

Abstract
A general method for designing proteins to bind and sense any small molecule of interest would be widely useful. Due to the small number of atoms to interact with, binding to small molecules with high affinity requires highly shape complementary pockets, and transducing binding events into signals is challenging. Here we describe an integrated deep learning and energy based approach for designing high shape complementarity binders to small molecules that are poised for downstream sensing applications. We employ deep learning generated psuedocycles with repeating structural units surrounding central pockets; depending on the geometry of the structural unit and repeat number, these pockets span wide ranges of sizes and shapes. For a small molecule target of interest, we extensively sample high shape complementarity pseudocycles to generate large numbers of customized potential binding pockets; the ligand binding poses and the interacting interfaces are then optimized for high affinity binding. We computationally design binders to four diverse molecules, including for the first time polar flexible molecules such as methotrexate and thyroxine, which are expressed at high levels and have nanomolar affinities straight out of the computer. Co-crystal structures are nearly identical to the design models. Taking advantage of the modular repeating structure of pseudocycles and central location of the binding pockets, we constructed low noise nanopore sensors and chemically induced dimerization systems by splitting the binders into domains which assemble into the original pseudocycle pocket upon target molecule addition.
One Sentence Summary
We use a pseuodocycle-based shape complementarity optimizing approach to design nanomolar binders to diverse ligands, including the flexible and polar methotrexate and thyroxine, that can be directly converted into ligand-gated nanopores and chemically induced dimerization systems.
The design of small molecule (SM) binding proteins is more challenging than the design of protein binders as there are fewer atoms to interact with; hence high shape complementary (SC) pockets which make contact with the majority of the available atoms are required for high affinity(1). Designing such complementary pockets requires high accuracy modeling of protein-ligand packing, hydrogen bonding, π-π and other interactions, and the generation of scaffolds that can harbor such close interactions surrounding a large fraction of the SM surface. These challenges are particularly critical for designing binders to flexible polar compounds: polar groups make hydrogen bonds to water in the unbound state, which must be replaced by hydrogen bonds to the protein for binding to be favorable, and flexible compounds lose considerable entropy upon binding which must be compensated by extensive favorable interactions. In nature, SM binding receptors not only specifically bind their targets but transduce binding events into downstream signals(2); a further design challenge is to devise general approaches for similarly coupling binding to sensing. Previous successes in SM binder design have focused on relatively rigid hydrophobic targets and often have required considerable experimental optimization to improve binding affinities from micromolar level to high nanomolar levels(3–8). Deep learning (DL) diffusion approaches have been extended to SM binder design, but successes thus far are all for large, bulky, rigid nonpolar molecules(9); the power of DL approaches may be limited by the significantly smaller amount of training data compared to the protein-protein interaction case(9, 10). Overall, generation of binders to polar and flexible small molecules, and systematic approaches for converting such binders into sensors, remain largely outstanding challenges for computational protein design.
We set out to develop a general method for designing SM binding proteins with high shape complementarity for their targets and with properties enabling facile conversion into sensors. We hypothesized that a design approach beginning with identification of protein scaffolds with high SC(1, 11) to the targeted SM would be able to ultimately achieve higher affinity binding than approaches based on fixed scaffolds(3, 5, 6, 8), and enable binding to flexible and polar targets. We reasoned further that if the scaffolds could be split into two or more independently folded domains such that SM binding energy drives association, binders could be readily converted into sensors. This strategy requires that the domains be folded prior to association to avoid aggregation and proteolysis of the split domains in the unbound state. Thus scaffold sets that can harbor binding pockets of widely ranging shapes and sizes, and at the same time can be readily split into multiple independently folded domains could provide a general solution to the sensor design problem(11).
We reasoned that designed pseudocyclic scaffolds consisting of a repeating structural unit surrounding a central pore or pocket could satisfy the above requirements(11) (Fig 1a). First, depending on the geometry of the structural unit and the number of units in the closed pseudocycle, the central pocket can have a very wide range of sizes and shapes. Second, since the interactions within the protein are almost entirely local–between residues close along the sequence within single repeat units or between adjacent repeat units, pseudocycles can be split into multiple domains that retain almost all of the stabilizing interactions present in the unsplit protein, and hence these split domains are likely to fold and be well behaved on their own(11). We set out to (Fig 1a & S1a–b) develop a general approach integrating DL and Rosetta(12) energy-based design methods(11, 13–16) to generate high SC pseudocycle based binding proteins for any desired small molecule (Fig 1b).
Figure 1. Pseudocycle-based SC optimizing design method and target SMs.
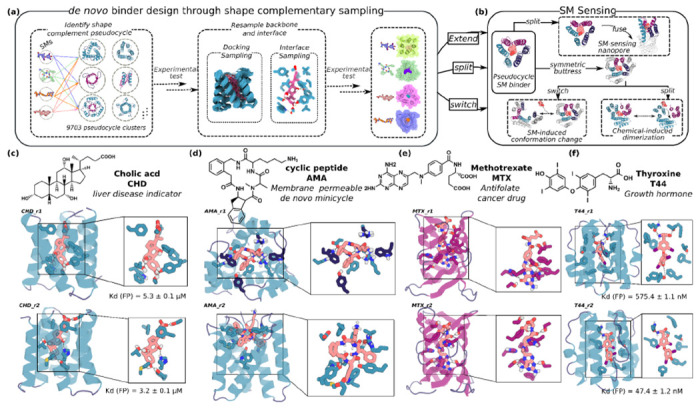
(a) Diverse conformers of the SM of interest are docked into deep learning generated pseudocycles containing a wide array of central pockets(11), and the interface sequence optimized for high affinity binding using Rosetta or LgandMPNN. Top ranked designs are tested experimentally, and the backbones of the best hits and the docked poses are extensively resampled. Following sequence design, top ranked second round designs are experimentally tested (see Fig S1). (b) Because pseudocycles are constructed from modular repeating units which surround the central binding pocket, the binders can be readily transformed into sensors through multiple strategies. (c-f) Examples of first round design models for each target ligand.
