

Abstract
Background
Functional inactivation of ATRX characterizes large subgroups of malignant gliomas in adults and children. ATRX deficiency in glioma induces widespread chromatin remodeling, driving transcriptional shifts and oncogenic phenotypes. Effective strategies to therapeutically target these broad epigenomic sequelae remain undeveloped.
Methods
We utilized integrated multiomics and the Broad Institute Connectivity Map (CMAP) to identify drug candidates that could potentially revert ATRX-deficient transcriptional changes. We then employed disease-relevant experimental models to evaluate functional phenotypes, coupling these studies with epigenomic profiling to elucidate molecular mechanism(s).
Results
CMAP analysis and transcriptional/epigenomic profiling implicated the Class III HDAC Sirtuin2 (SIRT2) as a central mediator of ATRX-deficient cellular phenotypes and a driver of unfavorable prognosis in ATRX-deficient glioma. SIRT2 inhibitors reverted Atrx-deficient transcriptional signatures in murine neuroepithelial progenitor cells (mNPCs), impaired cell migration in Atrx/ATRX-deficient mNPCs and human glioma stem cells (GSCs), and increased expression of senescence markers in glioma models. Moreover, SIRT2 inhibition impaired growth and increased senescence in ATRX-deficient GSCs in vivo. These effects were accompanied by genome-wide shifts in enhancer-associated H3K27ac and H4K16ac marks, with the latter in particular demonstrating compelling transcriptional links to SIRT2-dependent phenotypic reversals. Motif analysis of these data identified the transcription factor KLF16 as a mediator of phenotype reversal in Atrx-deficient cells upon SIRT2 inhibition.
Conclusions
Our findings indicate that SIRT2 inhibition selectively targets ATRX-deficient gliomas for senescence through global chromatin remodeling, while demonstrating more broadly a viable approach to combat complex epigenetic rewiring in cancer.
Keywords: astrocytoma, ATRX, epigenetics, glioma, SIRT2
Key Points.
1 SIRT2 expression correlates with unfavorable prognosis in ATRX-deficient glioma.
2 SIRT2 inhibition induces cellular senescence and impairs ATRX-deficient glioma growth.
3 SIRT2 inhibition modulates chromatin landscapes to impact transcriptional regulation.
Importance of the Study.
Epigenomic dysfunction is increasingly implicated in cancer, with molecular abnormalities in established epigenetic regulator genes defining many tumor types. Tractable therapeutic strategies addressing this often complex pathobiology remain elusive. In this study, we synthesize in silico analyses, in vitro and in vivo disease modeling, and global epigenomic profiling to characterize a viable approach for targeting malignant gliomas harboring inactivation of the chromatin remodeling gene ATRX. Our findings have translational implications for the treatment of currently incurable brain tumors and, in a broader sense, provide a workable framework for systematically targeting epigenomic dysfunction in cancer.
Malignant gliomas are the most common primary brain tumors and despite considerable molecular and clinical heterogeneity, are uniformly associated with refractivity to standard therapeutic modalities and poor overall survival.1 Innovative treatment strategies targeting the precise molecular mechanisms underlying glioma pathogenesis are urgently needed. Functional inactivation of the ATRX gene (α-thalassemia mental retardation X-linked), most commonly through loss-of-function mutations or deletion, defines major glioma subclasses also characterized by concurrent mutations in TP53, and in genes encoding either IDH1 or IDH2 metabolic enzymes in adults or histone H3 monomers in children.2–5ATRX encodes a SWI/SNF family chromatin remodeler and appears to play a central role in the mediation of chromatin state, gene expression, and genomic integrity in certain cell types.6–12 ATRX deficiency occurs at high levels across a range of neuroepithelial and mesenchymal neoplasms, where it is thought to induce alternative lengthening of telomeres (ALT), a telomerase-independent telomere maintenance mechanism.13,14 ATRX deficiency also promotes replication stress and DNA damage, likely through accumulation of G-quadruplex DNA secondary structures genome-wide and modulates developmentally relevant transcriptional programs through dysregulated chromatin marks and epigenomic landscapes.11,12,15,16 Strategies to therapeutically combat the broad physiological effects of ATRX deficiency remain unclear.
Recent work in multiple tumor variants has shown the feasibility of targeting epigenetic abnormalities to suppress oncogenic phenotypes in cancer cells, pointing to potential efficacy for analogous approaches in ATRX-deficient glioma.17 We previously demonstrated that ATRX deficiency induces cellular differentiation and motility phenotypes in primary murine neuroepithelial progenitor cells (mNPCs), recapitulating disease-defining characteristics of ATRX-mutant glioma through induction of genome-wide shifts in chromatin accessibility and underlying transcription.11 These findings suggest that sufficiently reverting the ATRX-deficient epigenomic state, along with its broad transcriptional sequelae, represents a tractable strategy for therapeutic development. To address this possibility, we coupled CMAP analysis with integrated epigenomic and transcriptional data to identify SIRT2 as a driver of ATRX-deficient oncogenic phenotypes. Targeting SIRT2 with small molecule inhibitors reverted ATRX-deficient transcriptional signatures, impairing cellular motility and promoting senescence in Atrx/ATRX-deficient mNPCs/human GSCs. Subsequent in vivo studies demonstrated that SIRT2 inhibition selectively impaired growth and promoted senescence in ATRX-deficient GSC xenografts. Finally, epigenomic profiling revealed that these therapeutically relevant effects were associated with shifts in H3K27ac and H4K16ac landscapes, along with underlying gene expression programs. Taken together, our findings demonstrate that SIRT2-dependent epigenomic rewiring promotes ATRX-deficient oncogenic phenotypes and can be effectively targeted by small molecular inhibitors to induce senescence in ATRX-mutant gliomas.
Materials and Methods
CLUE Data Analysis
CLUE (https://clue.io/) is a cloud-based hub designed to identify candidate drug compounds by way of transcriptional signatures.18 Genes differentially expressed between Atrx− and Atrx+ mNPCs, were mapped, ranked, and filtered for high fold changes (>1.5-fold change) and low p-values (<0.01) and matched to human orthologs using the DAVID conversion tool (https://david.ncifcrf.gov/conversion.jsp). These signatures were then correlated with transcriptional profiles generated by perturbagen exposure in nine human cancer cell lines (HEPG2, MCF7, HT29, HCC515, HA1E, A459, A375, VCAP, and PC3) using the LINCS (L1000) database. All results were then ranked and clustered by strength of anti-correlation with the Atrx-deficient transcriptional signature.
Cell Lines and Drugs
All cell lines used in this study were tested for mycoplasma contamination every three months and used at minimal passage number. mNPCs (Atrx+ and Atrx−) were generated as described previously11 and cultured in NeuroCult Basal Medium containing NeuroCult Proliferation Supplement, 20 ng/ml EGF, 10 ng/ml basic FGF, 2 μg/ml heparin (Stemcell Technologies). GS 522, TS 603, GSC 246, GSC 236, TS 543, and TS 543-shATRX have been described previously11,12 and were cultured in DMEM/F12 media with 20 ng/ml EGF, 10 ng/ml basic FGF, 2 μg/ml heparin (Stemcell Technologies). The status of ATRX, TP53 and IDH1 (R132H) were routinely assessed in all lines either western blot or Sanger sequencing. AGK2, AK7 and Thiomyristoyl were obtained from SelleckChem.
Quantitative Reverse Transcriptase PCR
Total RNA was extracted using Qiagen RNeasy Plus, followed by DNAse treatment, following manufacturer’s instructions. 1 μg of total RNA from each sample was then converted to cDNA using First Strand cDNA Synthesis Kit (#K1612, Thermo), followed by qPCR using Power SYBR Green PCR Master Mix (Applied Biosystems). Data were analyzed by the ΔΔCt method using GAPDH as a housekeeping gene. Primers for Quantitative Reverse Transcriptase PCR (RT-qPCR) are listed in Supplementary Table 1.
Cell Viability Assays
Assays incorporated 5000 cells/well with serial concentrations for AGK2 (1–10 μM), AK7 and Thiomyristoyl (10–60 μM) incubated for 48 hours in 96-well plates. Viability was then assessed with the CellTiter-Glo Luminescence assays (Promega) according to manufacturer-recommended procedures and quantified using a Tecan INFINITE M1000 PRO luminometer.
Cell Migration Assays
mNPCs and human GSC lines were seeded on laminin (20 μg/ml) coated 96 well dishes. Cells were grown into a dense monolayer and then scratched with the Wound Maker (Essen BioScience), following manufacturer instructions. Sirt2i (AGK2: 1 μM, AK7 and Thiomyristoyl: 10 μM) and DMSO were added with appropriate media and wound closure was monitored using an IncuCyte Zoom System (Essen BioSciences) for 48 hours. Images were analyzed with Incucyte Scratch Wound Cell Migration Software (Essen BioSciences).
RNA-seq and Data Analysis
Atrx- mNPCs were treated with Sirt2 inhibitors (AGK2: 1 μM, AK7 and Thiomyristoyl: 10 μM) or DMSO vehicle for 48 hours, followed by total RNA extraction using Qiagen RNeasy Plus kit (three biological replicates), according to manufacturer’s instructions. Subsequent processing and sequencing occurred at the MD Anderson Advanced Technology and Genomics Core, (ATGC). Following Agilent BioAnalyzer quality assessment, libraries were generated using Truseq preparation kits (Illumina) and samples run on the HiSeq4000 platform in a 76 bp pair-end sequencing format. Raw fastq files were subjected to FASTQC analysis for quality control, followed by alignment to the mouse mm9 genome using RNASTAR (version 2.7.8a). Raw transcript counts were generated using HTseq-count tool (version 0.9.1), followed by Principal Component Analysis (PCA). Differentially expressed genes (DEGs) were calculated using DESeq2 (version 2.11.40.7) and were later subjected to Gene Set Enrichment Analysis (GSEA) analysis using a desktop version of the tool.
Antibodies
All commercial antibodies utilized for ChIP-seq, western blotting, and immunofluorescence in this study are listed in Supplementary Table 3.
ChIP-seq Analysis
Atrx- mNPCs were treated with either AGK2 (1 μM) or DMSO vehicle for 48 hours, fixed with 37% formaldehyde, and quenched with 125 mM glycine. Samples were then sonicated using a Bioruptor Plus (Diagenode) and immunoprecipitated using antibodies to H3K4me1, H3K27ac, H3K27me3, and H4K16ac. DNA was reverse-crosslinked for 4 hours, followed by solid-phase reversible immobilization (SPRI) beads clean-up and eluted in Tris-EDTA buffer. Eluted DNA was assessed for quality, followed by library preparation and run on a Novaseq6000 using a 50 bp single-end read format at the ATGC. Generated ChIP-seq data underwent quality-control measures using the FASTQC tool and aligned to the mm9 reference genome with Bowtie2 (version 2.4.5). Only unique mapped reads were retained. MACS2 callpeak (version 2.2.7.1) was used for calling peaks from BAM files with default parameters. BED files were further subjected to gene annotation using GREAT (version 4) analysis with Basal plus extension parameters (Proximal: 5kb upstream, 1kb downstream, plus distal up to 1000kb). For MEME analysis, ChIP-MEME was utilized using default parameters. Peak annotation and visualization were performed using the PAVIS annotation tool. ChIP-seq heatmaps were generated using seqMINER. For Super-enhancer calling, ranking of super-enhancers (ROSE) algorithm was used on H3K27ac ChIP-seq data, followed by pathway prediction analysis using Enrichr.
