

Abstract
Pigment producing melanocytes overcome frequent oxidative stress in their physiological role of protecting the skin against deleterious effects of solar UV irradiation. This is accomplished by the activity of several endogenous antioxidant systems including the thioredoxin antioxidant system, in which, thioredoxin reductase 1 (TR1) plays an important part. To determine whether TR1 contributes to the redox regulation of melanocyte homeostasis, we have generated a selective melanocytic TR1 knockout mouse model (Txnrd1mel−/−), which exhibits a depigmentation phenotype consisting of variable amelanotic ventral spotting and reduced pigmentation on the extremities (tail tip, ears, and paws). The antioxidant role of TR1 was further probed in presence of acute neonatal UVB irradiation, which stimulates melanocyte activation and introduces a spike in oxidative stress in the skin microenvironment. Interestingly, we observed a significant reduction in overall melanocyte count and proliferation in absence of TR1. Furthermore, melanocytes exhibited an elevated level of UV-induced DNA damage in the form of 8-oxo-2’-deoxyguanosine following acute UVB treatment. We also saw engagement of compensatory antioxidant mechanisms through increased nuclear localization of transcription factor NRF2. Altogether, the above data indicate that melanocytic TR1 positively regulate melanocyte homeostasis and pigmentation during development and protects against UVB induced DNA damage and oxidative stress.
INTRODUCTION
Melanocytes are dendritic, pigment-producing cells in the skin tasked with its protection against the deleterious effects of solar ultraviolet (UV) radiation by producing two types of light absorbent melanin pigment: photoprotective, brown-black eumelanin and red-yellow pheomelanin. This synthetic process utilizes several enzymes including tyrosinase (TYR), the rate limiting enzyme, dopachrome tautomerase (DCT), and tyrosinase related protein 1 (TYRP1). Melanin-containing melanosomes are then shared with adjacent skin cells (e.g. keratinocytes) and situated around their nuclei to block UV-mediated DNA damage(Boissy, 2003).
During development, melanocytes originate in the neural crest as melanoblasts that begin to express microphthalmia-associated transcription factor (MITF), the master regulator of melanocyte differentiation and melanogenesis, around E10.5 (embryonic day). They then migrate dorsolaterally through the developing dermis to encompass the entire body(Jordan and Jackson, 2000). Other important transcription factors involved in this process include SOX10 and PAX3(Seberg et al., 2017). In the fur-bearing regions of mice, melanocytes invade the epidermis at E13.5 and begin to colonize the developing hair follicles (HFs) where two populations form. The first are quiescent melanocyte stem cells (MCSCs) that will occupy the stem cell niche in the HF bulge region and a second portion that migrate further down the HF to the bulb and differentiate into mature melanocytes capable of pigmenting the growing hair(Li, 2014, Mort et al., 2015). As the HF cycles, the differentiated population of melanocytes undergo apoptosis along with a significant portion of the HF(Muller-Rover et al., 2001). When new hairs grow, the MCSCs in the HF bulge will differentiate and repopulate the bulb region. Loss of MCSCs in the stem cell niche causes greying of the hair(Nishimura et al., 2002).
Melanocytes endure a large degree of oxidative stress not only due to exogenous insults to the skin like UV, but also endogenously as the melanogenesis process itself produces reactive oxygen species (ROS)(Denat et al., 2014). Therefore, melanocytes need to employ endogenous antioxidant systems to combat ROS including the thioredoxin and glutathione antioxidant systems. The thioredoxin antioxidant system is comprised of NADPH, thioredoxin reductase 1 (TR1), and thioredoxin (TRX). This system works by the transfer of electrons from NADPH to TR1 allowing it to reduce protein disulfides on TRX which can then go on and reduce its own substrates(Holmgren and Lu, 2010). TR1 is known to have elevated expression in the skin following UV(Cassidy et al., 2017, Oh et al., 2004) and is highly upregulated in many cancer types(Nguyen et al., 2006) including brain(Esen et al., 2015), breast(Cadenas et al., 2010), and colorectal (Raffel et al., 2003). In melanoma, we found that increasing TR1 expression correlates with disease progression suggesting that its antioxidant function is coopted(Cassidy et al., 2015). In the same report, we showed that a human melanoma cell line engineered to express a microRNA that targets TR1 was reliant on glycolysis for cell survival in vitro and readily formed highly metastatic tumors in a mouse xenograft model. However, when we combined the TR1 knockdown with the pharmacologic blockade of glycolysis, metastases in the mice were eliminated.
To better understand the role of TR1 in normal melanocyte physiology in vivo, we have generated a selective knockout (KO) mouse model in which homozygous floxed TR1 (Txnrd1fl/fl) has been specifically ablated in melanocytes when in the presence of Cre recombinase driven by a tyrosinase promoter (Tyr-cre). This mouse model exhibits a depigmentation phenotype consisting of variable ventral amelanotic spotting as well as reduced pigmentation on the extremities. In the present study, we characterized that phenotype as well as probed for the ability of melanocytes to respond to spikes of oxidative stress such as acute UVB irradiation elucidating a unique role of oxidoreductase TR1 in controlling pigmentation, melanocyte homeostasis and acute UVB induced oxidative stress.
RESULTS
Confirmation of in vivo melanocytic Txnrd1 knockout and generation of Txnrd1mel−/− mice
Ablation of melanocytic Txnrd1 was confirmed at the genome level via PCR genotyping. Two pairs of primers showed either the floxed status of the targeted gene region (+,L2) or the occurrence of cre-mediated recombination leading to ablation of the floxed region (L−) (Supplementary Figure 1a). The sex of the mice was also determined using a primer pair for sex determining region Y (SRY) as the transgene for Tyr-cre is x-linked necessitating the use of male mice to eliminate the confounding issue of x-linked inactivation. Using those primers, we only saw a Txnrd1 excision band (L−) in the presence of floxed Txnrd1 (L2) and cre allele which provides evidence of successful cre-mediated ablation of Txnrd1 (Supplementary Figure 1b). Furthermore, we collected DNA from embryonic dorsal skin at E17.5 and again saw a cre excision band in male fetuses only indicating embryonic ablation of Txnrd1 (Supplementary Figure 1c).
To confirm TR1 KO at the protein level, epidermal skin cell culture was prepared to separate out TR1 negative melanocytes from adjacent TR1 positive non-melanocyte epidermal skin cells. Co-immunocytochemistry (co-ICC) of Txnrd1L2/L2 epidermal skin cell culture for PAX3 and DCT demonstrated their co-expression enabling the use of PAX3 as a melanocyte marker in this context. The co-expression of PAX3 and DCT in TR1 KO melanocytes is shown in subsequent figures. KO of TR1 in melanocytes was then confirmed by co-ICC of PAX3/TR1 where PAX3+ Txnrd1mel−/− melanocytes lacked expression of TR1 and non-melanocyte skin cells continued to express TR1 (Figure 1a). We made all subsequent analyses in male Txnrd1L2/L2 and Txnrd1mel−/− mice.
Figure 1. Confirmation of melanocytic Txnrd1 knockout at the protein level in vivo.
