient samples for analysis received a calibrated score of ≥ 0.9 on the MNP classifier: 17/51(33%) were classified as IHG, 3/51 (6%) as pediatric-type diffuse HGG (H3 K27-altered diffuse midline glioma, DMGH3K27M, or pediatric-type diffuse HGG MYCN subtype, pHGGMYCN), 1/51 (2%) as pleomorphic xanthoastrocytoma (PXA), 4/51 (8%) as LGG (MYB/MYBL1-altered diffuse astrocytoma, DA MYB/MYBL1, pilocytic astrocytoma, PA, or ganglioglioma, GG), and 9/51 (18%) as CNS tumor with BCOR internal tandem duplication (CNS BCOR ITD), CIC-rearranged sarcoma (CNS SARC CIC), FOXR2-activated CNS neuroblastoma (CNS NB FOXR2), neuroepithelial tumor with PLAGL1-fusion (NET PLAGL1), neuroepithelial tumor with PATZ1 fusion (NET PATZ1), CNS embryonal tumor with PLAG family amplification (ET PLAG), or atypical embryonal tumor with multilayered rosettes, non-C19MC-altered (ETMR_Atyp) collectively categorized as “Other-CNS Tumors.” Of the 17/51(33%) non-classifiable (NC) tumors that had calibrated scores of < 0.9, 7/17 (40%) were classified according to their genetic alterations, including MYCN amplification (Supplemental Figure 2), gene fusions (TRIM25::MET, CLIP2::MET, GOPC::ROS1) (Supplemental Figure 3), or ALK rearrangements. In all, 22/51 (39%) tumors were classified as IHG, 6/51 (11%) were HGG, 4/51 (7%) were LGG, and 9/51(16%) were other CNS tumors (Figure 2).
Figure 2.
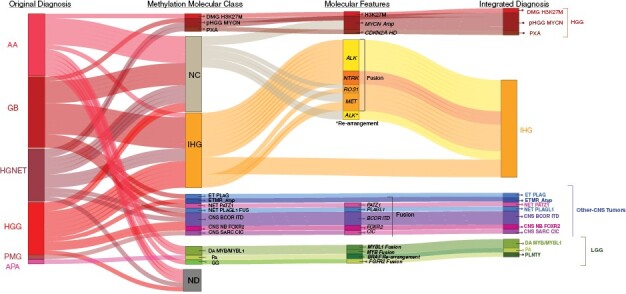
Molecular analysis and integrated diagnosis. (A) Sankey’s plot demonstrates the integration of histology at diagnosis, methylation class (MNP version 12.5), and molecular characteristics to formulate the integrated diagnosis. Abbreviations: Anaplastic astrocytoma, AA; Anaplastic pilocytic astrocytoma, APA; CNS embryonal tumor with PLAG-family amplification, ET PLAG; CNS tumor with BCOR internal tandem duplication, CNS BCOR ITD; CIC -rearranged CNS sarcoma SARC CIC; FOXR2-activated CNS neuroblastoma, CNS NB FOXR2; Embryonal tumor with multilayered rosettes, ETMR; Infant-type hemispheric glioma, IHG; Glioblastoma; Ganglioglioma, GG; High-Grade Glioma, HGG; H3K27M-mutant diffuse midline glioma, DMG H3K27M; High-grade neuroepithelial tumor, HGNET; Low-Grade Glioma, LGG; MYB/MYBL1-altered diffuse astrocytoma, DA MYB/MYBL1; Neuroepithelial tumor with PATZ1 fusion, NET PATZ; neuroepithelial tumor, PLAGL1-fused, NET PLAGL1; Non-classifiable, NC; Not done, ND; Pediatric diffuse high-grade glioma subtype MYCN, pHGGMYCN; Pilocytic astrocytoma, PA; Pilomyxoid glioma, PMG; Pleomorphic xanthoastrocytoma, PXA; Polymorphous low-grade neuroepithelial tumor of the young, PLNTY;
Clinical, Histopathologic, and Molecular Features by Integrated Diagnosis
Infant-type Hemispheric Glioma (IHG)
IHG patients (n = 22) were, on average, younger (median age at diagnosis = 0.36 years, range: 0.00–4.40 years) than non-IHG patients (median age at diagnosis = 1.83 years, range: 0.02–3.82 years, p = 0.0001). All were cerebral hemispheric tumors, and 90% (20/22) were nonmetastatic at diagnosis. As shown in Supplemental Figure 4, ependymoma-like architecture, with perivascular anucleate zones, was a frequent finding.10,21 In addition, areas with prominent desmoplasia resembling desmoplastic infantile astrocytoma were seen in some tumors.2,10 However, the high cellularity, readily apparent mitotic activity, high Ki-67 labeling, and necrosis, some in palisading forms, supported the high-grade designation. Most (19/22) patients were primarily treated with chemotherapy only (18 low-risk or low-like risk therapy, one high-risk), and three were treated with surgery and targeted therapy. Most patients (17/22, 77%) underwent GTR or NTR. More than 70% (16/22) tumors had pathogenic gene fusions involving an RTK—ALK (7), MET (4), ROS1 (2), NTRK1 (2), or NTRK3 (1)—with various partners (Figure 3A and B, Supplemental Figure 3). Two tumors did not have detectable gene fusions, but ALK rearrangement was detected by FISH. These tumors characteristically lacked additional somatic mutations and had a flat genome with few large-scale (>500 Kb) copy number variations. However, one tumor harbored a CDKN2A homozygous deletion, and another had a H3-3A K27M mutation.
