

Abstract
Upadacitinib is a selective Janus kinase (JAK) inhibitor which is approved by the US Food and Drug Administration, the European Medicines Agency, as well as other agencies around the world for the treatment of several chronic inflammatory diseases, including rheumatic, dermatologic, and gastrointestinal diseases. Through inhibition of JAK, upadacitinib inhibits phosphorylation of downstream effector proteins, which consequently inhibits cytokine signaling for key pathways involved in inflammatory diseases. Upadacitinib more potently inhibits JAK1 than other JAK isoforms. The pharmacokinetics, pharmacodynamics, efficacy, and safety of upadacitinib were characterized in many clinical trials, which demonstrated the superiority of upadacitinib treatment over placebo or an active comparator in rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, non‐radiographic axial spondyloarthritis, atopic dermatitis, Crohn's disease, and ulcerative colitis. The safety profile of upadacitinib supported a favorable benefit–risk profile across all the approved indications. In this article, we review the mechanism of action of upadacitinib and describe how the JAK–STAT (Janus kinase–signal transducers and activators of transcription) pathway is involved in the pathogenesis of several chronic and progressive immune‐mediated inflammatory diseases. In addition, this review also provides an overview of key clinical trials that were conducted as well as relevant data which supported the clinical development of upadacitinib and informed the recommended dose(s) in each of the approved indications.
CLINICAL AND TRANSLATIONAL CARD FOR UPADACITINIB
Mechanism of Action: Janus Kinase (JAK) Inhibitor.
Indication(s): Rheumatoid Arthritis, Psoriatic Arthritis, Ankylosing Spondylitis, Non‐radiographic Axial Spondyloarthritis, Atopic Dermatitis, Ulcerative Colitis, Crohn's Disease.
Summary of Dosage and Administration: Rheumatoid Arthritis, Psoriatic Arthritis, Ankylosing Spondylitis, and Non‐radiographic Axial Spondyloarthritis: 15 mg once‐daily*.
Atopic Dermatitis: 15 mg or 30 mg once‐daily*.
Ulcerative Colitis: 45 mg once‐daily for induction (8 weeks); 15 mg or 30 mg once‐daily for maintenance*.
Crohn's Disease: 45 mg once‐daily for induction (12 weeks); 15 mg or 30 mg once‐daily for maintenance*.
*Administered using oral extended‐release tablets. Please refer to local prescribing information for additional details including recommended dosage in special populations and drug–drug interactions.
Main Metabolic Pathway: CYP3A4
Key Pharmacokinetic Characteristics: AUCinf (Mean) 265 ng h/mL for 15 mg, 543 ng h/mL for 30 mg, 752 ng h/mL for 45 mg; C max (Mean) 31.6 ng/mL for 15 mg, 71.8 ng/mL for 30 mg, 90.7 ng/mL for 45 mg; T max (Median) 2–4 h; t 1/2 (Harmonic Mean) 8–14 h.
INTRODUCTION
Cytokines play a central role in the pathogenesis of many chronic and progressive immune‐mediated dermatologic (atopic dermatitis [AD]), rheumatic (rheumatoid arthritis [RA], ankylosing spondylitis [AS], psoriatic arthritis [PsA]), and gastrointestinal (ulcerative colitis [UC], Crohn's disease [CD]) diseases. Atopic dermatitis, characterized by pruritus and red swollen skin lesions, emanates from skin barrier dysfunction and immune dysregulation mediated by the dysfunctional release of several interleukins (ILs). 1 Increased levels of pro‐inflammatory cytokines, including several ILs, granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), and tumor necrosis factor (TNF) mediate the clinical progression of RA characterized by persistent joint inflammation, proliferative synovitis and destruction of articular cartilage, and erosion of subchondral bone resulting in structural damage. 2 Several ILs and TNF‐α play a detrimental role in the initiation and progression of AS characterized by inflammation and potential ankylosis of sacroiliac joints and the spine, which is also frequently associated with inflammation of the enthesis. 3 Likewise, TNF‐α, interferon‐γ (IFN‐γ), and several ILs underlie the musculoskeletal and dermatologic hallmarks of PsA. 4 Ulcerative colitis, characterized by inflammation and ulcers in the lining of the large colon and rectum, and CD, which can involve different areas of the digestive tract and typically manifests with abdominal pain, fatigue, prolonged diarrhea, and weight loss, are two inflammatory bowel diseases (IBDs) that have similar but unique pathogenesis driven by several ILs, IFN‐γ, and TNF‐α. 5
Drugs that target cytokines or cytokine receptors (e.g., “biologics”) are widely used in the treatment of immune‐mediated inflammatory disorders by blocking a unique cytokine at the extracellular level. Although effective, innovative strategies as well as selective therapies for immune‐mediated inflammatory diseases require novel mechanisms of actions.
