

Abstract
CYP2A6, a genetically variable enzyme, inactivates nicotine, activates carcinogens, and metabolizes many pharmaceuticals. Variation in CYP2A6 influences smoking behaviors and tobacco-related disease risk. This phenome-wide association study examined associations between a reconstructed version of our weighted genetic risk score (wGRS) for CYP2A6 activity with diseases in the UK Biobank (N = 395 887). Causal effects of phenotypic CYP2A6 activity (measured as the nicotine metabolite ratio: 3′-hydroxycotinine/cotinine) on the phenome-wide significant (PWS) signals were then estimated in two-sample Mendelian Randomization using the wGRS as the instrument. Time-to-diagnosis age was compared between faster versus slower CYP2A6 metabolizers for the PWS signals in survival analyses. In the total sample, six PWS signals were identified: two lung cancers and four obstructive respiratory diseases PheCodes, where faster CYP2A6 activity was associated with greater disease risk (Ps < 1 × 10−6). A significant CYP2A6-by-smoking status interaction was found (Psinteraction < 0.05); in current smokers, the same six PWS signals were found as identified in the total group, whereas no PWS signals were found in former or never smokers. In the total sample and current smokers, CYP2A6 activity causal estimates on the six PWS signals were significant in Mendelian Randomization (Ps < 5 × 10−5). Additionally, faster CYP2A6 metabolizer status was associated with younger age of disease diagnosis for the six PWS signals (Ps < 5 × 10−4, in current smokers). These findings support a role for faster CYP2A6 activity as a causal risk factor for lung cancers and obstructive respiratory diseases among current smokers, and a younger onset of these diseases. This research utilized the UK Biobank Resource.
Keywords: UK Biobank, nicotine metabolism, CYP2A6 weighted genetic risk score, PheWAS, lung cancer
Introduction
Cytochrome P450 2A6 (CYP2A6) is a genetically polymorphic enzyme that metabolically inactivates nicotine, activates tobacco-specific nitrosamine carcinogens, and is associated with numerous smoking behaviors [1–3]. CYP2A6 metabolizes many drugs including tegafur, letrozole, and ketamine [4, 5]. CYP2A6 has numerous star (*) variant alleles (described in PharmVar) [3]. Single nucleotide polymorphisms (SNPs), in addition to hybrid, duplicated, and deleted CYP2A6 alleles, alter CYP2A6 activity [6, 7]. Genome-wide association studies (GWASs) have increased our understanding of the SNPs-captured variability in CYP2A6 activity [8, 9].
A well-established robust biomarker of CYP2A6 activity is the ratio of 3′-hydroxycotinine/cotinine: the nicotine metabolite ratio (NMR) [10, 11]. Nicotine is oxidized to cotinine then to 3′-hydroxycotinine by CYP2A6 [11]; the latter is mediated exclusively by CYP2A6 [1]. The NMR in regular smokers has low temporal fluctuation due to the long half-life of cotinine (~16–19 h) and the formation dependent kinetics of 3′-hydroxycotinine [11, 12]. The NMR is highly correlated with nicotine clearance [11, 13]; as a result, the NMR is associated with smoking behavior. Faster CYP2A6 metabolizers (measured using the NMR or CYP2A6 genomics) generally smoke more cigarettes per day (CPD), smoke more intensely, inhale more deeply, have shorter time to first cigarette, are more dependent on nicotine, have lower unaided quit rates, and have altered response to cessation therapies [14–18]. Moreover, faster CYP2A6 activity is associated with increased lung [19, 20] and head and neck cancers risk [21].
We developed weighted CYP2A6 genetic risk scores (wGRSs) in smokers [22, 23]. The European ancestry wGRS incorporates seven CYP2A6 variants and explains 33.8% of NMR variation [22], while the African ancestry wGRS incorporates 11 variants and explains 32.4% of NMR variation [23]. The wGRSs also predict nicotine intake, smoking quantity, and response to cessation therapies [22, 23] and are particularly useful as the NMR cannot be calculated in non-regular or non-smokers, nor in biobanks where biomarker measurement is not feasible [11, 22].
Large-scale biobanks rich in phenotypic and genotypic data has enabled novel studies of disease risk. Phenome-wide association studies (PheWASs) use genotypic markers to identify associated phenotypes, including diseases, in a hypothesis-free manner [24]. The causal links of these phenotype-genotype associations can then be explored using Mendelian Randomization (MR), a causal inference approach which is less susceptible to reverse causation [25].
A previous PheWAS involving a single CYP2A6 variant (rs113288603C > T) found an association with reduced hearing loss symptoms in women aged > 60 years who had smoked > 100 cigarettes (lifetime) [26]. This study was small, used a specific population, and incorporated limited variation in CYP2A6. The current PheWAS aimed to examine CYP2A6 wGRS associations with diseases in the UK Biobank (N ~ 500 000). The top PheWAS associations were then evaluated in 1) MR analyses to estimate causal effects of CYP2A6 variation, and 2) survival analyses of age at diagnosis in faster versus slower CYP2A6 metabolizers.
