

Abstract
INTRODUCTION:
Targeted insertion of large genetic payloads without DNA double-strand breaks remains a major challenge for genome engineering. CRISPR-associated transposases (CASTs) represent a promising alternative to nuclease- and prime editing–based approaches and involve the repurposing of nuclease-deficient CRISPR effectors to facilitate RNA-guided transposition. Type V-K CASTs offer several potential upsides compared with other homologous systems because of their compact size, easy programmability, and unidirectional integration behavior.
RATIONALE:
Despite these desirable properties, type V-K CASTs exhibit poor fidelity compared with type I-F CASTs, and the molecular basis for this lack of specificity has remained elusive. We rationalized that determining the relative involvement of each CAST component during on-versus off-target insertion, including the guide RNA and Cas effector itself, would enable us to unravel the basis for this decreased specificity. To achieve this, we sought to monitor transposition using a combination of biochemistry and high-throughput sequencing, together with biophysical approaches, to visualize single transposase molecules using fluorescence and electron microscopy. We reasoned that a deeper understanding of target site selection and transpososome assembly would reveal new opportunities for technology engineering and improvement.
RESULTS:
Using biochemical and cellular transposition experiments, we found that a representative CAST system from Scytonema hofmannii (ShCAST) was highly prone to catalyzing untargeted transposition in a reaction that proceeded independently of Cas12k and the guide RNA. Gene deletion experiments identified the minimal necessary machinery as TnsB, TnsC, and TniQ, and a cryo–electron microscopy (cryo-EM) structure revealed a BCQ transpososome complex that resembled the Cas12k-containing transpososome, with TnsC playing a major role in defining the overall architecture. Additional biochemical experiments identified TnsC as the primary driver of untargeted integration but also showed that TnsB exhibits an integration preference for RNA-targeted sites over untargeted sites. Next, using single-molecule experiments and meta-analyses of genome-wide integration data, we discovered that AT-rich regions are preferred hotspots for untargeted transposition because of the binding specificity imparted by TnsC, and that TnsB also imposes local sequence bias to determine the precise insertion site. Knowledge of these motifs allowed us to direct untargeted transposition events to user-defined regions of a plasmid, and we confirmed the role of TnsC in mediating AT-rich preference by mutating akey DNA strand–contacting residue, K103. Finally, we harnessed knowledge of the role played by TnsC in directing untargeted transposition to design improved ShCAST vectors that suppressed RNA-independent transposition events and increased type V-K CAST specificity up to 98.1% in Escherichia coli without compromising the efficiency of on-target integration.
CONCLUSION:
Our results reveal that CRISPR-associated transposases can exhibit both RNA-guided and RNA-independent pathways and that the TnsC ATPase plays a major role in dictating target site selection. Whether both pathways are active in a native microbial context remains unknown, although we speculate that untargeted transposition likely represents the relic of an earlier, more primitive transposon lifestyle before CRISPR-Cas–targeting systems were acquired. This work highlights the importance of determining molecular mechanisms as an entry point to enable new opportunities for leveraging CASTs as an accurate, kilobasescale genome engineering tool.
CRISPR-associated transposases (CASTs) repurpose nuclease-deficient CRISPR effectors to catalyze RNA-guided transposition of large genetic payloads. Type V-K CASTs offer potential technology advantages but lack accuracy, and the molecular basis for this drawback has remained elusive. Here, we reveal that type V-K CASTs maintain an RNA-independent, “untargeted” transposition pathway alongside RNA-dependent integration, driven by the local availability of TnsC filaments. Using cryo–electron microscopy, single-molecule experiments, and high-throughput sequencing, we found that a minimal, CRISPR-less transpososome preferentially directs untargeted integration at AT-rich sites, with additional local specificity imparted by TnsB. By exploiting this knowledge, we suppressed untargeted transposition and increased type V-K CAST specificity up to 98.1% in cells without compromising on-target integration efficiency. These findings will inform further engineering of CAST systems for accurate, kilobase-scale genome engineering applications.
Graphical Abstract
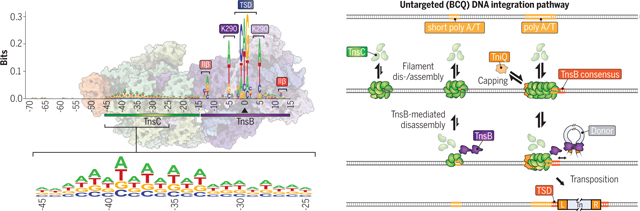
Mechanism of RNA-independent, untargeted integration by type V-K CASTs. We interrogated high-throughput sequencing datasets to reveal a consensus motif at untargeted integration events (left) characterized by TnsC- and TnsB-specific footprints. Together with single-molecule data and cryo-EM structures, these results revealed a BCQ transposition pathway (right) in which AT-rich sites are preferentially bound by TnsC filaments and capped by TniQ, leading to recruitment of TnsB-donor DNA complexes for downstream integration.
Bacteria encode diverse mobile genetic elements that exhibit a wide spectrum of transposition behaviors ranging from selective targeting of fixed attachment sites to promiscuous insertion into deenerate sequence motifs (1). Although insertion specificity is often dictated by a single recombinase enzyme (2, 3), some transposons encode heteromeric transposase complexes that distribute DNA target and integration activities across multiple distinct molecular components (4, 5). Tn7-like transposons are unique in this regard, in that they have evolved to exploit diverse molecular pathways for target site selection, including site-specific DNA-binding proteins (6), replication fork–specific DNA-binding proteins (7–9), CRISPR RNA– guided DNA binding complexes (10–12), and additional DNA targeting pathways that have yet to be characterized (13). CRISPR-associated transposases (CASTs), in particular, represent both a fascinating example of CRISPR-Cas exaptation and an opportune starting point for the development of next-generation tools for programmable, large-scale DNA insertion (14, 15).
CAST systems characterized to date fall within either type I or type V classes, which differ in their reliance on either Cascade or Cas12k effector complexes, respectively (10–12, 16, 17). Although the core transposition machinery is conserved across CAST families and includes a DDE-family transposase for integration (TnsB), an AAA+ ATPase for target site selection (TnsC), and anadaptor protein for CRISPR-transposition coupling (TniQ), key molecular features distinguish the integration behaviors of archetypal type I-F and type V-K systems. Whereas second-strand cleavage is catalyzed by the TnsA endonuclease in type I-F CASTs, leading to cut-and-paste transposition products, type V-K CASTs lack TnsA and instead mobilize through a copy-and-paste process, yielding cointegrate products (18–20). Type I-F CASTs achieve single-digit genomic integration efficiencies when expressed in mammalian cells, as opposed to low but detectable activity only on ectopic plasmid targets for an improved type V-K CAST homolog (20, 21). Additionally, heterologous expression of the CAST machinery from both systems yields vastly different integration specificities, with VchCAST (I-F) exhibiting mostly on-target activity in bacterial cells compared with an abundance of off-target insertions catalyzed by a representative V-K CAST system from Scytonema hofmannii (ShCAST) (11, 14, 15). Despite these differences, type V-K CASTs have a compact coding sequence composed of four components compared with type I-F CASTs (1666 versus 2748 amino acids) and integrate predominantly in a unidirectional orientation (11). The molecular basis underlying these distinguishing properties remains unexplored, particularly for type V-K CAST systems, limiting their practical application.
Recent structural studies have provided new insights into the overall architecture of RNA-guided, ShCAST transpososome complexes (22, 23). Target sites are marked by Cas12k binding (24, 25), in conjunction with TniQ and ribosomal protein S15, which engages the tracrRNA component (22), leading to stable R-loop formation reminiscent of other CRISPR effectors. In a key next step that is still poorly understood, TnsC assembles into filaments around double-stranded DNA, which can form adjacent to bound Cas12k-TniQ complexes (22) or on naked DNA (24, 26), acting as a platform for the subsequent recruitment of the TnsB transposase that is scaffolded along conserved binding sites in the transposon left and right ends. DNA integration then occurs through a concerted transesterification reaction at sites exposed by the TnsC filament, leading to transposons inserted at a fixed spacing downstream of the Cas12k-bound target site (11, 23). Whether a similar assembly pathway is operational at the many off-target integration events observed with ShCAST expression in cells, or if these represent an alternative transposition pathway, has not been systematically explored (Fig. 1A).