Figure 3.
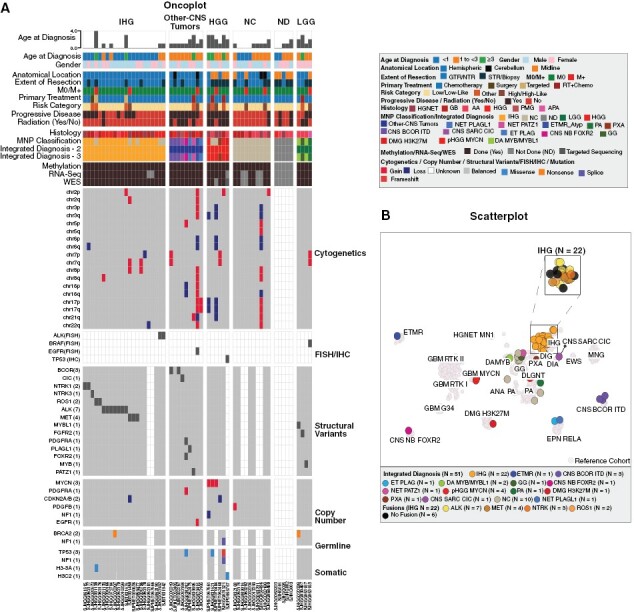
Molecular and Clinical Features by Integrated Diagnosis: (A) Oncoplot depicting molecular (Methylation Class, Structural Variants, Copy Number Variants, Somatic Mutations, and Cytogenetics) and clinical characteristics of tumors by newly defined integrated diagnosis. (B) Scatter Plot of the integrated diagnosis of the tumors (N = 51) against 2482 reference cases from Capper et al.17 and Clarke et al.10 The cohort samples appear as solid-colored circles over reference samples shown in faded grey color. Abbreviations: Anaplastic astrocytoma, AA; Anaplastic pilocytic astrocytoma, APA; CNS embryonal tumor with PLAG-family amplification, ET PLAG; CNS tumor with BCOR internal tandem duplication, CNS BCOR ITD; CIC -rearranged CNS sarcoma SARC CIC; FOXR2-activated CNS neuroblastoma, CNS NB FOXR2; Embryonal tumor with multilayered rosettes, ETMR; Ependymoma, EPN; Ewing’s Sarcoma, EWS; Infant-type hemispheric glioma, IHG; Glioblastoma, GB; Glioblastoma Multiforme, GBM; Ganglioglioma, GG; High-Grade Glioma, HGG; H3K27M-mutant diffuse midline glioma, DMG H3K27M; High-grade neuroepithelial tumor, HGNET; Low-Grade Glioma, LGG; meningioma, MNG; MYB/MYBL1-altered diffuse astrocytoma, DA MYB/MYBL1; Neuroepithelial tumor with PATZ1 fusion, NET PATZ; Neuroepithelial tumor, PLAGL1-fused, NET PLAGL1; Non-classifiable, NC; Not done, ND; Pediatric diffuse high-grade glioma subtype MYCN, pHGGMYCN; Pilocytic astrocytoma, PA; Pilomyxoid glioma, PMG; Pleomorphic xanthoastrocytoma, PXA; Polymorphous low-grade neuroepithelial tumor of the young, PLNTY.
Low-grade glioma (LGG)
Four tumors had molecular findings aligning best with LGG (Figure 3A and B). The median age at diagnosis was 2.46 years (1.90–3.42 years). All patients were female, and one had M + disease. Three were enrolled on SJYC07 (1 high-risk, 2 low-risk), and one was treated with focal radiation and adjuvant chemotherapy. These tumors were classified as HGG at enrollment due to their infiltrative growth, mitotic activity, and high Ki-67 proliferation index, which ranged from 5% to 10% in multiple areas. Alterations in BRAF, MYBL1, MYB, and FGFR2 were found in these tumors after retrospective molecular analysis (Supplemental Figure 5).