Materials and Methods
Study population
The main dataset was the UK Biobank [27]. The data for 502 505 individuals was accessed in 2020; we restricted analyses to those of European ancestry with genotype information (N = 395 887). The second dataset (the NMR dataset) included treatment-seeking smokers (≥ 10 CPD) from a clinical trial (described elsewhere (NCT01314001)) [28, 29]. This NMR dataset was used for validation of the reconstructed CYP2A6 wGRS and in MR analyses. We restricted analyses to those of European ancestry that had NMR and genotyping information (N = 922).
CYP2A6 weighted genetic risk score and genotyping sources
The wGRS construction from the NMR in European ancestry smokers is described in detail elsewhere [22]. Briefly, seven variants were selected from independent signals identified from conditional analyses in a GWAS of the NMR (i.e. CYP2A6 activity biomarker) and from common CYP2A6 functional alleles [8, 22]. These variants were tested in an additive model from which the effect alleles’ weights were multiplied by the standard deviation of the NMR. The number of alleles multiplied by the allele’s respective weight was summed to give an overall score in which a higher score is indicative of faster CYP2A6 activity [30]. The wGRS scale was constructed to be able to match the Clinical Pharmacogenetics Implementation Consortium guidelines for CYP activity scores, and to replicate previous metabolizer groupings [22]. For example, an individual without a function-altering allele would have a score of 2.0.
Genotyping, imputation, and quality control methods for the UK Biobank sample are described elsewhere [27, 31]. From the original seven variants in the European ancestry wGRS [22], three were available in the UK Biobank (rs56113850, rs1801272, and rs28399442 (CYP2A6*12)). Proxy variants rs2644891, rs61663607, and rs76112798 were used to replace rs2316204, rs113288603, and rs28399433 (CYP2A6*9), respectively. These proxy variants were chosen based on their highest linkage disequilibrium (R2 > 0.80) with their respective original variants in European ancestry individuals (determined by NIH’s LDlink open-source tool [32]), and each proxy variant had similar weights per allele on the NMR (Table 1). CYP2A6*4 was not available in the UK Biobank’s genotyping or imputation data. One variant, rs28399442, was directly genotyped in the UK Biobank, while the remaining variants were extracted from the third version of the imputation data [27]. Imputed variants passed standard quality control procedures in the UK Biobank with imputation quality scores 0.92–0.99.
Table 1.
CYP2A6 wGRS variants’ effect sizes on the phenotypic biomarker in the NMR dataset.
| CYP2A6 wGRS variants (El-Boraie et al. [22]) | Effect Allele | Other Allele | EAF in the NMR dataset | Weight per effect allele | Variants/proxy variants available in the UK Biobank | Effect Allele | Other Allele | EAF in the NMR dataset | EAF in the UK Biobank | Weight per effect allele | LD R2 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs56113850 | C | T | 0.562 | 0.135 | rs56113850 | C | T | 0.562 | 0.575 | 0.135 | 0.814 |
| rs2316204 | T | C | 0.649 | 0.080 | rs2644891 | C | T | 0.674 | 0.680 | 0.079 | 0.988 |
| rs113288603 | T | C | 0.087 | −0.025 | rs61663607 | C | T | 0.087 | 0.080 | −0.025 | 0.956 |
| *9 (rs28399433) | C | A | 0.063 | −0.159 | rs76112798 | T | C | 0.064 | 0.064 | −0.160 | – |
| *2 (rs1801272) | T | A | 0.025 | −0.250 | rs1801272 | T | A | 0.025 | 0.022 | −0.250 | – |
| *12 | CYP2A6/2A7 hybrid | – | 0.022 | −0.272 | rs28399442a | A | C | 0.022 | 0.021 | −0.249 | – |
| *4 | (CYP2A6 Deletion)b | – | 0.009 | −0.350 | – | – | – | – | – | – | – |
wGRS: weighted Genetic Risk Score; NMR: Nicotine Metabolite Ratio; EAF: Effect Allele Frequency; UK: United Kingdom; LD: Linkage Disequilibrium. R2 was obtained from NIH’s LDlink open-source tool to determine the degree of concordance between proxy (UK Biobank) variants and original variants included in the CYP2A6 wGRS.
aConcordance confirmed by PCR assay (Bloom et al. [76]).
bNot available in genotyped or imputed UK Biobank data.
Phenotypes sources
Health outcomes encoded as International Classification of Diseases (ICD), Ninth and Tenth Revisions codes in the UK Biobank, were mapped to the clinically representative phenotype codes (PheCodes) to collapse the granularity of ICD codes into focused diagnostic codes for research [33, 34] using the “createUKBPhenome” workflow. From 2973 ICD-9 and 7030 ICD-10 codes detected in the UK Biobank sample, 1692 PheCodes were generated after exclusion of sex-mismatched sex-specific diagnoses. After filtering out PheCodes with < 200 cases, 1029 PheCodes were included in the PheWAS.