Immunofluorescence Analysis
For immunostaining, mNPCs were grown on chamber slides treated with Sirt2i (AGK2: 1 μM, AK7 and Thiomyristoyl: 10 μM) for 48 hours and were fixed using 4% paraformaldehyde for 10 min. Cells were permeabilized using 0.5% Tween-20, 0.2% Triton X-100 in PBS for 10 min and blocked for 1 hour 5% BSA solution, followed by overnight incubation with primary antibody. Slides were washed 3 times with PBST solution and incubated with Alexa Fluor 488/594 secondary antibodies (Invitrogen) at room temperature for 1 hour. Slides were washed thrice and mounted with coverslips using DAPI counterstain (#H-1200-10, Vector laboratories). Imaging was done using an Olympus confocal microscope (FV-1000), and Z-stack images captured using a 60× objective with a pixel size of 0.1-µm and 0.5-µm depth. Images were processed using Fiji software.
Chromatin Condensation Parameter (CCP) Analysis
Measurement for chromatin condensation was performed using single nuclei DAPI stained images for mNPCs treated with Sirt2i (AGK2: 1 μM, AK7 and Thiomyristoyl: 10 μM) for 48 hours. CCP was developed in MATLAB and utilized the Sobel edge detection algorithm for analysis. Z-stack images were thresholded and processed using Fiji software followed by CCP analysis.
Protein Isolation and Western Blot:
mNPCs were treated with Sirt2i (AGK2: 1 μM, AK7 and Thiomyristoyl: 10 μM) for 48 hours, lysed in RIPA buffer (150 mM NaCl; 50 mM Tris pH 8.0; 1.0% IGEPAL CA-630; 0.5% sodium deoxycholate; 0.1% SDS; Sigma–Aldrich) supplemented with protease inhibitors (cOmplete mini, Roche Diagnostics; 1 mM PMSF; 10 mM NaF; 2.5 mM Na3VO4) and centrifuged at 10,000 × g at 4 ° C for 30 min. Resulting supernatants were quantified using BCA protein assays (#23225, Thermo) and immunoblotted with standard procedures.
Senescence β-Galactosidase Staining
Sirt2i (AGK2: 1 μM, AK7 and Thiomyristoyl: 10 μM) or vehicle-treated mNPCs and human GSCs were treated for 48 hours. The cells were fixed using 4% paraformaldehyde for 10 min using the Senescence β-Galactosidase Staining Kit and stained for β-galactosidase overnight following manufactures instructions (#9860, CST). Cells were then washed with PBS twice and β-galactosidase-positive constituents counted at low magnification (20x). Images were taken using Nikon ECLIPSE Ts2 inverted microscope and processed using Fiji software.
Generation of SIRT2 Knockdown Cell Lines
SIRT2 knockdown was achieved using a mammalian vector designed from Vectorbuilder. The vector carried shRNA against SIRT2 for human cells (shRNA1) and Sirt2 for mouse cells (shRNA2) (see Supplementary Table 2) with dual selection markers for EGFP and Puromycin. Murine NPCs and human GSC cells were transduced with the knockdown vectors, and subsequently, FACS sorting was performed based on EGFP expression. Cells that expressed EGFP were further selected using Puromycin (1 μg/ml) to ensure a high knockdown efficiency. To confirm SIRT2 depletion, RT-qPCR was performed using primers listed in Supplementary Table 1.
Mouse Xenograft Experiments
All animal procedures described herein were approved by Institutional Animal Care and Use Committee (IACUC) of M.D. Anderson Cancer Center (protocol # 00001597-RN02). TS543 and TS543-shATRX GSCs were grown in cell culture dishes, dissociated with Accutase (#07920, Stemcell), resuspended in DMEM/F12 media mixed 1:1 with Matrigel Growth Factor Reduced (GFR) Basement Membrane Matrix (#354230, Corning), and injected into nude mice flanks in a 50 μl aliquot containing 2 × 106 cells. Mice were randomly segregated into two groups: vehicle or AK-7-treated (40 mg/kg), with each administered via intraperitoneal injection daily until health-defined end points. Tumor size was measured with calipers. For growth curve analysis and survival studies, mice in study cohorts were sacrificed when tumors reached 2000 mm3 in total volume. The tumors were excised from sacrificed mice and subjected to routine histopathological processing.
Immunohistochemistry
Five-micron FFPE sections were deparaffinized and subjected to antigen retrieval with citrate buffer (10 mM). Sections were then blocked with 2% goat serum for 1 hour, followed by overnight incubation with primary antibody at 4° C. Sections were washed with 0.01% PBST, incubated with ImmPRESS-HRP Goat Anti-Rabbit IgG secondary antibody (Vector Laboratories) for 1 hour, and developed using Alexa Fluor 488/594 Tyramide SuperBoost Kit. Slides were washed thrice with 0.01% PBST, mounted with coverslips using DAPI counterstain (#H-1200-10, Vector laboratories), and imaged using an Olympus confocal microscope (FV-1000). Images were processed using Fiji software.
TCGA and CGGA Gene Expression Data
Level-3 gene expression and methylation data were obtained from TCGA along with the corresponding sample annotation file (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga). For CGGA, RNA-seq count data, DNA methylation, and clinical information were downloaded from the access portal (http://www.cgga.org.cn/download.jsp).
Statistics
All statistical analyses were performed with either GraphPad Prism 8.0, R (v3.5.0), or Microsoft Excel. All results, unless otherwise stated, represent at least three independent experiments and are plotted as mean ± SEM. All data were statistically analyzed using unpaired or paired two-tailed t-tests. For transcriptional and epigenome data, the hypergeometric distribution was utilized and followed default statistical parameters. P values are represented using * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001.
Results
Integrated Multiomic Analysis Identifies SIRT2 as a Targetable Candidate Driving ATRX-deficient Phenotypes in Glioma
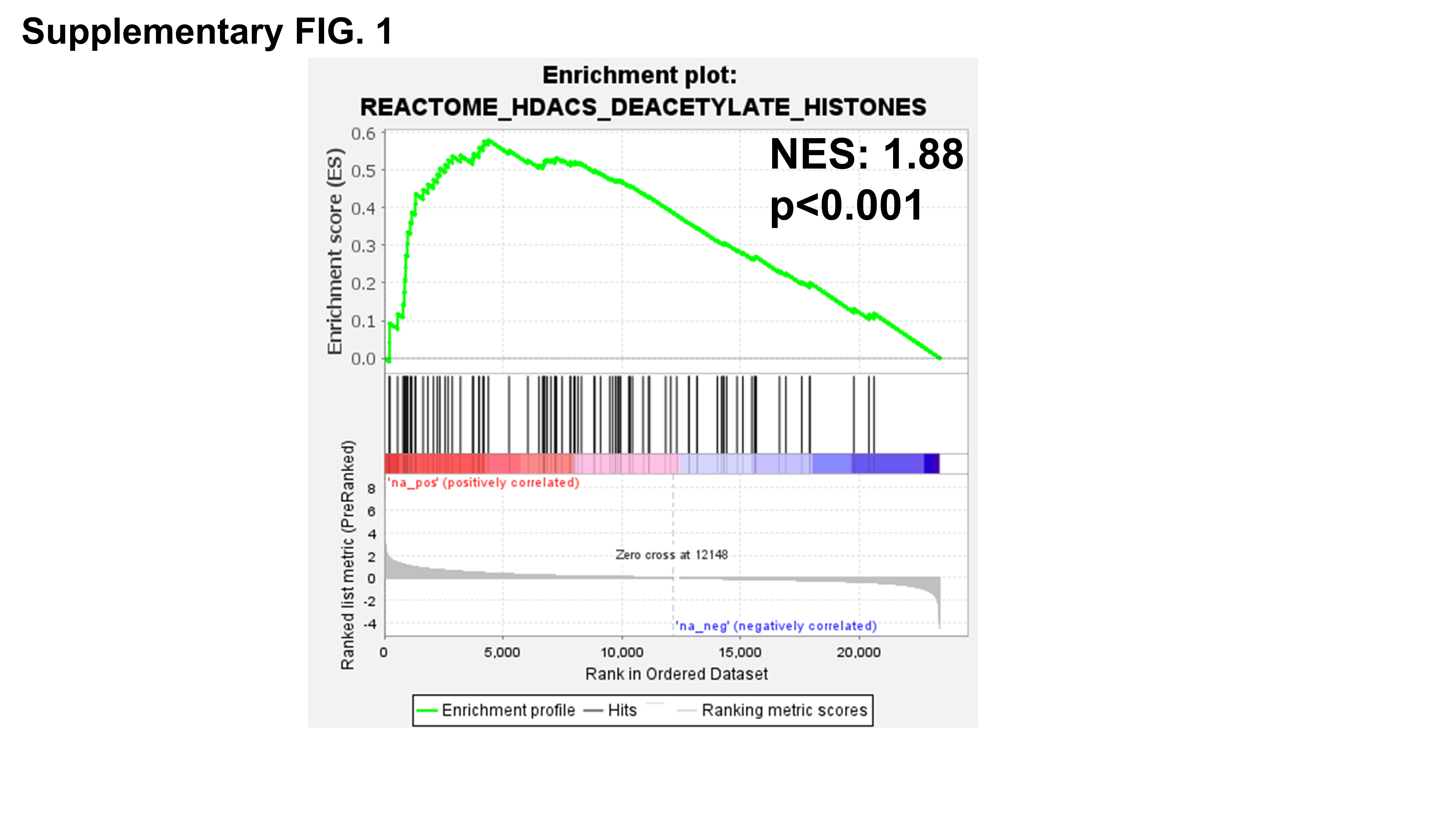
As ATRX deficiency has been repeatedly shown to alter chromatin architecture and phenotypically relevant gene expression, we hypothesized that effective pharmacological strategies to combat ATRX-deficiency would revert ATRX-deficient transcriptional signatures. To explore this possibility, we used the Broad Institute CMAP repository to interrogate our existing RNA-seq data derived from Atrx+ and Atrx− mNPCs. CMAP compiles transcriptional signatures from cell lines subjected to either genetic or pharmacologic perturbation.18 Using the CLUE algorithm and the LINCS (L1000) perturbagen database, we identified drug-treated gene expression signatures demonstrating strong negative correlations with Atrx-deficient transcriptional shifts in mNPCs. Of all perturbagens interrogated, 17 of the top 50 results were well-established HDAC inhibitors, including Vorinostat, Belinostat and Panobinostat (Figure 1A). Moreover, we found that HDAC-associated transcriptional signatures positively correlated by gene set enrichment analysis (GSEA) with transcriptional profiles of ATRX-deficient human gliomas from the Cancer Genome Atlas (TCGA) dataset (Supplementary Figure 1). In light of these findings, we integrated RNA-seq, ATAC-seq, and Atrx ChIP-seq data from mNPCs to determine the extent to which HDACs differentially expressed (fold change > 1.5, p < 0.05) in the Atrx-deficient setting were transcriptionally influenced through epigenetic mechanisms. While this analysis implicated multiple HDACs in some manner, only Sirtuin 2 (Sirt2) demonstrated positional association with an Atrx ChIP-seq peak and both increased gene expression and proximity (<2 kb) to a locus of increased chromatin accessibility—as determined by ATAC-seq—in the Atrx-deficient context (Figure 1B–C). Our prior work has shown that ATRX loss induces shifts in chromatin accessibility at vacant ATRX binding sites, mediated at least in part by misincorporation of H3.3 histone monomers.11 Sirt2 is an NAD+-dependent lysine deacetylase involved in a variety of biological processes and facilitates chromatin condensation by deacetylating lysine moieties on both histone 3 (H3K27) and histone 4 (H4K16) substrates.19–21
Figure 1.