More recent research from the past few decades has revealed that many of the cytokines that have been identified as fundamental drivers for autoimmune diseases signal through the JAK–STAT (Janus kinase–signal transducers and activators of transcription) pathway. There are four Janus kinase (JAK) enzymes: JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2). Both pan‐JAK and selective‐JAK inhibitors have emerged in recent years as a new family of drugs aimed at modulating distinct downstream cytokine signaling across various immune‐mediated inflammatory diseases. Depending on the disease state, JAKs may either act cooperatively to transduce cytokine signaling or exhibit the preferential use of one JAK over another; thus, the differential properties of currently approved JAK inhibitors (such as a unique chemical structure, discrete mode of binding, and affinities) give rise to unique efficacy and safety profiles. 6 , 7
Upadacitinib is an oral, selective, and reversible JAK inhibitor that has demonstrated a consistent favorable benefit/risk profile across multiple diseases leading to its respective approval. In RA, PsA and across the axial SpA spectrum, it can induce rapid and durable improvement in joint signs and symptoms, pain, and function, while leading to stringent targets of disease control. 8 , 9 , 10 Similarly in both UC and CD, upadacitinib demonstrated rapid clinical and endoscopic improvement at the end of induction, which was sustained to the end of maintenance, with a positive benefit/risk profile. 11 , 12 Lastly, in AD, upadacitinib achieved stringent disease control, improvement in the ubiquitous symptom of itch, as well as various patient‐reported quality of life outcomes. 13
DRUG REGULATORY APPROVAL
Upadacitinib was initially approved by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) for the treatment of RA in 2019. Currently, upadacitinib is also approved for PsA, AS, non‐radiographic axial spondyloarthritis (nr‐axSpA), CD, and UC in multiple markets for adults and for adults and children 12 years of age and older with AD. 14 Upadacitinib is also currently being evaluated for the treatment of other inflammatory diseases (NCT05843643, NCT04927975, NCT05889182, NCT06012240). Upadacitinib is available as an extended‐release once‐daily (q.d.) tablet in strengths of 15, 30, and 45 mg in the United States, Europe, and many other countries.
MECHANISM OF ACTION
The JAK–STAT pathways are composed of four JAK kinases and seven STATs (STAT1–6, including homologs STAT5a and STAT5b). A cytokine binding to its receptor initiates the signaling cascade as well as the subsequent association/rearrangement of the receptor subunits. This rearrangement enables JAK activation by transphosphorylation and, upon activation, JAKs phosphorylate the receptors. This phosphorylation allows STATs to bind to the receptor and become phosphorylated by activated JAKs (Figure 1). The phosphorylated STATs (pSTATs) are able to form either homo‐ or heterodimers and are then able to translocate into the nucleus where they bind their respective promoter elements. Binding to the promoter elements allows them to regulate transcription of target genes. Since each cytokine receptor recruits and employs a specific combination of JAK kinases, these combinations have important implications in the therapeutic targeting of JAKs in various diseases. 1 , 5 , 15
FIGURE 1.
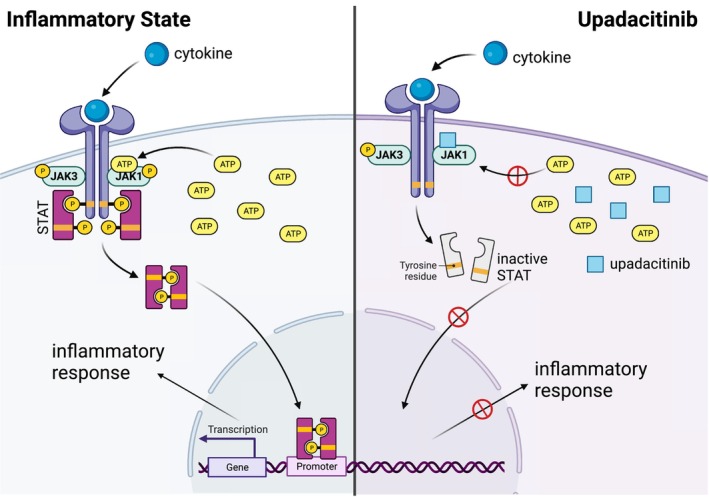
Upadacitinib mechanism of action.