Smoking status, age at recruitment, genetic sex, CPD, pack-years, and the first 10 genetic principal components (PCs) were extracted for use as covariates or for stratification. Smoking status was identified by self-report (current, former, or never smokers) at initial assessment. Genetic sex and PCs were generated by the UK Biobank quality control team [27, 31]. CPD originated from the touchscreen question “About how many cigarettes do you smoke on average each day?”. Pack-years was calculated in UK Biobank individuals who have ever smoked, using the following equation: Number of cigarettes per day/20 * (Age stopped smoking—Age start smoking).
Phenome-wide association study analyses
CYP2A6 wGRS associations with disease PheCodes were determined using logistic regression in “glm” function in R (v3.5.3) [35] in the total analytic sample (N = 395 887). The wGRS was not transformed to maintain the scale, and since the wGRS was used as the predictor in logistic regressions analyses, which are relatively robust to violations of the normality assumption for predictor variables. We adjusted the final ORs by exponentiating them to the power of 1 SD of the wGRS so that a unit change in the wGRS would correspond to 1 SD. Covariates included in all analyses were age at recruitment and the first 10 PCs. Genetic sex and smoking status were also included as covariates in analyses not stratified by these variables. We also performed a PheWAS stratified by smoking status (39 940 current, 139 292 former, and 215 274 never smokers) and further stratified current smokers by sex. Additionally, we conducted separate exploratory PheWAS in current smokers smoking ≤ 20 CPD. Manhattan plots were constructed using the “PheWAS” R package [24]. Signals surviving Bonferroni corrections (P < 0.05/the number of PheCodes) in each PheWAS were considered significant at the phenome-wide level, and referred to as phenome-wide significant (PWS) signals, as is convention [24, 36]. Signals surviving a False Discovery Rate (FDR) threshold of 5% using the Benjamini-Hochberg’s procedure (P < 0.05*the rank of association/the number of PheCodes) [37] in each PheWAS were considered nominal associations.
Mendelian Randomization of the PWS signals
The PWS signals were analyzed in two-sample MR to evaluate causal effects of faster CYP2A6 activity. The first sample was European ancestry smokers (N = 922) from the NMR dataset [28, 29]; this is the same sample that was used to derive the reconstructed wGRS variant weights, as described above. Linear regression was used to derive the instrument-exposure estimate in the NMR dataset. The second sample was current smokers of European ancestry from the UK Biobank (N = 39 940). Logistic regression was used to derive the instrument-outcome estimates in the UK Biobank. The Wald ratio estimator method in the “MendelianRandomization” R package was used to determine the causal estimates of the NMR—the CYP2A6 phenotypic biomarker as the exposure—effect on the PWS signals [38]. The wGRS was used as a single instrument; hence, heterogeneity assessment was not needed. MR Egger regression analyses were used to evaluate horizontal pleiotropy of the wGRS variants as individual instruments. A significant deviation in the MR Egger intercept would indicate potential horizontal pleiotropy (i.e. the variants may be influencing the outcome via a route other than CYP2A6). Leave-one-out sensitivity analysis was performed to assess the influence of each variant in the wGRS on the top PWS signal using the “mr_leaveoneout_plot” function in the “TwoSampleMR” R package [39, 40]. In this process, each variant was removed iteratively from the model and the fluctuation in MR estimates was assessed. Fluctuations would indicate the influence of that variant on the causal estimates. Steiger filtering was performed to ensure the validity of the variants comprising the wGRS and limit reverse causal bias for the top PWS signal using the “steiger_filtering” function in the “TwoSampleMR” R package [39, 40].
Overlapping diagnoses
The overlap between disease-positive cases for the PWS signals was examined. A combination of Euler and UpSet plots were used to highlight overlapping patterns in cases identified for the PWS signals in current smokers. For the PWS signals, the number of individuals with overlapping diagnoses was visualized using the “venneuler” and “UpSetR” R packages [41, 42].
Disease risk and survival analysis for age at diagnosis
Due to skewness in the wGRS distribution and to enhance clinical interpretation, we also ran a sensitivity analysis on the top PWS signals using a cut-point previously determined by the Youden index J statistic, aligning with the smoking cessation clinically verified NMR phenotype cut-point of 0.31 (slower: wGRS < 2.14; vs faster: wGRS ≥ 2.14 metabolizers) [22]. For survival analyses, time-to-event was used with age in years as the time and the first report of a disease diagnosis as the event. Survival time was defined as the age when the individual who smoked had a disease diagnosis or the end-of-follow-up (whichever came first) defined as the year when the data were accessed (2020). Individuals that did not attain the outcome (i.e. had no disease diagnosis) by 2020 were censored and had their age in 2020 recorded as T1. We employed the Kaplan Meier method to estimate survival curves according to CYP2A6 metabolizer group status, using a wGRS cut-point of 2.14. Cox proportional hazards models were used to estimate the hazard ratios for the associations between CYP2A6 metabolizer group and time to disease diagnosis. Cox proportional hazards models were estimated using the “survival” R package v3.2-7 [43].