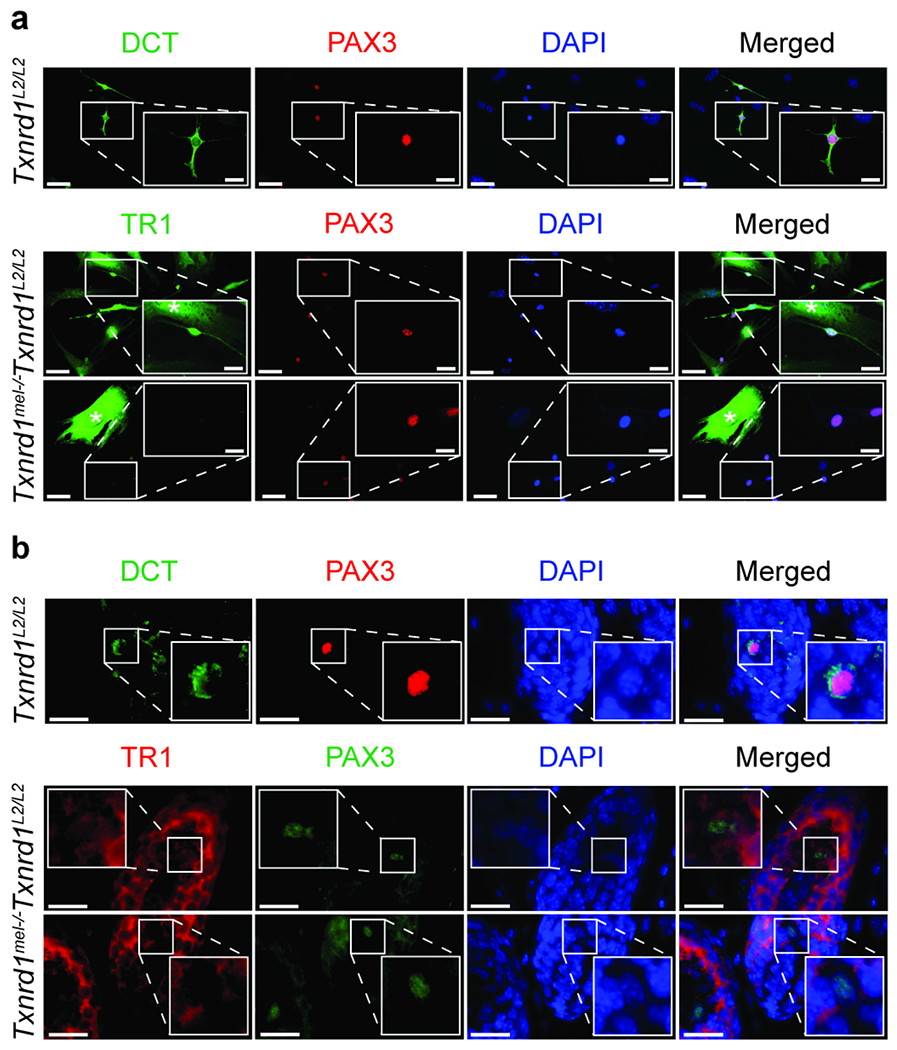
(a) ICC of isolated neonatal epidermal skin cell cultures or (b) IHC of neonatal epidermal skin co-labeled for melanocytes using either Pax3 (red)/DCT (green) or Pax3 (red, c; green, d) /TR1 (green, c; red, d). Scale bars are (a) 50 μm (inset 20 μm) or (b) 20 μm. * Indicates non-melanocyte epidermal skin cells.
To confirm TR1 KO in the intact skin, we examined TR1 expression using co-immunohistochemistry (co-IHC) in the follicular melanocytes in the bulb region apical to the dermal papilla as they were relatively separated from adjacent non-melanocyte TR1 positive cells. The co-expression of PAX3 and DCT in the HF bulb indicated that PAX3 could be used as a melanocyte marker in that context as well. We then performed co-IHC for PAX3 and TR1 and observed an absence of TR1 expression in Txnrd1mel−/− melanocytes (Figure 1b).
Txnrd1mel−/− mice exhibit pigmentation defects on the ventral torso and extremities
83% of the Txnrd1mel−/− mutant mice (45/54) displayed noticeable amelanotic spots on the ventral torso while none of the Txnrd1L2/L2control (54/54) exhibited any phenotype. These amelanotic patches varied in size from 10-20% of ventral fur to just a barely noticeable clustering of depigmented hairs. Furthermore, we observed reduced pigmentation on the extremities (Figure 2a). The depigmentation of the fore and hind paw skin (pes) was consistent; however, on the tail it was more variable but to a lesser degree than that seen on the ventral torso. The observed phenotype at P10 consistently correlated with the genotype determined via PCR.
Figure 2. Characterization of the phenotypic pigmentation defects associated with melanocytic ablation of Txnrd1.
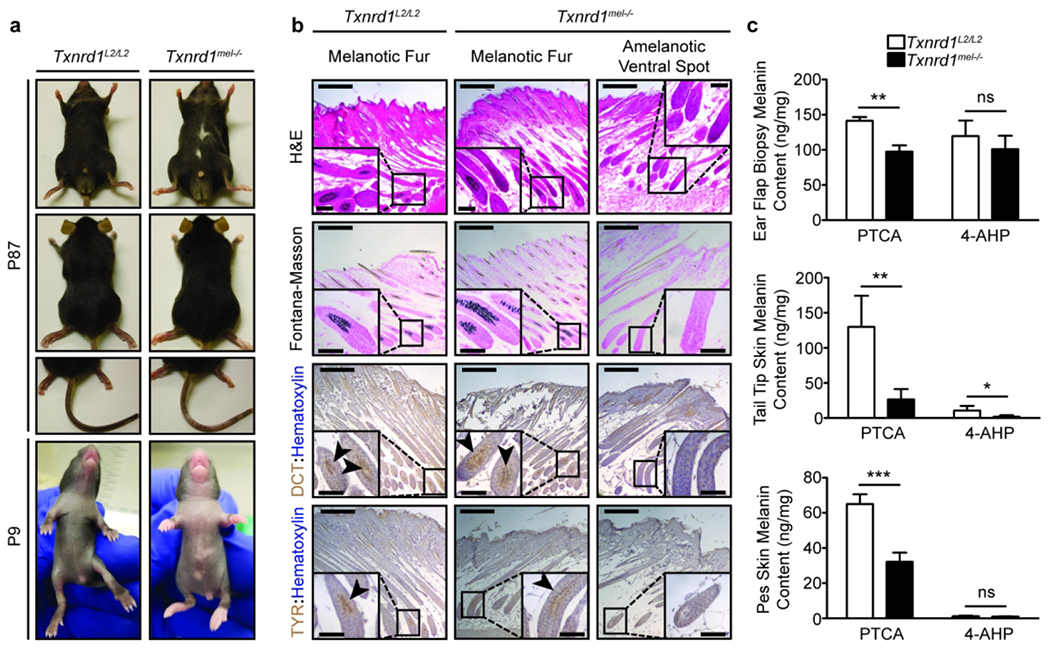
(a) Images of the depigmentation phenotype observed in Txnrd1mel−/− mice. (b) Histology of the amelanotic ventral spot as compared to pigmented dorsal hair in both Txnrd1mel−/− and Txnrd1L2/L2 mice. (c) Quantification of P64 (4 mm ear punch biopsy) and P21 (2 cm tail tip and left and right pes) skin melanin content via HPLC analysis of eumelanin marker pyrrole-2,3,5-tricar-boxylic acid (PTCA) or pheomelanin marker 4-amino-3-hydroxyphenylalanine (4-AHP) from aqueous skin homogenates subjected to either hydrogen peroxide oxidation or hydroiodic acid hydrolysis, respectively. PTCA HPLC utilized UV detection while 4-AHP HPLC used electrochemical detection. Melanin content was normalized to the dry weight of the sample. Data represent mean±SD with N = 4. Scale bars are 200 μm (50 μm inset). * p < 0.05, ** p < 0.01, *** p < 0.001
Fontana-Masson staining revealed the absence of melanin pigment and IHC for melanocyte markers DCT and TYR showed a complete absence of their expression in the HF bulb (Figure 2b). Further examination of the amelanotic ventral spots using co-IHC for PAX3/DCT showed the absence of follicular melanocytes around the bulge and in the bulb regions (Supplementary Figure 2a). Outside of the amelanotic ventral spots we did not see any difference in melanocyte density on the ventral skin of P4.5 neonates (Supplementary Figure 2b).