Upadacitinib functions as an adenosine triphosphate (ATP)‐competitive JAK inhibitor (Figure 1), competing with ATP and blocking nucleotide binding to inhibit kinase activity and the phosphorylation of downstream effectors. Thus, STAT dimers are not formed and the translocation to the nucleus and promotor binding are inhibited. Enzymatic assays demonstrated that upadacitinib inhibits JAK1 with a half‐maximal inhibitory concentration (IC50) of 0.043 μM, JAK2 (IC50 = 0.12 μM), JAK3 (IC50 = 2.3 μM), and TYK2 (IC50 = 4.7 μM). 16 In cellular assays, upadacitinib exhibited >40‐, 130‐, and 190‐fold greater selectivity for JAK1 versus JAK2, JAK3, and TYK2, respectively, in a set of engineered cell lines that was employed to study the cellular potency and selectivity of upadacitinib on each individual kinase. 7
The pharmacokinetic–pharmacodynamic analyses that characterized the relationships between upadacitinib plasma exposures and in vivo pharmacodynamic effects demonstrated greater potency of inhibition of IL‐6‐induced pSTAT3 (as a measure of JAK1 activity) than IL‐7‐induced pSTAT5 (as a measure of JAK1/3 activity) for upadacitinib. 17 The potency ratio for inhibition of IL‐6‐induced pSTAT3 relative to IL‐7‐induced pSTAT5 (based on ratio of estimated EC50 values) is 2.0, which is consistent with the higher potency towards inhibiting JAK1 than JAK3 observed in vitro. 7
Together, these findings support that upadacitinib potently inhibits JAK1 and is less potent against the other isoforms, JAK2, JAK3, and TYK2. 7 , 16 Because different receptors are associated with different JAKs, selective blockade of one JAK can inhibit a specific biologic function or key cytokines involved in each immune‐mediated inflammatory disease while allowing other JAK‐dependent cytokines to signal normally. However, despite upadacitinib exhibiting selectivity for JAK1, JAK enzymes work cooperatively, and may therefore have some biological effects on all pairings involving JAK1. 18
PHARMACOKINETICS/PHARMACODYNAMIC CHARACTERISTICS
Upadacitinib plasma exposures are dose‐proportional over the therapeutic dose range. Upadacitinib is absorbed with a median time to reach maximum plasma concentration (T max) of 2–4 h, following oral administration of upadacitinib extended‐release formulation. 19 With once‐daily administration of upadacitinib, steady‐state plasma concentrations are achieved, with minimal accumulation, within 4 days. 14 The mean terminal elimination half‐life of upadacitinib ranged from 8 to 14 h. Upadacitinib is eliminated predominantly as the unchanged parent drug in urine (24%) and feces (38%), while upadacitinib metabolites account for approximately 34% of drug excretion. 14 The pharmacokinetics of upadacitinib have been shown to be comparable across all different patient populations evaluated to date. The pharmacologic activity of upadacitinib is attributed to the parent molecule which accounts for ~79% of the total plasma while the main metabolite accounts for ~13% of the total plasma. No active metabolites have been identified for upadacitinib.
Upadacitinib mean AUCinf after a single dose administration of 15 mg upadacitinib was 18%, 33%, and 44% higher in patients with mild (estimated glomerular filtration rate [eGFR] 60 to <90 mL/min/1.73 m2), moderate (eGFR 30 to <60 mL/min/1.73 m2), and severe renal impairment (eGFR 15 to <30 mL/min/1.73 m2), respectively, compared with participants with normal renal function (eGFR ≥90 mL/min/1.73 m2). 20 Upadacitinib mean C max was similar among participants with normal and impaired renal function. 20 These changes in exposures resulted in no dose adjustment recommendations for patients with mild or moderate renal impairment. For IBD patients with severe renal impairment, the recommended induction and maintenance doses are 30 mg q.d. and 15 mg q.d., respectively. Patients with severe renal impairment for all other approved indications have a recommended dose of 15 mg q.d.. Following the administration of a single dose of 15 mg upadacitinib, patients with mild (Child‐Pugh A) and moderate (Child‐Pugh B) hepatic impairment, had 28% and 24% higher mean AUCinf, respectively, relative to participants with normal liver function. 21 Compared with participants with normal liver function, upadacitinib mean C max was unchanged in patients with mild hepatic impairment and 43% higher in patients with moderate hepatic impairment. 21 Upadacitinib was not studied in patients with severe hepatic impairment. No dose adjustment is recommended in patients who have mild or moderate hepatic impairment; however, this recommendation may vary by jurisdiction and the local approved label should be referred to for the recommended doses.
Upadacitinib metabolism is mediated by mainly CYP3A4 with a potential minor contribution from CYP2D6. 14 Administration of strong CYP3A inhibitors increase upadacitinib AUC by 75% and C max by 70% while strong inducers of CYP3A reduce upadacitinib plasma exposures by approximately half. The recommended dose for upadacitinib co‐administered with strong CYP3A inhibitors is 15 mg q.d. for all approved indications, except for the induction doses in IBD that are recommended to be 30 mg q.d. Upadacitinib is not recommended to be used concomitantly with strong CYP3A inducers as the reduction in plasma exposures could lead to reduced therapeutic benefit in patients.
Concomitant administration of strong CYP2D6 inhibitors or pH‐modifying medications has no effect on upadacitinib plasma exposures. 19 Administration of multiple 30 mg q.d. doses of upadacitinib to healthy subjects had a limited effect on midazolam (sensitive substrate for CYP3A; ~25% decrease in midazolam exposures), rosuvastatin (~20% to 30% decrease in exposures), atorvastatin (23% decrease in exposures), and no effect on plasma exposures for sensitive substrates for CYP1A2, CYP2B6, CYP2C9, CYP2C19, or CYP2D6. No dose adjustment of the co‐administered drugs is recommended based on the lack of clinically relevant effects by upadacitinib on the pharmacokinetics of these drugs.