Results
CYP2A6 weighted genetic risk score
We computed the CYP2A6 wGRS for all UK Biobank participants from available genotypes for the wGRS variants (or proxy variants, see methods). The reconstructed six-variant wGRS (Table 1) explained 30.1% of log-transformed NMR variance in the European ancestry sample from which the score was derived (N = 922), similar to the 33.8% captured using the original seven-variant wGRS [22] (allele frequencies are in Supplementary Table 1). The reconstructed six-variant wGRS mean was 2.22 ± 0.18 (N = 395 857), compared to 2.20 ± 0.19 for the original seven-variant wGRS [22].
Phenome-wide association study
Phenome-wide associations were determined through logistic regression using the reconstructed CYP2A6 wGRS (untransformed continuous score) as predictor. Diseases surpassing Bonferroni and FDR thresholds were deemed PWS signals and nominal associations, respectively (see methods). In our total analytic sample, six PWS signals were detected across two disease clusters after adjusting for age, sex, 10 genetic principal components (PCs), and smoking status (Fig. 1A, Table 2, Supplementary Table 2). The two signals in neoplasms were lung cancers (cancer of bronchus (lung), and cancer within the respiratory system), while the four signals in respiratory diseases were obstructive respiratory diseases (emphysema, obstructive chronic bronchitis, chronic bronchitis, and chronic airway obstruction). Three additional nominal associations included abnormal findings examination of lungs, and two iron deficiency anemias PheCodes. Without adjustment for smoking status in the total sample, there were three PWS signals (cancer of bronchus (lung), cancer within the respiratory system, and emphysema), and six additional nominal associations (obstructive chronic bronchitis, chronic bronchitis, tobacco use disorder, and chronic airway obstruction, and two iron deficiency anemias (Fig. 1B, Table 2, Supplementary Table 2)).
Figure 1.

Neoplasms and respiratory diseases clusters associated with CYP2A6 wGRS in the total UK Biobank sample. Manhattan plots for PheWAS results adjusted for age at recruitment, genetic sex, 10 PCs, with (A); and without (B) smoking status (current vs former vs never-smokers). Each data point represents one phenotype in its respective disease category on the x-axis plotted against the association significance (−log10 P). The horizontal solid lines represent the phenome-wide significant threshold using Bonferroni correction (P = 0.05/1029 = 4.86 × 10−5), while the blue horizontal dashed lines represent the nominal association thresholds (5% FDR equivalent to P = 4.37 × 10−4). PheWAS sample (N = 305 224–395 887); cases and controls sample sizes are found in Supplementary Table 2.
Table 2.
Top phenome-wide signals in main analysis and their respective effect sizes across conditions.
| Phenotype | ORa | 95% CI lowera | 95% CI uppera | P | N Cases | N Controls |
|---|---|---|---|---|---|---|
| All adjusted for smoking status b | ||||||
| Cancer of bronchus (lung) | 1.114 | 1.073 | 1.157 | 1.78E−08 | 2926 | 391 072 |
| Emphysema | 1.127 | 1.079 | 1.176 | 5.56E−08 | 2236 | 356 292 |
| Cancer within the respiratory system | 1.100 | 1.062 | 1.139 | 8.55E−08 | 3397 | 391 072 |
| Obstructive chronic bronchitis | 1.055 | 1.034 | 1.076 | 1.70E−07 | 10 649 | 356 292 |
| Chronic bronchitis | 1.052 | 1.032 | 1.074 | 4.24E−07 | 10 856 | 356 292 |
| Chronic airway obstruction | 1.046 | 1.028 | 1.065 | 8.40E−07 | 13 261 | 356 292 |
| All unadjusted for smoking | ||||||
| Cancer of bronchus (lung) | 1.099 | 1.059 | 5.633 | 6.79E−07 | 2945 | 392 431 |
| Emphysema | 1.108 | 1.062 | 10.633 | 2.56E−06 | 2252 | 357 462 |
| Cancer within the respiratory system | 1.087 | 1.050 | 6.633 | 2.31E−06 | 3419 | 392 431 |
| Obstructive chronic bronchitis | 1.039 | 1.019 | 7.633 | 1.39E−04 | 10 747 | 357 462 |
| Chronic bronchitis | 1.036 | 1.017 | 8.633 | 2.92E−04 | 10 956 | 357 462 |
| Chronic airway obstruction | 1.032 | 1.014 | 9.633 | 4.19E−04 | 13 372 | 357 462 |
| Current smokers | ||||||
| Cancer of bronchus (lung) | 1.222 | 1.144 | 1.306 | 2.97E−09 | 1007 | 38 856 |
| Emphysema | 1.170 | 1.090 | 1.257 | 1.40E−05 | 847 | 33 802 |
| Cancer within the respiratory system | 1.206 | 1.131 | 1.285 | 9.53E−09 | 1081 | 38 856 |