Next, we examined extremities for their melanin content by taking skin samples from the ears, paws, and tail tips. We observed a significant reduction in eumelanin content in the KO skin samples as well as generally low levels of pheomelanin in both WT and KO mice (Figure 2c). The low levels of pheomelanin are likely due to the genetic background of our mice being C57BL/6. The above results indicate an important role of melanocytic-TR1 in regulating skin pigmentation in vivo.
TR1 knockout melanocytes exhibit increased UVB sensitivity
To probe into the TR1 KO pigmentation phenotype, we employed UVB irradiation to perturb melanocyte stem cells and induce them to differentiate and migrate out of the HF niche. This treatment also introduces a spike of oxidative stress into the skin microenvironment as well as inducing DNA damage, towards which, TR1 plays a mitigating role. Acute neonatal UVB irradiation is a commonly used model to examine how melanocytes respond in both the short and long term as acquired DNA damage can lead to melanomagenesis later in life given a conducive mutational background.
We subjected P3.5 neonates to a 350 mJ/cm2 dose of UVB and then collected dorsal skin samples 24 or 72 hours later (Figure 3a). These timepoints are to observe the initial impact of irradiation on melanocytes and the 72-hour timepoint is also when activated MCSCs have migrated to the interfollicular epidermis. We found that there were significantly less DCT+/PAX3+ melanocytes around the hair follicle bulge region at both timepoints and no difference in migrating melanocytes in the interfollicular epidermis at 72 hours (Figure 3b).
Figure 3. Txnrd1mel−/− mouse skin exhibits fewer melanocytes with reduced proliferation and increased nuclear localization of NRF2 following acute neonatal UVB irradiation.
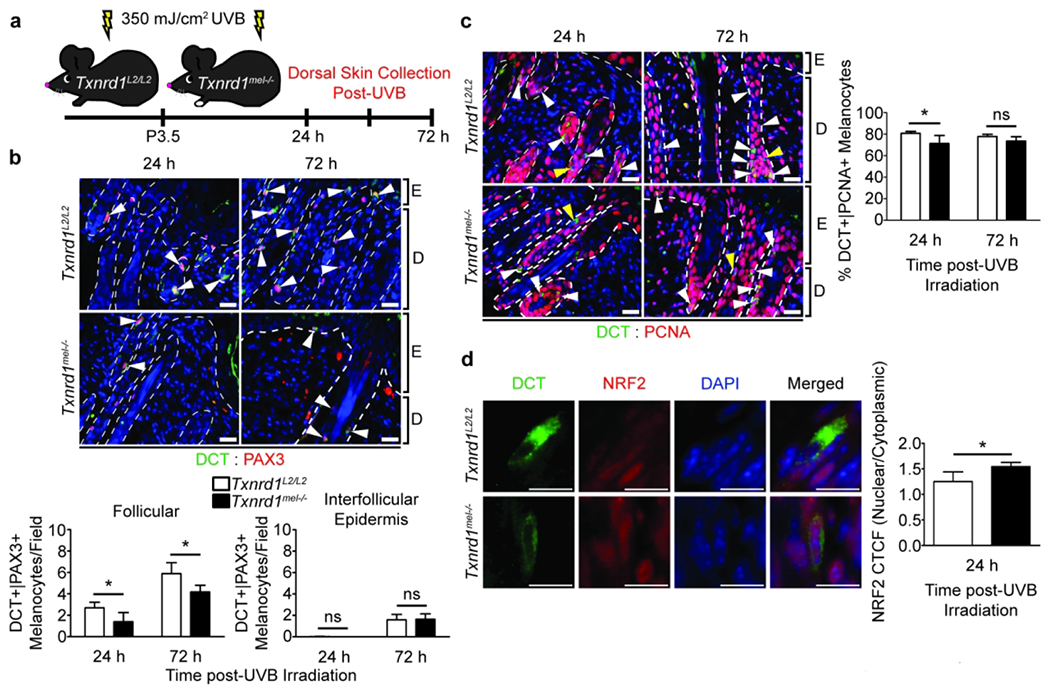
(a) Schematic of the UVB dosage regimen and dorsal skin sample collection. Co-IHC and quantification for (b) DCT+ (green)/Pax3+ (red) melanocytes and (c) DCT+ (green)/PCNA+ (red) proliferating melanocytes. Scale bars are 20 μm. Arrowheads indicate DCT single positive cells (yellow) or double positive cells (white). (d) Analysis of DCT+ (green)/NRF2+ (red) melanocytes for nuclear localization of NRF2 via quantification of the nuclear to cytoplasmic corrected total cellular fluorescence (CTCF) ratio using ImageJ. Scale bars are 10 μM. Data represent mean±SD with N = 4. ns = nonsignificant, E = epidermis, D = dermis. * = p < 0.05
The UVB irradiated skin samples were also probed for proliferating melanocytes by co-IHC using antibodies against PCNA and DCT. We found that there were significantly fewer proliferating melanocytes in the TR1 KO skin 24 hours after irradiation; however, the difference was reduced after 72 hours (Figure 3c).
We were then interested in examining the expected activation of compensatory antioxidant mechanisms in melanocytes lacking functional TR1 activity. We first sought to do so by measuring glutathione (GSH) concentration in vitro using PIG1 immortalized human melanocytes in which TR1 activity had been knocked down (TR1lo) using a microRNA (miRNA) and comparing them to control cells with a miRNA targeted to a non-eukaryotic gene (TR1high). We found that TR1lo cells had a significantly increased GSH concentration of 8.3 nmol/106 cells as compared to TR1high cells at 6.4 nmol/106 cells GSH (Supplementary figure 3a). The concentration of GSH could be depleted in both cell lines by treatment with buthionine sulfoximine (BSO), a glutamate cysteine ligase (GCL) inhibitor. These data suggest that even in the absence of an exogenous spike in oxidative stress that compensatory mechanisms assist cells in dealing with decreased TR1 function in vitro.