Upadacitinib has been demonstrated to reversibly inhibit IL‐6 and IL‐7 cytokine signaling in vivo in a concentration‐dependent manner, which was assessed through IL‐6‐induced STAT3 phosphorylation (pSTAT3) and IL‐7‐induced STAT5 phosphorylation (pSTAT5) ex vivo. 17 The time course for these signaling events by upadacitinib follows the plasma concentration levels with maximum inhibition coinciding with the maximum plasma concentration, and STAT phosphorylation levels returning close to baseline level by the end of the dosing interval. 17 These pharmacokinetic–pharmacodynamic analyses were utilized to inform dose selection of the initial phase II studies conducted in the overall upadacitinib development program.
KEY CLINICAL TRIALS
Upadacitinib has a well‐established and comprehensive clinical program in the approved indications of RA, PsA, AS, nr‐axSpA, AD, CD, and UC. A summary of key clinical trials for approved indications with upadacitinib is provided in Table 1.
TABLE 1.
Summary of upadacitinib key registrational trials supporting approved indications and summary of upadacitinib efficacy compared to placebo for key efficacy endpoints from representative registrational clinical trials across approved indications.
| Patient population | UPA approved dose | Study | Doses evaluated | Comparator | Efficacy endpoint | Efficacy timepoint | Percentage of subjects achieving efficacy endpoint | ||
|---|---|---|---|---|---|---|---|---|---|
| UPA | Placebo | Active comparator | |||||||
| RA | 15 mg q.d. | SELECT‐COMPARE (NCT02629159) | 15 mg q.d. | Placebo/Adalimumab 40 mg eow | ACR20 | Week 12 | 459/651 (71%) | 237/651 (36%) | 206/327 (63%) |
| RA | 15 mg q.d. |
SELECT‐MONOTHERAPY |
15 and 30 mg q.d. | Methotrexate | ACR20 | Week 14 | 147/217 (68%) | – | 89/216 (41%) |
| RA | 15 mg q.d. |
SELECT‐NEXT |
15 and 30 mg q.d. | Placebo | ACR20 | Week 12 | 141/221 (64%) | 79/221 (36%) | – |
| RA | 15 mg q.d. |
SELECT‐BEYOND |
15 and 30 mg q.d. | Placebo | ACR20 | Week 12 | 106/164 (65%) | 48/169 (28%) | – |
| RA | 15 mg q.d. |
SELECT‐EARLY |
7.5, 15, and 30 mg q.d. | Methotrexate | ACR50 | Week 12 | 165/317 (52%) | – | 89/314 (28%) |
| PsA | 15 mg q.d. |
SELECT‐PsA 1 |
15 and 30 mg q.d. | Placebo/Adalimumab 40 mg eow | ACR20 | Week 12 | 303/429 (71%) | 153/423 (36%) | 279/429 (65%) |
| PsA | 15 mg q.d. |
SELECT‐PsA 2 |
15 and 30 mg q.d. | Placebo | ACR20 | Week 12 | 120/211 (57%) | 51/212 (24%) | – |
| AS | 15 mg q.d. |
SELECT‐AXIS 1 |
15 mg q.d. | Placebo | ASAS 40 | Week 14 | 48/93 (52%) | 24/94 (26%) | – |
| AS | 15 mg q.d. |
SELECT‐AXIS 2 Study 1 |
15 mg q.d. | Placebo | ASAS 40 | Week 14 | 94/211 (45%) | 38/209 (18%) | – |
| nr‐axSpA | 15 mg q.d. |
SELECT‐AXIS 2 Study 2 |
15 mg q.d. | Placebo | ASAS 40 | Week 14 | 70/156 (45%) | 35/157 (23%) | – |
| AD | 15 mg q.d. |
MEASURE UP 1 |
15 and 30 mg q.d. | Placebo | EASI75 | Week 16 | 196/281 (70%) | 46/281 (16%) | – |
| AD | 30 mg q.d. |
MEASURE UP 1 |
15 and 30 mg q.d. | Placebo | EASI75 | Week 16 | 227/285 (80%) | 46/281 (16%) | – |
| AD | 15 mg q.d. |
MEASURE UP 2 |
15 and 30 mg q.d. | Placebo | EASI75 | Week 16 | 166/276 (60%) | 37/278 (13%) | – |
| AD | 30 mg q.d. |
MEASURE UP 2 |
15 and 30 mg q.d. | Placebo | EASI75 | Week 16 | 206/282 (73%) | 37/278 (13%) | – |
| AD | 15 mg q.d. |
AD UP |
15 and 30 mg q.d. + TCS | Placebo | EASI75 | Week 16 | 194/300 (65%) | 80/304 (26%) | – |
| AD | 30 mg q.d. |
AD UP |
15 and 30 mg q.d. + TCS | Placebo | EASI75 | Week 16 | 229/297 (77%) | 80/304 (26%) | – |
| AD | 30 mg q.d. |
HEADS UP |
30 mg q.d. | Dupilumab | EASI75 | Week 16 | 247/348 (71%) | – | 210/344 (61%) |
| UC |
45 mg q.d. (Induction) |
U‐ACHIEVE |
45 mg q.d. | Placebo | Clinical remission | Week 8 | 83/319 (26%) | 7/154 (5%) | – |
| UC |
45 mg q.d. (Induction) |
U‐ACCOMPLISH |
45 mg q.d. | Placebo | Clinical remission | Week 8 | 114/341 (34%) | 7/174 (4%) | – |
| UC |
15 mg q.d. (Maintenance) |
U‐ACHIEVE Maintenance |
15 and 30 mg q.d. | Placebo | Clinical remission | Week 52 | 63/148 (42%) | 8/149 (12%) | – |
| UC |
30 mg q.d. (Maintenance) |
U‐ACHIEVE Maintenance |
15 and 30 mg q.d. | Placebo | Clinical remission | Week 52 | 80/154 (52%) | 8/149 (12%) | – |
| CD |
45 mg q.d. (Induction) |
U‐EXCEL |
45 mg q.d. | Placebo | Clinical remission | Week 12 | 173/350 (50%) | 51/176 (29%) | – |
| CD |
45 mg q.d. (Induction) |