| Obstructive chronic bronchitis | 1.098 | 1.060 | 1.137 | 1.65E−07 | 3802 | 33 802 |
| Chronic bronchitis | 1.095 | 1.058 | 1.134 | 2.90E−07 | 3846 | 33 802 |
| Chronic airway obstruction | 1.089 | 1.053 | 1.126 | 5.71E−07 | 4179 | 33 802 |
| Former smokers | ||||||
| Cancer of bronchus (lung) | 1.072 | 1.016 | 1.130 | 1.12E−02 | 1423 | 137 624 |
| Emphysema | 1.090 | 1.027 | 1.156 | 4.44E−03 | 1161 | 123 841 |
| Cancer within the respiratory system | 1.066 | 1.015 | 1.120 | 1.11E−02 | 1654 | 137 624 |
| Obstructive chronic bronchitis | 1.051 | 1.022 | 1.081 | 5.57E−04 | 5195 | 123 841 |
| Chronic bronchitis | 1.050 | 1.021 | 1.080 | 6.16E−04 | 5270 | 123 841 |
| Chronic airway obstruction | 1.044 | 1.017 | 1.071 | 1.16E−03 | 6312 | 123 841 |
| Ever smokers c | ||||||
| Cancer of bronchus (lung) | 1.109 | 1.065 | 1.156 | 8.55E−07 | 2430 | 176 468 |
| Emphysema | 1.100 | 1.051 | 1.150 | 3.86E−05 | 2008 | 157 632 |
| Cancer within the respiratory system | 1.099 | 1.057 | 1.143 | 1.92E−06 | 2735 | 176 468 |
| Obstructive chronic bronchitis | 1.048 | 1.026 | 1.071 | 2.14E−05 | 8997 | 157 632 |
| Chronic bronchitis | 1.047 | 1.024 | 1.069 | 3.28E−05 | 9116 | 157 632 |
| Chronic airway obstruction | 1.042 | 1.021 | 1.063 | 7.11E−05 | 10 491 | 157 632 |
| Never smokers | ||||||
| Cancer of bronchus (lung) | 1.043 | 0.954 | 1.141 | 3.53E−01 | 496 | 214 592 |
| Emphysema | 1.154 | 1.008 | 1.322 | 3.83E−02 | 228 | 198 649 |
| Cancer within the respiratory system | 1.032 | 0.955 | 1.114 | 4.28E−01 | 662 | 214 592 |
| Obstructive chronic bronchitis | 0.983 | 0.937 | 1.032 | 4.89E−01 | 1652 | 198 649 |
| Chronic bronchitis | 0.980 | 0.935 | 1.027 | 3.95E−01 | 1740 | 198 649 |
| Chronic airway obstruction | 0.996 | 0.960 | 1.035 | 8.49E−01 | 2770 | 198 649 |
OR, Odds ratio from the logistic regression estimates of the wGRS effects on each phenotype; CI, Confidence interval; P, significance value of the wGRS estimate; N, sample Number as cases and controls. Italicized text indicates different groups. All analyses are adjusted for age at recruitment, genetic sex, and first ten genetic principal components.
aThe OR was calculated as the exponent of the regression beta, the 95% CI lower was calculated as: exp(beta − 1.96*SE); while the 95% CI upper was calculated as: exp(beta + 1.96*SE). The ORs and CIs were then transformed by exponentiating them to the power of 1 SD of the wGRS.
bN = 1435 excluded for ‘prefer not say’ in smoking status.
cEver smokers defined by combining current and former smokers’ groups.
Since all six PWS signals were smoking-related diseases, as variation in CYP2A6 activity is associated with smoking [14–18], and as we found a significant interaction between smoking status and the wGRS on disease risk (Fig. 2, Supplementary Table 3), we next stratified by smoking status. In current smokers, the same six PWS signals in the total analytical sample (adjusted for smoking status) were observed, albeit with larger estimates and lower P values in current smokers (except emphysema (Fig. 3A, Table 2, Supplementary Tables 2 and 4). After adjusting the current smokers PheWAS for pack-years or CPD, the estimates for the top six PWS signals were weaker, suggesting a role of smoking in the CYP2A6 effect on these diseases (Supplementary Tables 5 and 6). Nominal associations found in current smokers included constipation, other symptoms of respiratory system, secondary malignant neoplasm, appendiceal conditions, respiratory insufficiency, calculus of ureter, and abnormal findings examination of lungs.
Figure 2.

Smoking modifies the CYP2A6 wGRS effects on disease risk. Smoking status (current, former, versus never smokers) interactions plotted as predicted disease probability with confidence intervals for the six phenome-wide significant signals: (A) cancer of bronchus (lung), (B) obstructive chronic bronchitis, (C) cancer within the respiratory system, (D) chronic airway obstruction, (E) emphysema, and (F) chronic bronchitis. Interaction term significance denoted as P < 0.001 ‘***’, <0.01 ‘**’, <0.05 ‘*’). The never smokers group is the reference. Interaction sample (N = 358 528–394 469); cases and controls sample sizes are found in Supplementary Table 2.