High-grade glioma (HGG)
Six patients had HGG more commonly seen in older children (Figure 3A and B). The median age at diagnosis was 2.73 years (1.70–3.82 Years). One had M + disease. Four were enrolled on SJYC07 (2 high-risk, 2 low-risk). All tumors demonstrated typical features of HGG, including infiltrative growth, prominent mitotic activity, high Ki-67 labeling, and necrosis. In addition, these tumors had frequent large-scale chromosomal gains and losses. Focal CNVs were noted in MYCN (amplifications, 3) and CDKN2A/B (loss, 1) (Supplemental Figures 2A-D). Single tumors had mutations in TP53 (n = 1), H3F3A (n = 1), and NF1 (n = 1) (Figure 3B).
Other-CNS tumors
This group consisted of tumors that did not align well with a diagnosis of IHG, LGG, or HGG, including CNS BCOR ITD (3), CNS SARC CIC (1), NET PATZ1 (1), CNS NB FOXR2 (1), NET PLAGL1 (1), ET PLAG (1), ETMR (1). The median age at diagnosis was 1.83 years (1.02-3.63 years). All except one (ETMR) had M0 disease. All were enrolled on SJYC07 low-risk, except one high-risk (ETMR) and 1 NET PATZ1 treated on NPTP other risk. NGS testing showed BCOR ITD, CIC::DUX4 fusion, MN1::PATZ1 fusion, L1 promoter::FOXR2 fusion,22 EWSR1::PLAGL1 fusion, PLAG1 amplification, or gene fusion involving C19MC in these tumors (Supplemental Figure 6).
Clinical Outcome by Integrated Diagnosis
Survival analyses were conducted on the 41 patients with classifiable tumors. EFS and OS were significantly better for IHG when compared with HGG (p = 0.0043, p = 0.00013) and other CNS tumors (p = 0.011, p = 0.014). However, the EFS and OS were not significantly different between IHG and LGG (p = 0.95, p = 0.43) (Figure 4A).
Figure 4.
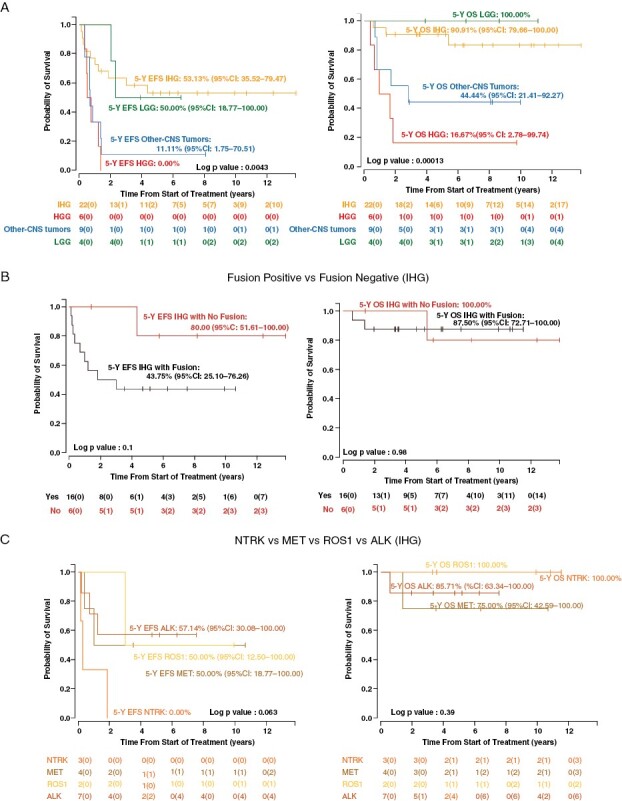
Event Free (EFS) and Overall Survival (OS) based on Integrated Diagnosis (A) EFS and OS by integrated diagnosis Tier 3 (IHG vs. LGG vs. HGG vs. Other-CNS Tumors). (B) EFS and OS in IHG based on the presence of RTK fusions (Fusion-positive vs. fusion-negative cases). (C) EFS and OS of the fusion-positive IHG patients based on the fusion type.