To confirm the above observation in vivo, we examined nuclear factor erythroid 2-related factor 2 (NRF2) for its nuclear localization in melanocytes 24 h after acute neonatal UVB irradiation via co-IHC. We determined that there was a significant increase in the nuclear to cytoplasmic corrected total cellular fluorescence (CTCF) signal ratio, indicating elevated nuclear localization of transcription factor NRF2 in Txnrd1mel−/− mice as compared to Txnrd1L2/L2 mice (Figure 3d, Supplementary Figure 3b). We also observed that the ratio for nuclear to whole cell NRF2 CTCF was also significantly higher in KO melanocytes as compared to WT (data not shown). Furthermore, a one sample t-test was performed to determine if the nuclear to cytoplasmic CTCF ratio of WT or KO melanocytes was significantly higher than 1.000 which would suggest NRF2 nuclear localization occurred. We found significance only with TR1 KO melanocytes suggesting nuclear localization is correlated with TR1 ablation (WT p-value = 0.0792 and KO p-value = 0.0009). Together, these data suggest that ablation of TR1 has a short-term effect on melanocytic response to UVB due to the engagement of compensatory mechanisms such as the glutathione antioxidant system. Indeed, this observation was further corroborated when we allowed UVB irradiated neonates to persist until 10 weeks of age and, after induced hair cycling using Nair cream depilation, there were not significantly more depigmented hairs present in the dorsal fur of TR1 KO mice (Supplementary Figure 4). Altogether, the above results suggest a critical role of melanocytic-TR1 in mediating UV-induced melanocyte homeostasis in vivo.
UVB induced DNA damage is elevated in TR1 knockout melanocytes
One of the most deleterious consequences of UVB irradiation of the skin is DNA damage. We were interested to see if ablation of melanocytic TR1 would lead to an elevation of DNA damage that results as both a direct consequence from UV irradiation and that which arises from increased oxidative stress. To do so, we examined DCT+ melanocytes via co-IHC for the presence of either 8-oxo-2’-deoxyguanosine (8-oxo-dG) or cyclobutane pyrimidine dimers (CPDs). There was a significant induction in DNA damage resulting from oxidative stress as seen by staining for 8-oxo-dG (Figure 4a) in KO skin as compared to WT at both timepoints. On the other hand, there was a trend in elevated CPD formation at both timepoints; however, the difference was not significant (24 hour p-value = 0.065 and 72 hour p-value = 0.066) (Figure 4b). We also took higher magnification images highlighting singular melanocytes to better exhibit the positive signal corresponding to DNA damage (Supplementary Figure 5). The above results highlight a critical role of melanocytic-TR1 in protecting against UV-induced oxidative stress in vivo.
Figure 4. Ablation of Txnrd1 induces elevated DNA damage in response to UVB irradiation.
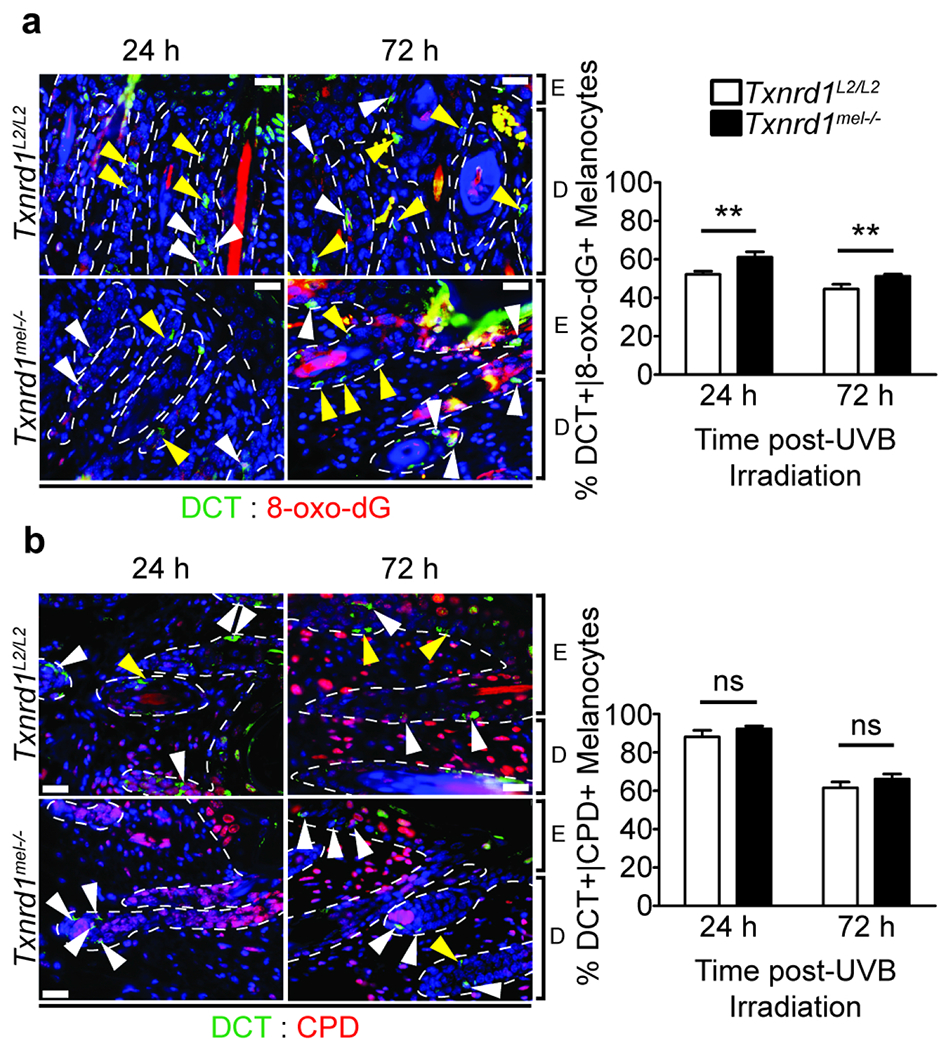
Co-IHC of neonatal dorsal skin sections 24 or 72 hours following acute UVB irradiation for markers corresponding to DCT+ (green) melanocytes exhibiting UVB induced DNA damage in the form of 8-hydroxyguanine (8-oxo-dG) (red, a) and cyclobutane pyrimidine dimers (CPDs) (red, b). Scale bars are 20 μm. Arrowheads indicate DCT single positive cells (yellow) or double positive cells (white). Data represent mean±SD with N = 4. E = epidermis, D = dermis. * p < 0.05, ** p < 0.01
DISCUSSION
Ablation of Txnrd1 in this mouse model highlights the important role of thioredoxin reductase 1 in melanocyte development and physiology. In this present study we show, as has not been previously reported to our knowledge, that genetic KO of the antioxidant oxidoreductase TR1 in melanocytes leads to a depigmentation phenotype in vivo(Baxter et al., 2019). Searching the Mouse Genome Database (Blake et al., 2021) for oxidoreductase genes associated with a pigmentation phenotype yielded one result, Sod2, which has been shown to be involved in pigmentation of the eye due to changes in the morphology of the retinal pigment epithelium(Sandbach et al., 2001). Recently, another oxidoreductase, Nicotinamide Nucleotide Transhydrogenase (NNT), expressed in the mitochondria has been shown to modulate skin pigmentation. NNT acts through a MITF and UV independent mechanism tied to tyrosinase stability and melanosome maturation(Allouche et al., 2021). Interestingly, Allouche et al. observed the inverse effect on pigmentation from NNT disruption than we saw from TR1 knockout. It is likely that both oxidoreductases interact with pigmentary pathways in distinct ways based on their sub-cellular localization since NNT was reported to be primarily expressed in the mitochondria whereas we and others have shown only modest mitochondrial TR1 expression with the preponderance localized to the cellular cytosol (Branco et al., 2014, Cassidy et al., 2015). We also note that in zebrafish treated with the TR1 inhibitor auranofin, significant hypopigmentation is observed in the skin and the eyes of the fish, and expression of zebrafish homologs of pigmentation genes mitfb and trp-1a are decreased(Gao et al., 2017).