Figure 3.

Smoking status stratified PheWAS suggesting a role of smoking on disease risk. Manhattan plots for PheWAS results in current smokers (A); former smokers (B); and never smokers (C) adjusting for age at recruitment, genetic sex, and 10 PCs. Each data point represents one phenotype in its respective disease category on the x-axis plotted against the association significance (−log10 P). The horizontal solid lines represent the phenome-wide significant threshold using Bonferroni correction (P = 0.05/1029 = 4.86 × 10−5), while the blue horizontal dashed lines represent the nominal association thresholds (5% FDR equivalent to P = 6.80 × 10−4 for (A); 4.86 × 10−5 for (B) and (C)). Constipation surpassed the nominal association threshold in current smokers but was omitted by the plotting software. PheWAS sample (N = 27 822–215 274); cases and controls sample sizes are found in Supplementary Table 2.
In former and never smokers, no PWS signals or nominal associations were found (Fig. 3B and C, Table 2 and Supplementary Table 2). Findings in ever smokers (i.e. current and former smokers together) were similar to those in current smokers (Supplementary Table 2).
Disease risk, CYP2A6 activity, and smoking behaviors vary by sex [28]. Since the strongest PWS signals were identified in current smokers, we further stratified current smokers by sex to discover potential sex-specific novel associations and sex-differentiated effects of CYP2A6 on the six PWS signals identified. In female current smokers (CPD mean ± SD = 14.32 ± 7.28), two PWS signals were detected (Fig. 4A, Supplementary Table 7). The top PWS signal, also seen in current smokers, was cancer of bronchus (lung). Constipation was the second PWS signal; as constipation is currently not included in the PheWAS R package, it is not depicted on the Manhattan plot. There was a significant sex-by-wGRS interaction effect only on constipation in current smokers (ORinteraction = 0.883; 95% CI = 0.800–0.975; P = 0.014).
Figure 4.

Lung-related cancers among the top CYP2A6 wGRS PheWAS signals in both female, and male current smokers. (A) Female only PheWAS. (B) Male only PheWAS. Sex-stratified PheWAS adjusted for age at recruitment, and 10 PCs. Each data point represents one phenotype in its respective disease category on the x-axis plotted against the association significance (−log10 P). The horizontal solid lines represent the phenome-wide significant (PWS) threshold using Bonferroni correction (P = 0.05/934 = 5.35 × 10−5 for (A); and 0.05/834 = 5.60 × 10−5 for (B)), while the blue horizontal dashed lines represent the nominal association thresholds (5% FDR equivalent to P = 4.28 × 10−4 for (A); 3.00 × 10−4 for (B)). Constipation surpassed the PWS threshold in females but was omitted by the plotting software. PheWAS sample (N = 13 640–21 494); cases and controls sample sizes are found in Supplementary Table 7.
In male current smokers (CPD mean ± SD = 17.49 ± 9.20), two PWS signals were detected (Fig. 4B, Supplementary Table 7). The top PWS signal, also seen in the total analytic sample and current smokers, was cancer within the respiratory system, followed by diseases of the larynx and vocal cords. There was a significant sex-by-wGRS interaction effect only on diseases of the larynx and vocal cords in current smokers (ORinteraction = 1.284; 95% CI = 1.078–1.530; P = 0.005).
In a case–control study, CYP2A6 variation (four alleles assessed) was associated with lung cancer risk, an effect that appeared stronger in lighter smokers (≤ 20 CPD) compared to the total sample, likely as heavier smoking overshadows CYP2A6 effects [44]. In current smokers, we explored whether the six PWS signals were more pronounced in relatively lighter smokers (≤ 20 CPD). Slightly higher ORs were observed in lighter smokers (Supplementary Fig. 1), consistent with the direction previously observed [44].
PheWAS model diagnostics
The wGRS distribution was negatively skewed but did not differ by smoking status (Supplementary Fig. 2). We ran model diagnostics on the top PWS signal in current smokers. The wGRS displayed a linear pattern against the logit function of the PWS signal, albeit with high variability (Supplementary Fig. 3). In the model adjusted for age, sex, and 10 PCs, we found some evidence of heteroscedasticity detected from the funneling shape of the binned residuals and a number of points with a standardized residual values >3 (Supplementary Fig. 4).