U‐EXCEED |
45 mg q.d. | Placebo | Clinical remission | Week 12 | 126/324 (39%) | 36/171 (21%) | – |
| CD |
15 mg q.d. (Maintenance) |
U‐ENDURE |
15 and 30 mg q.d. | Placebo | Clinical remission | Week 52 | 63/169 (37%) | 25/165 (15%) | – |
| CD |
30 mg q.d. (Maintenance) |
U‐ENDURE |
15 and 30 mg q.d. | Placebo | Clinical remission | Week 52 | 80/168 (48%) | 25/165 (15%) | – |
Abbreviations: ACR20, 20% improved in disease activity defined by the American College of Rheumatology; AD, atopic dermatitis; AS, ankylosing spondylitis; ASAS40, 40% improvement from baseline defined by theAssessment in Spondyloarthritis International Society; CD, Crohn’s disease; EASI75, 75% reduction from baseline in the Eczema Area and Severity Index; e.o.w., every other week; nr‐axSpA, non‐radiographic axial spondyloarthritis; PsA, psoriatic arthritis; q.d., once‐daily; RA, rheumatoid arthritis; TCS, topical corticosteroid; UC, ulcerative colitis; UPA, upadacitinib.
Ongoing clinical trials for new indications
Upadacitinib is currently being evaluated in ongoing clinical trials for the treatment of giant cell arteritis, Takayasu arteritis, hidradenitis suppurativa, nonsegmental vitiligo, and systemic lupus erythematosus. Pediatric clinical trials are currently ongoing for evaluation of upadacitinib for the treatment of pediatric AD, polyarticular course juvenile idiopathic arthritis, systemic juvenile idiopathic arthritis, and pediatric UC.
SUMMARY OF CLINICAL EFFICACY AND SAFETY
Efficacy
The efficacy of upadacitinib compared with placebo or active comparator was evaluated in clinical trials across the different approved immune‐mediated inflammatory diseases. A high‐level summary of upadacitinib's efficacy across key registrational trials for the approved indications is provided in Table 1. 14 Upadacitinib met the primary end points and most secondary end points across all clinical trials and demonstrated superiority over placebo with standard of care background therapies in RA, PsA, AS, nr‐axSpA, AD, UC, and CD. 8 , 9 , 10 , 11 , 12 Additionally, upadacitinib demonstrated superiority to active comparators of adalimumab and abatacept in RA and to dupilumab in AD, as well as non‐inferiority to adalimumab in PsA. 8 , 9 , 13 , 22 These results demonstrate the robustness of upadacitinib clinical efficacy profile and the significant additional value it provides over previously approved treatments. Favorable real‐world effectiveness and safety for upadacitinib have also been demonstrated. 23 , 24 , 25 , 26
The relationships between upadacitinib plasma exposures and efficacy varied between different clinical conditions, which consequently resulted in the approved doses being specific to different immune‐mediated inflammatory diseases. This could be driven by differences in inflammatory burden, site of inflammation, and the implicated cytokine pathways between different diseases. 1 , 2 , 3 , 4 , 5 Examples of the exposure–response relationships for upadacitinib average plasma concentrations (C avg) and select efficacy end points are shown in Figure 2. For RA and PsA, efficacy approached a plateau at plasma exposures associated with the 15 mg q.d. dosing regimen of upadacitinib (approximate median C avg of 15 ng/mL). In AS, conversely, there was no clear trend towards higher response rates with increasing upadacitinib plasma exposures, indicating that plasma exposures associated with 15 mg q.d. maximized the response. Analyses for induction data in UC and CD demonstrated that exposures associated with 45 mg q.d. regimen (approximate median C avg of 40 ng/mL) maximized upadacitinib efficacy at the end of the induction period. For AD as well as for maintenance in UC and CD, the exposures associated with 30 mg q.d. regimen (approximate median C avg of 29 ng/mL) provided additional benefit compared with the 15 mg q.d. regimen for chronic use, resulting in the approval of both regimens for these indications. Additionally, dosing recommendations in special populations (e.g., in patients with renal or hepatic impairment) or with strong CYP3A inhibitors or inducers in different indications were based on the range of exposures evaluated in the pivotal phase III trials, as well as the exposure–response relationships for efficacy and safety for each indication. 27 , 28 , 29 , 30
FIGURE 2.