The depigmentation phenotype seen in Txnrd1mel−/− mice suggests defective development of the melanocyte lineage and we hypothesize that there are three potential mechanisms through which this might occur: (1) reduced migration of melanoblasts from the neural crest cells in absence of TR1, (2) reduced proliferation and survival of the relatively few melanoblasts that migrate furthest, and/or (3) reduced differentiation of melanocytes from Schwann cell precursors (Adameyko et al., 2009).
Much of the focus in the literature on the impact of TR1 on cellular migration is in the context of anticancer treatment. Indeed, TR1 has been shown to contribute to migration in breast(Bhatia et al., 2016), colorectal(Zheng et al., 2019), and salivary adenoid cystic carcinoma(Jiang et al., 2015). Furthermore, TR1, acting via its substrate Trx, has been directly implicated in contributing to cancer cell motility in melanoma cells with induced L-plastin (LPL) expression as well as in LPL expressing prostate cancer cells via redox modifications of LPL at the Cys101 residue(Balta et al., 2019). In the context of normal development of the melanocyte lineage, melanoblasts begin migration with induced MITF expression in neural crest cells at E10.5 and have migrated to encompass almost the entire embryo by E16.5 with only the hindlimb foot excluded (Jordan and Jackson, 2000).It has been previously shown that the Tyr-Cre gene used in this study is expressed as early as E11.5 (Delmas et al., 2003), and therefore, we expect that TR1 KO occurs relatively early in melanoblast migration. In this study, we have shown Txnrd1 embryonic knockout occurs by E17.5 in our mouse model. As such, we cannot make an exact determination on whether TR1 contributes solely to efficient melanoblast or melanocyte migration pre or postnatally. However, the depigmentation phenotype seen here is mainly exhibited in the feet with only variable ventral spotting thus indicating a modest contribution of TR1 to melanoblast migration if any. Moreover, our data show no effect of TR1 KO on UV-activated MCSC migration into the interfollicular epidermis suggesting an impact might only be seen on further migrating cells, such as melanoblasts during development, or an alternative reason such as reduced survival and/or proliferation might account for the observed depigmentation phenotype.
Regarding proliferation, our data indicate that a predominant effect of TR1 ablation is in the ability of KO cells to withstand spikes of oxidative stress. This is evident from our DCT/PAX3 and DCT/PCNA dual staining of UVB irradiated neonatal skin where after 24 hours, there were significantly less of either double-positive stained melanocytes in the mutant skin lacking TR1. However, after 72 hours proliferation of TR1 KO melanocytes recovered likely due compensatory mechanisms such as increased glutathione synthesis(Prigge et al., 2017, Wall et al., 2019) or paracrine effects of Trx secreted by UV-irradiated keratinocytes(Funasaka and Ichihashi, 1997). This is supported by our findings on GSH concentration in vitro and NRF2 nuclear localization in vivo. NRF2 is a key transcriptional regulator of numerous antioxidant genes including Txnrd1 by binding to antioxidant response elements (AREs) in target gene promoter regions (Hawkes et al., 2014). It also similarly regulates expression of GSH synthesis enzyme glutamate cysteine ligase (GCL), a heterodimer of modifier (GCLM) and catalytic (GCLC) protein subunits with AREs in their respective gene promoter regions (Lu, 2013). We observed in vitro that knockdown of TR1 activity in melanocytes elevated cellular GSH concentration thus indicating the compensatory engagement of the glutathione antioxidant system via increased GSH synthesis, occurring even in the absence of exogenous oxidative stress. We show evidence for this in vivo where we observed a likely increase in activation of NRF2 via its disassociation from its cytoplasmic negative regulator kelch-like ECH-associated protein 1 (KEAP1) and subsequent nuclear localization in TR1 KO mice after UVB irradiation. Interestingly, it has been previously reported that UVB does not activate NRF2 in melanocytes as compared to UVA (Marrot et al., 2008). Our data support those findings as we did not see significant NRF2 nuclear localization in WT mice with functional TR1 activity in the presence of UVB. What we observed is further reinforced by similar observations by others regarding TR1’s importance in the development of the T-cell lineage and the normal physiology of myeloid cells(Muri et al., 2020). In that study, mice with conditional TR1 KO were subjected to emergency hematopoiesis via a sublethal dose of lipopolysaccharide and monitored for induced proliferation of myeloid cells. They found that compensatory mechanisms, specifically the glutathione antioxidant system, were overwhelmed and not sufficient to prevent reduced myeloid cell counts in the absence of TR1. What these observations might mean during development is that in the absence of TR1 the furthest migrating melanoblasts are unable to engage in the rapid proliferation needed to sufficiently populate the regions most distant from the neural crest.
Those data also correlate to the DNA damage observed in TR1 KO melanocytes when subjected to a large dose of UVB where we saw a large increase in formation of 8-oxo-dG. This is similar to what has been previously seen in case of TR1 knockout where cells lacking TR1 are sensitive to spikes in oxidative stress with compensatory mechanisms initially being overwhelmed, however, given enough time homeostasis can be regained. Interestingly, it appears that TR1 contributes most towards preventing DNA damage that occurs via oxidative stress as opposed to other UV mediated damage as seen by our results regarding CPDs. We also speculate that TR1 ablation did not inhibit the rate of DNA repair within melanocytes as the decrease in both 8-oxo-dG and CPDs between timepoints was similar in both WT and KO mice.
TR1 contributes to intracellular redox signaling by providing reducing equivalents to its substrate Trx. Redox signaling is activated by many sources of ROS, including production of superoxide by membrane associated NADPH oxidases which can be stimulated by growth receptor activation(Parvez et al., 2018). These ROS cause reversible redox modifications to sensitive cysteine residues in many different proteins, including phosphatases, kinases and transcription factors. Trx can directly participate in reversing redox signals, or by reducing other thiol peroxidases including the peroxiredoxins(Parvez et al., 2018). One redox regulated transcription factor controlled by the TR1/Trx system is NF-κB(Hayashi et al., 1993, Lillig and Holmgren, 2007). It has been shown that the absence of TR1 influences DNA binding activity of NF-κB in myeloid cells (Muri et al., 2020). Reduction of a disulfide bond involving Cys62 of p50 subunit by Trx has been shown to mediate this phenomenon(Matthews et al., 1992).
The TR1/Trx system also has a known function in survival of melanoma cells that is driven by interactions with reduced Trx with apoptosis-stimulating kinase 1 (ASK-1 also known as MAP3K5). A mutation in the Trx binding domain (R256C) of MAP3K5 increases affinity for Trx and increases proliferation and anchorage-independent growth of melanoma cells in culture. The R256C mutation was found in 7 tumors in three different whole exome/genome analyses of a total of 288 melanomas(Prickett et al., 2014).
Finally, there is a separate pathway for melanocyte development that does not include melanoblasts directly migrating from the neural crest. This second wave of melanocytes differentiate from Schwann cell precursors that disassociate from the peripheral nervous system later in development(Adameyko et al., 2009). Melanocytes of this origin mainly populate the extremities and ventral torso, matching where we observed the pigmentation defects in Txnrd1mel−/− mice. A possible mechanism for this developmental pathway could be that TR1 modulate redox status of transcription factors regulating melanocyte differentiation critical for populating and pigmenting the ventral trunk and extremities.