Mendelian Randomization of PWS signals
We confirmed the impact of CYP2A6 activity on disease outcomes using MR. We used a single-instrument approach with Wald ratio estimator to test for causation in two-sample MR. The instrument was the CYP2A6 wGRS as a continuous score, the exposure was the NMR (CYP2A6 activity phenotype), and the outcomes were the six PWS signals. Estimates for all six PWS signals in current smokers were significant in the MR, suggesting faster CYP2A6 activity may causally increase the risk for lung cancers and obstructive respiratory diseases (Fig. 5). The MR estimates in the total sample were also significant, albeit weaker (Supplementary Fig. 5). We did not detect evidence of horizontal pleiotropy; the intercepts of MR Egger were not significant (P > 0.1). Leave-one-out sensitivity analysis for the top PheWAS signal suggested the estimates were not driven by any individual CYP2A6 variant. Omitting rs2644891, rs61663607, rs76112798, rs28399442, or rs56113850 individually rendered the MR model insignificant, suggesting that each of these variants is important in the effect of CYP2A6 activity on the outcome (Supplementary Fig. 6). In contrast, omitting CYP2A6*2 (rs1801272) did not weaken the significance of the estimate, suggesting a weak influence of this variant on the CYP2A6 effect on the outcome. As expected, rs56113850—the top GWAS signal for the NMR [8] used to construct the wGRS [22]—had the strongest influence on the MR model. We used six CYP2A6 variants in our wGRS and only one was weak; thus, our MR analyses were unlikely to suffer from weak instrument bias [45, 46]. All wGRS variants passed Steiger filtering indicating they are more strongly associated with the exposure than the outcome (Supplementary Table 8), further supporting instrument validity and direction of causation. The F-statistic for the wGRS and each of the variants included were all >10 (Supplementary Table 9). We also identified a putatively causal association between iron deficiency anemia and faster CYP2A6 activity (OR = 1.071; 95%CI: 1.033–1.113), but no smoking-by-wGRS interaction effect (wGRS-by-smoking status: ORinteraction = 1.001; 95% CI = 0.973–1.030; P = 0.927, wGRS-by-CPD: ORinteraction = 0.976; 95% CI = 0.987–1.004; P = 0.313).
Figure 5.
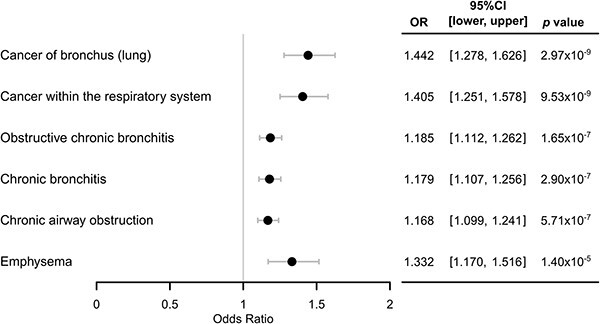
Significant MR causal estimates of faster CYP2A6 activity on the PWS signals in current smokers. Two-sample MR using the NMR dataset as the first sample to determine instrument-exposure (i.e. wGRS-NMR) effects adjusting for age, genetic sex, and the first four genetic principal components; and current smokers of the UK Biobank as the second sample to determine instrument-outcome (i.e. wGRS-PWS signal) effects adjusting for age, genetic sex, and the first 10 genetic principal components. OR: Odds ratio of MR estimates; CI: Confidence intervals of the odds ratio. Ordered from top to bottom by significance in current smokers stratified PheWAS (Fig. 3A). Using the total analytic sample of the UK Biobank as the second sample also resulted in significant MR causal estimates with some dilution of the effects (Supplementary Fig. 5).
Overlapping diagnoses
We examined the overlap in the six PWS signals cases in current smokers. For the two neoplasms, cancer of bronchus (lung), and cancer within the respiratory system, there was nearly 100% overlap. For the four obstructive respiratory diseases, the largest overlap was between obstructive chronic bronchitis, chronic bronchitis, and chronic airway obstruction, which are nested categories; emphysema had less overlap and appeared as a slightly more distinct cluster. Between the top signal of each of the two main clusters, 28% of cases of cancer of bronchus (lung) overlapped with obstructive chronic bronchitis, suggesting shared underlying mechanisms (Fig. 6).
Figure 6.

Overlapping number of cases in the top phenotypes associated with CYP2A6 wGRS in current smokers. Euler diagram circle sizes are reflective of sample size with each phenotype. Each vertical bar in the UpSet plot represents the overlap of cases exclusively shared between the phenotypes marked by the closed black circles connected by the black solid lines.
Faster CYP2A6 metabolizers: Disease risk and age of disease
To improve clinical interpretability, we dichotomized the wGRS using a cut-point of 2.14 (based on the NMR 0.31 clinical cut-point which predicts smoking level and smoking cessation success) [22]. In current smokers, faster metabolizers (wGRS ≥ 2.14) had a significantly higher risk to develop lung cancers and obstructive respiratory diseases compared to slower metabolizers (wGRS < 2.14) (Table 3).
Table 3.