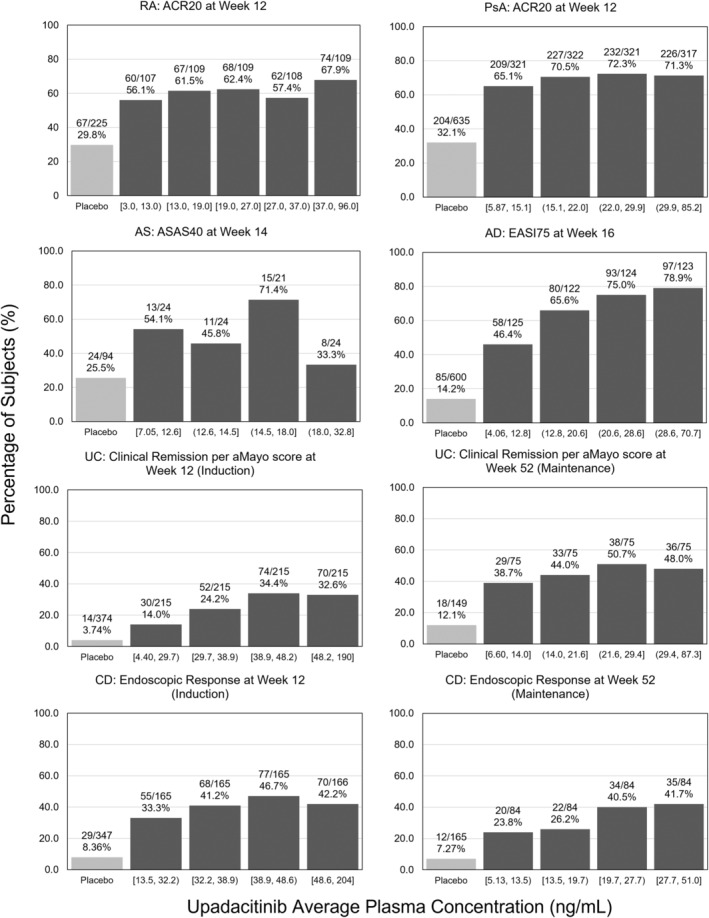
Upadacitinib exposure–response relationships for select efficacy end points by indication. ACR20, 20% improved in disease activity defined by the American College of Rheumatology; AD, atopic dermatitis; aMayo, adapted Mayo; AS, ankylosing spondylitis; ASAS40, 40% improvement from baseline defined by the Assessment in Spondyloarthritis International Society; CD, Crohn's disease; EASI75, 75% reduction from baseline in the Eczema Area and Severity Index; PsA, psoriatic arthritis; RA, rheumatoid arthritis; UC, ulcerative colitis.
Safety
The safety of upadacitinib has been evaluated in an extensive clinical development program, demonstrating a consistent safety profile across RA, PsA, axSpA, UC, CD, and AD. 31 Upadacitinib is generally well‐tolerated with observed differences in safety profiles likely reflective of varying patient characteristics across different populations and comorbidities that impact background risk. 31 The most common adverse events reported across the upadacitinib clinical trials were similar and included events such as upper respiratory tract‐related infection, nasopharyngitis, and increase in blood creatine phosphokinase levels while acne was a more common adverse event reported in the AD trials. 31 As reported by Burmester et al., the pattern, characteristics and incidence of COVID‐19 infections, including frequency of events and those leading to hospitalization, observed in patients receiving upadacitinib were generally similar to what has been observed in the general population. 31 There was no significant change in rate of malignancies (excluding non‐melanoma skin cancer [NMSC]) over time of exposure to upadacitinib across diseases and doses. 31 Rates of herpes zoster (HZ), NMSC, and elevations in creatine phosphokinase levels were higher with upadacitinib than adalimumab in the RA and PsA populations. The majority of HZ events with upadacitinib were mild or moderate, infrequently led to discontinuation and involved a single dermatome, with no events involving the central nervous system or internal organs. Rates of NMSC were generally consistent across diseases for upadacitinib with no events observed in AS. Slightly higher rates of NMSC were also observed with upadacitinib 30 mg versus 15 mg in AD. Events of NMSC were generally non‐serious and did not lead to treatment discontinuation. 31 Deaths, serious infections, major adverse cardiovascular events (MACE), venous thromboembolism, and malignancies were observed, with rates generally lowest in AS and AD; however, there was no evidence suggesting that the number of deaths in RA, PsA, or AD exposed to upadacitinib were higher than what would have been expected for the general population. The number of deaths reported in the IBD program was too limited to enable comparison with the general population and no deaths reported in the AS program. 31
In the context of the recent safety outcomes of the Oral Rheumatoid Arthritis triaL (ORAL) Surveillance study 32 that compared a different JAK inhibitor, tofacitinib, to TNF inhibitor therapy, a post‐hoc analysis evaluating the safety profile of upadacitinib in RA patients with higher risk of cardiovascular disease from the SELECT‐RA clinical program including those from the SELECT‐COMPARE trial, a head‐to‐head study of upadacitinib versus adalimumab, was conducted. The analysis showed an increased risk of MACE, malignancy (excluding NMSC), and venous thromboembolism in the higher risk patients compared with the overall population, but the risk for these events was comparable between upadacitinib and adalimumab‐treated patients. Higher rates of NMSC and HZ were observed with upadacitinib across all populations, and increased rates of serious infections were also detected in upadacitinib‐treated patients at higher cardiovascular risk. 33 While the interpretation of this post‐hoc analysis may be associated with certain limitations, the data may also suggest the uniqueness of upadacitinib compared with other JAK inhibitors.