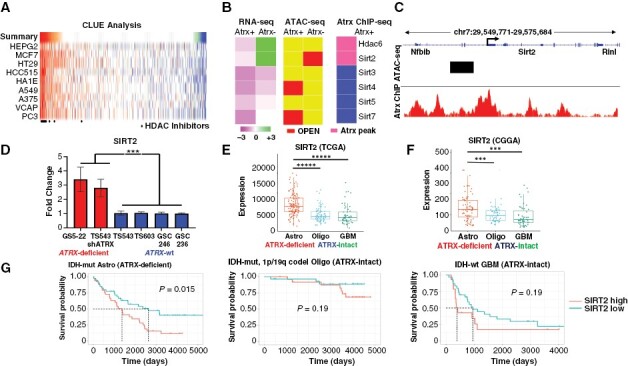
SIRT2 is upregulated in ATRX-deficient glioma. (A) Heatmap of CMAP analysis of Atrx-deficient transcriptional changes in mNPCs demonstrating strong negative correlations with several HDAC inhibitors. (B) Heatmaps showing transcript levels (RNA-seq), proximity to regions of increased chromatin accessibility (ATAC-seq; OPEN), and proximity to ATRX binding sites (Atrx ChIP-seq), of various HDACs in mNPCs (Atrx status shown). (C) Schematic of the Sirt2 locus showing associated Atrx binding peaks (Atrx ChIP) and region of increased chromatin accessibility in Atrx− mNPCs (ATAC-seq). (D) RT-PCR analysis showing increased SIRT2 expression in ATRX-mutant (GS5-22), and ATRX-knockdown (TS543-shATRX) GSCs, compared to ATRX-wildtype (TS543, TS603, GSC 246, and GSC 236) GSCs. Expression analysis in TCGA (E) and CGGA (F) datasets revealed increased SIRT2 levels ATRX-deficient gliomas relative to ATRX-intact counterparts. (G) Kaplan–Meier analysis showing that high SIRT2 expression is associated with poor prognosis in ATRX-deficient gliomas but not in ATRX-intact counterparts. * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001.
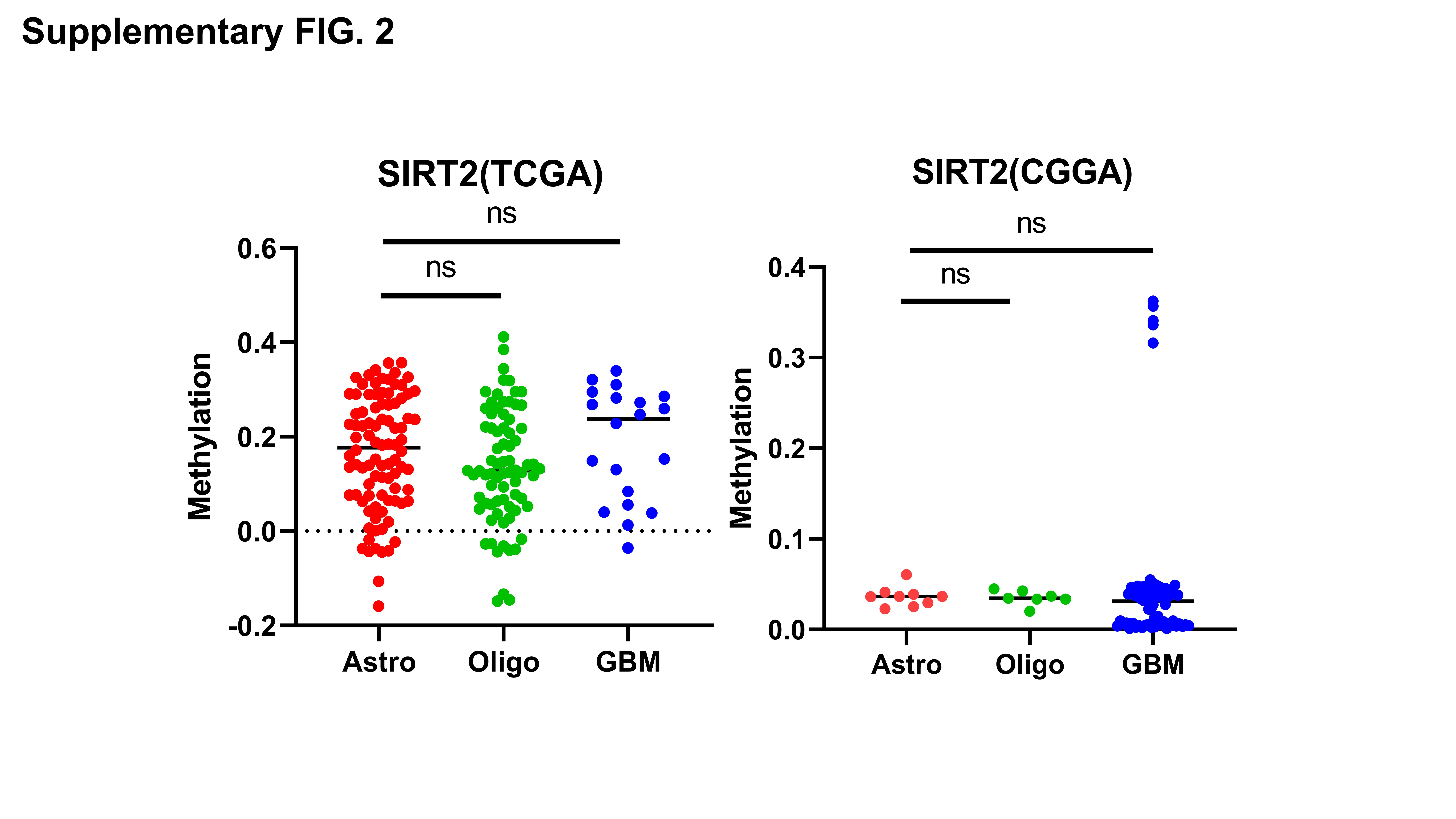
After implicating Sirt2 in ATRX-deficient gliomagenesis with our isogenic mNPCs, we sought to explore the potential clinical relevance of SIRT2 in human tumors. We first evaluated the expression of SIRT2 in a panel of patient-derived GSCs, featuring ATRX-mutant (GS5-22), ATRX-wildtype (TS543, GSC 246, GSC 236, and TS603), and ATRX-knockdown (TS543-shATRX) backgrounds. In total, these models feature both IDH-mutant (GS5-22 and TS603) and IDH-wildtype (TS543, TS543-shATRX, GSC 246, and GSC 236) gliomas, with ATRX-deficient (GS5-22 and TS543-shATRX) and ATRX-wildtype (TS543, TS603, GSC246, and GSC 236) variants represented in each. Using RT-PCR, we found that SIRT2 transcript levels were notably elevated in both ATRX-mutant and ATRX-knockdown GSCs relative to ATRX-wildtype isogenic and non-isogenic counterparts (Figure 1D). We also utilized publicly available and clinically annotated gene expression data from TCGA and the Chinese Glioma Genome Atlas (CGGA). ATRX-deficient gliomas are almost exclusively found in the IDH-mutant astrocytoma subclass of adult glioma—identified by mutations in either IDH1 or IDH2 and the absence of chromosome 1p/19q codeletion (IDH-mutant astrocytomas)—where they constitute the vast majority of cases. We found that SIRT2 expression was significantly higher in this glioma subclass relative to the other major adult glioma variants (IDH-mutant and 1p/19q codeleted oligodedroglioma and IDH-wildtype glioblastoma; Figure 1E–F). Moreover, when each glioma subclass was stratified by median SIRT2 transcript levels (CGGA data), high SIRT2 expression correlated with significantly reduced overall survival only in ATRX-deficient gliomas with no outcome differences discerned for other disease subtypes (Figure 1G). Levels of CpG island methylation at the SIRT2 locus were not significantly different between the three glioma subclasses in either TCGA or CCGA datasets, supporting alternative epigenetic mechanisms underlying SIRT2 overexpression and increased chromatin accessibility at the SIRT2 locus in the ATRX-deficient context (Supplementary Figure 2). Taken together, these data identify SIRT2 overexpression as a specific epigenetic consequence of ATRX deficiency in glioma and suggest that its therapeutic targeting may be a viable strategy for combating ATRX-deficient oncogenic phenotypes in glioma cells.
Sirt2 Inhibition Attenuates Atrx-deficient Transcriptional Profiles Impacting both Cellular Motility and Senescence.