Fig. 1. Type V-K CASTs direct frequent Cas12k- and RNA-independent transposition events.

(A) Schematic of type V-K CAST transposition occurring at on-target sites (RNA-dependent) and untargeted sites (RNA-independent). (B) Experimental TagTn-seq pipeline used for in vitro and genomic samples. (C) Fraction of total genome-mapping integration reads detected at on-target and untargeted sites for the WT pHelper expression plasmid across multiple sgRNAs (top), plotted above on-target transposition efficiencies for the same sgRNAs as measured by Taqman qPCR (bottom). (D) Total genome-mapping reads detected for WT pHelper or pHelper with the indicated deletions, normalized and scaled. (E) Magnified view of integration reads comprising ≤1% of E. coli genome-mapping reads in an experiment performed without Cas12k and guide RNA. (F) Cryo-EM reconstruction of the untargeted transpososome revealing the assembly of TniQ (orange), TnsC (green), and TnsB (purple) in a strand-transfer complex (STC). The target DNA and transposon DNA are represented in light blue and dark blue, respectively. For visualization, a composite map was generated using two local resolution-filtered reconstructions from the focused refinements. Magnified and cutaway views show TnsC forming a helical assembly on the target DNA, positioning residues K103 and T121 (pink) adjacent to one strand of the target DNA (dark blue). The 5′ and 3′ ends of the TnsC-interacting DNA strand are indicated. Two turns of TnsC and TnsB footprint on DNA until TSD cover ~25 and 13 bp, respectively. Only selected TnsC monomers are represented in the cutaway for clarity. (G) Cas12k and the sgRNA were cloned onto a separate vector, and the promoter driving Cas12k expression was varied. Reads detected at on-target and untargeted sites during transposition assays were normalized and scaled. For (C), (D), (E), and (G), the mean is shown from N = 2 independent biological replicates.
Here, we set out to investigate the mechanism of target site selection for the archetypal type V-K CAST system from S. hofmannii, focusing special attention on the role of TnsC in regulating fidelity. We found that ShCAST is prone to extensive, RNA-independent transposition through a pathway that requires only TnsB, TnsC, and TniQ. Although these untargeted integration events initially appear random, analysis of high-throughput sequencing data revealed a bias for AT-rich sites, which was corroborated by single-molecule biophysical studies of TnsC DNA-binding behavior. By modulating DNA substrates in biochemical transposition assays, we demonstrated that the preference for AT-rich sequences could lead to predictable reaction outcomes. Furthermore, we found that transposition specificity could be substantially improved by limiting cytoplasmic TnsC levels, further highlighting the role of TnsC filament formation in pathway choice between RNA-dependent and RNA-independent transposition. Collectively, our results underscore the value of mechanistic studies in revealing new opportunities to engineer and leverage CAST systems as a potent DNA insertion technology.
Results
Type V-K CASTs perform RNA-dependent and RNA-independent transposition
Previous studies of the type V-K ShCAST system from S. hofmannii revealed that a considerable proportion of genomic integration events occurs at sites distant from the target site dictated by the guide RNA (11, 14, 15). To understand the molecular basis of these events, we applied a high-throughput sequencing approach to unbiasedly capture genome-wide integration events upon ShCAST expression with various genetic perturbations (Fig. 1B and materials and methods). After testing five distinct single-guide RNAs (sgRNAs), we found that the fraction of on-target integration events ranged from 12 to 76%, and that most events occurred elsewhere, with DNA insertions seemingly randomly distributed across the genome at low individual frequencies (Fig. 1C and fig. S1, A and B). We analyzed their proximal genetic neighborhood and failed to detect enriched sequence similarity to the guide RNA (fig. S1, C to E), suggesting that these events were not mismatched off-targets aberrantly targeted by RNA-guided Cas12k, but rather were the consequence of RNA-independent transposition; therefore, we tentatively referred to these as untargeted integration events (Fig. 1C). When we deleted cas12k and the sgRNA from the original pHelper expression plasmid, the CRISPR-lacking ShCAST system still produced efficient genome-wide transposition products (Fig. 1, D and E) (11). These results establish that type V-K CAST systems are capable of both RNA-dependent targeted DNA integration and RNA-independent untargeted DNA integration.
We performed additional control experiments and confirmed that TnsC, an AAA+ regulator, and TnsB, the DDE-family transposase, are essential for both RNA-dependent and RNA-independent transposition, because their deletion completely abrogated integration (Fig. 1D). We initially hypothesized that TnsB and TnsC would comprise the minimum necessary protein components for RNA-independent transposition, similar to the reliance of phage Mu transposition on two homologous gene products, MuA and MuB (27). However, tniQ deletion had a severe effect on untargeted transposition (Fig. 1D), suggesting a crucial role in stabilizing and/or interacting with the TnsBC transpososome. Recent structures revealed that DNA-bound TnsC oligomers are capped on the N-terminal face by one or more TniQ protomers (22, 26), and our biochemical experiments similarly demonstrated that TniQ only stably associated with DNA in the presence of TnsC, as reflected by fluorescence polarization experiments (fig. S1F). Thus, much like the requirement for TnsB, TnsC, and TniQ in transposition by Tn5053 (28), we conclude that ShCAST—and perhaps type V-K CAST systems more generally—maintain a prominent BCQ pathway that facilitates CRISPR RNA–independent, untargeted transposition.
To capture the architecture of components contributing to untargeted integration, we used cryo–electron microscopy (cryo-EM) to visualize TnsB, TnsC, and TniQ in a strand-transfer complex (STC). Although the cryo-EM density of TniQ was less well resolved (~8 Å) compared with other subunits (fig. S2 and table S1), likely due to heterogeneity of binding configurations, we were able to unambiguously dock atomic models of all protein components and DNA into the map (Fig. 1F). The overall structure of the BCQ transpososome resembled the Cas12k-containing transpososome (fig. S3A), with two turns of TnsC filaments preferentially selected even with free DNA available at the TniQ end. Further, DNA-interacting residues of TnsC (K103 and T121) were positioned to follow the helical symmetry of duplex DNA, as in the structure of helical TnsC filaments (22–24, 26). However, in contrast to the Cas12k-containing transpososome (23), the polarity of the interacting DNA strand in the BCQ transpososome was 3′ to 5′, following the direction of TniQ- to TnsB-binding face of TnsC, which was also noted in the structure of TniQ-TnsC (22, 26) (fig. S3B). Therefore, the BCQ transpososome structure, which represents a low-energy configuration of TnsC, reveals that DNA contacts in TnsC filaments are maintained differently at on-target and untargeted sites. Yet, the overall architecture of the BCQ transpososome, comprising the TnsB STC, two turns of a TnsC minifilament [spanning a DNA-binding footprint of 25 base pairs (bp)] and TniQ, resembles the on-target Cas12k-bound transpososome.
We next sought to determine whether the presence of Cas12k and an appropriate sgRNA would reduce the frequency of untargeted transposition events by sequestering protein components at the on-target site. After cloning cas12k onto a separate expression plasmid and systematically varying its promoter strength, we found that on-target integration events were proportionally increased, although without a reduction in the frequency of untargeted integration (Fig. 1G and fig. S1G). These results indicate that, at least under these expression conditions, the availability of Cas12k-sgRNA complexes limits RNA-guided DNA integration efficiency but does not directly affect the BCQ pathway. Type V-K CAST systems often encode a MerR-family transcriptional regulator adjacent to the Cas12k gene (12, 29), and a recent study demonstrated that these Cas V-K repressor (CvkR) proteins down-regulate both Cas12k and TnsB expression, although with distinct effects (30). Thus, although our present knowledge about CAST activity is largely limited to comparative genomics and artificial heterologous overexpression, it appears likely that CAST transposition in native contexts is regulated to modulate the frequency of RNA-dependent and RNA-independent target pathways.