Within the IHG cohort, gender and extent of tumor resection were not associated with differences in EFS (p = 0.32, p = 0.55) or OS (p = 0.14, p = 0.4) (Supplemental Figure 7). Metastatic disease, only present in 2/22 patients, was also not associated with EFS differences but its presence was associated with OS (p = 0.048 Supplemental Figure 6). Ten IHG patients had an event, and three died: one of secondary AML five years after therapy and two from tumor progression. Of the 2 patients who died of tumor, 1 refused treatment post-relapse, and 1 was treated with surgery followed by molecular targeted therapy. Seven patients were alive after progression or recurrence: 3 remain alive > 10 years after being treated with surgery and/or chemotherapy; 2 remain on molecular targeted therapy > 2 years after relapse; 1 progressed on molecular targeted therapy, was treated with surgery and focal RT, and remains alive > 3 years from radiation; 1 was treated only with surgery and remains alive, yet very neurologically compromised, 4 years later without any cancer-directed therapy (Supplemental Figure 8, Supplemental Table 1). EFS and OS for fusion-positive tumors compared to fusion-negative tumors were not significantly different (p = 0.1, p = 0.98) (Figure 4B). Also, no significant difference in survival was observed based on the type of RTK fusions in the tumor (Figure 4C, p = 0.063, p = 0.39).
Among the LGG, there were two events and no deaths. One patient with MYB-fused DA progressed on SJYC07 low-risk arm and was salvaged with surgery and focal radiation (Supplemental Figure 8). The other patient had BRAF-altered PA treated on SJYC07 high-risk arm, relapsed after 16 months from therapy completion, and was salvaged by MEK inhibitor (Supplemental Table 1).
On the other hand, all patients with HGG had disease progression, and five died of their disease despite being treated with radiation therapy post-relapse (Supplemental Table 1, Supplemental Figure 8). Similarly, eight of the nine patients with other CNS tumors had disease progression, and five died of disease while three were salvaged (Supplemental Table 1, Supplemental Table 7).
Intellectual Outcome
Given the vulnerable age group and high survival of this population, neurocognitive data were analyzed to see how patients and, particularly, survivors fared after therapy. Twenty-four patients (10 IHG and 14 non-IHG) had their intelligence quotient (IQ) tested and recorded at at least two longitudinal time points. Of the non-IHG cases, 1 (6.6%) had HGG, 2 (13.3%) had LGG, 4 (26.6%) had other CNS tumors, and 7/15 (46.6%) had NC tumors. Only 4 of the 24 patients received radiation, all belonging to the non-IHG group (1 HGG, 1 LGG, 1 other CNS tumor, and 1 NC). The entire cohort’s IQ showed an average progressive decline of 3.18 points per year (Figure 5A). No difference was noted in the rate of IQ decline between IHG and non-IHG (p = 0.7796). More details on neurocognitive analysis are available in the supplemental section.
Figure 5.
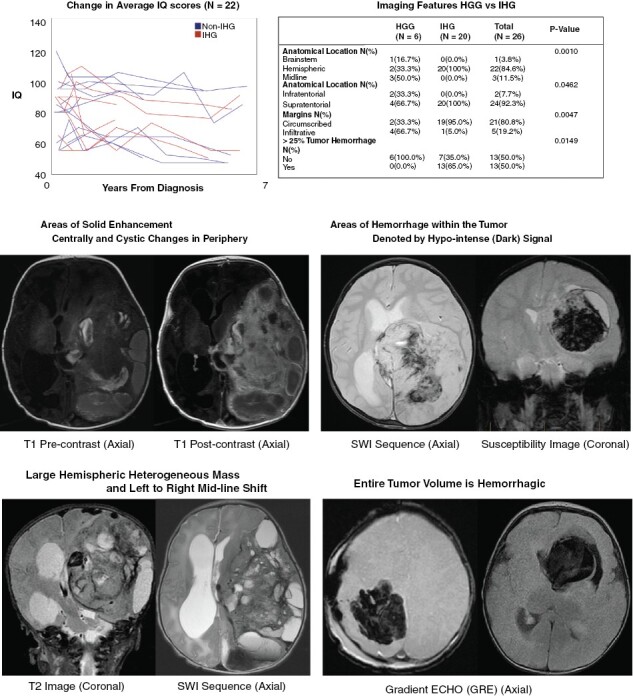
Neurocognitive and radiographic features of HGG in young children: (A) Change in IQ in 22 patients enrolled on SJYC07. (B) Comparison of Imaging features (anatomical location, tumor margin, and hemorrhagic changes within the tumor) in IHG vs. HGG. (C-F) Representative images of IHG.