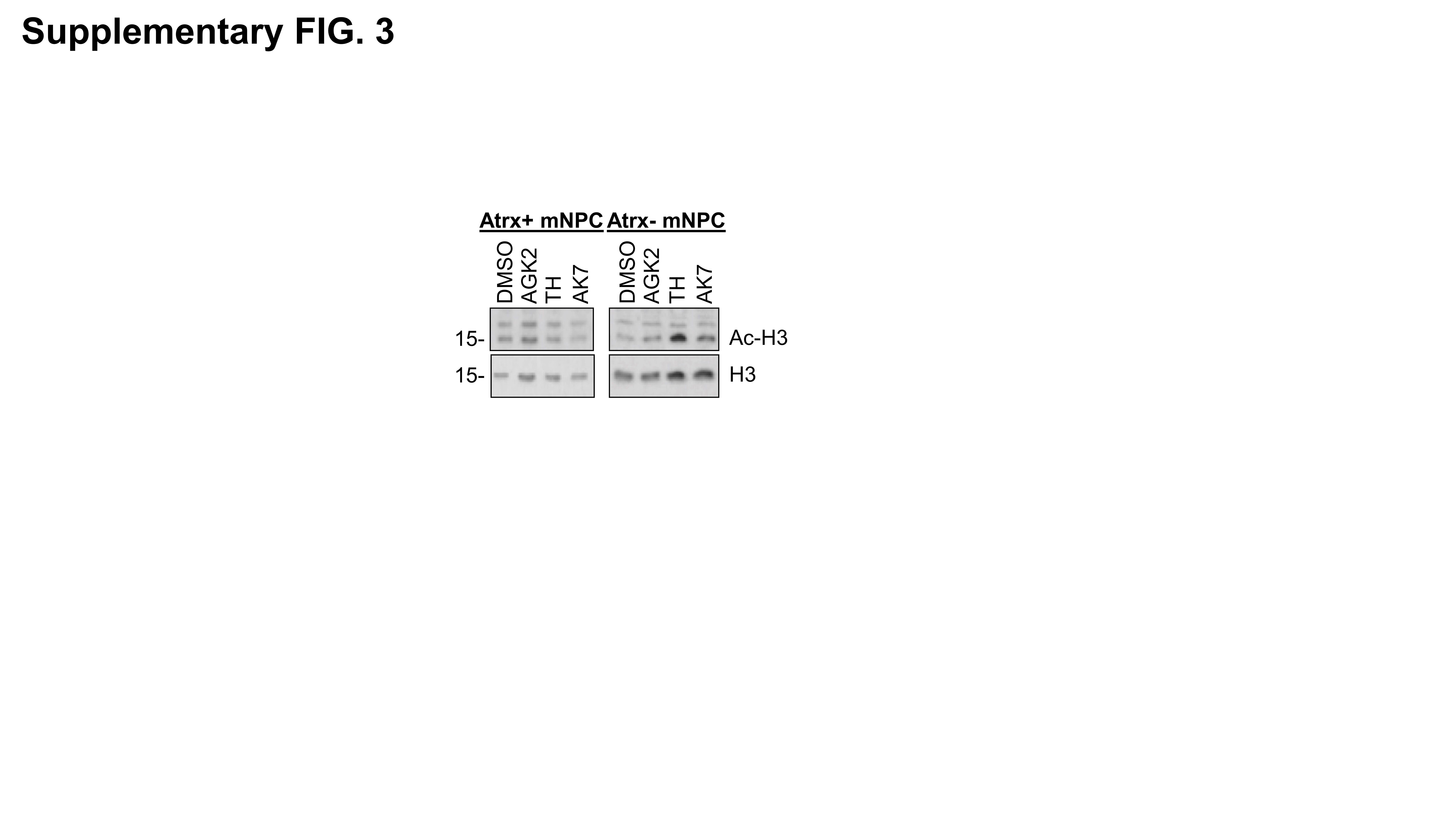
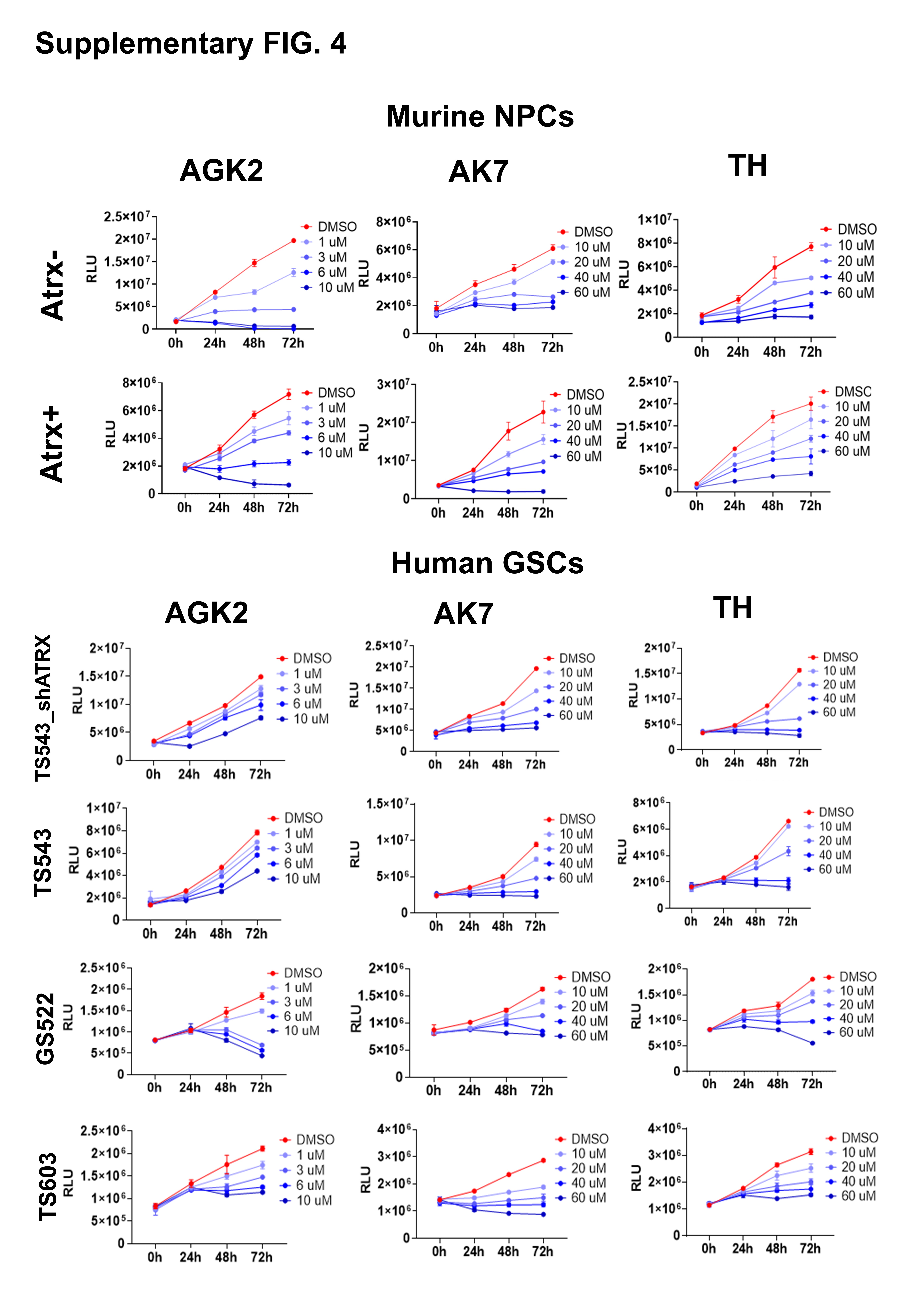
To determine the extent to which Sirt2 activity impacts gene expression and downstream cellular phenotypes in the Atrx-deficient context, we utilized three well-characterized small molecule SIRT2 inhibitors (Sirt2i), AGK2, AK7, and Thiomyristoyl (TH). We first demonstrated that micromolar levels of Sirt2i (1 μm for AGK2, and 10 μm for AK7 and TH) effectively increased H3 acetylation levels by western blot in Atrx− mNPCs, an effect not seen in Atrx + counterparts, which we had previously shown (see above) not to overexpress Sirt2 (Supplementary Figure 3). We also found by CellTiter-Glo proliferation assays that these concentrations of Sirt2is had minimal effects on cytotoxicity (Supplementary Figure 4). We then used RNA-seq to assess the impact of Sirt2i on transcriptional profiles in Atrx− mNPCs, documenting significant shifts in gene expression. Importantly, treatment with each Sirt2i effectively reverted the transcriptional signature of Atrx deficiency in mNPCs by GSEA (Figure 2A). Sirt2i-induced gene expression shifts also demonstrated associations with cytoskeletal mobilization, cell adhesion, cell motility, and senescence in Atrx− mNPCs (Figure 2B). These latter two biological processes echo our earlier work, which identified altered cell migration and differentiation as disease-relevant phenotypes characterizing ATRX-deficient glioma.11
Figure 2.
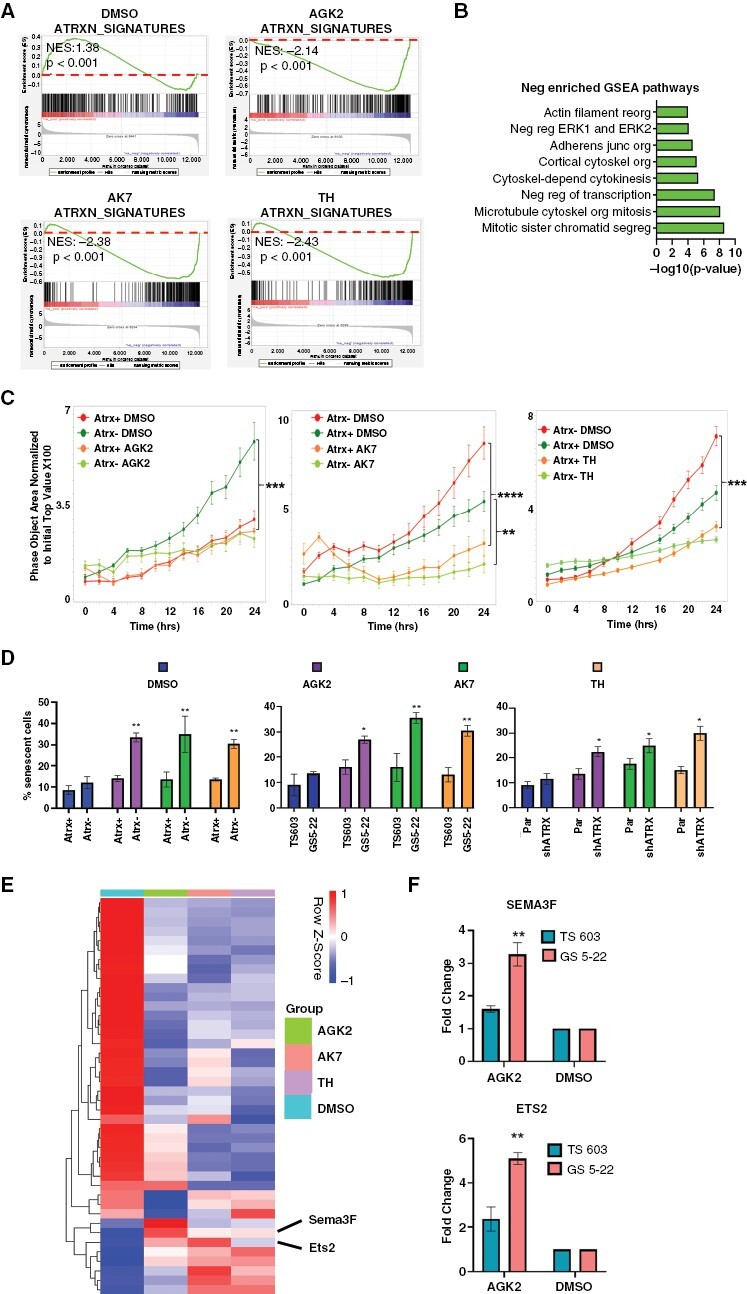
Sirt2i reverts Atrx-deficient transcriptional profiles, impacting motility and senescence. (A) GSEA plots documenting reversion of Atrx-deficient transcriptional signatures upon Sirt2i (AGK2: 1uM, AK7 and Thiomyristoyl: 10 µM) in Atrx− mNPCs. Dotted lines indicate the positioning of 0.0 enrichment scores. (B) Schematic of significantly enriched, negatively correlated GSEA pathways noted upon Sirt2i in Atrx- mNPCs. (C) 24-hour transwell migration assays for Atrx + and Atrx- mNPCs treated with Sirt2i (AGK2: 1 µM, AK7 and Thiomyristoyl: 10 µM) or vehicle control. (D) Quantified β-Galactosidase staining data for mNPCs (left) and GSCs (middle: TS603 and GS5-22, and right:TS543 and TS543 shATRX) demonstrating increased senescence with Sirt2i (AGK2: 1 µM, AK7 and Thiomyristoyl: 10 µM) in Atrx/ATRX-deficient contexts. (E) Heatmap showing expression levels of SenMayo database genes in Atrx− mNPCs treated with either DMSO or Sirt2i (AGK2: 1 µM, AK7 and Thiomyristoyl: 10 µM). (F) RT-PCR for senescence-associated transcripts ETS2 and SEMA3F showing larger increases upon 1uM AGK2 treatment in ATRX-mutant GSCs (GS 5-22) than in ATRX-wildtype GSCs (TS 603). Where applicable, error bars reflect SEM; p values determined by unpaired, two-tailed t-test. * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001