We chose four SMs as binding targets: cholic acid (CHD), methotrexate (MTX), thyroxine (T44), and a de novo designed cell permeable cyclic tetrapeptide called AMA (Fig. 1, S4 a–d, & Table S4). CHD is the primary bile acid, and detection of its free form is important for liver disease determination(17). MTX is an anti-folate cancer treatment agent which requires regular blood monitoring to reduce adverse outcomes for patients(18). T44 is a critical human hormone regulating energy usage and other functions; at home monitoring to detect free T44 levels is used for patient thyroid condition diagnosis. The current commercially available detection methods for CHD, MTX, and T44 are either time-consuming, chromatography-based analytical methods, or immunoassays(17–21), which cannot distinguish individual molecules from variants (CHD, T44) or bound from free forms (T44, MTX); higher affinity and specificity binders are needed for rapid at-home SM sensing devices. From the design perspective, T44 and MTX are significantly more flexible and polar than previous SM targets for computational protein design(3–8), and MTX is particularly challenging due to its high polarity and flexibility (Fig S4e & Table S4). To test the ability of the method to design binders to larger ligands, we also included AMA (Fig 1 & S4a), a de novo designed cell membrane-permeable tetrapeptidic macrocycle(22); designed binders for such compounds could be turned into chemical-induced dimerization (CID) systems enabling bioorthogonal control for adoptive cell therapies and other applications. We obtained or synthesized fluorescein (FITC, see ‘Preparation of the FITC-labeled SM target’ in Methods) or biotin labeled versions of all four ligands for experimental screening (Fig S4).
Identification of pseudocycle scaffolds with high shape complementary to target
We begin by identifying for a given ligand of interest the most shape complementary pockets present in AlphaFold2 and ProteinMPNN generated pseudocycle scaffolds(11) with a wide range of pocket shapes and sizes (Fig 1a & S1a). We first used RDKit(23) to generate hundreds of physically allowed rotamers for each ligand, clustered them based on all-atom root-mean-square deviation (r.m.s.d), and selected a low Rosetta energy conformer to represent each cluster (it is not sufficient to only consider the lowest energy conformer as this overly limits the maximal shape complementarity that can be achieved; many native SM binders bind the ligand in a conformation different from its free form). For each ligand, we docked one to a few hundred rotamers (Table S1) to the pockets in 9,703 pseudocycles(11) using Rifgen/Rifdock(3), generating in total 1-10 million docks. To rapidly identify docks with relatively high SC between the protein and SM, we developed an in silico two-step ‘predictor’ (Fig S1a) which benchmarks showed highly correlated with the SC after the much more expensive full sequence design (Fig S5). The ‘predictor’ first uses a quick Rosetta protocol (~10 dock/CPU second, see ‘Predictor to select best docks’ in Methods) to pack the ligand-protein interface and estimate the protein-ligand contacts (using ‘contact_molarcular_surface’, CMS); The best solutions found in this screen were subjected to a more thorough interface design and scoring protocol (~1 dock/CPU second), and additional features including the predicted binding energy and the number of hydrogen bonds to the ligand were used to select promising docks (see ‘Step-wise SM binder design pipeline in detail’ in Methods). Full fixed-backbone sequence design was carried out for the top selected ~100,000 docks (Table S1) using position specific scoring matrix (PSSM)-based Rosetta design(12) (see “PSSM-based Rosetta design protocol” in Methods) or using a new deep learning based ligandMPNN sequence design method which enables direct optimization of interactions with small molecules (see “Iterative ligandMPNN design protocol” in Methods)(16) (Table S2). 5-10,000 designs for each target predicted to fold to the designed structures by AlphaFold2(14) (AF2) and make low free energy interactions with the ligand (as computed by Rosetta) were selected for experimental characterization (Table S1).
The selected designs were encoded in oligonucleotide libraries, displayed on the yeast cell surface and binding was assessed using fluorescence-activated cell sorting (FACS), and deep sequencing (see ‘High-throughput methods for binder identification’ in Methods). For CHD (Fig 1c & S2a–b) we obtained two binders based from the same scaffold with Kds of 3.2 ± 0.1 and 5.3 ±0.1 μM by fluorescence polarization (FP, see ‘Fluorescence polarization studies’ in Methods); in the design models all ligand polar atoms are contacted by sidechains, with these key polar residues buttressed by hydrogen-bonding networks (Fig 1c & S2a–b). For T44, we again obtained two binders based on the same scaffold with Kds of 575.4 ± 1.1 nM and 47.4 ± 2 nM using FP (Fig 1f & S2c–d). For MTX and AMA, we identified one and two potential binders from each two and four hit pseudocycle scaffolds, respectively (Fig 1d–e, S3 & S6); these binders were weaker than the CHD and T44 binders but still showed clear ligand-specific binding signals on FACS using yeast surface display (Fig S3). In all four cases, there were 100 or fewer designs from the scaffolds that gave rise to binders (for example, only 32 designs were tested from the scaffold for CHD which yielded 2 binders), so had we been able to identify the most appropriate scaffold for each compound in advance, the protocol would have yielded binders with small scale screening (the contact molecular surface with ligand was generally higher for the designs with binding activity, but more data is needed to set thresholds; Fig S1c). Binders were obtained using both the PSSM-based Rosetta design protocol and the ligandMPNN design protocol (Table S2), suggesting that when there is close shape complementarity between protein and ligand, the details of the sequence design method become less critical.
We were able to obtain a co-crystal structure of one of the CHD binders in complex with CHD (CHD_r1, Fig 2 & S7a–c). The co-crystal structure agrees closely with the computational design model with a 0.80 Å Cα-r.m.s.d between the two. All ligand-protein hydrogen bond interactions and hydrogen bond networks were accurately recapitulated (Fig 2b, d), including two hydrogen bonds to the side hydroxyl groups of CHD and a hydrogen bonding network around the CHD head hydroxyl group (CHD_r1, Fig 2c). This structure demonstrates that our interface design pipeline can accurately design detailed protein-ligand interactions.
Figure 2. X-ray crystallography demonstrates accuracy of design approach.