Relative TnsB and TnsC stoichiometry determines the transposition pathway choice
Many other bacterial transposons encode transposition proteins homologous to ShCAST, including type I CASTs, Tn7, Tn5053, IS21, and Mu (4, 8, 10, 27, 28). The TnsBC module is common to all, and in the case of Mu, the AAA+ ATPase component known as MuB plays a dominant role in directing untargeted, genome-wide transposition by recruiting the MuA transposase to potential integration sites (5). Moreover, structural studies have demonstrated that MuB and ShTnsC both form continuous, non-specific filaments on double-stranded DNA (dsDNA) (5, 24, 26), in contrast to the discrete closed or semiclosed rings formed by TnsC from VchCAST (Tn6677) and Escherichia coli Tn7 (31, 32). Therefore, we set out to experimentally investigate the role of TnsC in target site selection and the effect of variable stoichiometries of TnsB, TnsC, and TniQ on untargeted integration. However, one of the major hindrances that we encountered while trying to vary the expression of transposon components in cells was the associated toxicity, particularly with the overexpression of TnsC (Fig. 2A and fig. S4, A and B). When we inoculated liquid cultures with a strain expressing tnsC alone from a strong promoter and induced overexpression in the lag phase, we observed a complete growth arrest for most of the clones, with only a few strains undergoing delayed growth, likely due to suppressor mutations in the plasmid or genome (fig. S4A). This cellular toxicity was completely rescued with a mutation to the arginine finger motif, which abrogates TnsC filamentation (33) and transposition, or was partially rescued by coexpression of TnsC and TnsB (Fig. 2A and fig. S4C). These results implicate nonspecific DNA filamentation as a likely source of cellular toxicity, which can be relieved in part by the ability of TnsB to disassemble TnsC filaments, as demonstrated from in vitro experiments (24, 26, 34).
Fig. 2. Biochemical reconstitution of transposition reveals distinct efficiencies at on-target and untargeted sites.

(A) Growth curves upon induction of WT or mutant TnsC with or without TnsB. Data are shown as mean ± SD for N = 2 independent biological replicates inoculated from individual colonies. (B) Assay schematic for probing in vitro plasmid-to-plasmid transposition events using recombinantly expressed CAST components. (C) In vitro integration reads mapping to pTarget from experiments in which TnsC was titrated from 0.1 to 2 μM. Data were normalized and scaled to highlight untargeted integration events relative to on-target insertions. (D) On-target specificity from biochemical transposition assays at varying TnsC concentrations, calculated as the fraction of on-target reads divided by total plasmid-mapping reads (bottom). Total integration activity also decreased as a function of TnsC concentration, as seen by the normalized plasmid-mapping reads (top). (E) Scatter plot showing reproducibility between untargeted integration reads observed in vitro at two high TnsC concentrations; each data point represents transposition events mapping to a single base-pair position within pTarget. The Pearson linear correlation coefficient is shown (two-tailed P < 0.0001); on-target events were masked. (F) Normalized integration reads detected at a representative untargeted site (left) and at the on-target site (right), with 1 μM TnsC and the indicated TnsB concentration. Note the differing y-axis ranges. (G) On-target specificity from biochemical transposition assays at 1 μM TnsC and the indicated TnsB concentration, shown as in (D).
To modulate the stoichiometries of CAST components contributing to untargeted integration, while avoiding confounding factors such as toxicity, we adopted a biochemical approach. After recombinantly expressing and purifying ShCAST components and testing the activity of TnsC and TnsB in vitro (fig. S4, D to G), we established a plasmid-to-plasmid (pDonor-to-pTarget) transposition assay (Fig. 2B). In initial experiments, we amplified on-target products by targeted polymerase chain reaction (PCR), thereby revealing molecular requirements for each of the transpososome components and the expected distance separating the target and integration site (fig. S5, A to C). Next, we coupled our biochemical experiments with tagmentation-based high-throughput sequencing to unbiasedly map DNA transposition events regardless of their insertion site (Fig. 2B and materials and methods). We found that, at low (0.1 μM) concentrations of TnsC, transposition was highly accurate, with >99% of reads representing on-target integration events, defined as occurring within a 100-bp window downstream of the target site (Fig. 2, C and D). However, when we systematically increased the concentration of TnsC while keeping all other components constant, the frequency of untargeted integration events increased, approaching levels similar to those observed in cellular transposition assays (Fig. 2D and fig. S5E). Substantial untargeted integration events also occurred in the absence of Cas12k and sgRNA under these conditions, in agreement with in vivo experiments (fig. S5D). TnsC concentrations of 1 μM or higher resulted in a decrease in both on-target and untargeted integration, which may be have been caused by the prohibitive coating of DNA by TnsC filaments (see below). When interpreted together with structural data, these results suggest that RNA-independent transposition is likely initiated by the formation of dsDNA-bound TnsC filaments. Untargeted integration events were not randomly distributed across pTarget but instead were clustered into specific and reproducible hotspot regions (Fig. 2E), suggesting a selectivity for certain, as-yet-undetermined sequence features (see below).
We next tested the impact of other protein components on in vitro transposition activity. Ribosomal protein S15, a recently described host factor that stimulates ShCAST transposition by binding the sgRNA (22), substantially increased the frequency of on-target integration events, as measured both by deep sequencing and quantitative PCR (qPCR), but had no discernible effect on untargeted integration events (figs. S5, G and H). However, increasing the concentration of TniQ led to a monotonic increase in the frequency of untargeted integration events without a major effect on on-target integration (fig. S5F), suggesting that the RNA-independent pathway may be more sensitive to limited TniQ availability.
The TnsB transposase has been previously shown to disassemble TnsC filaments from dsDNA (24, 26, 34), so it is possible that titrating excess amounts of TnsB would lead to partial or full disassembly of TnsC filaments necessary for transposition, regardless of their molecular context. However, when we varied the amount of the TnsB transposase, we observed distinct effects at on-target and untargeted sites (Fig. 2F and fig. S5, I to K). Increasing TnsB led to a notable increase in RNA-guided integration but resulted in a slight decrease in untargeted events (fig. S5, I to K), leading to an overall rescue of specificity at on-target sites with high TnsC concentrations. This observation suggests that TnsC filaments at targeted versus untargeted sites are differentially susceptible to TnsB-induced disassembly and/or react to undergo strand transfer with distinct kinetics. Transpososome structures reveal that TnsB interacts with TnsC filaments on only one face (23, 34), and no major structural changes are associated with TnsC filaments at on-target and untargeted sites (Fig. 1F). Therefore, we suggest that the distinct nature of DNA interactions made by TnsC at both of these sites determines filament stabilization versus disassembly.
Collectively, these results suggest that the natural propensity of TnsC to form long filaments on dsDNA exerts a fitness cost on cells in the absence of accessory transposase machinery and is a driver of RNA-independent, untargeted transposition. We next sought to investigate whether TnsC exhibits any bias when selecting RNA-independent sites for transposition.
TnsC preferentially targets AT-rich DNA during RNA-independent transposition
We developed a single-molecule approach to visualize DNA binding by TnsC using DNA curtains (Fig. 3A), in which λ-phage genomic DNA molecules are tethered between chrome patterns on a quartz slide and imaged by total internal reflection fluorescence microscopy (35). Fluorescently labeled TnsC remained fully active for RNA-guided transposition, albeit with slightly increased specificity relative to wild-type (WT) (fig. S6A), suggesting that the N-terminal appendage may subtly affect DNA binding and/or transpososome assembly. In DNA curtains experiments, TnsC exhibited stable and high-affinity binding in the presence of ATP, and the data furthermore revealed a marked preference for the 3′ half of the genome (Fig. 3B). The λ-phage genome is known to be divided into a GC-rich half and an AT-rich half (36), and our analyses revealed a significant correlation between AT content and TnsC localization (Fig. 3C), indicating that TnsC filaments preferentially accumulate on the AT-rich half of the λ-phage genome. Time-course experiments further revealed that TnsC binds to AT-rich regions at a faster rate before saturating the entire λ-DNA substrate within 5 to 10 min of incubation (Fig. 3D and fig. S6B and movie S1). A preference for AT-rich regions has been previously observed for MuB in both single-molecule microscopy experiments and in vivo transposition studies (37, 38), supporting the idea that this property is likely to be broadly conserved across AAA+ regulators from other transposon families. Incubation of DNA curtains with high TnsC concentrations resulted in complete coating of the λ-DNA substrate (fig. S6C and movie S2), which could explain the decrease in both on-target and untargeted integration observed in biochemical transposition assays at similarly high TnsC concentrations (Fig. 2C and fig. S5E).