Imaging Review
Of the 41 cases that had integrated diagnosis of IHG, HGG, LGG and Other CNS tumors pre-operative contrast- and non-contrast-enhanced cross-sectional MR and CT images of 35 cases—HGG (n = 6), LGG (n = 4), IHG (n = 20), and other CNS tumors (n = 5)—were available for review. Anatomical location (brainstem vs. hemispheric vs. midline) (p = 0.0011), tumor margin (circumscribed vs. infiltrative) (p = 0.0073), restricted diffusion (p = 0.0057), T1 hypointensity (p = 0.0207), and ≥ 25% tumor hemorrhage (p = 0.036) were significantly different among the diagnosis categories (Supplemental Table 3). As compared to HGG, IHGs were more likely to be circumscribed (95%, p = 0.0047) and localized to the hemispheres (100%, p = 0.0010, Figure 5B-D), whereas HGGs were mostly infiltrative (66.7%) and found in multiple locations—hemisphere (33.3%), midline (50.0%), and brainstem (16.7%). Furthermore, a majority (65%; 13/20) of IHG had at least 25% of the tumor volume occupied by hemorrhage, while none of the HGGs were noted to be hemorrhagic (Figure 5E-F). Five of the 13 hemorrhagic IHGs were > 75% hemorrhagic.
Surgery Review
Given the imaging differences described, we reviewed surgeries to explore differences in surgical outcomes by integrated diagnosis. Indeed, 77% (17/22) of IHGs had a GTR/NTR as compared to 17% (1/6) of HGG (p = 0.013). Detailed surgical reports were available for 16 out of 22 (72%) of the IHG patients but a direct comparison of surgical morbidity with other entities could not be made due to limited numbers. Nevertheless, of these 16 patients, a total of 30 craniotomies were recorded: 6 (37.5%) had one craniotomy, 6 (37.5%) had two craniotomies, and 4 (25%) had 3 craniotomies. Forty-three percent (13/30) of the craniotomies were complicated by blood volume loss requiring massive colloidal and crystalloid transfusion during surgery. Thirteen percent (4/30) required resuscitation during surgery due to hypovolemic shock leading to a temporary cessation of surgery. Ten patients required a second surgery, and of them, 4 (40%) were performed in response to postoperative complications from the first surgery. These were to mitigate increasing intracranial pressure from postsurgical inflammation, postsurgical intratumoral hemorrhage, and large-volume blood loss from resection bed hemorrhage. There were no reports of pre-surgical embolization in any of the reviewed operative reports (Supplemental Table 4).
Discussion
In this study, we evaluated young children diagnosed with HGG and treated with similar strategies. The tumors were analyzed by DNA methylation profiling and next-generation sequencing, and the resulting integrated diagnosis was correlated with histopathology, clinical characteristics, imaging, and outcomes. The survival of the entire cohort was similar to previous studies and higher than what is expected for older children with HGG.2,10
We found histologically diagnosed HGGs in young children can be categorized by integrated diagnosis into four categories: IHG, HGG, LGG, and other CNS tumors.
About 40% of tumors belonged to the IHG group and occurred in very young patients as large, hemispheric, and, generally, nonmetastatic tumors. RTK gene fusions were common and present in > 70% of IHG in our cohort.23–25 The OS was excellent (5 yr ≈ 91%) and consistent with the high survival rates of young children with HGG,1,4–7,9 however, the EFS (5 yr ≈ 53%) showed that close to half of these tumors still progressed after initial therapy. Despite this observation we were not able to identify any significant risk factors for progression among extent of resection, protocol therapy vs NPTP, presence or absence of fusion, except for metastatic disease which only occurred in < 10% of patients with IHG. Yet, remarkably, despite this relatively high progression rate, most patients were salvaged by either surgery, chemotherapy or use of targeted therapy and did not require radiation therapy. This suggests that an optimal treatment strategy has yet to be defined and merits further study.
About 12% of tumors in this cohort aligned with HGGs that are more commonly seen older children (i.e. pHGGMYCN, DMG H3K27M, PXA). The average age was older, and the survival was much poorer (17% OS) than patients with IHG. This is important since it suggests survival is more closely associated with biologic type of disease rather than the age at which a patient is diagnosed. In other words, even though young patients with HGG generally have a favorable prognosis, if they are diagnosed with a DMG H3K27M they will have a poor prognosis regardless of their age at diagnosis.
About 8% of the tumors showed molecular characteristics of LGG, having been classified when current molecular analyses were unavailable and histopathologic features prompted a diagnosis of HGG, according to WHO criteria of the time for diffuse gliomas. Despite some high-risk clinical features and progressive disease in two of four patients, they exhibited excellent outcomes (100% OS), suggesting that the increased mitotic activity and high Ki-67 immunolabeling observed in these tumors do not correlate with inferior patient outcomes. Although, given the limited number of patients with LGG within our cohort, these results require further verification in larger a cohort.