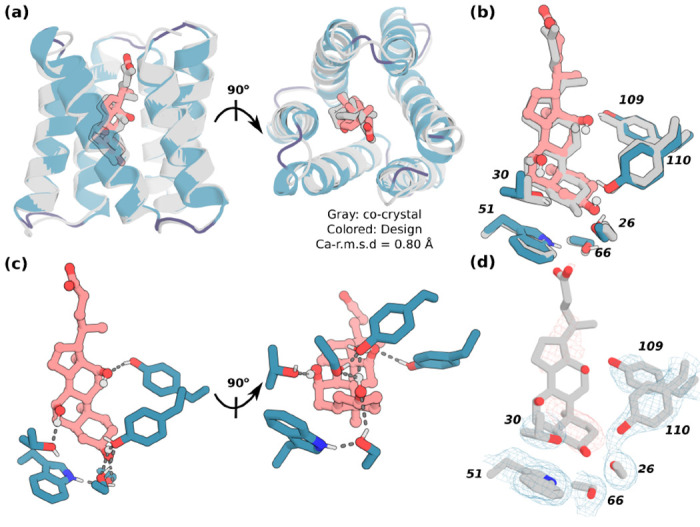
(a) The crystal structure of CHD_r1 (gray) is very similar to its computational design model (teal). (b-c) The designed sidechain interactions with the three CHD hydroxyl groups and the buttressing hydrogen bonding network are closely recapitulated in the crystal structure (design is colored, crystal structure is in gray). (d) The ligand and the key interacting residues were well resolved with clear electron density. The protein backbone is shown in cartoons, and CHD and the key interacting side chains in sticks. Pink, ligand carbon atoms; red, oxygen; blue, nitrogen; white, polar hydrogen. The residue numbers of the key residues are labeled. Also see Fig S7a–c.
Generating higher affinity designs by scaffold resampling
We next sought to generate higher affinity designs by sampling new backbones around the pseudocycle scaffolds which gave rise to the first round hits. We generated ~5000 sequences for each scaffold using proteinMPNN(13) and predicted their structures using AF2(14); this generates backbones which range from 0.5 to 3 Å Ca-r.m.s.d from the starting template (Fig S1b). The pre-generated ligand conformers were docked against the corresponding resampled scaffolds using RifDock(3) generating 10-30 million new docks. After two-step predictor selection, 50,000-500,000 docks were selected for PSSM-based Rosetta design protocol or ligandMPNN design protocols, as described above. 5,000-15,000 de novo designs (Table S1) were selected for a second round of binder screening (see “High-throughput methods for binder identification” in Methods). As expected, the designs from the second round showed significantly higher SC to the ligand compared to the designs from the first round (Fig S1c).
We observed significant improvement in binder quality and quantity in the second round design round (Table S1 & S2). For CHD, the highest affinity binders among all the 37 verified binders (as measured by FP) improved by ~700 fold from 3.2 and 5.3 μM (Fig 1c & S2a–b) in the first round to 4.68 nM in the 2nd round (Fig 3a–d & see full binder list S8–10). Site saturation mutagenesis (SSM) of the five highest affinity 2nd round binders (CHD-d1 to d5) confirmed that the key interactions in the design model were essential for binding (Fig S11). For T44, the affinity by FP improved from 47.4 nM to 18.2 nM (Fig 3g–h, S14–15) . For AMA and MTX, the binding affinities in the first round were too weak to be measured off the yeast surface (where there is very high avidity), likely in the high micromolar to millimolar range. In the second round, we obtained 870 nM binders (measured by surface plasmon resonance) for AMA (Fig 3e & S12, SPR, see ‘SPR studies’), and for MTX, the most challenging target (Fig 1e & S6a), we obtained a 6.9 μM binder (MTX-d1) from one of the β barrel-like scaffolds (Fig 3f), a first example of a de novo binder to a highly polar and flexible ligand (Fig S4e & Table S4). The designed binding mode of MTX-d1 is supported by SSM analysis and competition assays (Fig S16). As in the first round, binders were obtained using both the Rosetta-based and ligandMPNN-based interface design protocols (Table S2) but the ligandMPNN based method generated generally tighter binders. Control experiments for CHD and MTX in which the backbone and docking pose were fixed did not yield similar or any improvements in affinity(16), highlighting the importance of resampling for increasing shape complementarity and binding affinity.
Figure 3. Experimental characterization of selected designed binders from the affinity improving round.
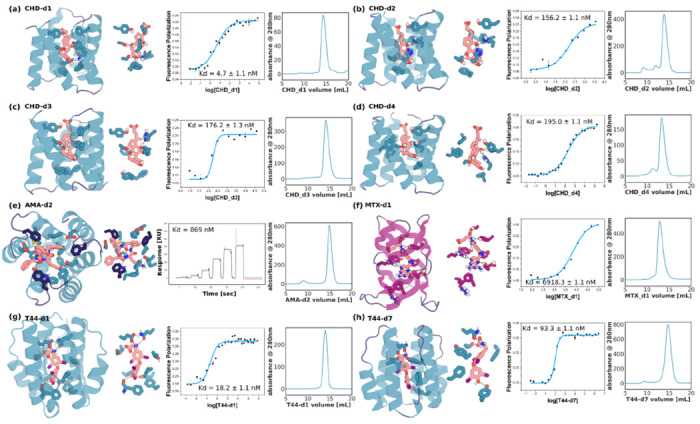
(a-d) Nanomolar affinity CHD binders CHD_d1 to d4 (full list in Fig S9), (e) nanomolar binder for AMA (see Fig S12); (f) micromolar methotrexate binder, (g-h) two nanomolar T44 binders (full list at Fig S14). For each panel, from left to right, the design model, zoom in on the sidechain-ligand interactions, FP (or SPR in the case of AMA) binding measurements, and SEC traces. Kd values and error bars are from two independent experiments. Interacting side chains and ligands are shown in sticks, with oxygen, nitrogen, iodine, and polar hydrogen colored in red, blue, purple, and white, respectively. Key interactions are indicated by gray dashed lines. The cartoon of and sticks from helixes, sheets, and loops are colored in teal, magenta, and dark blue, respectively.
Converting pseudocycle binders into sensors
Because in pseudocycles the stabilizing interactions are primarily within repeat units and between adjacent units, all of the designs by construction can be split into two or more chains that are likely to fold at least in part in isolation (Fig 4a, g, this contrasts with most globular proteins, where splitting is likely to disrupt the central hydrophobic core). Because of this, ligand binding can be coupled to reconstitution of the full pseudocycle.
Figure 4. Conversion of pseudocycle binders into ligand gated channels and CID systems.