Fig. 3. RNA-independent integration events occur at preferred sequence motifs.

(A) Schematic for single-molecule DNA curtains assay to visualize TnsC binding. λ-phage DNA substrates are double-tethered between chrome pedestals and visualized used total internal reflection fluorescence microscopy. (B) mNG-labeled TnsC preferentially binds AT-rich sequences on the λ-DNA substrate near the 3′ (pedestal) end (movie S1). (C) Correlation between AT content and mNG-TnsC fluorescence intensity visualized along the length of λ-DNA. The Pearson linear correlation coefficient is shown (two-tailed P < 0.0001). Data are shown as mean ± SD for N = 66 molecules. (D) Binding kinetics for mNG-TnsC at AT-rich and AT-poor regions of the λ-DNA substrate. Apparent kobs at AT-rich sites ≈0.37 min−1, 95% confidence interval (CI) = 0.35 to 0.39, and at AT-poor sites ≈0.28 min−1, 95% CI = 0.27 to 0.30. Data are shown as mean ± SD for N = 87 molecules (thick line, shaded region). Binding kinetics for AT- and GC-rich sites when compared gave a P value of 0.017 upon bootstrapping. (E) Cumulative frequency distributions for the AT content within a 100-bp window flanking integration events using ShCAST with WT TnsC and sgRNA-1 (N = 5505 unique integration events), compared with random sampling of the E. coli genome (N = 50,000 counts). The distributions were significantly different on the basis of results of a Mann-Whitney U test (P = 1.48 × 10−135). (F) Cumulative frequency distribution comparison as in (E) but with a K103A TnsC mutant (N = 1932 unique integration events), which revealed a loss of AT bias (P = 0.1349). (G) Meta-analysis of untargeted transposition specificity was performed by extracting sequences from a 140-bp window flanking the integration site and generating a consensus logo. (H) WebLogo from a meta-analysis of untargeted genomic transposition (N = 5855 unique integration events) with a modified pHelper lacking Cas12k and sgRNA. The site of integration is noted with a maroon triangle. An AT-rich sequence spanning ~25 bp likely reflects the footprint of two turns of a TnsC filament (black), whereas motifs within/near the TSD represent TnsB-specific sequence motifs (green). Specific TnsB residues and domains contacting the indicated nucleotides are shown. The magnified inset highlights periodicity in the sequence bound by TnsC. (I) Schematic showing the relative spacing of sequence features bound by Cas12k, TnsC, and TnsB in both on-target (RNA-dependent) and untargeted (RNA-independent) DNA transposition. In both cases, the TnsC footprint covers ~25 bp of DNA and directs polarized, unidirectional integration downstream in a L-R orientation. (J) Magnified view of the ShCAST transpososome structure highlighting sequence-specific contacts between TnsB and the target DNA observed in (H). The Protein Data Bank identification number is 8EA3 (23).
Given our observation that untargeted transposition events in biochemical assays preferred certain hotspot regions of pTarget and were reproducible between independent experiments (Fig. 2E), we hypothesized that AT content might be an underlying feature explaining these data. We analyzed the nucleotide composition surrounding all unique integration events on pTarget and found that they were indeed skewed toward more AT-rich DNA (fig. S6, D and E, and materials and methods). Direct visual superposition of AT content and DNA integration data further revealed that hotspot regions for untargeted transposition in pTarget generally correlated with regions of higher AT content (fig. S6F). We observed the same phenomenon after performing transposition assays with a λ-DNA substrate and repeating similar analyses to assess AT bias in the location of untargeted integration sites (fig. S6, G to I). Finally, we analyzed untargeted integration events in the E. coli genome from experiments performed without Cas12k and sgRNA and found that these were also highly enriched at local regions of high AT content (Fig. 3E and fig. S6J). These results provide evidence that AT-rich sites on DNA are preferentially bound by TnsC, and thus are preferentially “targeted” for RNA-independent transposition.
We next sought to uncover additional sequence features common to CRISPR-independent ShCAST transposition products. We performed a meta-analysis of all genome-wide integration sites after orienting the flanking sequences based on the asymmetric transposon ends, and then generated a consensus sequence logo of the resulting alignment (Fig. 3G and materials and methods). This analysis revealed two notable clusters of sequence features: nucleotide preferences directly within and surrounding the target-site duplication (TSD), and an AT-rich nucleotide cluster located upstream of the integration site (Fig. 3H). The AT-rich region spans ~25 bp and could thus accommodate two turns of a dsDNA-bound TnsC filament, similar to the TnsC architecture and foot-print observed within the context of Cas12k-containing and Cas12k-lacking transpososomes (Fig. 1F) (23). The observation that this region is located on only one side of all integration sites suggests that RNA-independent integration events result from binding of TnsC filaments to AT-rich DNA, followed by directional recruitment of TnsB to define downstream sites for transposon insertion in the same left-right (L-R) orientation as occurs at RNA-guided target sites (11) (Fig. 3I). In general, we refer to this orientation as TnsC-LR, which could be applicable to other systems using a AAA+ ATPase for integration. We found that higher sequence conservation was located farthest from the site of integration, proximal to the presumed region where TnsC filaments are capped by TniQ (Fig. 1F), and the observed dinucleotide periodic trend is reminiscent of structures demonstrating that TnsC monomers contact every two bases of DNA (24, 26).
The greatest conservation in the sequence logo corresponds to sequences contacted by TnsB within the BCQ transpososome. As with prior library-based experiments for both type I-F and V-K CAST systems, our results indicate that TnsB preferentially integrates into sites containing GCWGC within the TSD (Fig. 3H) (11, 39). However, we also uncovered a bias for (A/T) at symmetric positions located ±5 bp from the TSD center, which is contacted by residue K290 of two TnsB monomers within the transpososome (Fig. 3J). Nucleotide preferences ±12 bp from the TSD could also be explained by the proximity of these residues with the TnsB IIβ DNA-binding domain (R416, T417, Q425, and N428), which also makes similar sequence contacts to the penultimate TnsB-binding sites located within the transposon left and right ends (23, 34, 40). When we analyzed untargeted integration events from our previously published ShCAST data (14), in which NGS libraries were generated and sequenced using an alternative strategy, we observed the same sequence features, confirming the robustness of this observation (fig. S7A). The absence of any conserved sequence features upstream of the AT-rich region, where the target site would normally be located during Cas12k-mediated transposition, corroborated our earlier interpretation that most of the cellular transposition events were RNA-independent.
Previously, it was shown that a K103A point mutation, one of the two TnsC residues that contact DNA, increased the number of untargeted events without compromising the ability of TnsC to bind DNA (26). In agreement with these results, when we tested the same mutant in cellular transposition assays, we observed a severe loss of on-target events but a preservation of untargeted events, which were enriched near the E. coli origin of replication (figs. S4C and S6K). When we performed a meta-analysis of untargeted integration events, we found that the TnsC K103A mutant no longer exhibited an A/T preference, in contrast to WT TnsC (Fig. 3F and fig. S7B). This observation, together with the loss of on-target integration (fig. S4C), suggests that the K103A mutation results in a more promiscuous mode of DNA binding that supports integration anywhere in the genome without specific sequence requirements.