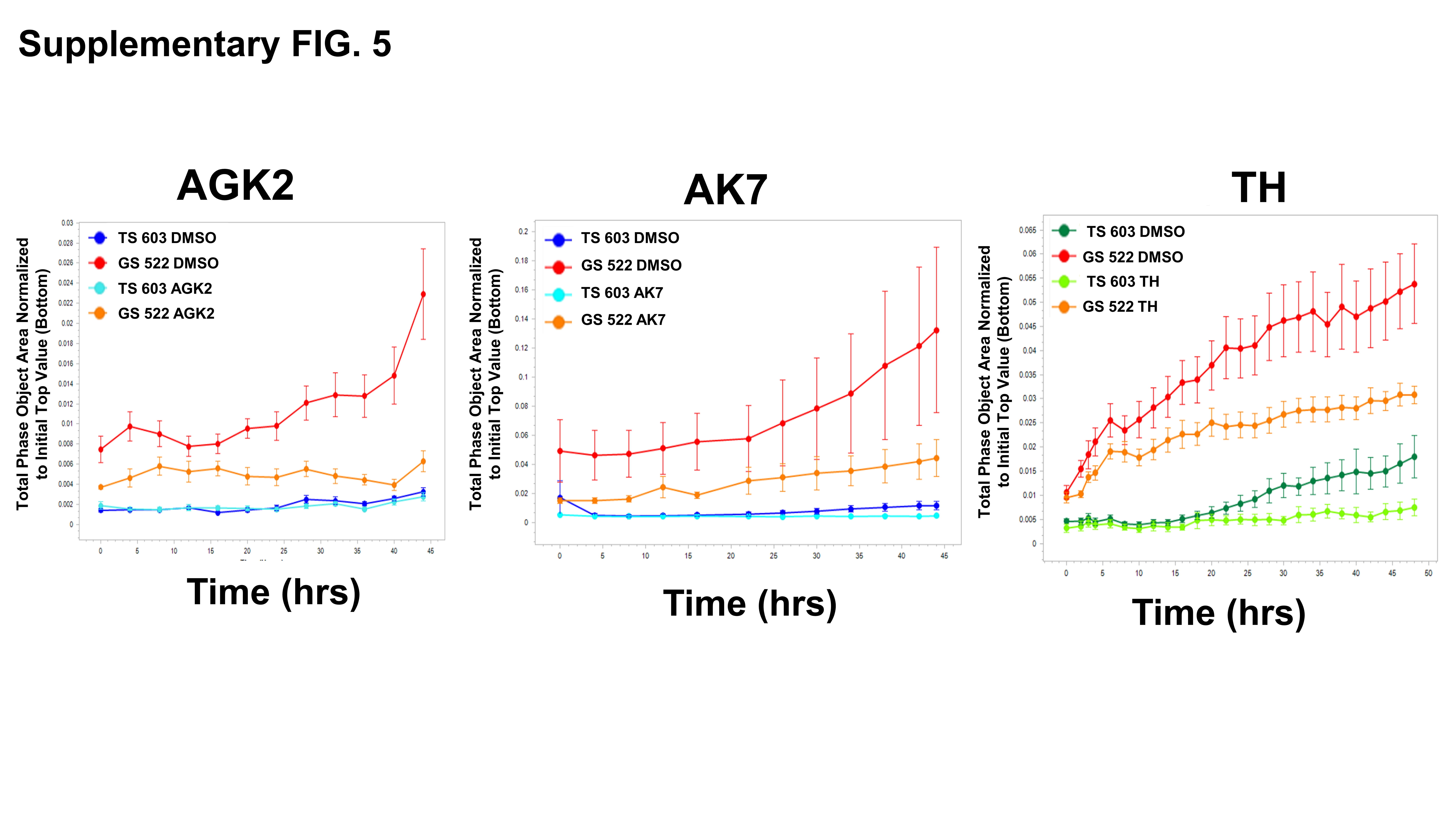
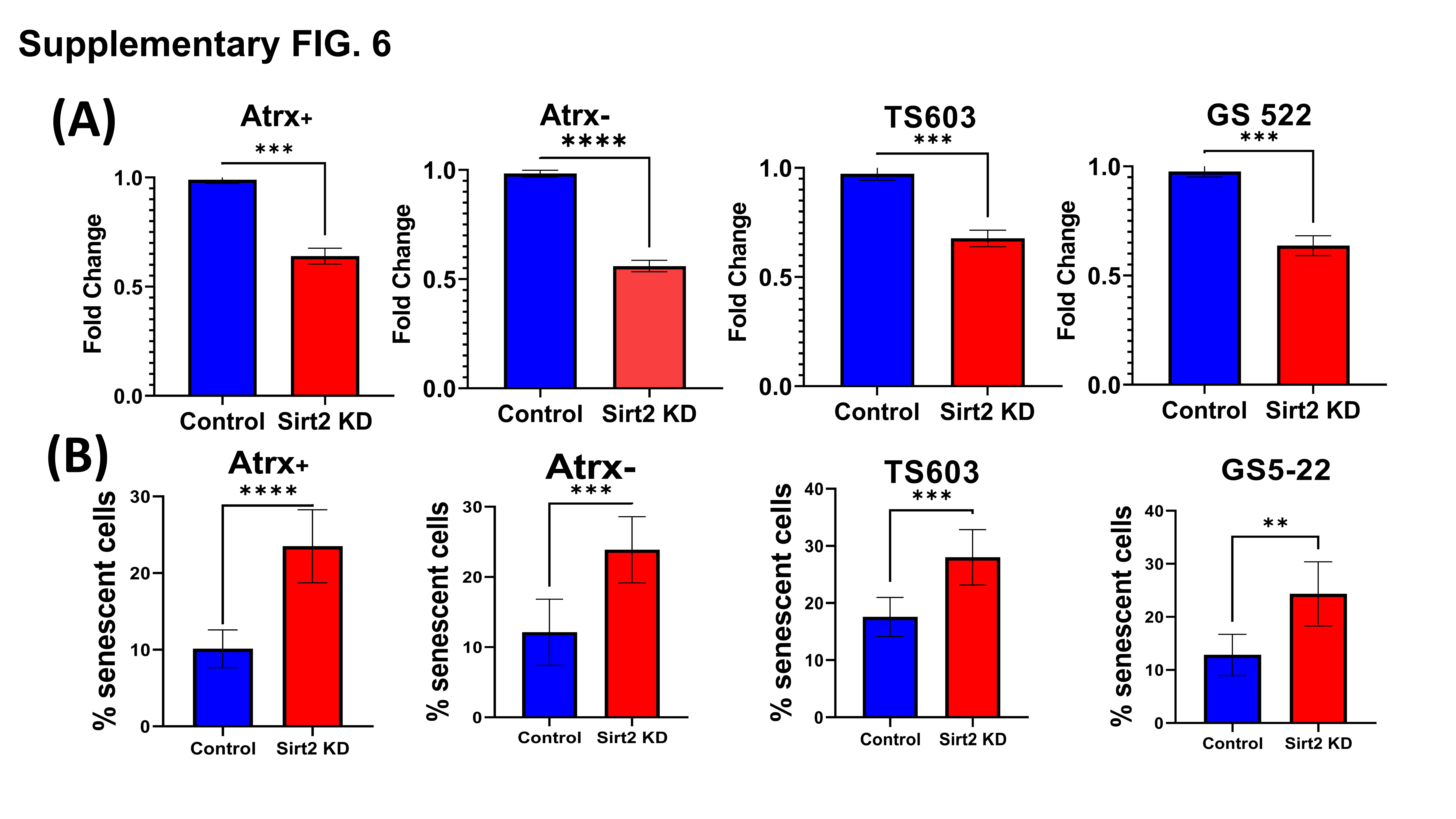
We then functionally assessed the consequences of Sirt2i on cellular motility and differentiation, specifically interrogating migration and senescence in murine and human experimental systems. Sirt2i profoundly impaired transwell migration of both Atrx− mNPCs and ATRX-mutant GSCs, with less pronounced although statistically significant effects on Atrx+/ATRX-wildtype counterparts (Figure 2C and Supplementary Figure 5). Sirt2i also induced β-galactosidase, a well-established marker of senescence, to a significantly greater extent in the Atrx/ATRX-deficient context for both mNPCs and GSCs (Figure 2D). ShRNA-based SIRT2 knockdown yielded similar results with regard to senescence, with strong effects across Atrx/ATRX-deficient and Atrx/ATRX-intact cell lines (Supplementary Figure 6). Congruent with these findings, Sirt2i treatment in mNPCs dramatically altered levels of key senescence-associated transcripts from the SenMayo database (Figure 2E). SenMayo is a publically available transcriptional repository that includes signatures identifying the senescent cell state across tissues and species with high fidelity.22 While the majority of SenMayo genes normally inhibit senescence and were repressed by Sirt2i, a smaller subset of senescence-promoting transcripts, including Ets2 and Sema3f, were upregulated. Subsequent RT-PCR documented significantly increased SEMA3F and ETS2 expression in AGK2-treated ATRX-mutant, but not ATRX-wildtype, GSCs (Figure 2F). Taken together, these findings document the outsized effects of SIRT2 and Sirt2i on ATRX-deficient gene expression in glioma and associated motility and senescence phenotypes.
Sirt2i Impairs Glioma Growth and Induces Senescence In Vivo
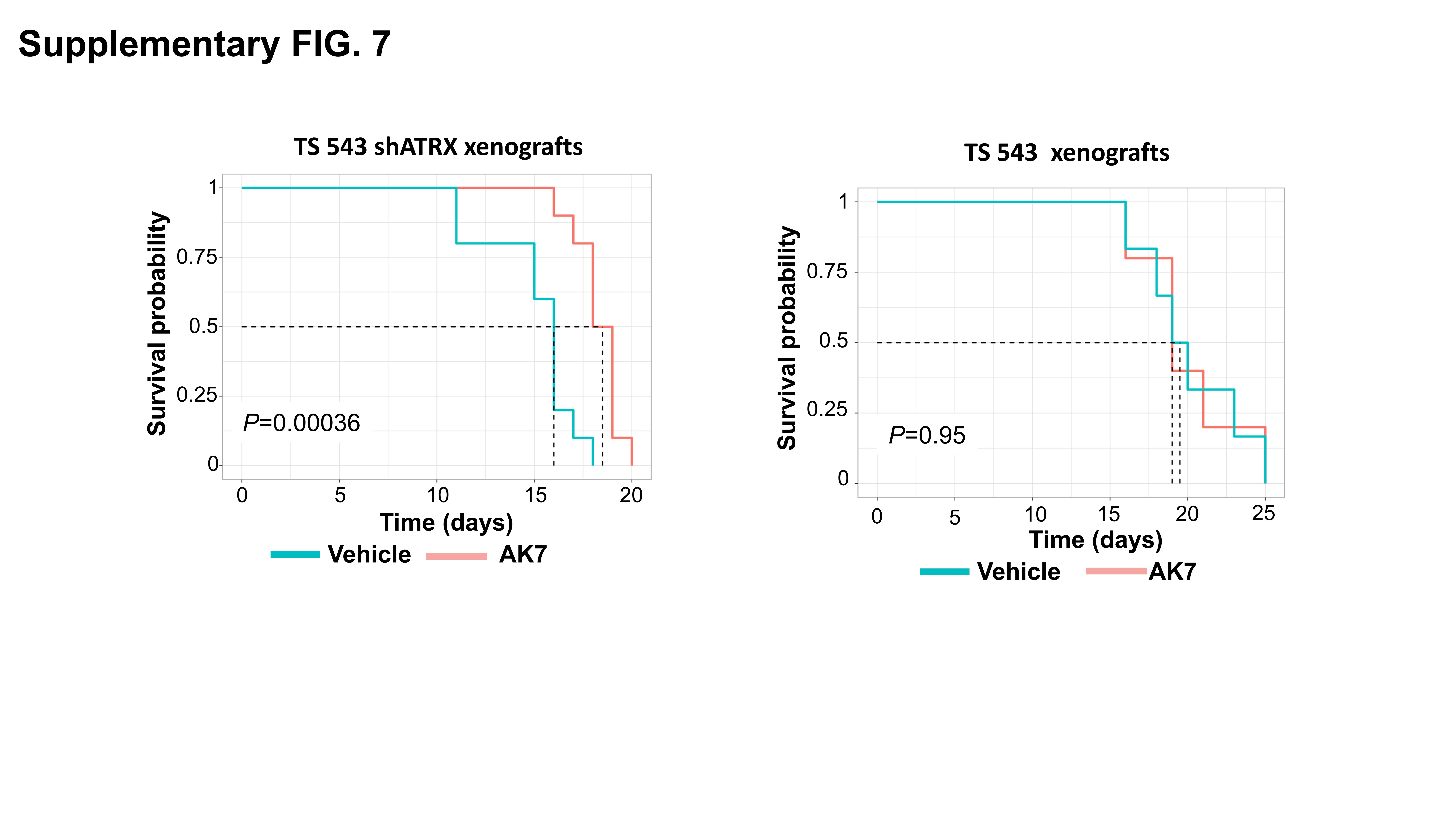
To evaluate the impact of Sirt2i on ATRX-deficient gliomagenesis in vivo, we generated TS543 and TS543-shATRX flank xenografts in Nu/Nu mice, as in our prior work evaluating therapeutic approaches selectively targeting ATRX-deficient glioma.12 Following GSC inoculation, mice were treated daily with either vehicle (DMSO) or 40 mg/kg AK7. We found that AK7 significantly slowed xenograft growth in the ATRX-deficient context and prolonged murine survival relative to vehicle control (Figure 3A and Supplementary Figure 7). Moreover, immunofluorescence analysis of tumors extracted after mouse sacrifice demonstrated reduced proliferation by MIB1 (Ki67) index and increased expression of the senescence markers p16 and SEMA3F (Figure 3B and E). Analogous findings of significance were inapparent in similarly treated isogenic TS543 ATRX-intact xenografts (Figure 3C–E). These data demonstrate in principle the potential of targeting SIRT2 to selectively promote senescence and combat tumor growth in ATRX-deficient gliomas.