Finally, about 18% belonged to other CNS tumors (i.e. CNS BCOR ITD, CNS NB FOXR2, CNS SARC CIC, NET PATZ1, ET PLAG, ETMR, NET PLAGL1). These patients were older at diagnosis and had poor survival on therapy intended for young children with HGG.
Our findings underscore the importance of integrating layers of information, as recommended by the current 5th edition of the WHO classification of CNS tumors, to reach an integrated diagnosis.26 This approach is essential for improving diagnostic accuracy and prognostication, selecting the appropriate therapy, and comparing data across studies. Given the rarity of these tumors, we have constructed a visually interactive and easy-to-use portal (https://viz.stjude.cloud/st-jude-childrens-research-hospital/visualization/infant-high-grade-gliomas~879), which allows users to select their cohort of interest, search relevant molecular findings, explore treatment, and generate survival outcomes. Links to the raw molecular data are also available to facilitate the comparison of our data to additional samples.
We also showed that the IHGs have distinguishing imaging features that differentiated them from other infantile tumors. When compared to other tumors in our cohort, IHGs always occurred in the cerebral hemispheres and had well-circumscribed margins with large tumor volumes being occupied by hemorrhage. Consequently, identifying these radiographic features early in the course of the disease may prove valuable for clinical decision-making particularly regarding the immediate needs for extensive surgical resection versus diagnostic sampling via biopsy.
In addition, through serial neurocognitive evaluations on a subset of patients, we observed, that the consequences of surgery and chemotherapy are not trivial. Likely owing to the young age at diagnosis and the extensive surgery required, children with these tumors experienced a significant neuro-cognitive decline, even though most of the patients did not receive radiation therapy. Therefore, although > 90% of patients with IHG survived their disease, the morbidity was high, calling into question the merit of continuing the current approach of maximal surgical resection and chemotherapy.
Promising activity in refractory brain tumors for agents that target NTRK1/2/3, ALK, and ROS1 (i.e. entrectinib, larotectinib, and lorlatinib)23–25 highlights the need to consider using these agents in frontline therapy in formulations amenable to young children. Could targeted therapy replace surgery and chemotherapy altogether or reduce the tumor and decrease surgical morbidity? Despite this temptation, an abundance of caution is advised. Little is known about the long-term effects of these medicines on children, and we strongly recommend any novel approach be implemented in controlled and well-monitored clinical trials. While notable responses have been observed on these targeted agents, the disease often returned upon discontinuation of the drug such that the drug is more of a chronic controller than a cure, as we have seen with other RTKIs in hematologic, solid and CNS malignancies, thereby rendering the patient dependent upon the inhibitor indefinitely.,23–25 With weight gain, hypercholesterolemia, and bone fractures already being reported and long-term effects on development and cognition remaining unknown, this long-term dependence remains concerning.23–25 Currently, a pilot study is testing the efficacy of larotrectinib in newly diagnosed HGG with NTRK1/2/3 fusions (NCT04655404). However, this approach excludes IHG patients with other RTK fusions or fusion-negative tumors and larger international collaborative studies that include all IHG patients appears to be the only viable way toward defining the best approach for these rare tumors.
Limitations of this study include the small sample size, use of a prospective clinical trial cohort (SJYC07) combined with a retrospective cohort (NPTP), and the challenge of handling changing diagnoses over time as diagnostic testing improved. Consequently, our results should be interpreted with caution and as evidenced by the wide confidence interval may contain findings that will only be identified through exploration of larger datasets. We included NPTP patients with a slightly higher age cutoff (0–5 years) in order to capture more subjects and although there were no significant differences noted in the EFS and OS for NPTP vs SJYC07 patients the difference in data collection lends itself to bias. Nevertheless, with such improvements in discriminating tumor types, we hope to have demonstrated the necessity of re-evaluating available cohorts using these integrated approaches to better inform treatment decisions. In addition, although our results with surgery and chemotherapy are encouraging, the surviving patients are mainly from the IHG and LGG groups. Given that other retrospective studies have reported similar trends with a mixture of therapies, our results suggest that the outcome is biologically driven, at least in part. Hence, further reduction of treatment-related toxicity needs to be explored. Furthermore, given the rarity of IHG, international collaboration and data sharing are critical for future clinical trials if we expect to advance cure rates while minimizing toxicity.
Supplementary Material
Acknowledgment
The authors would like to thank Matthew Lear at St. Jude Biorepository, Julie Justice in the Department of Pathology, and Emily Walker and Geoffrey Neale in the Hartwell Center for their assistance in the molecular characterization of the tumors. This study was previously presented at the 2022 Annual Meeting of the American Association of Neuropathologists and at the 2023 Society for Neuro-Oncology's 7th Biennial Pediatric Neuro-Oncology Research Conference.