Altogether, the thioredoxin antioxidant system provides a fascinating system to examine redox regulation of physiological processes and our data provides compelling firsthand in vivo evidence for the role of an oxidoreductase in positively regulating skin pigmentation, melanocyte homeostasis, and UV-induced oxidative stress. Future research into TR1’s role in melanomagenesis and disease progression, using an inducible cre model to knockout TR1 at different disease stages, will provide insight into TR1 as a therapeutic target for melanoma treatment in addition to its contribution to melanocyte physiology. Furthermore, the well-studied pigmentation process itself is a useful model to study such redox regulation by providing visually distinct clues into how seemingly disparate biological factors intermingle to establish a phenotype in vivo.
MATERIALS AND METHODS
Mice
Generation of the Txnrd1mel−/− line was done through breeding previously described Txnrd1fl/fl mice(Bondareva et al., 2007) with those harboring Tyr::Cre (Tyr-cre) Delmas et al., 2003). The genotyping primers used are listed in Supplementary Table 1. Mice were housed in our approved University Animal Facility and were kept on 12-hour light cycle and provided food/water ad libitum. All the experiments performed in this study were in accordance with our institution approved Animal Care and Use protocol (ACUP).
Neonatal Epidermal Skin Cell Isolation and Culture
To develop epidermal skin cell cultures, post-natal day 3.5 (P3.5) neonates were sacrificed via decapitation, washed in 70% EtOH, then PBS, and torso skin collected. Separation of the epidermis from dermis was initiated using a 1 hour 45 min sterile-filtered 4mg/mL in PBS dispase II (MilliporeSigma; St. Louis, MO) incubation at 37°C. The epidermis was then peeled away from dermis with forceps, minced with scissors, placed into 0.025% Tyrpsin-0.01% EDTA (ThermoFisher; Waltham, MA), vortexed vigorously for 1 min, and then incubated at 37°C for 10 min with rocking. Trypsinized epidermis was then quenched in DMEM (VWR; Radnor, PA) supplemented with 10% FBS (R&D Systems; Minneapolis, MN). Cells were counted, centrifuged at 300 x g, and then resuspended into melanocyte growth medium consisting of MCDB-153 medium supplemented with 14.05 mM sodium bicarbonate (Fisher Scientific; Pittsburg PA), penicillin-streptomycin 100 U/mL (Thermofisher), amphoteracin B 0.5 ug/mL (ThermoFisher), α-tocopherol 1 μg/mL (Sigma-Aldrich), 4% FBS (Cytiva Life Sciences, Marlborough, MA), 5 μg/mL insulin (Sigma-Aldrich), 0.6 ng/mL bFGF (Sigma-Aldrich), 80 nM TPA (Sigma-Aldrich), 13 μg/mL BPE (ThermoFisher), 260 ng/μL endothelin 3 (Lonza; Basel, Switzerland). Cells were then seeded at 1x105 cells/cm2. The cells were passaged four times before ICC.
Immunocytochemistry
Epidermal skin cell cultures were grown on poly-d-lysine coated coverslips (Neuvitro Corporation; Camas, WA) and then fixed in cold 1:1 Acetone/Methanol. ICC was performed as previously described(Carpenter et al., 2019). A list of the antibodies used overnight at 4°C can been seen in Supplementary Table 2.
Histological Analyses
For all histology experiments formalin-fixed paraffin embedded (FFPE) skin samples were sectioned at 5 μM thickness using an RM2255 microtome (Leica Microsystems; Wetzlar, Germany) affixed with D554X microtome blades (C. L. Sturkey Inc.; Lebanon, PA) onto SuperFrost Plus slides (VWR). Hematoxylin and eosin (H&E) staining was performed as previously described(Carpenter et al., 2018).
Immunohistochemistry
FFPE skin samples were rehydrated, and antigen retrieval performed using citrate buffer (pH 6.0) with a microwave. In sections developed with DAB, melanin was bleached prior to antigen retrieval using 5% H2O2 in PBS in a 60°C water bath for 15 min. Blocking was done at RT in 10% normal goat serum in PBS-T. For primary antibodies of mouse origin, a mouse-on-mouse block was done using Goat α-Mouse IgG (Jackson Immunolabs; West Grove, PA) 1:50 in PBS-T at RT for 1 hour. Sections were washed before and after each blocking step with PBS-T. A list of the primary antibodies used overnight at 4°C as well as secondary antibodies used at RT for 2 hours can been seen in Supplementary Table 2. Pax3 antibody was received from the Developmental Studies Hybridoma Bank (DSHB) and described by Venters et al. (Venters et al., 2004). DAB staining was developed using Streptavidin-Peroxidase R.T.U (Vector Labs; Burlingame, CA) and the DAB Substrate Kit (Vector Labs) in accordance with manufacturer’s instructions. Slides were counterstained at RT with either 5 min hematoxylin for DAB or 10 min 0.2 ng/mL DAPI in PBS-T for fluorescence. Slides were dehydrated and mounted with DPX.
Melanin Quantification
Eumelanin and pheomelanin were quantified as previous described(Ito et al., 2011, Tadokoro et al., 2003). Briefly, left ear biopsies were taken from P64 mice using 4 mm punch biopsy after euthanasia by CO2 inhalation followed by cervical dislocation. In euthanized P21 mice, tail biopsies consisted of a 2 cm section of the tail skin (starting from the tip) collected by making an incision along the tail with a razorblade and peeling the skin from the tail bone. P21 hind paw (pes) biopsies were taken by severing both right and left pes from the rest of the leg just after the ankle joint and then making an incision along the pes in order to peel the skin off like a glove. Collected skin samples were then freeze dried overnight using a FreeZone Plus 6 L (Labconco; Kansas City, MO), and weighed. Samples were then homogenized in 400 μL DI water using a Ten-Broeck homogenizer and 100 μL aliquots subjected to either alkaline hydrogen peroxide oxidation or hydroiodic acid hydrolysis. Determination of eumelanin and pheomelanin content was done via HPLC quantification of markers PTCA and 4-AHP, respectively. For pes skin melanin quantification, the values from right and left rear paws were averaged together for each mouse. For PTCA analysis, the HPLC system consisted of a JASCO 880-PU liquid pump (JASCO Co., Tokyo, Japan), a C18 column (Capcell Pak MG, Osaka Soda, Osaka, Japan) and a JASCO UV detector (JASCO Co.) at 272 nm. For 4-AHP analysis, the HPLC system consisted of a JASCO 880-PU liquid pump (JASCO Co.), a JASCO C18 column (JASCO Catecholpak), and an EC detector SI-2 (3005C, Osaka Soda) at 500 mV.
Neonatal UVB Irradiation
Acute neonatal UVB irradiation was performed as previously described(Chagani et al., 2016). Briefly, male P3.5 neonates were subjected to 350 mJ/cm2 UVB measured with a IL1400A photometer (International Light Technologies; Peabody, MA) from a UV box with four TL-20 W/12RS UV-B bulbs (Philips; Amsterdam, Netherlands).
Histology Imaging and Quantification
Light microscopy was capture using a Leica DM E (Leica Microsystems). Fluorescent microscopy was done using a Zeiss AXIO Imager.Z1 (Carl Zeiss; Oberkochen, Germany). For each biological replicate, triplicate technical replicates were performed and consisted of 10 fields each. Slides were quantified by 2 independent investigators blinded from the experimental groups of the samples.