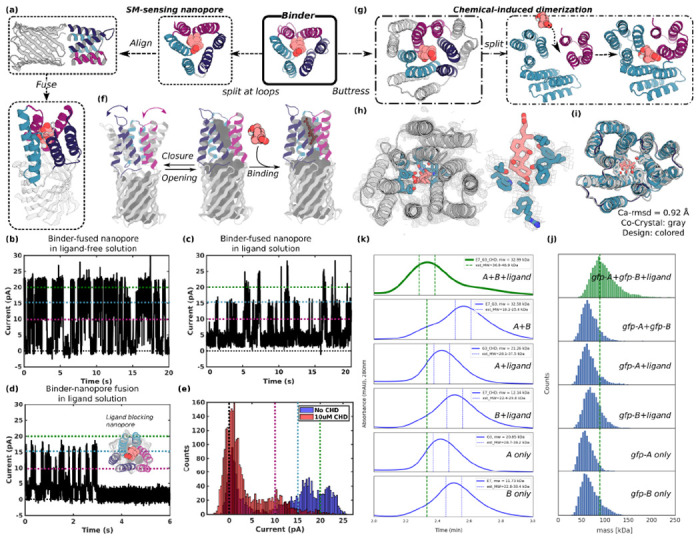
(a-f) Ligand sensing de novo nanopore construction. (a) The three structural repeat units of CHD_r1 were inserted into three different loops in a 12 stranded de novo nanopore using inpainting to join the chains such that the central axes of binder and nanopore are aligned. The conductance of the original nanopore is ~220 pS (Fig S17a), and is not influenced by CHD. The conductance of binder-fused nanopore in the absence (b) and presence of CHD (c-e): In the absence of CHD (c), the pore fluctuates between a state with high conductance very similar to the unmodified pore and a low conductance state (c); in the presence of CHD the duration of the low conductance states is greatly increased (c-e); the longer record (c) and a single closure event (d) are shown for clarity, and the histogram of the current with and without ligand is shown in (e). Different currents, 0, 10, 15, 20 pA are marked out for clarity using dashlines in black, magenta, teal, and green, respectively in b-e. The gated nanopores are robust through multiple cycles of opening and closure (c), upon reversal of the voltage, the original high conductance state is restored (Fig S17b). (f) The conductance fluctuations of the binder-fused pore in absence of ligand likely reflect transient association of the 3 subunits; ligand binding stabilizes the associated state leading to prolonged blocking of the pore. (g-k) CID system construction. CHD binder, CHD_r1, was buttressed by diffusion of an outer ring of helices to increase the stability of split protein fragments (g & Fig S18). The crystal structure of the buttressed binder with ligand (h & Fig S7d–f) is in close agreement with the design model (i). To create a CID system, we split the buttressed binders into halves and redesign the protein-protein interface to increase solubility of the fragments and disfavor association in the absence of ligand. Characterization of CHD induced association of the split fragments by size exclusion chromatography (k) and mass photometry (j). Dimerization of the two split domains (A and B) in presence (first trace from top), but not the absence, (second trace from top) of ligand. The individual monomers do not dimerize in the presence(third and forth trace from top) or absence (fifth and sixth traces from the top) of ligands. N terminal gfp tags were fused to the monomers to facilitate detection by mass photometry.
We first took advantage of this property by integrating a CHD binder, CHD_r1, consisting of 3 repeats of a helical hairpin around the central binding site, into a de novo designed 12 stranded beta barrel nanopore(24) such that reconstitution of the complete pseudocycle from the split domains would block ion conductance through the pore (Fig 4a). We aligned the central axis of CHD_r1 with the central axis of the designed nanopore (TMB12_3)(24), and inserted the three helical hairpins of the binder between strands 3 and 4, 7 and 8, and 11 and 12 of the beta barrel using RosETTAfold joint inpainting(25) to build short connectors (5-8 Å) between the binder hairpins and the nanopore (Fig 4a, see ‘Design of SM binder fused nanopore’ in Methods).
We expressed 12 integrated CHD binder-nanopore fusions in E. coli and purified them from inclusion bodies (see “Design of SM binder fused nanopore” in Methods). Five designs with monomeric SEC peaks were integrated into planar lipid bilayers, and ionic conductances were measured under an applied voltage of 100 mV. The open-pore conductance of the original nanopore is constant at ~220 pS and was not affected by CHD (Fig S17a). Two of the binder-nanopore fusions had similar open-pore conductances (Fig 4b) to the original nanopore (Fig 17Sa), but showed frequent transitions to two lower conductance states which likely reflect transient association of 2 or 3 helical hairpins in the absence of the ligand (Fig 4f left). In the presence of CHD, the dwell time of the nanopore in the closed state significantly increased for the most sensitive design (Fig 4c–e), likely reflecting stabilization of the trimeric pseudocycle by the ligand (Fig 4f right). The current amplitudes approached near zero for ~0.4-3 seconds on average before returning to higher conductance (Fig 4c–e); each fluctuation to a low conductance state likely corresponds to an independent CHD binding event indicating that the pores are robust to multiple cycles of CHD binding and release (Fig 4c). The limited number of observed states enabled quantification of the pore blocking and release dwell time distributions, which were consistent with the binding affinity of the unfused CHD binder (FigS 17c–d). In some cases the blocked state was very long lived (Fig 4d), switching back to fluctuating between open and closed states only upon reversal of the applied voltage polarity without compromising the stability or the conductance of the nanopore (Fig S17b). The lower noise and considerable increase in duration of the closed state conductances at longer timescales (100 ms to s) in the presence of the ligand (the fraction of transitions to closed states with dwell times of one second or more increased from 1% to 6.5% upon addition of CHD) leads to lower sampling frequency requirements compared to previously engineered sensors(26–28). The simplicity of construction of nanopore sensors by incorporating designed pseudocycles binding targets of interest into de novo designed quiet nanopores should enable the generation of a wide variety of new sensors with superior properties.
We next sought to convert the pseudocycle binders into chemically-induced dimerization (CID) systems (Fig 4g). There has been considerable interest in CID systems in protein engineering and synthetic biology for SM inducible switches for regulating protein association(29), signaling or enzymatic activity(30). Despite the potential, almost all work has utilized a small number of CID systems, such as rapamycin-FKBP-FRB(31), and involved natural proteins as one(32) or both partners(33). Synthetic biology approaches would benefit considerably from systematic approaches for designing CID systems for new ligands, for example for feedback control based on product levels in metabolic engineering.