Figure 3.
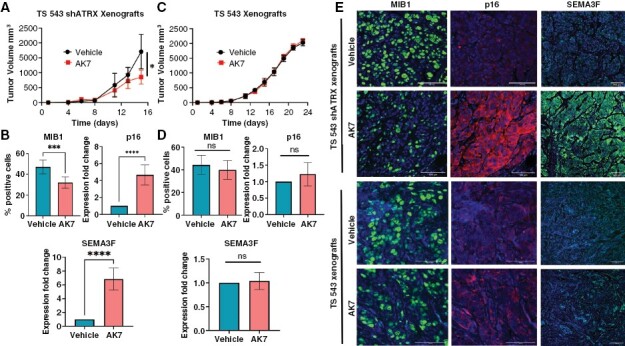
Sirt2i selectively promotes cellular senescence and impairs ATRX-deficient GSC tumor growth in vivo. (A) Growth curves for TS543 shATRX flank xenografts in mice treated with vehicle or AK7 (40 mg/kg daily following engraftment) (N=5 per group). (B) Quantified immunofluorescence staining showing decreased proliferation (MIB1) and increased expression of senescence markers (p16 and SEMA3F) in AK7-treated explanted TS543 shATRX xenografts versus vehicle-treated controls. (C) Growth curves for TS543 flank xenografts in mice treated with vehicle or AK7 (40 mg/kg daily following engraftment) (N=5 per group). (D) Quantified immunofluorescence staining showing expression of MIB1 and senescence markers (p16 and SEMA3F) in AK7 or vehicle-treated explanted TS543 xenografts. (E) Representative immunofluorescence images of immunofluorescence studies in xenografts (60x magnification). Where applicable, error bars reflect SEM; p values determined by unpaired, two-tailed t-test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and ns = not significant).
Sirt2i Induces Epigenomic Reprogramming in ATRX-deficient Glioma Models through Mobilization of both H3K27ac and H4K16ac Histone Marks
Having documented the impact of Sirt2i on functionally relevant ATRX-deficient transcriptional shifts, we sought to delineate epigenomic mechanisms underlying these effects. As histone acetylation is typically associated with chromatin accessibility at enhancer regions, we reasoned that Sirt2i should decrease the extent of nuclear chromatin condensation. We confirmed this conjecture using single nuclei chromatin condensation parameter (CCP) analysis in mNPCs after a 48-hour treatment, observing more pronounced effects in the Atrx− context (Figure 4A–B). To more fully delineate alterations in enhancer landscapes genome-wide, we then performed chromatin immunoprecipitation-high throughput sequencing (ChIP-seq) on isogenic mNPCs treated with either vehicle (DMSO) or 1 μM AGK2 for 48 hours, assessing H3K27ac and H3K4me1, two marks typically associated with active enhancers, along with H3K27me3, a polycomb repression mark whose levels often demonstrate reciprocal behavior to those of H3K27ac. Sirt2i increased the size and number of H3K4me1 and H3K27ac-marked peaks with concomitant decreases in the size and number of H3K27me3-marked peaks (Figure 4C). Once again, these effects were stronger in the Atrx− context. Applying Rank Ordering of Super-Enhancers (ROSE) to our ChIP-seq data,23,24 we found that super-enhancer (SE) calls increased more dramatically in AGK2-treated Atrx− mNPCs than in Atrx+ counterparts (3266/1158 = 2.82% vs 790/441 = 1.79%; Figure 4D). Pathway analysis of gene sets associated with these mobilized SEs implicated a range of relevant biological processes, including cell motility, adhesion, and death (Figure 4E–F). However, the subtle distinctions between these pathway correlations with regard to Atrx status, along with the absence of associated senescence signatures, raised the possibility that alternative molecular mechanisms were playing major roles in Sirt2i-induced cellular phenotypes.
Figure 4.
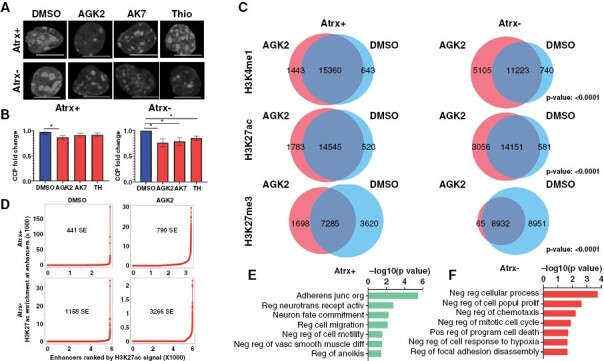
Sirt2i modulates super-enhancer landscapes in Atrx− mNPCs: (A-B) Single cell nuclear chromatin condensation (CCP) analysis demonstrating reduced chromatin condensation in Atrx-deficient mNPCs with more modest effects in Atrx + counterparts. (C) Venn diagrams showing shared and unique ChIP-seq peaks for enhancer-associated H3K4me1 and H3K27ac and repressive H3K27me3 marks in Atrx + and Atrx− mNPCs deficient NPCs. Hypergeometric P values are shown. (D) Rank Ordering of Super-Enhancers (ROSE) analysis for H3K27ac showing larger increases in super-enhancer number following 1 µM AGK2 treatment in Atrx− mNPCs than in Atrx + mNPCs. (E-F) GSEA correlations for all gene sets significantly associated with Sirt2i-mobilized super-enhancers in Atrx + and Atrx− mNPCs. Where applicable, error bars reflect SEM; P values determined by unpaired, two-tailed t-test (*P < 0.05)
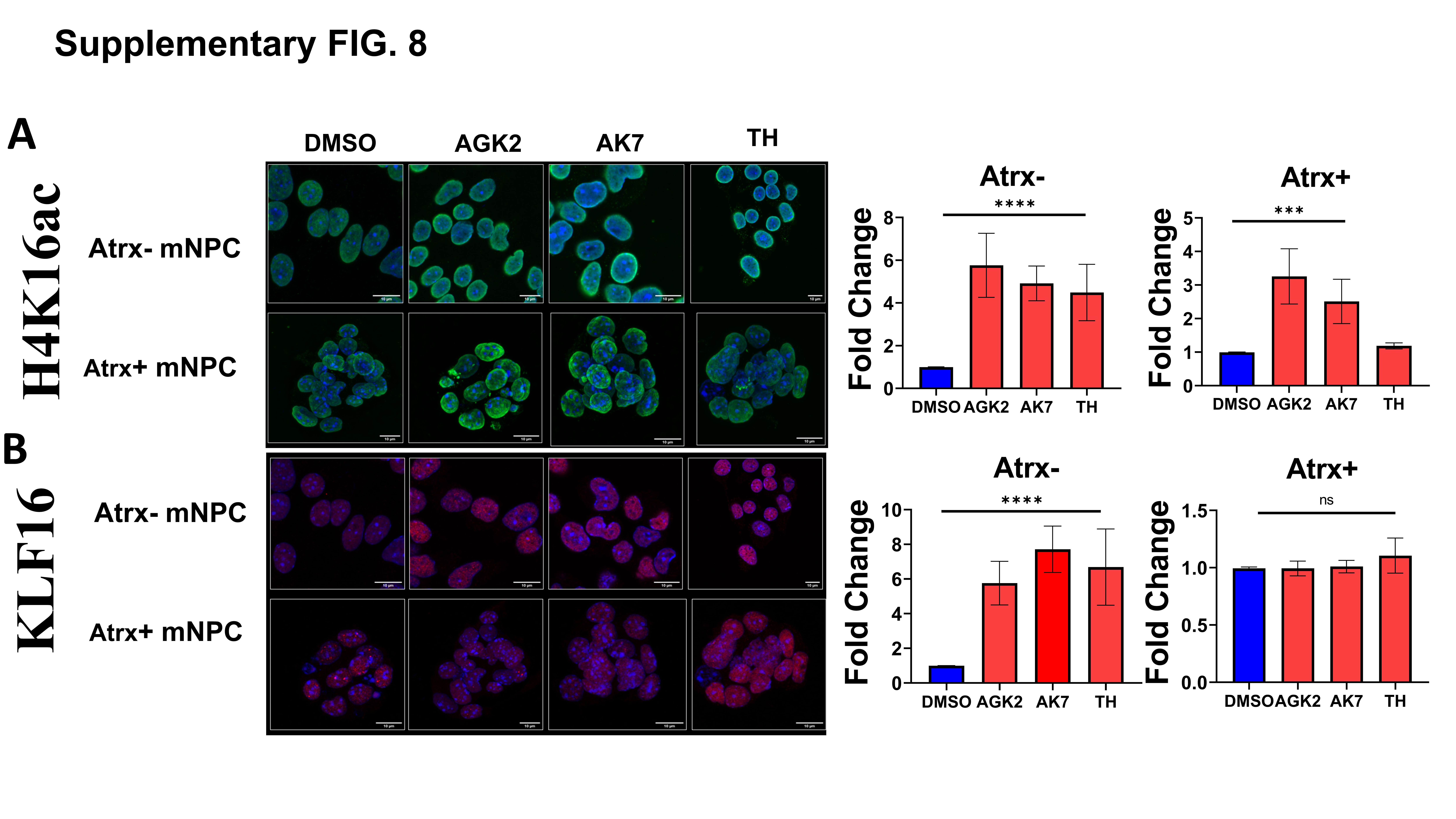
H4K16 acetylation (H4K16ac) is an enhancer-associated chromatin mark that influences both transcriptional activation and repression.25,26 Intriguingly, SIRT2 has been shown to directly target H4K16ac for deacetylation during specific phases of the cell cycle.21 To evaluate the impact of Sirt2i on H4K16ac, we first employed immunofluorescence in mNPCs, delineating increased nuclear expression in response to drug treatment for all three inhibitors, accentuated in the Atrx-deficient setting (Supplementary Figure 8A). We then employed H4K16ac ChIP-seq in vehicle or AGK2-treated mNPCs, assessing enrichment peak number and distribution. We found that while Sirt2i had relatively modest effects in Atrx+ mNPCs, Atrx− counterparts exhibited both increased peak number and altered peak distribution (Figure 5A). GSEA analysis of H4K16ac-marked genes arising with AGK2 treatment in Atrx− mNPCs demonstrated associations with cellular senescence, apoptosis, and adhesion not present for Atrx+ isogenics, which instead exhibited correlations with metabolic pathways (Figure 5B).