To quantify NRF2 localization via IHC we used ImageJ (Schneider et al., 2012). First, the dilution for PEP8 (DCT) antibody was decreased from 1:250 to 1:100 to yield enhanced cytoplasmic staining of melanocytes. Following imaging, regions of interest (ROIs) for the DCT+ whole cell and fully encompassed DAPI+ nucleus were manually drawn for each melanocyte in the field with suitable DCT staining. The measure function was used to calculate pixel area, mean grey value, integrated density (IntDen), and RawIntDen (the sum of all grey values in selection) for the ROIs corresponding to whole cell, nuclear, and two background selections drawn in the dermis where minimal cy3 signal was detected. The histogram function was used to calculate total pixel count for whole cell and nuclear ROIs. Cytoplasmic corrected total cellular fluorescence (CTCF) was then back-calculated by subtracting the whole cell ROI values for pixel count, pixel area, and RawIntDen by the corresponding nuclear ROI values.
These new values were then used to calculate cytoplasmic mean grey value and cytoplasmic IntDen. CTCF was then calculated for the nucleus and cytoplasm using the following formula:
After nuclear and cytoplasmic CTCF was determined, the ratio of nuclear to cytoplasmic CTCF was then calculated for each melanocyte. Drawing of ROIs and ImageJ fluorescence quantification was done on unprocessed images. IHC for NRF2 was done in technical duplicates that consisted of 10 fields each.
Software and Statistics
The software for light and fluorescence microscopy image capturing was Leica Application Suite 4 (Leica Microsystems) and AxioVision 4.8 (Carl Zeiss), respectively. Images were processed using Photoshop (Adobe Inc.; San Jose, CA). Quantification of NRF2 IHC CTCF was performed using the ImageJ program ver. 1.53f51 (National Institute of Health; Bethesda MD). Prism 5 (GraphPad Software Inc.; San Diego, CA) was used to conduct statistical analysis. Significance was determined using Student’s two-tailed t-test or one sample t-test.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Indra lab members past and present as well as work done by Gail Kent in the Leachman-Cassidy Lab. We also thank Dr. Lionel Larue from Institut Curie, France for the Tyr-Cre mice used in this study. Research reported in this publication was supported in part by National Institute of Environmental Health Sciences (NIEHS) of the National Institutes of Health (NIH) under the award number 1R01ES016629-01A1 (PI: AI), the OSU/OHSU College of Pharmacy Pilot Project Grant (PI: AI) and Training grant from National Center for Complementary and Integrative Health (NCCIH) of the National Institutes of Health under award number T32AT010131.
Abbreviations
- TR1
thioredoxin reductase 1
- TRX
thioredoxin
- UVB
ultraviolet B
- HF
hair follicle
- MCSC
melanocyte stem cell
- CTCF
corrected total cellular fluorescence
Footnotes
ANIMAL STUDIES
All animal studies were carried out in accordance with the guidelines approved by The Oregon State University Institutional Animal Care and Use Committee (IACUC).
CONFLICT OF INTEREST
The authors state no conflict of interest.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study can be made available upon request by the corresponding author as per institutional guidelines. No datasets were generated or analyzed in this study.
References
- Adameyko I, Lallemend F, Aquino JB, Pereira JA, Topilko P, Muller T, et al. Schwann cell precursors from nerve innervation are a cellular origin of melanocytes in skin. Cell 2009;139(2):366–79. [DOI] [PubMed] [Google Scholar]
- Allouche J, Rachmin I, Adhikari K, Pardo LM, Lee JH, McConnell AM, et al. NNT mediates redox-dependent pigmentation via a UVB- and MITF-independent mechanism. Cell 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balta E, Hardt R, Liang J, Kirchgessner H, Orlik C, Jahraus B, et al. Spatial oxidation of L-plastin downmodulates actin-based functions of tumor cells. Nat Commun 2019;10(1):4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter LL, Watkins-Chow DE, Pavan WJ, Loftus SK. A curated gene list for expanding the horizons of pigmentation biology. Pigment Cell Melanoma Res 2019;32(3):348–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia M, McGrath KL, Di Trapani G, Charoentong P, Shah F, King MM, et al. The thioredoxin system in breast cancer cell invasion and migration. Redox Biol 2016;8:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake JA, Baldarelli R, Kadin JA, Richardson JE, Smith CL, Bult CJ, et al. Mouse Genome Database (MGD): Knowledgebase for mouse-human comparative biology. Nucleic Acids Res 2021;49(D1):D981–D7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissy RE. Melanosome transfer to and translocation in the keratinocyte. Exp Dermatol 2003;12Suppl 2:5–12. [DOI] [PubMed] [Google Scholar]
- Bondareva AA, Capecchi MR, Iverson SV, Li Y, Lopez NI, Lucas O, et al. Effects of thioredoxin reductase-1 deletion on embryogenesis and transcriptome. Free Radic Biol Med 2007;43(6):911–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco V, Godinho-Santos A, Goncalves J, Lu J, Holmgren A, Carvalho C. Mitochondrial thioredoxin reductase inhibition, selenium status, and Nrf-2 activation are determinant factors modulating the toxicity of mercury compounds. Free Radic Biol Med 2014;73:95–105. [DOI] [PubMed] [Google Scholar]
- Cadenas C, Franckenstein D, Schmidt M, Gehrmann M, Hermes M, Geppert B, et al. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res 2010;12(3):R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter EL, Chagani S, Nelson D, Cassidy PB, Laws M, Ganguli-Indra G, et al. Mitochondrial complex I inhibitor deguelin induces metabolic reprogramming and sensitizes vemurafenib-resistant BRAF(V600E) mutation bearing metastatic melanoma cells. Mol Carcinog 2019;58(9):1680–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter EL, Le MN, Miranda CL, Reed RL, Stevens JF, Indra AK, et al. Photoprotective Properties of Isothiocyanate and Nitrile Glucosinolate Derivatives From Meadowfoam (Limnanthes alba) Against UVB Irradiation in Human Skin Equivalent. Front Pharmacol 2018;9:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy PB, Honeggar M, Poerschke RL, White K, Florell SR, Andtbacka RH, et al. The role of thioredoxin reductase 1 in melanoma metabolism and metastasis. Pigment Cell Melanoma Res 2015;28(6):685–95. [DOI] [PubMed] [Google Scholar]
- Cassidy PB, Liu T, Florell SR, Honeggar M, Leachman SA, Boucher KM, et al. A Phase II Randomized Placebo-Controlled Trial of Oral N-acetylcysteine for Protection of Melanocytic Nevi against UV-Induced Oxidative Stress In Vivo. Cancer Prev Res (Phila) 2017;10(1):36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagani S, Kyryachenko S, Yamamoto Y, Kato S, Ganguli-Indra G, Indra AK. In Vivo Role of Vitamin D Receptor Signaling in UVB-Induced DNA Damage and Melanocyte Homeostasis. J Invest Dermatol 2016;136(10):2108–11. [DOI] [PubMed] [Google Scholar]
- Delmas V, Martinozzi S, Bourgeois Y, Holzenberger M, Larue L. Cre-mediated recombination in the skin melanocyte lineage. Genesis 2003;36(2):73–80. [DOI] [PubMed] [Google Scholar]