We first stabilized the pseudocycles by building a second ring of stabilizing structural elements around the inner ring which forms the binding interface (Fig 4g & S18a): the interactions between the inner and outer rings should contribute sufficient stabilization to make up for the loss in interactions between the N and C terminal repeats upon splitting. We chose the CHD binder, CHD_r1, as a proof-of-concept, and incorporated a second ring of helices using RFDiffusion(10); the inner and outer helices packed closely around a hydrophobic core (Fig S18a). We obtained a co-crystal structure of the buttressed binder with CHD bound (Fig 4h,i & S7d–f) with the outer buttressing ring of helices, the ligand conformation and key interacting sidechains very similar to the design model (Fig 4h–i & S7e–f). The buttressed binder was more stable and bound to CHD more tightly, likely due to rigidification of the target binding conformation (Fig S18b). This approach of testing binding first at the single layer stage and then adding an outer layer by buttressing to stabilize the highest affinity binders has the considerable advantage of reducing the difficulty and cost of design testing: the single ring structures are less than 120 aa and can be encoded on two 250-300 nt commercially available oligonucleotides from an oligo array, whereas the two ring structures are longer than 200aa for which gene synthesis is far more expensive and lower throughput.
To generate a CID system, we split the buttressed binder into two parts (Fig 4g). Using tied-position proteinMPNN design (see “CID design and testing” Methods), we optimized the newly created interfaces to increase the solubility of the split domains while retaining the ability to form the holo-complex in the presence of the ligand. We expressed, purified, and tested 34 designed CID protein pairs and determined their oligomerization state by SEC (Fig 4k) and mass photometry (Fig 4j) in the presence and absence of CHD. For the best behaving of these designs, the individual domains were monomeric in isolation but associated to form the heterodimer in the presence of 10 uM CHD in both experiments. Because the individual subdomains (A is 9.8 kDa, B is 18.9 kDa) of our designed CID system are smaller than the detection limit of mass photometry (around 30 kDa), we expressed A and B with a N-terminus green fluorescent protein (gfp). By both SEC and mass photometry, dimerization was observed with ligand (Fig 4j, first lane from top) but not without ligand (Fig 4j, second lane from top). Individual proteins with (Fig 4j, third and forth lane from top) or without (Fig 4j, fifth and sixth lane from top) ligands did not show dimerization. The mass of the dimer peak derived from mass photometry, 89.0 ± 22.0 Da, was close to the expected 83 kDa for the gfp-A-ligand-gfp-B complex. As protein association can be readily coupled to site specific transcriptional activation (for example by linking a DNA binding module with an activator module) and other cellular readouts, the ability to generate CID systems for potentially any SM of interest could have very broad impact for metabolic pathway engineering by enabling feedback control based on levels of the desired product and/or unwanted or intermediate species.
Conclusion
Our SC pseudocycle-based design approach goes considerably beyond previous design studies(3–9) in generating binders to large, polar, and/or flexible SMs, such as MTX, T44, and AMA, with affinities in the range for use in diagnostics without experimental affinity maturation. The crystal structures of the CHD binders with the intricate network of side chain-ligand interactions nearly identical to the computational design model highlights the accuracy of the design approach. While the importance of SC for binding has been long understood(1), our direct optimization of SC by design would have been very difficult prior to the recent advances in DL-based protein structure prediction and design(13–15), which enabled rapid de novo sampling and evaluation of pseudocycle topologies(11) , resampling of the best solutions for a given ligand, and design of interactions with the ligand (with the new LIgandMPNN); the ability to iteratively resample the backbones and docking poses was essential for binding affinity improvement.
Our pseudocycle-based approach has a number of advantages for ligand binder and sensor design. First, centrally located and high SC binding pockets can be generated with high affinity for very diverse SMs. The variety of possible individual repeat unit structures is almost unlimited, and the number of repeat units can be readily varied, leading to vast numbers of possible pseudocycle structures all harboring central pockets. Because the structures are largely locally encoded with few long range contacts, interactions with each portion of the ligand can subsequently be optimized independently. Second, the ability to test small genetic footprint single rings forming the binding pocket in a first step, and then buttress the best designs in a second step, enables rapid and low cost gene synthesis for exploring diverse design solutions without compromising the robustness and stability of the final binding modules. Third, because of the local encoding of the structure, designed binders can be readily integrated into ligand gated channels and CID systems by splitting into subdomains that retain most of the interactions present in the full protein. The power of de novo design is highlighted by our ability to integrate the three repeat units of the CHD binder into three loops of a robust designed nanopore to create a CHD gated nanopore with almost complete gating of current by CHD; because of the high signal to noise ratio, minimal post processing of the acquired signal is required compared to previously engineered pores based on native proteins, which typically exhibit high level of noise in the absence of ligand due to the presence of a multitude of partially occluded states(26, 34). Here we used RFdiffusion(10) to build the outer pseudocycle to buttress the inner ring; moving forward it should be possible to adapt RFdiffusion All-Atom(9) to generate single ring pseudocycles directly around target ligands. We anticipate that our SC pseudocycle-based approach should enable generation of robust ligand responsive channels and sensors for a wide variety of molecules of biological interest.
Supplementary Material
Acknowledgments
We thank Dr. Sam Pellock and Dr. Indrek Kalvet for providing many general discussions for laboratory settings. We thank Avi Swartz and Dr. Patrick Erickson for providing chromatography assistance. We thank Dr. Buwei Huang and Dr. Robert Ragotte for providing SPR assistance. Crystallographic diffraction data was collected at the Northeastern Collaborative Access Team beamlines at the Advanced Photon Source and at CBMS/NSLS2. NECAT is funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The Center for Bio-Molecular Structure (CBMS) is primarily supported by the NIH-NIGMS through a Center Core P30 Grant (P30GM133893), and by the DOE Office of Biological and Environmental Research (KP1607011). NSLS2 is a U.S.DOE Office of Science User Facility operated under Contract No. DE-SC0012704. This publication resulted from the data collected using the beamtime obtained through NECAT BAG proposal # 311950. Dr. Meerut Said, Long Tran, and Dr. Sgardip Majumder are co-second authors, they contributed equally, and everyone agrees their authorship orders can be exchanged to benefit their own career development.