Figure 5.
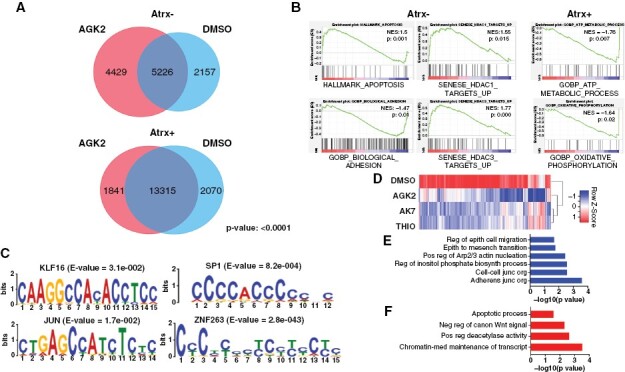
Sirt2i impacts H4K16ac levels to influence phenotypically relevant gene expression in the Atrx-deficient context. (A) Venn diagrams showing shared and unique ChIP-seq peaks for H4K16ac in Atrx + and Atrx− mNPCs, with hypergeometric P value. (B) GSEA correlations for gene sets associated with Sirt2i-mobilized H4K16ac peaks in Atrx + and Atrx− mNPCs. (C) H4K16ac MEME analysis in 1uM AGK2 treated Atrx- mNPCs implicating KLF16, JUN, SP1, and zinc finger family members as key transcriptional cofactors. (D) Heatmap showing broad shifts in the expression levels of MEME-associated KLF16 target genes in Sirt2i-treated Atrx− mNPCs. GSEA associations with Sirt2i-modulated KLF16 target transcripts showing negative (E) and positive (F) correlations with relevant motility and apoptotic networks.
We also performed MEME analysis on AGK2-induced H4K16ac peak sites in Atrx− mNPCs to identify coregulators potentially mediating downstream transcriptional and functional consequences. This approach revealed several transcription factors (TFs) with significant motif correlations including KLF16, JUN, SP1, and multiple zinc finger family members (ZNF263, ZNF384) (Figure 5C). Examining our RNA-seq data, we found that KLF16, a member of Krüppel-like factor (KLF) TF family and a previously reported tumor suppressor in gliomas,27 was specifically upregulated in Sirt2i-treated Atrx− mNPCs. Immunofluorescence in Atrx+ and Atrx− mNPCs confirmed this finding (Supplementary Figure 8B). KLF TFs bind consensus CACCC or GT boxes, induce chromatin remodeling and either activate or repress transcription based on heterodimerization partners and biological context.28,29 Further emphasizing the downstream impact of KLF16 engagement by Sirt2i, we found that MEME-associated KLF16 target genes were broadly downregulated by Sirt2i in Atrx− mNPCs (Figure 5D). GSEA analysis of these genes revealed strong negative associations with cellular motility processes and positive associations with apoptosis (Figure 5E–F). Taken together, these findings implicate perturbed H3K27ac and H4K16ac landscapes in the pharmacological impact of Sirt2i on Atrx/ATRX-deficient glioma models. In particular, H4K16ac-based molecular mechanisms likely underlie significant effects on cell motility and senescence induced by Sirt2i in Atrx− mNPCs and ATRX-mutant/knockdown GSCs.
Discussion
The discovery of epigenetic abnormalities as core features in many cancers continues to motivate efforts to characterize and target the key molecular events underlying oncogenesis in these diverse contexts. Such studies in malignant glioma are of particular need, given the dismal survival profile associated with all disease subtypes. Significant numbers of both adult and pediatric glioma feature ATRX inactivation as a defining molecular feature,5,30 signaling the importance of this driver event to disease pathogenesis. However, the numerous and varied consequences of ATRX deficiency—ranging from complex chromatin remodeling to replication stress and genomic instability to telomeric dysfunction—necessarily complicate strategies for therapeutic development in affected tumors.6,8,9,12,31,32 Our prior studies demonstrated that ATRX deficiency profoundly impacts chromatin accessibility genome-wide in putative glioma cells of origin, with master regular-like influence on global transcriptional profiles mediating disease-defining differentiation and motility phenotypes.11 In light of these findings, we reasoned that reverting the epigenomic rewiring induced by ATRX deficiency, along with downstream transcriptional sequelae, could potentially hamper its gliomagenic effects. Recent work highlights the potential of epigenetic therapies to elegantly modulate the multi-component gene sets governing complex cellular states in cancer.33
Leveraging gene expression profiles for Atrx+ and Atrx- mNPC isogenics, we used the Broad Institute CMAP to identify HDAC inhibitors as promising candidates to combat ATRX-deficient epigenomic and transcriptional rewiring. Chromatin condensation by way of histone deacetylation impacts a wide range of biological processes including cell cycle, metabolism, angiogenesis, apoptosis, and differentiation.34–36 Small molecule HDAC inhibitors have received FDA approval for the treatment of selected hematological malignancies and have progressed through clinical trials for multiple solid tumor variants.37 Moreover, reported links between HDAC inhibition, terminal differentiation, and senescence induction in the glioma context were particularly intriguing in light of earlier findings from our group and others implicating ATRX deficiency in developmental disruptions within the neuroepithelial lineage.11,38
Multiomic analyses integrating our mNPC findings with large, publicly available datasets derived from human tumors revealed the class III HDAC SIRT2 as selectively upregulated in ATRX-deficient glioma, where high expression levels selectively conferred unfavorable prognosis. Much like other HDACs, SIRT2 exhibits broad functionality across many cancer-relevant molecular networks, actively shuttling between nuclear and cytoplasmic compartments.39,40 As indicated above, SIRT2 deacetylates the epigenomically active H3K27 and H4K16 histone residues, but also directly modifies a range of other targets, including α-tubulin complexes and the anaphase-promoting complex/cyclosome (APC/C).19–21,41,42 To date, SIRT2 has been implicated in breast, nonsmall cell lung, hepatocellular, and colorectal carcinomas, where high expression levels are associated with aggressive clinical behavior.40,43 Of note, SIRT2-mediated deacetylation of KRAS induces cell proliferation, colony formation, and tumor growth in lung and colorectal cancer models.44 SIRT2 also appears to regulate epithelial-to-mesenchymal transition in basal-like breast cancer by deacetylating K116 of the Slug protein.45 These findings speak to the multifaceted mechanisms by which SIRT2 promotes oncogenesis, as well as their intriguing cell type specificity.
Targeting SIRT2 with small molecule inhibitors has demonstrated promising results for multiple agents across several cancer models. For instance, AGK2 enhanced the anticancer effects of chemotherapeutic drugs in TP53-mutant colorectal cancer cells, SirReal2 induced acetylation of PEPCK1 and suppression of the RAS/ERK/JNK/MMP-9 pathway in gastric cancers, and AK7 hampered the growth of intracranial glioblastoma xenografts through increased α-tubulin acetylation.46 In the present study, we found that Sirt2i reverted ATRX-deficient transcriptional profiles and cell motility phenotypes, while also selectively promoting senescence in the ATRX-deficient context. These observations, initially made in mNPC isogenics, effectively translated to patient-derived GSCs. SIRT2 knockdown achieved similar results in both human and murine experimental models, albeit without ATRX-deficient selectivity. This latter finding likely reflects imprecision of the shRNA-based approach relative to titrated pharmacological inhibition with regard to refined therapeutic windows. Moreover, genetic knockdown may not fully recapitulate the extent to which Sirt2i differentially impairs the activity of SIRT2 on its multiple downstream targets (discussed further below). Importantly, Sirt2i also selectively hampered ATRX-deficient GSC tumor growth in vivo, impacting both cellular proliferation and senescence. Taken together, our results demonstrate the potential clinical utility of Sirt2i in the selective targeting of ATRX-deficient epigenomic dysfunction in both adult and pediatric malignant glioma. The induction by Sirt2i of terminal differentiation in particular echoes analogous established approaches employing epigenetic therapies in myeloid malignancies and recapitulates reported effects of HDAC inhibitors in sarcoma, lung, and prostate cancer models.47,48
Impairing SIRT2 activity through pharmacological inhibition would be expected to affect a number of the protein’s downstream targets, notably impacting epigenetically influential histone modifications. Indeed, the profound shifts in global transcriptional profiles we observed with Sirt2i in Atrx− mNPCs are consistent with foundational remodeling of underlying chromatin architecture. HDAC inhibition increases chromatin acetylation levels genome-wide, directly modulating transcription factor access,49–51 and HDACi has been shown to exert physiological effects in cancer models in large part through effects on histone acetylation.34,50,52 Consistent with this earlier literature, we found that Sirt2i lead to genome-wide increases in H3K27ac and decreases H3K27me3 in isogenic mNPCs. While these effects were more pronounced in Atrx− mNPCs, impacted genes exhibited significant correlations with similar pathways—cell adhesion and motility, apoptosis, and differentiation—in both Atrx-intact and -deficient contexts. Moreover, senescence-associated molecular networks were not implicated by GSEA analysis. Such findings were not entirely consistent with the observed phenotypic consequences of Sirt2i, particularly in Atrx−/ATRX− mNPCs and GSCs. Accordingly, we suspected that alternative epigenomic mechanisms were being engaged by Sirt2i in the Atrx-deficient context.
As mentioned earlier, SIRT2 preferentially deacetylates H4K16 during the G2/M cell cycle phase, which coincides with SIRT2 localization to cytoskeleton structures.19–21 Loss of H4K16ac is seen in many human cancers and aged cell states, co-occurring with site-specific DNA hypomethylation.53–55 H4K16ac appears to serve both enhancer and repressor functions with regard to transcription, working with the nucleolar remodeling complex (NoRC) to recruit both histone acetylases and DNA methyltransferases.26 We found that Sirt2i significantly and selectively enhanced H4K16ac in Atrx- mNPCs, with ChIP-seq enrichment peaks demonstrating GSEA associations with cellular senescence, apoptosis, and adhesion not present for Atrx+ counterparts. Moreover, MEME analysis implicated KLF16 as a key transcriptional regulator mediating the effects of altered H4K16ac profiles on cell motility and apoptotic gene sets. Taken together, these findings suggest that dysregulated H4K16ac promotes oncogenesis. While this unusual histone modification has yet to be extensively implicated in cancer, one very recent study demonstrated that increasing H4K16ac levels with either Ex-547, a Sirtuin 1 (SIRT1) inhibitor, or panobinostat decreased proliferative capacity in the human myeloid leukemia cells.56
In summary then, we identify and characterize novel molecular mechanisms underlying the oncogenic phenotypes induced by ATRX deficiency in putative glioma cells of origin and effectively target underlying epigenomic dysfunction in murine isogenic and human GSC models, demonstrating efficacy in vitro and in vivo. Our work reveals a viable pathway towards precision therapeutic development for epigenetically driven cancers that leverages transcriptional signatures, experimental and in silico multiomic analyses, and repurposed pharmaceuticals. We expect that analogous approaches could be effectively implicated in other deadly tumors.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We would like to graciously thank Drs. Frederick Lang and Erik Sulman for providing access to GSC lines. We would also like to thank Jacob Mattia for assistance with CCP experiments. Finally, we would like to acknowledge the MD Anderson Genomics Core Facility (GCF).