We previously reported transposition activity for a type V-K CAST homolog also found in S. hofmannii, ShoCAST (previously referred to as ShoINT), which is diverged from ShCAST and more similar to AcCAST (11, 14). We were curious as to whether untargeted ShoCAST transposition events would exhibit similar sequence preferences as ShCAST, so we performed meta-analyses on published transposition data (14). Highly similar motifs emerged in the resulting sequence logo, but with a major difference in the window of AT-rich DNA located upstream of the integration site, which spanned only ~10 bp compared with ~25 bp observed with ShCAST (fig. S7C). This difference is consistent with the finding that ShoCAST and AcCAST integrate ~10 bp closer to the target site than ShCAST (11, 14), suggesting that the transpososomes from this subfamily of CAST systems, for both RNA-dependent and RNA-independent transposition pathways, may comprise a shorter TnsC filament spanning only one turn of DNA.
Altogether, these observations demonstrate how subtle sequence motifs at RNA-independent integration sites can be gleaned from meta-analyses of genome-wide integration data. They furthermore reveal that RNA-independent transposition is not random, but rather, that the BCQ transposition pathway preferentially selects certain genomic regions over others.
Preferred sequence motifs lead to semi-targeted, RNA-independent transposition
To test the importance of TnsBC-specific sequence motifs more directly for CRISPR-independent integration, we designed biochemical transposition assays using a series of isogenic pTarget substrates that differed only in the sequence content of a select region that was poorly targeted in previous experiments (Fig. 4A, substrate pT-1). We hypothesized that we could generate targeted, RNA-independent insertions within this region if an optimal sequence were designed to include both the poly-A stretch and flanking TnsB consensus motifs observed in the sequence logo described above (Fig. 3H, substrate pT-2). As further controls, we substituted the poly-A stretch with either poly-AT or poly-GC, mutagenized the TnsB consensus, or replaced both motifs (Fig. 4A, substrates pT-3 through pT-6). We then tested each substrate in biochemical transposition assays and plotted the normalized integration frequency within this window of interest.
Fig. 4. Artificial induction of semi-targeted RNA-independent transposition at preferred motifs.

(A) A region on pTarget exhibiting low integration activity (original, blue) was substituted with rationally engineered sequences (colored lines) based on TnsC- and TnsB-binding preferences, generating the indicated pTarget variants (pT-1 to pT6). (B) After performing biochemical transposition assays with the indicated pTarget substrates, integration reads were normalized and mapped to either the forward strand (fwd, red) or reverse strand (rev, black). The intended untargeted integration site based on optimized poly-A and TnsB consensus motifs is marked with a maroon triangle and dotted line; the representative region at right (850 to 900 bp) is shown to highlight consistency in integration events observed elsewhere on pTarget.
The resulting data demonstrate that RNA-independent integration events occur in predictable ways depending on the sequence features uncovered through our analyses (Fig. 4B). Substrate pT-2 exhibited a predominant integration product precisely at the engineered site and in the expected T-LR orientation, whereas this integration product was entirely absent when the poly-A was replaced with poly-GC, strengthening our conclusion that favorable TnsC filamentation is important for RNA-independent integration (Fig. 4B, substrates pT-4 and pT-6). When we retained the poly-A stretch but mutated the consensus motif favored by TnsB, integration products were more heterogeneously positioned (Fig. 4B, substrate pT-5), suggesting that preferred TnsB-DNA interactions play an important role in dictating the exact insertion site, as similarly concluded by our recent study on the type I-F VchCAST system (39). When we replaced the poly-A sequence with poly-AT (thus increasing the A content on the opposite strand), the intended integration event was diminished in frequency and accompanied by an increase in upstream integration events on the opposite strand (Fig. 4B, substrate pT-3), demonstrating that nucleotide composition can modulate the preferred directionality of TnsC filament formation and thus integration.
These experiments reveal that TnsC prefers to filament unidirectionally on A-rich DNA stretches, leading to downstream integration in the T-LR orientation. The efficiency and exact site of integration is thus a combination of TnsC filament formation propensity and local TnsB sequence preferences.
TnsC availability controls the specificity of cellular ShCAST transposition activity
Beyond highlighting the role of TnsC in biasing untargeted integration events to occur at select hotspot regions of the genome, our results more generally implicate TnsC filament formation as a major driver of RNA-independent transposition activity. Because our in vitro results suggest that TnsB differentially selects TnsC filaments at on-target versus untargeted sites, we hypothesized that this difference could be exploited to increase the overall on-target integration accuracy. To test this hypothesis, we designed perturbations intended to repress TnsC filament formation at non–Cas12k-bound target sites either by fusing TnsC directly to CRISPR effector proteins, or by lowering overall TnsC expression levels.
When we fused Cas12k and TnsC, we observed an increase in on-target accuracy (fig. S8A), as was recently reported by Tou et al. (20). We initially hypothesized that this effect might result from local seeding of TnsC filaments upon Cas12k target binding, but coexpression of unfused Cas12k had no adverse effect on specificity, suggesting instead that TnsC filamentation may be partially impaired with an N-terminal adduct. We also replaced Cas12k with dCas9 and generated dCas9-TnsC fusions, hoping to similarly seed TnsC filaments at target sites bound by dCas9-sgRNA complexes. However, we noted no observable on-target integration and severely diminished untargeted integration events (fig. S8B), suggesting that these designs were nonfunctional. These experiments suggested that fusion strategies may be poorly suited to increase the probability of TnsC filament formation at RNA-dependent target sites without extensive further engineering and mutagenesis.
Next, we pursued an alternative strategy, motivated by our biochemical observation that increasing TnsC concentration tilted the balance between RNA-dependent (on-target) and RNA-independent (untargeted) transposition toward the latter pathway (Fig. 2, C and D). To determine whether the same feature was applicable in cellular experiments, we relocated tnsC from the original high-copy pHelper plasmid to a separate, medium-copy plasmid, where it was controlled by its own promoter (Fig. 5A). When we tested genomic integration activity under various promoter strengths, we observed considerable differences in on-target specificity (Fig. 5, A and B). Consistent with our in vitro results, low TnsC expression from a lac promoter resulted in 98% of integration events occurring on-target, whereas high TnsC expression with a T7 promoter resulted in considerably lower accuracy (57%), akin to the original pHelper vector (Fig. 5, A to C, and fig. S8, C and D). Cells expressing TnsC under control of a T7 promoter also showed a significant enrichment for insertion events across the T7 RNAP gene, suggesting that these clones were likely enriched within the population as a way of escaping TnsC-induced toxicity (fig. S8, C and E).
Fig. 5. The fidelity of RNA-guided DNA integration is controlled by TnsC concentration.

(A) Schematic of alternative ShCAST expression strategy in which TnsC was encoded on a separate plasmid (pTnsC) driven by a Lac or T7 promoter. Distinct cellular expression levels were confirmed by Western blot against a 3xFLAG epitope tag fused to TnsC (bottom). (B) Fraction of total genome-mapping integration reads detected at on-target and untargeted sites upon TnsC expression with a Lac or T7 promoter. (C) Genome-wide view of E. coli genome-mapping reads for the original WT ShCAST system compared with a modified ShCAST system with low TnsC expression. The magnified view visualizes reads comprising ≤1% of genome-mapping reads. The target site is marked with a green triangle. (D) Fraction of total genome-mapping integration reads detected at on-target and untargeted sites, with the original ShCAST system or modified ShCAST system with low TnsC expression. Data for five sgRNAs are shown. For (B) and (D), the mean is shown from N = 2 independent biological replicates. (E) Model for target-site selection and transpososome assembly during on-target, RNA-dependent transposition (right) or untargeted, RNA-independent transposition (left) by type V-K CAST systems. Within the untargeted pathway, TnsC preferentially forms filaments at AT-rich regions and is capped by TniQ, leading to the downstream site being selected by TnsB for integration. Cas12k-bound targets may better nucleate TnsC filament formation, and we hypothesize that TnsC filaments loaded at Cas12k-bound targets serve as better substrates for DNA integration, compared with untargeted sites. All structures of TnsC filaments representing untargeted sites (22–24, 26), including the BCQ transpososome, reveal K103 residues of the TnsC monomers forming the filament proximal to TnsB, contacting DNA with opposite strand polarity compared with on-target structures (fig. S3B) (22, 23). This could be decisive for the distinct efficiencies observed at these sites.