Funding
This research was supported by various sources of funding. L.A., M.S., S.M., I.G., G.R.L., D.J., J.D., I.A., B.C., A.B., A.K., PL., V.A., P.S., D.Z., D.H., XL., M.G., D.K.V. and D.B. thank the Audacious Project at the Institute for Protein Design for their generous support. L.A. also acknowledges the Washington Research Foundation, Innovation Fellows Program and Translational Research Fund, and Bill and Melinda Gates Foundation #OPP1156262. M.S. also thanks the Higgins Family and the Defense Threat Reduction Agency (DTRA) Grant HDTRA1-19-1-0003. S.M. also thanks the Air Force Office of Scientific Research under award number FA9550-22-1-0506. G.R.L. also thanks the Washington Research Foundation, Innovation Fellows Program. A.P. thanks the Washington Research Foundation. D.J. also acknowledges a gift from Microsoft and Schmidt Futures funding from Eric and Wendy Schmidt by recommendation of the Schmidt Futures program. J.D. also thanks a gift from Microsoft and the Open Philanthropy Project Improving Protein Design Fund. I.A. also acknowledges Spark Therapeutics/Computational Design of a Half Size Functional ABCA4 project, a gift from Microsoft, the Defense Threat Reduction Agency (DTRA) Grant HDTRA1-21-1-0007, the SSGCID which is supported by NIAID Federal Contract # HHSN272201700059C, and NIH Grant 75N93022C00036 under NIAID contract. B.C. also thanks NIH Grant R01AG063845, the Open Philanthropy Project Improving Protein Design Fund, and Howard Hughes Medical Institute (HHMI). A.B. also acknowledges NSF Grant CHE-1629214, NIH Grant R0AI160052, DTRA Grant HR0011-21-2-0012 under HEALR program, and HHMI. A.K. also thanks DTRA Grant HR0011-21-2-0012 under HEALR program and The Bill and Melinda Gates Foundation #OPP1156262. PL. also acknowledges DTRA Grant HDTRA1-19-1-0003. P S. also thanks The Juvenile Diabetes Research Foundation International (JDRF) grant # 2-SRA-2018-605-Q-R. C.N. acknowledges Novo Nordisk Foundation Grant NNF18OC0030446. D.F. also acknowledges a gift from Microsoft. D.Z. also thanks The Audacious Project at the Institute for Protein Design. D.H. also acknowledges NIH Grants U19AG065156 and R01AG063845 and HHMI. X L. also thanks DTRA Grant HR0011-21-2-0012 under HEALR program, the Juvenile Diabetes Research Foundation International (JDRF) grant # 2-SRA-2018-605-Q-R, the Helmsley Charitable Trust Type 1 Diabetes (T1D) Program Grant # 2019PG-T1D026, and the Bill and Melinda Gates Foundation #OPP1156262. D.B. is also supported by HHMI.
Footnotes
Competing interests
L.A., M.S., L.T., S.M., and D.B. are the authors of the patent application (DE NOVO DESIGNED SMALL MOLECULE BINDERS VIA EXTENSIVE SHAPE COMPLIMENTARY SAMPLING, 49962.01US1, filing date: 2023/12/05) submitted by the University of Washington for the design, composition, and function of the binders and sensors created in this study. V.A. is the author of a patent application (EP 23218330.1, filing date: 2023/12/19) submitted by the VIB-VUB Center for Structural Biology, composition, and function of the nanopore (TMB12_3) used in this study. P.J.S. is the author of a patent application (xx, filing data: yy) submitted by the University of Washington for the design and functional characterization of peptidic minicycles.
Data and Materials availability
All Data and scripts are available online. All scripts for stepwise sampling are available at github (repository will be public after BiorXiv release): https://github.com/LAnAlchemist/Pseudocycle_small_molecule_binder. All scripts for CID design are available at github https://github.com/iamlongtran/pseudocycle_paper (repository will be public after BiorXiv release). All sequencing data were analyzed using an in-house script, and the git repository is at: https://github.com/feldman4/ngs_app.
Reference
- 1.Gao M., Skolnick J., The distribution of ligand-binding pockets around protein-protein interfaces suggests a general mechanism for pocket formation. Proc. Natl. Acad. Sci. U. S. A. 109, 3784–3789 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takeuchi J., Fukui K., Seto Y., Takaoka Y., Okamoto M., Ligand–receptor interactions in plant hormone signaling. Plant J. 105, 290–306 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Dou J., Vorobieva A. A., Sheffler W., Doyle L. A., Park H., Bick M. J., Mao B., Foight G. W., Lee M. Y., Gagnon L. A., Carter L., Sankaran B., Ovchinnikov S., Marcos E., Huang P.-S., Vaughan J. C., Stoddard B. L., Baker D., De novo design of a fluorescence-activating β-barrel. Nature 561, 485–491 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dou J., Doyle L., Greisen P. Jr., Schena A., Park H., Johnsson K., Stoddard B. L., Baker D., Sampling and energy evaluation challenges in ligand binding protein design: Computational Protein Design. Protein Sci. 26, 2426–2437 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bick M. J., Greisen P. J., Morey K. J., Antunes M. S., La D., Sankaran B., Reymond L., Johnsson K., Medford J. I., Baker D., Computational design of environmental sensors for the potent opioid fentanyl. eLife 6, e28909 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Polizzi N. F., DeGrado W. F., A defined structural unit enables de novo design of small-molecule-binding proteins. Science 369, 1227–1233 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tinberg C. E., Khare S. D., Dou J., Doyle L., Nelson J. W., Schena A., Jankowski W., Kalodimos C. G., Johnsson K., Stoddard B. L., Baker D., Computational design of ligand-binding proteins with high affinity and selectivity. Nature 501, 212–216 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee G. R., Pellock S. J., Norn C., Tischer D., Dauparas J., Anishchenko I., Mercer J. A. M., Kang A., Bera A., Nguyen H., Goreshnik I., Vafeados D., Roullier N., Han H. L., Coventry B., Haddox H. K., Liu D. R., Yeh A. H.-W., Baker D., Small-molecule binding and sensing with a designed protein family. doi: 10.1101/2023.11.01.565201 (2023). [DOI] [Google Scholar]