Contributor Information
Prit Benny Malgulwar, Department of Translational Molecular Pathology, University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Carla Danussi, Sanofi, Research and Development, Cambridge, Massachusetts, USA.
Sharvari Dharmaiah, Department of Translational Molecular Pathology, University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
William Johnson, Department of Translational Molecular Pathology, University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Anand Singh, Department of Genomic Medicine, Division of Cancer Medicine, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Kunal Rai, Department of Genomic Medicine, Division of Cancer Medicine, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Arvind Rao, Departments of Biostatistics, Computational Medicine and Bioinformatics, and Radiation Oncology, University of Michigan, Ann Arbor, Michigan, USA.
Jason T Huse, Department of Translational Molecular Pathology, University of Texas MD Anderson Cancer Center, Houston, Texas, USA; Department of Pathology, University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Funding
National Institutes of Health/National Cancer Institute (NIH/NCI, R01 CA240338, JTH); the American Cancer Society (RSG-16-179-01-DMC, JTH, RSG-16-005-01, AR); the Ben and Catherine Ivy Foundation (Ivy Foundation Translational Adult Glioma Award, JTH); the Brockman Foundation (JTH), Career Development Award from the MD Anderson Brain Tumor SPORE (AR); Cancer Prevention and Research Institute of Texas GCC Center for Advanced Microscopy and 1mage 1nformatics Award (RP170719; AR); University of Texas MD Anderson NIH/NCI Cancer Center Support Grant (P30 CA016672); MD Anderson Moonshot program.
Conflict of interest statement
None declared.
Authorship statement
Conceptualization: PBM, CD, JTH. Methodology: PBM, CD, SD, AR, AS, KR, JTH. Investigation: PBM, CD, SD, AR, AS, KR, JTH. Funding acquisition: JTH. Project administration: PBM, JTH. Supervision: PBM, JTH. Writing—original draft: PBM, JTH. Writing—review & editing: PBM, JTH.
Data Availability
All transcriptional and epigenomic data have been deposited in the Gene Expression Omnibus (GEO) repository: GSE234320.
References
- 1. Omuro A, DeAngelis LM.. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310(17):1842–1850. [DOI] [PubMed] [Google Scholar]
- 2. Jiao Y, Killela PJ, Reitman ZJ, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3(7):709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kannan K, Inagaki A, Silber J, et al. Whole exome sequencing identified ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget. 2012;3(10):1194–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu XY, Gerges N, Korshunov A, et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol. 2012;124(5):615–625. [DOI] [PubMed] [Google Scholar]
- 5. Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–231. [DOI] [PubMed] [Google Scholar]
- 6. Berube NG, Mangelsdorf M, Jagla M, et al. The chromatin-remodeling protein ATRX is critical for neuronal survival during corticogenesis. J Clin Invest. 2005;115(2):258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clynes D, Higgs DR, Gibbons RJ.. The chromatin remodeller ATRX: a repeat offender in human disease. Trends Biochem Sci. 2013;38(9):461–466. [DOI] [PubMed] [Google Scholar]
- 8. Law MJ, Lower KM, Voon HP, et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell. 2010;143(3):367–378. [DOI] [PubMed] [Google Scholar]
- 9. Ratnakumar K, Duarte LF, LeRoy G, et al. ATRX-mediated chromatin association of histone variant macroH2A1 regulates alpha-globin expression. Genes Dev. 2012;26(5):433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Valle-Garcia D, Qadeer ZA, McHugh DS, et al. ATRX binds to atypical chromatin domains at the 3’ exons of zinc finger genes to preserve H3K9me3 enrichment. Epigenetics. 2016;11(6):398–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Danussi C, Bose P, Parthasarathy PT, et al. Atrx inactivation drives disease-defining phenotypes in glioma cells of origin through global epigenomic remodeling. Nat Commun. 2018;9(1):1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang Y, Yang J, Wild AT, et al. G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat Commun. 2019;10(1):943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heaphy CM, de Wilde RF, Jiao Y, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science (New York, N.Y). 2011;333(6041):425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lovejoy CA, Li W, Reisenweber S, et al. ; ALT Starr Cancer Consortium. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012;8(7):e1002772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koschmann C, Calinescu AA, Nunez FJ, et al. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci Transl Med. 2016;8(328):328ra328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oppel F, Tao T, Shi H, et al. Loss of atrx cooperates with p53-deficiency to promote the development of sarcomas and other malignancies. PLoS Genet. 2019;15(4):e1008039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cheng Y, He C, Wang M, et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther. 2019;4:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Corsello SM, Bittker JA, Liu Z, et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med. 2017;23(4):405–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Inoue T, Hiratsuka M, Osaki M, et al. SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene. 2007;26(7):945–957. [DOI] [PubMed] [Google Scholar]
- 20. Serrano L, Martinez-Redondo P, Marazuela-Duque A, et al. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 2013;27(6):639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vaquero A, Scher MB, Lee DH, et al. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006;20(10):1256–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saul D, Kosinsky RL, Atkinson EJ, et al. A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues. Nat Commun. 2022;13(1):4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Loven J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Taylor GC, Eskeland R, Hekimoglu-Balkan B, Pradeepa MM, Bickmore WA.. H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction. Genome Res. 2013;23(12):2053–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou Y, Grummt I.. The PHD finger/bromodomain of NoRC interacts with acetylated histone H4K16 and is sufficient for rDNA silencing. Curr Biol. 2005;15(15):1434–1438. [DOI] [PubMed] [Google Scholar]
- 27. Chen X, Li S, Ke Y, et al. KLF16 suppresses human glioma cell proliferation and tumourigenicity by targeting TFAM. Artif Cells Nanomed Biotechnol. 2018;46(sup1sup1):608–615. [DOI] [PubMed] [Google Scholar]
- 28. Pearson R, Fleetwood J, Eaton S, Crossley M, Bao S.. Kruppel-like transcription factors: a functional family. Int J Biochem Cell Biol. 2008;40(10):1996–2001. [DOI] [PubMed] [Google Scholar]
- 29. Ilsley MD, Gillinder KR, Magor GW, et al. Kruppel-like factors compete for promoters and enhancers to fine-tune transcription. Nucleic Acids Res. 2017;45(11):6572–6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brat DJ, Verhaak RG, Aldape KD, et al. ; Cancer Genome Atlas Research Network. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372(26):2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Clynes D, Jelinska C, Xella B, et al. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat Commun. 2015;6:7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Clynes D, Jelinska C, Xella B, et al. ATRX dysfunction induces replication defects in primary mouse cells. PLoS One. 2014;9(3):e92915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sato T, Cesaroni M, Chung W, et al. Transcriptional selectivity of epigenetic therapy in cancer. Cancer Res. 2017;77(2):470–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li Y, Seto E.. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med. 2016;6(10):a026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peng L, Seto E.. Deacetylation of nonhistone proteins by HDACs and the implications in cancer. Handb Exp Pharmacol. 2011;206:39–56. [DOI] [PubMed] [Google Scholar]
- 36. Ropero S, Esteller M.. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007;1(1):19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ho TCS, Chan AHY, Ganesan A.. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J Med Chem. 2020;63(21):12460–12484. [DOI] [PubMed] [Google Scholar]
- 38. Babikir H, Wang L, Shamardani K, et al. ATRX regulates glial identity and the tumor microenvironment in IDH-mutant glioma. Genome Biol. 2021;22(1):311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. North BJ, Verdin E.. Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS One. 2007;2(8):e784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang L, Kim S, Ren X.. The clinical significance of SIRT2 in malignancies: a tumor suppressor or an oncogene? Front Oncol. 2020;10:1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li T, Garcia-Gomez A, Morante-Palacios O, et al. SIRT1/2 orchestrate acquisition of DNA methylation and loss of histone H3 activating marks to prevent premature activation of inflammatory genes in macrophages. Nucleic Acids Res. 2020;48(2):665–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim HS, Vassilopoulos A, Wang RH, et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell. 2011;20(4):487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang W, Chen W, Su H, et al. Recent advances in the development of histone deacylase SIRT2 inhibitors. RSC Adv. 2020;10(61):37382–37390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang MH, Laurent G, Bause AS, et al. HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol Cancer Res: MCR. 2013;11(9):1072–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhou W, Ni TK, Wronski A, et al. The SIRT2 deacetylase stabilizes slug to control malignancy of basal-like breast cancer. Cell reports. 2016;17(5):1302–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Funato K, Hayashi T, Echizen K, et al. SIRT2-mediated inactivation of p73 is required for glioblastoma tumorigenicity. EMBO Rep. 2018;19(11):e45587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bar-Hai N, Ishay-Ronen D.. Engaging plasticity: Differentiation therapy in solid tumors. Front Pharmacol. 2022;13:944773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stubbins RJ, Karsan A.. Differentiation therapy for myeloid malignancies: beyond cytotoxicity. Blood Cancer J. 2021;11(12):193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Slaughter MJ, Shanle EK, Khan A, et al. HDAC inhibition results in widespread alteration of the histone acetylation landscape and BRD4 targeting to gene bodies. Cell reports. 2021;34(3):108638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sanchez GJ, Richmond PA, Bunker EN, et al. Genome-wide dose-dependent inhibition of histone deacetylases studies reveal their roles in enhancer remodeling and suppression of oncogenic super-enhancers. Nucleic Acids Res. 2018;46(4):1756–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gatla HR, Zou Y, Uddin MM, Vancurova I.. Epigenetic regulation of interleukin-8 expression by class I HDAC and CBP in ovarian cancer cells. Oncotarget. 2017;8(41):70798–70810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Beacon TH, Delcuve GP, Lopez C, et al. The dynamic broad epigenetic (H3K4me3, H3K27ac) domain as a mark of essential genes. Clin Epigenetics. 2021;13(1):138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Meloni T, Cutillo S, Testa U, Luzzatto L.. Neonatal jaundice and severity of glucose-6-phosphate dehydrogenase deficiency in Sardinian babies. Early Hum Dev. 1987;15(6):317–322. [DOI] [PubMed] [Google Scholar]
- 54. Dang W, Steffen KK, Perry R, et al. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature. 2009;459(7248):802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rai TS, Cole JJ, Nelson DM, et al. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 2014;28(24):2712–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pessoa Rodrigues C, Akhtar A.. Differential H4K16ac levels ensure a balance between quiescence and activation in hematopoietic stem cells. Sci Adv. 2021;7(32):eabi5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All transcriptional and epigenomic data have been deposited in the Gene Expression Omnibus (GEO) repository: GSE234320.