To determine whether this increased specificity effect was generalizable, we tested a range of guides previously shown to exhibit low on-target accuracy when tested with pHelper and pDonor. In all cases, we observed a substantial increase in the relative frequency of on-target integration events with the modified CAST construct (Fig. 5D). This effect did not come at the expense of efficiency, as qPCR measurements revealed that on-target integration occurred with an equal or higher efficiency under low-TnsC conditions compared with our original pHelper design (fig. S8F). It is possible that at low TnsC conditions, the decreased availability of TnsC at untargeted sites presumably titrates fewer TnsB-DNA complexes away from on-target sites and might be the cause of the increased on-target efficiency.
Collectively, these results highlight the importance of relative expression levels for distinct components when delivering CAST machineries into target cells of interest and further confirm the key role of TnsC in driving RNA-independent transposition events.
Discussion
Recent structures have shed light on the assembly of transpososome components for RNA-guided integration by CAST systems (22, 23). However, a major proportion of integration events for type V-K CASTs occurs at untargeted sites across the genome, for which there was no known mechanistic basis. Combining structural and functional evidence, we establish here that type V-K CASTs maintain a distinct RNA-independent pathway facilitated by TnsB, TnsC, and TniQ (Fig. 5E). Our experiments revealed that the ability of TnsC to promiscuously form filaments on AT-rich DNA is a major driver of untargeted insertions. The role of TnsB in transposition, particularly at untargeted sites, is somewhat paradoxical, given its ability to disassemble TnsC and simultaneously facilitate integration. We speculate that stochastic TniQ binding might stabilize a specific configuration of TnsC filaments at untargeted sites, making them resistant to TnsB-mediated dissociation and instead promoting strand transfer. TnsC disassembly may therefore be less efficient in cellular contexts than originally observed in vitro at high protein concentrations (24, 26, 34). Our results highlight the competition between TnsB recruitment at TnsC-bound, RNA-guided target sites versus AT-rich untargeted sites, and indicate that TnsB preferentially reacts with Cas12k-bound on-target sites compared with untargeted sites (Fig. 5E). Future work will be necessary to resolve more precise kinetics of TnsC filamentation/disassembly in the presence of TniQ and TnsB and differential TnsB transposition kinetics as a function of TnsC assembly state.
The structure of the BCQ strand-transfer complex reveals two turns of a TnsC filament, an overall architecture reminiscent of the Cas12k-containing on-target transpososome (23), with no major structural differences associated with TnsC in either of these assemblies. However, TnsC residues K103 and T121 proximal to TnsB in the BCQ transpososome contact the DNA in 3′ to 5′ strand polarity, following the direction of TniQ- to TnsB-binding face of TnsC (Fig. 1F). The same strand is contacted in the case of random TnsC-DNA filaments (24, 26) and the nonproductive Cas12k transpososome (22), whereas in the productive on-target Cas12k transpososome, TnsC monomers proximal to TnsB contact the opposite strand (5′ to 3′). This interaction is thought to be a consequence of TnsC filament nucleation by Cas12k-TniQ and stabilization by additional DNA interactions (R182 and K119) (22, 23). TnsC variants with mutations to the DNA-contacting residues retain the ability to filament on DNA (24, 26) and maintain a substantial proportion of integration at untargeted sites accompanied by a drop in on-target integration (fig. S4C). This suggests that these residues may not be a prerequisite for TnsC-DNA binding. Rather, we propose that these residues may serve as an intrinsic regulatory feature to ensure that random TnsC filaments default to contacting in the 3′ to 5′ strand polarity. This interaction mode could represent an energetically less favored or passive TnsC configuration for TnsB-mediated integration, thereby permitting only a subset of sites scanned by TnsC in the genome to be licensed for untargeted transposition.
It is well known that poly-A tracts in the genome represent regions of altered DNA curvature (41), and our single-molecule experiments reveal that TnsC filamentation exhibits inherent affinity for AT-rich locations. Further meta-analyses of integration data revealed a preference for AT-rich sequences across a ~25-bp window spanning about two turns of a TnsC filament upstream of features recognized by TnsB. We suggest that AT-rich genomic regions with altered DNA curvature may resemble the bending of DNA observed between unproductive and productive Cas12k transpososomes (22), leading to preferential TnsC recruitment and a more favorable DNA-binding mode that promotes TnsB-based DNA integration. To our surprise, the TnsC K103A mutation led to a complete loss of AT preference in the integration profile, suggesting that this mutant may achieve an energetically more favorable filamentation state regardless of nucleotide composition. The loss of on-target integration for K103A may result from mutant TnsC filaments titrating TnsB-donor DNA complexes to untargeted sites in the genome more effectively than WT TnsC filaments.
The type V-K BCQ pathway, although not exactly similar, resembles the TnsE-mediated pathway in Tn7-like transposable elements, in which structural features associated with DNA replication are recognized to primarily drive widespread mobilization into plasmids (7, 8). A gain-of-function TnsC mutant (A225V) was also identified for E. coli Tn7, and it was capable of transposition in the absence of either of the two targeting factors, TnsD and TnsE (42), and facilitated integration at AT-rich sequences (43). It is possible that in a native setting, type V-K CASTs exhibit low-frequency insertion at AT-rich sites that might be triggered by certain stimuli specific to cyanobacteria. Such a model would imply a transient selfish behavior by CASTs, possibly when the availability of plasmid-targeting guide RNAs are limiting for its proliferation or in situations when mobilization to a new AT-rich site is beneficial for propagation of the element. Although there is currently no direct evidence to support this hypothesis, recent experiments with a native type V-K CAST system in cyanobacteria indicate that expression of Cas12k and TnsB are regulated by a CvkR transcriptional repressor (30), a feature that could be important in modulating the choice between targeted and untargeted transposition pathways. Considering the highly conserved operonic nature of TnsB, TnsC, and TniQ in Tn7-like elements, Tn5053, and type V-K CASTs, tight regulation in the stoichiometry of these proteins could be important for accessing an RNA-independent untargeted pathway (13). Future studies will be necessary to investigate this hypothesis further by deep sequencing bacteria with native type V-K elements to ensure that these rare events are not missed. Alternatively, the BCQ pathway may be an evolutionary relic of a primitive selfish pathway before these transposons acquired CRISPR-Cas–based targeting modules.
From a technology perspective, type V-K CASTs are among the most compact type of CRISPR-associated transposases, in terms of coding size, and thus offer a major potential opportunity relative to type I CAST systems. However, two key properties that limit their use for genome engineering applications are low on-target specificity and the generation of cointegrate transposition products due to lack of TnsA (14, 15, 18, 44). Recent efforts substantially decreased cointegrate formation by fusing an endonuclease to TnsB and improved specificity using chimeric fusion proteins or supplementing additional components such as pir (20). However, these strategies also compromise on-target integration efficiency and do not address the root cause of promiscuity. Our results provide a deeper molecular understanding of how type V-K CAST components undergo both RNA-guided and RNA-independent transposition. We identified TnsC filamentation on AT-rich DNA sequences as being the primary driver of untargeted integration and showed that under limiting TnsC concentrations, RNA-guided transposition becomes the primary pathway of choice biochemically and in cells. This rescue in specificity was generalizable for all the guides that we tested and resulted in equal or higher on-target integration efficiency compared with the original ShCAST pHelper. Our combined use of biochemical, structural, and single-molecule experiments reveals the mechanistic intricacies associated with target site selection by type V-K CAST and offers new opportunities for targeted DNA integration applications.