- 9.Krishna R., Wang J., Ahern W., Sturmfels P., Venkatesh P., Kalvet I., Lee G. R., Morey-Burrows F. S., Anishchenko I., Humphreys I. R., McHugh R., Vafeados D., Li X., Sutherland G. A., Hitchcock A., Hunter C. N., Baek M., DiMaio F., Baker D., Generalized Biomolecular Modeling and Design with RoseTTAFold All-Atom. doi: 10.1101/2023.10.09.561603 (2023). [DOI] [PubMed] [Google Scholar]
- 10.Watson J. L., Juergens D., Bennett N. R., Trippe B. L., Yim J., Eisenach H. E., Ahern W., Borst A. J., Ragotte R. J., Milles L. F., Wicky B. I. M., Hanikel N., Pellock S. J., Courbet A., Sheffler W., Wang J., Venkatesh P., Sappington I., Torres S. V., Lauko A., De Bortoli V., Mathieu E., Barzilay R., Jaakkola T. S., DiMaio F., Baek M., Baker D., “Broadly applicable and accurate protein design by integrating structure prediction networks and diffusion generative models” (preprint, Biochemistry, 2022); bioRxiv. [Google Scholar]
- 11.An L., Hicks D. R., Zorine D., Dauparas J., Wicky B. I. M., Milles L. F., Courbet A., Bera A. K., Nguyen H., Kang A., Carter L., Baker D., Hallucination of closed repeat proteins containing central pockets. Nat. Struct. Mol. Biol., doi: 10.1038/s41594-023-01112-6 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leaver-Fay A., Tyka M., Lewis S. M., Lange O. F., Thompson J., Jacak R., Kaufman K. W., Renfrew P. D., Smith C. A., Sheffler W., Davis I. W., Cooper S., Treuille A., Mandell D. J., Richter F., Ban Y.-E. A., Fleishman S. J., Corn J. E., Kim D. E., Lyskov S., Berrondo M., Mentzer S., Popović Z., Havranek J. J., Karanicolas J., Das R., Meiler J., Kortemme T., Gray J. J., Kuhlman B., Baker D., Bradley P., “Rosetta3” in Methods in Enzymology (Elsevier, 2011; https://linkinghub.elsevier.com/retrieve/pii/B9780123812704000196) vol. 487, pp. 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dauparas J., Anishchenko I., Bennett N., Bai H., Ragotte R. J., Milles L. F., Wicky B. I. M., Courbet A., de Haas R. J., Bethel N., Leung P. J. Y., Huddy T. F., Pellock S., Tischer D., Chan F., Koepnick B., Nguyen H., Kang A., Sankaran B., Bera A. K., King N. P., Baker D., Robust deep learning-based protein sequence design using ProteinMPNN. Science 378, 49–56 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., Bridgland A., Meyer C., Kohl S. A. A., Ballard A. J., Cowie A., Romera-Paredes B., Nikolov S., Jain R., Adler J., Back T., Petersen S., Reiman D., Clancy E., Zielinski M., Steinegger M., Pacholska M., Berghammer T., Bodenstein S., Silver D., Vinyals O., Senior A. W., Kavukcuoglu K., Kohli P., Hassabis D., Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baek M., DiMaio F., Anishchenko I., Dauparas J., Ovchinnikov S., Lee G. R., Wang J., Cong Q., Kinch L. N., Schaeffer R. D., Millán C., Park H., Adams C., Glassman C. R., DeGiovanni A., Pereira J. H., Rodrigues A. V., van Dijk A. A., Ebrecht A. C., Opperman D. J., Sagmeister T., Buhlheller C., Pavkov-Keller T., Rathinaswamy M. K., Dalwadi U., Yip C. K., Burke J. E., Garcia K. C., Grishin N. V., Adams P. D., Read R. J., Baker D., Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duparas J., Lee G. R., Pecoraro R., An L., Anishchenko I., Glasscock C., Baker D., Atomic context-conditioned protein sequence design using LigandMPNN. Biorxiv. [Google Scholar]
- 17.Zhao X., Liu Z., Sun F., Yao L., Yang G., Wang K., Bile Acid Detection Techniques and Bile Acid-Related Diseases. Front. Physiol. 13, 826740 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shadeed A., Kattach L., Sam S., Flora K., Farah Z., Examining the safety of relaxed drug monitoring for methotrexate in response to the COVID-19 pandemic. Rheumatol. Adv. Pract. 6, rkac100 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spencer C. A., Assay of Thyroid Hormones and Related Substances (Endotext [Internet], South Dartmouth (MA), 2017; https://www.ncbi.nlm.nih.gov/books/NBK279113/). [Google Scholar]
- 20.Welsh K. J., Soldin S. J., DIAGNOSIS OF ENDOCRINE DISEASE: How reliable are free thyroid and total T3 hormone assays? Eur. J. Endocrinol. 175, R255–R263 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tripathy N. K., Mishra S. K., Nathan G., Srivastava S., Gupta A., Lingaiah R., A Rapid Method for Determination of Serum Methotrexate Using Ultra-High-Performance Liquid Chromatography-Tandem Mass Spectrometry and Its Application in Therapeutic Drug Monitoring. J. Lab. Physicians 15, 344–353 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salveson P., De novo design f macrocycles. [Google Scholar]
- 23.Landrum G., Tosco P., Kelley B., Ric, Cosgrove D., Gedeck Sriniker, Vianello R., NadineSchneider, Kawashima E., Jones D. N, G., Dalke A., Cole B., Swain M., Turk S., AlexanderSavelyev, Vaucher A., Wójcikowski M., Take Ichiru, Probst D., Ujihara K., Scalfani V. F., Godin G., Lehtivarjo J., Walker R., Pahl A., Berenger Francois, Jasondbiggs, Strets123, rdkit/rdkit: 2023_03_3 (Q1 2023) Release, version Release_2023_03_3, Zenodo; (2023); 10.5281/ZENODO.591637. [DOI] [Google Scholar]
- 24.Sagadip, De novo nanopore. Biorxiv. [Google Scholar]