Overall, the safety profile of upadacitinib has been well‐characterized across a wide array of immune‐mediated inflammatory diseases and along with the associated efficacy as described above, support its favorable benefit–risk profile for use in the approved indications.
AUTHOR CONTRIBUTIONS
M.‐E.F.M. wrote the manuscript and designed the research. S.B. wrote the manuscript and analyzed the data. J.M.P., P.N., and P.W. wrote the manuscript.
FUNDING INFORMATION
AbbVie provided financial support for the study and participated in the design, study conduct, analysis, and interpretation of data, as well as the writing, review, and approval of the manuscript.
CONFLICT OF INTEREST STATEMENT
All authors are employees of AbbVie and may hold AbbVie stock or stock options.
ACKNOWLEDGMENTS
Medical writing support was provided by Wesley Wayman, PhD, an employee of AbbVie.
Mohamed M‐E, Bhatnagar S, Parmentier JM, Nakasato P, Wung P. Upadacitinib: Mechanism of action, clinical, and translational science. Clin Transl Sci. 2024;17:e13688. doi: 10.1111/cts.13688
DATA AVAILABILITY STATEMENT
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial‐level data (analysis data sets), as well as other information (eg, protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the US and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://vivli.org/ourmember/abbvie/ then select "Home".
REFERENCES
- 1. Huang IH, Chung WH, Wu PC, Chen CB. JAK‐STAT signaling pathway in the pathogenesis of atopic dermatitis: an updated review. Front Immunol. 2022;13:1068260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ciobanu DA, Poenariu IS, Crînguș LI, et al. JAK/STAT pathway in pathology of rheumatoid arthritis (review). Exp Ther Med. 2020;20:3498‐3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Raychaudhuri SK, Saxena A, Raychaudhuri SP. Role of IL‐17 in the pathogenesis of psoriatic arthritis and axial spondyloarthritis. Clin Rheumatol. 2015;34:1019‐1023. [DOI] [PubMed] [Google Scholar]
- 4. Mease P, Hall S, FitzGerald O, et al. Tofacitinib or adalimumab versus placebo for psoriatic arthritis. N Engl J Med. 2017;377:1537‐1550. [DOI] [PubMed] [Google Scholar]
- 5. Virtanen AT, Haikarainen T, Raivola J, Silvennoinen O. Selective JAKinibs: prospects in inflammatory and autoimmune diseases. BioDrugs. 2019;33:15‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guschin D, Rogers N, Briscoe J, et al. A major role for the protein tyrosine kinase JAK1 in the JAK/STAT signal transduction pathway in response to interleukin‐6. EMBO J. 1995;14:1421‐1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Parmentier JM, Voss J, Graff C, et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT‐494). BMC Rheumatol. 2018;2:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fleischmann R, Pangan AL, Song IH, et al. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III, double‐blind randomized controlled trial. Arthritis Rheumatol. 2019;71:1788‐1800. [DOI] [PubMed] [Google Scholar]
- 9. McInnes IB, Anderson JK, Magrey M, et al. Trial of upadacitinib and adalimumab for psoriatic arthritis. N Engl J Med. 2021;384:1227‐1239. [DOI] [PubMed] [Google Scholar]
- 10. van der Heijde D, Baraliakos X, Sieper J, et al. Efficacy and safety of upadacitinib for active ankylosing spondylitis refractory to biological therapy: a double‐blind, randomised, placebo‐controlled phase 3 trial. Ann Rheum Dis. 2022;81:1515‐1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Loftus EV Jr, Panés J, Lacerda AP, et al. Upadacitinib induction and maintenance therapy for Crohn's disease. N Engl J Med. 2023;388:1966‐1980. [DOI] [PubMed] [Google Scholar]
- 12. Danese S, Vermeire S, Zhou W, et al. Upadacitinib as induction and maintenance therapy for moderately to severely active ulcerative colitis: results from three phase 3, multicentre, double‐blind, randomised trials. Lancet. 2022;399:2113‐2128. [DOI] [PubMed] [Google Scholar]
- 13. Blauvelt A, Teixeira HD, Simpson EL, et al. Efficacy and safety of upadacitinib vs dupilumab in adults with moderate‐to‐severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2021;157:1047‐1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. RINVOQ™ (Upadacitinib) [US package insert]. North Chicago, IL: AbbVie Inc., 2019.