Contributor Information
Jason Chiang, Department of Pathology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Aditi Bagchi, Department of Oncology, Division of Neuro-Oncology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Xiaoyu Li, Department of Pathology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Sandeep K Dhanda, Department of Oncology, Division of Neuro-Oncology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Jie Huang, Department of Biostatistics, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Soniya N Pinto, Department of Diagnostic Imaging, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Edgar Sioson, Department of Computational Biology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
James Dalton, Department of Pathology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Ruth G Tatevossian, Cancer Biomarkers Laboratory, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Sujuan Jia, Cancer Biomarkers Laboratory, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Sonia Partap, Department of Neurology and Neurological Sciences, Stanford University, Stanford, CA, USA.
Paul G Fisher, Department of Neurology and Neurological Sciences, Stanford University, Stanford, CA, USA.
Daniel C Bowers, Division of Pediatric Hematology-Oncology, University of Texas Southwestern Medical School, Dallas, TX, USA.
Timothy E G Hassall, Queensland Children’s Hospital, Brisbane, QLD, Australia.
Congyu Lu, Department of Computational Biology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Airen Zaldivar-Peraza, Department of Computational Biology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Karen D Wright, Dana Farber/Boston Children’s Cancer and Blood Disorders Center, Boston, MA, USA.
Alberto Broniscer, Department of Oncology, Division of Neuro-Oncology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Ibrahim Qaddoumi, Department of Oncology, Division of Neuro-Oncology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Santhosh A Upadhyaya, Department of Pediatrics, Michigan Medicine, University of Michigan, Ann Arbor, MI, USA.
Anna Vinitsky, Department of Oncology, Division of Neuro-Oncology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Noah D Sabin, Department of Diagnostic Imaging, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Brent A Orr, Department of Pathology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Paul Klimo, Jr, Department of Surgery, St Jude Children’s Research Hospital, Memphis, TN, USA; Department of Neurosurgery, University of Tennessee Health and Science Center, Memphis, TN, USA; Le Bonheur Neuroscience Institute, Le Bonheur Children’s Hospital, Memphis, TN, USA.
Frederick A Boop, Department of Surgery, St Jude Children’s Research Hospital, Memphis, TN, USA; Department of Neurosurgery, University of Tennessee Health and Science Center, Memphis, TN, USA; Le Bonheur Neuroscience Institute, Le Bonheur Children’s Hospital, Memphis, TN, USA.
Jason M Ashford, Department of Psychology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Heather M Conklin, Department of Psychology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Arzu Onar-Thomas, Department of Biostatistics, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Xin Zhou, Department of Computational Biology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
David W Ellison, Department of Pathology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Amar Gajjar, Department of Oncology, Division of Neuro-Oncology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Giles W Robinson, Department of Oncology, Division of Neuro-Oncology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Conflict of Interest
S.K.D.- Monitoring or Advisory board: Kiromic. D.W.E.-Patent: Sole inventor of U.S. patent no. 9,005,907. A.G. - Monitoring or Advisory board: Day One Biopharmaceuticals. T.E.G.H. - Leadership or fiduciary role in advocacy group: Chair of Brain Child (not for profit charity). AOT - Contracts/Research Grants: Roche, Novocure, Apexigen, Senhwa, Incyte, Y-mAbs, MimiVax, National Brain Tumor Foundation; Monitoring or Advisory board: Hospital for Sick Kids, The Brain Tumor Charity. S.P. - Grants: Alex's Lemonade Stand, Pediatric Brain Tumor Consortium; Honoraria: Society for Neuro-Oncology (speaker), American Academy of Neurology (speaker). I.Q. - Monitoring or Advisory Board: Springworks, AztraZeneca. S.A.U. - Monitoring or Advisory Board: Nationwide Children's Oncology DSMB. The remaining authors declare no conflicts of interest.
Authorship
Experimental design: AG, GWR
Implementation: JC, AB, XL, SKD, JH, JD, DWE, AOT, GWR
Analysis and interpretation of the data: JC, AB, XL, SKD, JH, JD, AOT, DWE, GWR
Writing of the manuscript: all authors
All authors have read and approved the final version.