- 25.Wang J., Lisanza S., Juergens D., Tischer D., Watson J. L., Castro K. M., Ragotte R., Saragovi A., Milles L. F., Baek M., Anishchenko I., Yang W., Hicks D. R., Expòsit M., Schlichthaerle T., Chun J.-H., Dauparas J., Bennett N., Wicky B. I. M., Muenks A., DiMaio F., Correia B., Ovchinnikov S., Baker D., Scaffolding protein functional sites using deep learning. Science 377, 387–394 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahmad M., Ha J.-H., Mayse L. A., Presti M. F., Wolfe A. J., Moody K. J., Loh S. N., Movileanu L., A generalizable nanopore sensor for highly specific protein detection at single-molecule precision. Nat. Commun. 14, 1374 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fahie M. A., Chen M., Electrostatic Interactions between OmpG Nanopore and Analyte Protein Surface Can Distinguish between Glycosylated Isoforms. J.Phys. Chem. B 119, 10198–10206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foster J. C., Pham B., Pham R., Kim M., Moore M. D., Chen M., An Engineered OmpG Nanopore with Displayed Peptide Motifs for Single-Molecule Multiplex Protein Detection. Angew. Chem. Int. Ed Engl. 62, e202214566 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greene B. L., Kang G., Cui C., Bennati M., Nocera D. G., Drennan C. L., Stubbe J., Ribonucleotide Reductases: Structure, Chemistry, and Metabolism Suggest New Therapeutic Targets. Annu. Rev. Biochem. 89, 45–75 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rappas M., Niwa H., Zhang X., Mechanisms of ATPases - A Multi-Disciplinary Approach. Curr. Protein Pept. Sci. 5, 89–105 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Banaszynski L. A., Liu C. W., Wandless T. J., Characterization of the FKBP·Rapamycin·FRB Ternary Complex. J.Am. Chem. Soc. 127, 4715–4721 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Foight G. W., Wang Z., Wei C. T., Greisen P. Jr, Warner K. M., Cunningham-Bryant D., Park K., Brunette T. J., Sheffler W., Baker D., Maly D. J., Multi-input chemical control of protein dimerization for programming graded cellular responses. Nat. Biotechnol. 37, 1209–1216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stanton B. Z., Chory E. J., Crabtree G. R., Chemically induced proximity in biology and medicine. Science 359, eaao5902 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen J., Gu Y., Ke L., Zhang Q., Cao Y., Lin Y., Wu Z., Wu C., Mu Y., Wu Y.-L., Ren C., Zeng H., Cholesterol-stabilized membrane-active nanopores with anticancer activities. Nat. Commun. 13, 5985 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holm L., Laiho A., Törönen P., Salgado M., DALI shines a light on remote homologs: One hundred discoveries. Protein Sci. Publ. Protein Soc. 32, e4519 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B. A., Thiessen P. A., Yu B., Zaslavsky L., Zhang J., Bolton E. E., PubChem 2023 update. Nucleic Acids Res. 51, D1373–D1380 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Boyle N. M., Banck M., James C. A., Morley C., Vandermeersch T., Hutchison G. R., Open Babel: An open chemical toolbox. J. Cheminformatics 3, 33 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Humphrey W., Dalke A., Schulten K., VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Park H., Zhou G., Baek M., Baker D., DiMaio F., Force Field Optimization Guided by Small Molecule Crystal Lattice Data Enables Consistent Sub-Angstrom Protein-Ligand Docking. J. Chem. Theory Comput. 17, 2000–2010 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qi X., Loiseau F., Chan W. L., Yan Y., Wei Z., Milroy L.-G., Myers R. M., Ley S. V., Read R. J., Carrell R. W., Zhou A., Allosteric Modulation of Hormone Release from Thyroxine and Corticosteroid-binding Globulins. J. Biol. Chem. 286, 16163–16173 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoover D. M., DNAWorks: an automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res. 30, 43e–443 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chao G., Lau W. L., Hackel B. J., Sazinsky S. L., Lippow S. M., Wittrup K. D., Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 1, 755–768 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Cao L., Coventry B., Goreshnik I., Huang B., Sheffler W., Park J. S., Jude K. M., Marković I., Kadam R. U., Verschueren K. H. G., Verstraete K., Walsh S. T. R., Bennett N., Phal A., Yang A., Kozodoy L., DeWitt M., Picton L., Miller L., Strauch E.-M., DeBouver N. D., Pires A., Bera A. K., Halabiya S., Hammerson B., Yang W., Bernard S., Stewart L., Wilson I. A., Ruohola-Baker H., Schlessinger J., Lee S., Savvides S. N., Garcia K. C., Baker D., Design of protein-binding proteins from the target structure alone. Nature 605, 551–560 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dang B., Mravic M., Hu H., Schmidt N., Mensa B., DeGrado W. F., SNAC-tag for sequence-specific chemical protein cleavage. Nat. Methods 16, 319–322 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.An L., Cogan D. P., Navo C. D., Jiménez-Osés G., Nair S. K., van der Donk W. A., Substrate-assisted enzymatic formation of lysinoalanine in duramycin. Nat. Chem. Biol. 14, 928–933 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vorobieva A. A., De novo design of nanopores. Biorxiv. [Google Scholar]
- 47.Vorobieva A. A., White P., Liang B., Horne J. E., Bera A. K., Chow C. M., Gerben S., Marx S., Kang A., Stiving A. Q., Harvey S. R., Marx D. C., Khan G. N., Fleming K. G., Wysocki V. H., Brockwell D. J., Tamm L. K., Radford S. E., Baker D., De novo design of transmembrane β barrels. Science 371, eabc8182 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Varongchayakul N., Song J., Meller A., Grinstaff M. W., Single-molecule protein sensing in a nanopore: a tutorial. Chem. Soc. Rev. 47, 8512–8524 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pillai A., Idris A., Philomin A., Weidle C., Skotheim R., Leung P. J. Y., Broerman A., Demakis C., Borst A. J., Praetorius F., Baker D., “De novo design of allosterically switchable protein assemblies” (preprint, Biochemistry, 2023); 10.1101/2023.11.01.565167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kabsch W., XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Overview of the CCP 4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX : a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emsley P., Cowtan K., Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Williams C. J., Headd J. J., Moriarty N. W., Prisant M. G., Videau L. L., Deis L. N., Verma V., Keedy D. A., Hintze B. J., Chen V. B., Jain S., Lewis S. M., Arendall W. B., Snoeyink J., Adams P. D., Lovell S. C., Richardson J. S., Richardson D. C., MolProbity: More and better reference data for improved all-atom structure validation: PROTEIN SCIENCE.ORG. Protein Sci. 27, 293–315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.