Methods summary
All plasmid constructs used for this study were cloned using a combination of Gibson assembly, inverse (around-the-horn) PCR, restriction digestion, and ligation. Transposition assays in E. coli were performed in BL21 (DE3) cells based on a previously described method (14). For biochemical reconstitution of CAST transposition, proteins were expressed as N-terminal His6-SUMO-TEV fusions and purified according to previous protocols (11, 24). Cryo-EM structure determination of the BCQ transpososome was performed by incubating purified TnsB, TnsC, and TniQ proteins with a synthetic DNA substrate as previously described (26), preparing and imaging grids using a 200-kV Talos Arctica (Thermo Fisher), and performing downstream image analysis. TnsC-binding activity was tested using a fluorescence polarization assay with a 5′ fluorescein-tagged DNA substrate, and ATP hydrolysis was measured using a Malachite green phosphate assay. Biochemical plasmid-to-plasmid transposition reactions were incubated for 2 hours at 37°C and then quenched by flash-freezing in liquid nitrogen. On-target transposition efficiency for biochemical and E. coli transposition assays was measured using qPCR, and specificity measurements were made using tagmentation-based transposon insertion sequencing (TagTn-seq), with library preparation performed using Nextera XT DNA Library Preparation Kit (Illumina). Next-generation sequencing was performed on an Illumina NextSeq platform with a NextSeq high-output kit. Custom Python scripts were used for mapping transposon end–containing reads to the E. coli genome or plasmid substrates used in biochemical transposition. Genetic neighborhood and AT-enrichment analyses at transposon insertion sites were performed using various custom Python scripts available online. Motifs for untargeted transposition were determined by extracting and comparing a window of sequences adjacent to each integration site, and sequence consensus logos showing per residue conservation were plotted. Single-molecule double-tethered dsDNA curtain experiments were performed as previously described (35) using a fluorescent mNeonGreen-TnsC and were analyzed to examine TnsC binding to AT- and GC-rich regions. Western blots were performed with FLAG epitope-tagged TnsC variants expressed with a T7 or lac promoter and anti-FLAG antibody. A detailed materials and methods section for this study is provided in the supplementary materials.
Supplementary Material
ACKNOWLEDGMENTS
We thank N. Jaber and S. R. Pesari for laboratory support; P.A. Sims for helpful discussions on deep sequencing; M. Jovanovic for help with mass spectrometry; R. T. King for help with Taqman qPCR; D. R. Gelsinger for help with TagTn-seq; G. D. Lampe, F. T. Hoffmann, and S. Tang for critical feedback on the manuscript; J. E. Peters for useful discussions; L. F. Landweber for qPCR instrument access; the J. P. Sulzberger Columbia Genome Center for NGS support; and the Cornell Center for Materials Research facility, K. Spoth, and M. Silvestry-Ramos for maintenance of the electron microscopes used for this research (NSF MRSEC program, DMR-1719875).
Funding:
J.T.G. is supported by International Human Frontier Science Program postdoctoral fellowship LT001117/2021-C. E.C.G. is supported by National Institutes of Health (NIH) grant R35GM118026. E.H.K. is supported by NIH grant R01GM144566 and a Pew Biomedical Scholarship. S.H.S. is supported by NIH grants DP2HG011650, R21AI68976, and R01EB031935; a Pew Biomedical Scholarship; a Sloan Research Fellowship; an Irma T. Hirschl Career Scientist Award; and a generous startup package from the Columbia University Irving Medical Center Dean’s Office and the Vagelos Precision Medicine Fund.
Footnotes
Competing interests: Columbia University has filed a patent application related to this work for which J.T.G. and S.H.S. are inventors. S.H.S. is a cofounder and scientific adviser to Dahlia Biosciences, a scientific adviser to CrisprBits and Prime Medicine, and an equity holder in Dahlia Biosciences and CrisprBits. The remaining authors declare no competing interests.
SUPPLEMENTARY MATERIALS
Data and materials availability:
Cryo-EM reconstructions of the BCQ transpososome are available through the Electron Microscopy Data Bank with accession code EMD-41280. Next-generation sequencing data are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive with BioProject accession code PRJNA1010381. Custom Python scripts used for computational analysis of next-generation sequencing data are available at Zenodo (45).
REFERENCES AND NOTES
- 1.. Craig NL, Target site selection in transposition. Annu. Rev. Biochem 66, 437–474 (1997). doi: 10.1146/annurev.biochem.66.1.437; pmid: 9242914 [DOI] [PubMed] [Google Scholar]
- 2.Siguier P, Gourbeyre E, Chandler M, Bacterial insertion sequences: Their genomic impact and diversity. FEMS Microbiol. Rev 38, 865–891 (2014). doi: 10.1111/1574-6976.12067; pmid: 24499397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siguier P, Gourbeyre E, Varani A, Ton-Hoang B, Chandler M, Everyman’s guide to bacterial insertion sequences. Microbiol. Spectr 3, A3–A0030, 2014 (2015). doi: 10.1128/microbiolspec.MDNA3-0030-2014; pmid: 26104715 [DOI] [PubMed] [Google Scholar]
- 4.Arias-Palomo E, Berger JM, An atypical AAA+ ATPase assembly controls efficient transposition through DNA remodeling and transposase recruitment. Cell 162, 860–871 (2015). doi: 10.1016/j.cell.2015.07.037; pmid: 26276634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizuno N et al. , MuB is an AAA+ ATPase that forms helical filaments to control target selection for DNA transposition. Proc. Natl. Acad. Sci. U.S.A 110, E2441–E2450 (2013). doi: 10.1073/pnas.1309499110; pmid: 23776210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi KY, Spencer JM, Craig NL, The Tn7 transposition regulator TnsC interacts with the transposase subunit TnsB and target selector TnsD. Proc. Natl. Acad. Sci. U.S.A 111, E2858–E2865 (2014). doi: 10.1073/pnas.1409869111; pmid: 24982178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters JE, Craig NL, Tn7 recognizes transposition target structures associated with DNA replication using the DNA-binding protein TnsE. Genes Dev. 15, 737–747 (2001). doi: 10.1101/gad.870201; pmid: 11274058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters JE, Craig NL, Tn7: smarter than we thought. Nat. Rev. Mol. Cell Biol 2, 806–814 (2001). doi: 10.1038/35099006; pmid: 11715047 [DOI] [PubMed] [Google Scholar]
- 9.Finn JA, Parks AR, Peters JE, Transposon Tn7 directs transposition into the genome of filamentous bacteriophage M13 using the element-encoded TnsE protein. J. Bacteriol 189, 9122–9125 (2007). doi: 10.1128/JB.01451-07; pmid: 17921297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peters JE, Makarova KS, Shmakov S, Koonin EV, Recruitment of CRISPR-Cas systems by Tn7-like transposons. Proc. Natl. Acad. Sci. U.S.A 114, E7358–E7366 (2017). doi: 10.1073/pnas.1709035114; pmid: 28811374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strecker J et al. , RNA-guided DNA insertion with CRISPR-associated transposases. Science 365, 48–53 (2019). doi: 10.1126/science.aax9181; pmid: 31171706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klompe SE, Vo PLH, Halpin-Healy TS, Sternberg SH, Transposon-encoded CRISPR-Cas systems direct RNA-guided DNA integration. Nature 571, 219–225 (2019). doi: 10.1038/s41586-019-1323-z; pmid: 31189177 [DOI] [PubMed] [Google Scholar]