- Denat L, Kadekaro AL, Marrot L, Leachman SA, Abdel-Malek ZA. Melanocytes as instigators and victims of oxidative stress. J Invest Dermatol 2014;134(6):1512–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esen H, Erdi F, Kaya B, Feyzioglu B, Keskin F, Demir LS. Tissue thioredoxin reductase-1 expression in astrocytomas of different grades. J Neurooncol 2015;121(3):451–8. [DOI] [PubMed] [Google Scholar]
- Funasaka Y, Ichihashi M. The effect of ultraviolet B induced adult T cell leukemia-derived factor/thioredoxin (ADF/TRX) on survival and growth of human melanocytes. Pigment Cell Res 1997;10(1-2):68–73. [DOI] [PubMed] [Google Scholar]
- Gao XY, Li K, Jiang LL, He MF, Pu CH, Kang D, et al. Developmental toxicity of auranofin in zebrafish embryos. J Appl Toxicol 2017;37(5):602–10. [DOI] [PubMed] [Google Scholar]
- Hawkes HJ, Karlenius TC, Tonissen KF. Regulation of the human thioredoxin gene promoter and its key substrates: a study of functional and putative regulatory elements. Biochim Biophys Acta 2014;1840(1):303–14. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Ueno Y, Okamoto T. Oxidoreductive regulation of nuclear factor kappa B. Involvement of a cellular reducing catalyst thioredoxin. J Biol Chem 1993;268(15):11380–8. [PubMed] [Google Scholar]
- Holmgren A, Lu J. Thioredoxin and thioredoxin reductase: current research with special reference to human disease. Biochem Biophys Res Commun 2010;396(1):120–4. [DOI] [PubMed] [Google Scholar]
- Ito S, Nakanishi Y, Valenzuela RK, Brilliant MH, Kolbe L, Wakamatsu K. Usefulness of alkaline hydrogen peroxide oxidation to analyze eumelanin and pheomelanin in various tissue samples: application to chemical analysis of human hair melanins. Pigment Cell Melanoma Res 2011;24(4):605–13. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Feng X, Zheng L, Li SL, Ge XY, Zhang JG. Thioredoxin 1 mediates TGF-beta-induced epithelial-mesenchymal transition in salivary adenoid cystic carcinoma. Oncotarget 2015;6(28):25506–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan SA, Jackson IJ. A late wave of melanoblast differentiation and rostrocaudal migration revealed in patch and rump-white embryos. Mech Dev 2000;92(2):135–43. [DOI] [PubMed] [Google Scholar]
- Li A The biology of melanocyte and melanocyte stem cell. Acta Biochim Biophys Sin (Shanghai) 2014;46(4):255–60. [DOI] [PubMed] [Google Scholar]
- Lillig CH, Holmgren A. Thioredoxin and related molecules--from biology to health and disease. Antioxid Redox Signal 2007;9(1):25–47. [DOI] [PubMed] [Google Scholar]
- Lu SC. Glutathione synthesis. Biochim Biophys Acta 2013;1830(5):3143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrot L, Jones C, Perez P, Meunier JR. The significance of Nrf2 pathway in (photo)-oxidative stress response in melanocytes and keratinocytes of the human epidermis. Pigment Cell Melanoma Res 2008;21(1):79–88. [DOI] [PubMed] [Google Scholar]
- Matthews JR, Wakasugi N, Virelizier JL, Yodoi J, Hay RT. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res 1992;20(15):3821–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mort RL, Jackson IJ, Patton EE. The melanocyte lineage in development and disease. Development 2015;142(7):1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Rover S, Handjiski B, van der Veen C, Eichmuller S, Foitzik K, McKay IA, et al. A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J Invest Dermatol 2001;117(1):3–15. [DOI] [PubMed] [Google Scholar]
- Muri J, Thut H, Feng Q, Kopf M. Thioredoxin-1 distinctly promotes NF-kappaB target DNA binding and NLRP3 inflammasome activation independently of Txnip. Elife 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P, Awwad RT, Smart DD, Spitz DR, Gius D. Thioredoxin reductase as a novel molecular target for cancer therapy. Cancer Lett 2006;236(2):164–74. [DOI] [PubMed] [Google Scholar]
- Nishimura EK, Jordan SA, Oshima H, Yoshida H, Osawa M, Moriyama M, et al. Dominant role of the niche in melanocyte stem-cell fate determination. Nature 2002;416(6883):854–60. [DOI] [PubMed] [Google Scholar]
- Oh JH, Chung AS, Steinbrenner H, Sies H, Brenneisen P. Thioredoxin secreted upon ultraviolet A irradiation modulates activities of matrix metalloproteinase-2 and tissue inhibitor of metalloproteinase-2 in human dermal fibroblasts. Arch Biochem Biophys 2004;423(1):218–26. [DOI] [PubMed] [Google Scholar]
- Parvez S, Long MJC, Poganik JR, Aye Y. Redox Signaling by Reactive Electrophiles and Oxidants. Chem Rev 2018;118(18):8798–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prickett TD, Zerlanko B, Gartner JJ, Parker SCJ, Dutton-Regester K, Lin JC, et al. Somatic mutations in MAP3K5 attenuate its proapoptotic function in melanoma through increased binding to thioredoxin. J Invest Dermatol 2014;134(2):452–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigge JR, Coppo L, Martin SS, Ogata F, Miller CG, Bruschwein MD, et al. Hepatocyte Hyperproliferation upon Liver-Specific Co-disruption of Thioredoxin-1, Thioredoxin Reductase-1, and Glutathione Reductase. Cell Rep 2017;19(13):2771–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffel J, Bhattacharyya AK, Gallegos A, Cui H, Einspahr JG, Alberts DS, et al. Increased expression of thioredoxin-1 in human colorectal cancer is associated with decreased patient survival. J Lab Clin Med 2003;142(1):46–51. [DOI] [PubMed] [Google Scholar]
- Sandbach JM, Coscun PE, Grossniklaus HE, Kokoszka JE, Newman NJ, Wallace DC. Ocular pathology in mitochondrial superoxide dismutase (Sod2)-deficient mice. Invest Ophthalmol Vis Sci 2001;42(10):2173–8. [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9(7):671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seberg HE, Van Otterloo E, Cornell RA. Beyond MITF: Multiple transcription factors directly regulate the cellular phenotype in melanocytes and melanoma. Pigment Cell Melanoma Res 2017;30(5):454–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadokoro T, Kobayashi N, Zmudzka BZ, Ito S, Wakamatsu K, Yamaguchi Y, et al. UV-induced DNA damage and melanin content in human skin differing in racial/ethnic origin. FASEB J 2003;17(9):1177–9. [DOI] [PubMed] [Google Scholar]
- Venters SJ, Argent RE, Deegan FM, Perez-Baron G, Wong TS, Tidyman WE, et al. Precocious terminal differentiation of premigratory limb muscle precursor cells requires positive signalling. Dev Dyn 2004;229(3):591–9. [DOI] [PubMed] [Google Scholar]
- Wall SB, Wood R, Dunigan K, Li Q, Li R, Rogers LK, et al. Thioredoxin Reductase-1 Inhibition Augments Endogenous Glutathione-Dependent Antioxidant Responses in Experimental Bronchopulmonary Dysplasia. Oxid Med Cell Longev 2019;2019:7945983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Chen Y, Bai M, Liu Y, Xu B, Sun R, et al. The antimetastatic effect and underlying mechanisms of thioredoxin reductase inhibitor ethaselen. Free Radic Biol Med 2019;131:7–17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study can be made available upon request by the corresponding author as per institutional guidelines. No datasets were generated or analyzed in this study.