Funding
National Cancer Institute (P30CA021765 to JC, AG, GWR, P01CA096832 to JC; CCSG 5P30CA021765-43 Developmental Funds to GWR, XZ); Press On Fund (to GWR);
American Lebanese Syrian Associated Charities (to JC, AG, GWR)
References
- 1. Duffner PK, Krischer JP, Burger PC, et al. Treatment of infants with malignant gliomas: the Pediatric Oncology Group experience. J Neurooncol. 1996;28(2–3):245–256. [DOI] [PubMed] [Google Scholar]
- 2. Guerreiro Stucklin AS, Ryall S, Fukuoka K, et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun. 2019;10(1):4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mackay A, Burford A, Carvalho D, et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell. 2017;32(4):520–537.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dufour C, Grill J, Lellouch-Tubiana A, et al. High-grade glioma in children under 5 years of age: a chemotherapy only approach with the BBSFOP protocol. Eur J Cancer. 2006;42(17):2939–2945. [DOI] [PubMed] [Google Scholar]
- 5. Geyer JR, Finlay JL, Boyett JM, et al. Survival of infants with malignant astrocytomas. A Report from the Childrens Cancer Group. Cancer. 1995;75(4):1045–1050. [DOI] [PubMed] [Google Scholar]
- 6. Grundy RG, Wilne SH, Robinson KJ, et al. ; Children's Cancer and Leukaemia Group (formerly UKCCSG) Brain Tumour Committee. Primary postoperative chemotherapy without radiotherapy for treatment of brain tumours other than ependymoma in children under 3 years: results of the first UKCCSG/SIOP CNS 9204 trial. Eur J Cancer. 2010;46(1):120–133. [DOI] [PubMed] [Google Scholar]
- 7. Duffner PK, Horowitz ME, Krischer JP, et al. The treatment of malignant brain tumors in infants and very young children: an update of the Pediatric Oncology Group experience. Neuro Oncol. 1999;1(2):152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Macy ME, Birks DK, Barton VN, et al. Clinical and molecular characteristics of congenital glioblastoma. Neuro Oncol. 2012;14(7):931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. El-Ayadi M, Ansari M, Sturm D, et al. High-grade glioma in very young children: a rare and particular patient population. Oncotarget. 2017;8(38):64564–64578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clarke M, Mackay A, Ismer B, et al. Infant high-grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov. 2020;10(7):942–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu G, Diaz AK, Paugh BS, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46(5):444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Robinson GW, Rudneva VA, Buchhalter I, et al. Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol. 2018;19(6):768–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Upadhyaya SA, Robinson GW, Onar-Thomas A, et al. Molecular grouping and outcomes of young children with newly diagnosed ependymoma treated on the multi-institutional SJYC07 trial. Neuro Oncol. 2019;21(10):1319–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chiang JCH, Harreld JH, Tanaka R, et al. Septal dysembryoplastic neuroepithelial tumor: a comprehensive clinical, imaging, histopathologic, and molecular analysis. Neuro Oncol. 2019;21(6):800–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chiang J, Diaz AK, Makepeace L, et al. Clinical, imaging, and molecular analysis of pediatric pontine tumors lacking characteristic imaging features of DIPG. Acta Neuropathol Commun. 2020;8(1):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. He C, Xu K, Zhu X, et al. Patient-derived models recapitulate heterogeneity of molecular signatures and drug response in pediatric high-grade glioma. Nat Commun. 2021;12(1):4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ali JS, Ashford JM, Swain MA, et al. Predictors of cognitive performance among infants treated for brain tumors: findings from a multisite, prospective, longitudinal trial. J Clin Oncol. 2021;39(21):2350–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou X, Edmonson MN, Wilkinson MR, et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet. 2016;48(1):4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803–820. [DOI] [PubMed] [Google Scholar]
- 21. Olsen TK, Panagopoulos I, Meling TR, et al. Fusion genes with ALK as recurrent partner in ependymoma-like gliomas: a new brain tumor entity? Neuro Oncol. 2015;17(10):1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Flasch DA, Chen X, Ju B, et al. Somatic LINE-1 promoter acquisition drives oncogenic FOXR2 activation in pediatric brain tumor. Acta Neuropathol. 2022;143(5):605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bagchi A, Orr BA, Campagne O, et al. Lorlatinib in a child with ALK-fusion-positive high-grade glioma. N Engl J Med. 2021;385(8):761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Desai AV, Robinson GW, Gauvain K, et al. Entrectinib in children and young adults with solid or primary CNS tumors harboring NTRK, ROS1, or ALK aberrations (STARTRK-NG). Neuro Oncol. 2022;24(10):1776–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Doz F, van Tilburg CM, Geoerger B, et al. Efficacy and safety of larotrectinib in TRK fusion-positive primary central nervous system tumors. Neuro Oncol. 2022;24(6):997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Louis DN, Perry A, Wesseling P, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23(8):1231–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.