- 13.Faure G et al. , Modularity and diversity of target selectors in Tn7 transposons. Mol. Cell 83, 2122–2136.e10 (2023). doi: 10.1016/j.molcel.2023.05.013; pmid: 37267947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.. Vo PLH et al. , CRISPR RNA-guided integrases for highefficiency, multiplexed bacterial genome engineering. Nat. Biotechnol 39, 480–489 (2021). doi: 10.1038/s41587-020-00745-y; pmid: 33230293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubin BE et al. , Species- and site-specific genome editing in complex bacterial communities. Nat. Microbiol 7, 34–47 (2022). doi: 10.1038/s41564-021-01014-7; pmid: 34873292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsieh S-C, Peters JE, Discovery and characterization of novel type I-D CRISPR-guided transposons identified among diverse Tn7-like elements in cyanobacteria. Nucleic Acids Res. 51, 765–782 (2023). doi: 10.1093/nar/gkac1216; pmid: 36537206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saito M et al. , Dual modes of CRISPR-associated transposon homing. Cell 184, 2441–2453.e18 (2021). doi: 10.1016/j.cell.2021.03.006; pmid: 33770501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vo PLH, Acree C, Smith ML, Sternberg SH, Unbiased profiling of CRISPR RNA-guided transposition products by long-read sequencing. Mob. DNA 12, 13 (2021). doi: 10.1186/s13100-021-00242-2; pmid: 34103093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rice PA, Craig NL, Dyda F, Comment on “RNA-guided DNA insertion with CRISPR-associated transposases”. Science 368, eabb2022 (2020). doi: 10.1126/science.abb2022; pmid: 32499410 [DOI] [PubMed] [Google Scholar]
- 20.Tou CJ, Orr B, Kleinstiver BP, Precise cut-and-paste DNA insertion using engineered type V-K CRISPR-associated transposases. Nat. Biotechnol 41, 968–979 (2023). doi: 10.1038/s41587-022-01574-x; pmid: 36593413 [DOI] [PubMed] [Google Scholar]
- 21.Lampe GD et al. , Targeted DNA integration in human cells without double-strand breaks using CRISPR-associated transposases. Nat. Biotechnol, 1–12 (2023). doi: 10.1038/s41587-023-01748-1; pmid: 36991112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmitz M, Querques I, Oberli S, Chanez C, Jinek M, Structural basis for the assembly of the type V CRISPR-associated transposon complex. Cell 185, 4999–5010.e17 (2022). doi: 10.1016/j.cell.2022.11.009; pmid: 36435179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park J-U et al. , Structures of the holo CRISPR RNA-guided transposon integration complex. Nature 613, 775–782 (2023). doi: 10.1038/s41586-022-05573-5; pmid: 36442503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Querques I, Schmitz M, Oberli S, Chanez C, Jinek M, Target site selection and remodelling by type V CRISPR-transposon systems. Nature 599, 497–502 (2021). doi: 10.1038/s41586-021-04030-z; pmid: 34759315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao R et al. , Structural basis of target DNA recognition by CRISPR-Cas12k for RNA-guided DNA transposition. Mol. Cell 81, 4457–4466.e5 (2021). doi: 10.1016/j.molcel.2021.07.043; pmid: 34450043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park J-U et al. , Structural basis for target site selection in RNA-guided DNA transposition systems. Science 373, 768–774 (2021). doi: 10.1126/science.abi8976; pmid: 34385391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harshey RM, Transposable phage mu. Microbiol. Spectr 2, 2.5.31 (2014). doi: 10.1128/microbiolspec.MDNA3-0007-2014; pmid: 26104374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ya G. Kholodii et al. , Four genes, two ends, and a res region are involved in transposition of Tn5053: A paradigm for a novel family of transposons carrying either a mer operon or an integron. Mol. Microbiol 17, 1189–1200 (1995). doi: 10.1111/j.1365-2958.1995.mmi_17061189.x; pmid: 8594337 [DOI] [PubMed] [Google Scholar]
- 29.Hou S et al. , CRISPR-Cas systems in multicellular cyanobacteria. RNA Biol. 16, 518–529 (2019). doi: 10.1080/15476286.2018.1493330; pmid: 29995583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ziemann M et al. , CvkR is a MerR-type transcriptional repressor of class 2 type V-K CRISPR-associated transposase systems. Nat. Commun 14, 924 (2023). doi: 10.1038/s41467-023-36542-9; pmid: 36801863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen Y et al. , Structural basis for DNA targeting by the Tn7 transposon. Nat. Struct. Mol. Biol 29, 143–151 (2022). doi: 10.1038/s41594-022-00724-8; pmid: 35173349 [DOI] [PubMed] [Google Scholar]
- 32.Hoffmann FT et al. , Selective TnsC recruitment enhances the fidelity of RNA-guided transposition. Nature 609, 384–393 (2022). doi: 10.1038/s41586-022-05059-4; pmid: 36002573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanson PI, Whiteheart SW, AAA+ proteins: Have engine, will work. Nat. Rev. Mol. Cell Biol 6, 519–529 (2005). doi: 10.1038/nrm1684; pmid: 16072036 [DOI] [PubMed] [Google Scholar]
- 34.Park J-U, Tsai AW-L, Chen TH, Peters JE, Kellogg EH, Mechanistic details of CRISPR-associated transposon recruitment and integration revealed by cryo-EM. Proc. Natl. Acad. Sci. U.S.A 119, e2202590119 (2022). doi: 10.1073/pnas.2202590119; pmid: 35914146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.. Meir A, Kong M, Xue C, Greene EC, DNA llight on complex molecular systems during homologous recombination. J. Vis. Exp (160): (2020). doi: 10.3791/61320 [DOI] [PubMed] [Google Scholar]
- 36.Skalka A, Burgi E, Hershey AD, Segmental distribution of nucleotides in the DNA of bacteriophage lambda. J. Mol. Biol 34, 1–16 (1968). doi: 10.1016/0022-2836(68)90230-1; pmid: 4999721 [DOI] [PubMed] [Google Scholar]
- 37.Greene EC, Mizuuchi K, Direct observation of single MuB polymers: Evidence for a DNA-dependent conformational change for generating an active target complex. Mol. Cell 9, 1079–1089 (2002). doi: 10.1016/S1097-2765(02)00514-2; pmid: 12049743 [DOI] [PubMed] [Google Scholar]
- 38.Ge J, Lou Z, Cui H, Shang L, Harshey RM, Analysis of phage Mu DNA transposition by whole-genome Escherichia coli tiling arrays reveals a complex relationship to distribution of target selection protein B, transcription and chromosome architectural elements. J. Biosci 36, 587–601 (2011). doi: 10.1007/s12038-011-9108-z; pmid: 21857106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walker MWG, Klompe SE, Zhang DJ, Sternberg SH, Novel molecular requirements for CRISPR RNA-guided transposition. Nucleic Acids Res. 51, 4519–4535 (2023). doi: 10.1093/nar/gkad270; pmid: 37078593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tenjo-Castaño F et al. , Structure of the TnsB transposase-DNA complex of type V-K CRISPR-associated transposon. Nat. Commun 13, 5792 (2022). doi: 10.1038/s41467-022-33504-5; pmid: 36184667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haran TE, Mohanty U, The unique structure of A-tracts and intrinsic DNA bending. Q. Rev. Biophys 42, 41–81 (2009). doi: 10.1017/S0033583509004752; pmid: 19508739 [DOI] [PubMed] [Google Scholar]
- 42.Stellwagen AE, Craig NL, Gain-of-function mutations in TnsC, an ATP-dependent transposition protein that activates the bacterial transposon Tn7. Genetics 145, 573–585 (1997). doi: 10.1093/genetics/145.3.573; pmid: 9055068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biery MC, Stewart FJ, Stellwagen AE, Raleigh EA, Craig NL, A simple in vitro Tn7-based transposition system with low target site selectivity for genome and gene analysis. Nucleic Acids Res. 28, 1067–1077 (2000). doi: 10.1093/nar/28.5.1067; pmid: 10666445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen W et al. , Targeted genetic screening in bacteria with a Cas12k-guided transposase. Cell Rep. 36, 109635 (2021). doi: 10.1016/j.celrep.2021.109635; pmid: 34469724 [DOI] [PubMed] [Google Scholar]
- 45.Custom Python scripts for: George JT et al. , Mechanism of target site selection by type V-K CRISPR-associated transposases, Zenodo: (2023); doi: 10.1101/2023.07.14.548620 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM reconstructions of the BCQ transpososome are available through the Electron Microscopy Data Bank with accession code EMD-41280. Next-generation sequencing data are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive with BioProject accession code PRJNA1010381. Custom Python scripts used for computational analysis of next-generation sequencing data are available at Zenodo (45).