- 15. Mikami Y, Kanai T. Network approaches to uncover pathogenesis and therapeutic targets of inflammatory bowel diseases. Keio J Med. 2023;72:29‐43. [DOI] [PubMed] [Google Scholar]
- 16. European Medicines Agency . Rinvoq (upadacitinib) assessment report. Procedure No. EMEA/H/C/004760/0000 (EMA/608624/2019 Corr. 1). 2020.
- 17. Mohamed MF, Beck D, Camp HS, Othman AA. Preferential inhibition of JAK1 relative to JAK3 by upadacitinib: exposure–response analyses of ex vivo data from 2 phase 1 clinical trials and comparison to tofacitinib. J Clin Pharmacol. 2019;60:188‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taylor PC, Choy E, Baraliakos X, et al. Differential properties of Janus kinase inhibitors in the treatment of immune‐mediated inflammatory diseases. Rheumatology (Oxford). 2023;kead448. doi: 10.1093/rheumatology/kead448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mohamed MF, Klünder B, Othman AA. Clinical pharmacokinetics of upadacitinib: review of data relevant to the rheumatoid arthritis indication. Clin Pharmacokinet. 2020;59:531‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mohamed MF, Trueman S, Feng T, et al. Characterization of the effect of renal impairment on upadacitinib pharmacokinetics. J Clin Pharmacol. 2019;59:856‐862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trueman S, Mohamed MF, Feng T, et al. Characterization of the effect of hepatic impairment on upadacitinib pharmacokinetics. J Clin Pharmacol. 2019;59:1188‐1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rubbert‐Roth A, Enejosa J, Pangan AL, et al. Trial of upadacitinib or abatacept in rheumatoid arthritis. N Engl J Med. 2020;383:1511‐1521. [DOI] [PubMed] [Google Scholar]
- 23. Boneschansker L, Ananthakrishnan AN. Comparative effectiveness of upadacitinib and tofacitinib in inducing remission in ulcerative colitis: real‐world data. Clin Gastroenterol Hepatol. 2023;21:2427‐2429. e2421. [DOI] [PubMed] [Google Scholar]
- 24. Boesjes CM, Van der Gang LF, Zuithoff NPA, et al. Effectiveness of upadacitinib in patients with atopic dermatitis including those with inadequate response to dupilumab and/or baricitinib: results from the BioDay Registry. Acta Derm Venereol. 2023;103:adv00872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chugh R, Braga‐Neto MB, Fredrick TW, et al. Multicentre real‐world experience of upadacitinib in the treatment of Crohn's disease. J Crohns Colitis. 2023;17:504‐512. [DOI] [PubMed] [Google Scholar]
- 26. Diederik DC, Durez P, Lenaerts J, et al. AB0421 real‐world effectiveness of upadacitinib in patients with rheumatoid arthritis. Ann Rheum Dis. 2023;82:1398‐1399. [Google Scholar]
- 27. Nader A, Mohamed MF, Winzenborg I, et al. Exposure–response analyses of upadacitinib efficacy and safety in phase II and III studies to support benefit–risk assessment in rheumatoid arthritis. Clin Pharmacol Ther. 2020;107:994‐1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ponce‐Bobadilla AV, Stodtmann S, Eckert D, Zhou W, Liu W, Mohamed MEF. Upadacitinib population pharmacokinetics and exposure–response relationships in ulcerative colitis patients. Clin Pharmacokinet. 2023;62:101‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Muensterman E, Engelhardt B, Gopalakrishnan S, Anderson JK, Mohamed MF. Upadacitinib pharmacokinetics and exposure–response analyses of efficacy and safety in psoriatic arthritis patients – analyses of phase III clinical trials. Clin Transl Sci. 2022;15:267‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ismail M, Doelger E, Eckert D, et al. Population pharmacokinetic and exposure–response modelling to inform upadacitinib dose selection in adolescent and adult patients with atopic dermatitis. Br J Clin Pharmacol. 2023;89:3139‐3151. [DOI] [PubMed] [Google Scholar]
- 31. Burmester GR, Cohen SB, Winthrop KL, et al. Safety profile of upadacitinib over 15 000 patient‐years across rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis and atopic dermatitis. RMD Open. 2023;9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ytterberg SR, Bhatt DL, Mikuls TR, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med. 2022;386:316‐326. [DOI] [PubMed] [Google Scholar]
- 33. Fleischmann R, Curtis JR, Charles‐Schoeman C, et al. Safety profile of upadacitinib in patients at risk of cardiovascular disease: integrated post hoc analysis of the SELECT phase III rheumatoid arthritis clinical programme. Ann Rheum Dis. 2023;82:1130‐1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial‐level data (analysis data sets), as well as other information (eg, protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the US and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://vivli.org/ourmember/abbvie/ then select "Home".