PWS signals in current smokers regressed against dichotomized CYP2A6 wGRS (slower metabolizer group <2.14 is the reference group).
| Phenotype | ORa | 95% CI lowera | 95% CI uppera | P |
|---|---|---|---|---|
| Cancer of bronchus (lung) | 1.592 | 1.374 | 1.843 | 5.68E−10 |
| Cancer within the respiratory system | 1.560 | 1.347 | 1.805 | 2.64E−09 |
| Obstructive chronic bronchitis | 1.236 | 1.146 | 1.332 | 3.42E−08 |
| Chronic bronchitis | 1.249 | 1.155 | 1.350 | 2.59E−08 |
| Chronic airway obstruction | 1.238 | 1.148 | 1.335 | 2.92E−08 |
| Emphysema | 1.324 | 1.130 | 1.551 | 5.06E−04 |
aThe OR was calculated as the exponent of the regression beta, the 95% CI lower was calculated as: exp(beta—1.96*SE); while the 95% CI upper was calculated as: exp(beta + 1.96*SE).
Next, we used survival analyses to investigate the association between CYP2A6 activity and time to acquiring a diagnosis of cancer of bronchus (lung) and obstructive chronic bronchitis (Fig. 7), the top PWS signal from each cluster (Fig. 6). We selected survival analysis for its unique capabilities in handling time-to-event data, thus capturing the dynamic nature of the risk factor. Those in the CYP2A6 faster, versus slower metabolizer group, had an increased risk of a younger age of cancer of bronchus (lung) and obstructive chronic bronchitis diagnosis (Fig. 7). The other four PWS signals showed similar patterns (Supplementary Fig. 7, Supplementary Table 10).
Figure 7.

Current smokers with a faster CYP2A6 metabolizer status were at a higher risk for a younger age of diagnosis. Kaplan-Meier curves for: (A) cancer of bronchus (lung), and (B) obstructive chronic bronchitis. Faster metabolizers were defined as those with a genetic risk score of ≥2.14 while slower metabolizers were defined as those with a genetic risk score of <2.14. Number at risk were participants remaining in the study at each time point who did not yet attain a diagnosis nor were censored. No covariates were used in this analysis.
Discussion
This study is the largest CYP2A6 PheWAS and MR study conducted to date and the first to utilize a comprehensive genetic score for CYP2A6. We present novel evidence supporting a causal relationship between faster CYP2A6 activity and an elevated risk for lung cancers and obstructive respiratory diseases in the total analytical sample (adjusted for smoking) and in current smokers. We further showed that faster (vs. slower) CYP2A6 metabolizers were at a higher risk of developing these diseases at a younger age.
In previous candidate gene studies using various ancestral populations (e.g. Japanese, N = 1705 [47]; Chinese, N = 681 [48]; African, N = 494 [49]; and European, N = 860 [44]), faster CYP2A6 activity, measured by the NMR or CYP2A6 genomics, was associated with an increased risk for lung cancer. Similarly, faster CYP2A6 genotype groups were more prevalent in COPD patients compared to healthy non-smokers [50]. Furthermore, rs56113850T > C, the top variant associated with increased CYP2A6 activity in NMR GWASs [8, 51], was significantly associated with the risk for non-small [52] and squamous cell lung carcinomas [20]. Other GWASs have linked CYP2A6 with COPD [53] and emphysema [54]. Our study extended the relationships between CYP2A6 activity and these two disease clusters, using hypothesis-free methodology that avoids some of the issues inherent in candidate gene studies and provides evidence for causation.
One mechanism explaining the association between faster CYP2A6 activity and increased lung disease risk is via increased smoking. Faster CYP2A6 activity is associated with increased CPD in clinical studies [14, 16, 17]. Variants in the chromosome 19q13 locus, where CYP2A6 resides, were among the top genome-wide signals associated with CPD [55]. Heavier smoking is a known risk factor for lung cancer and COPD [44, 56, 57]. A recent PheWAS-MR study using a genetic risk score for CPD identified these same six PWS signals; the risk score for CPD did not incorporate any CYP2A6 variants [58]. Higher urinary levels of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) in urine, a biomarker of tobacco consumption, is found among faster CYP2A6 metabolizers [17]. In turn, higher NNAL levels have been associated with more severe COPD symptoms [59]. In our interaction analysis, smoking status significantly modified the CYP2A6 association with disease: the CYP2A6 effect was strongest in current smokers for all six PWS signals, weaker but significant in former smokers for the obstructive respiratory diseases signals (except emphysema), and absent in never smokers (Fig. 2, Supplementary Table 3). Smoking-related declines in lung function persist after smoking cessation [60]. While lung-related symptoms and mortality generally improve after cessation, they do not decline to those of never smokers [61]. Unlike COPD, lung cancer was not one of the top signals in former smokers, perhaps since the risk for lung cancer drops dramatically within five years of cessation [56, 62]. The cancer of bronchus (lung) phenotype was the top PWS signal in current smokers, while it was the 16th in former and the 353rd in never smokers.
In our previous case-control study in European ancestry smokers, the association between faster CYP2A6 activity and lung cancer risk was greater in lighter smokers (≤ 20 CPD) than in the overall smokers group [44]. In the current study, while a similar trend for higher CYP2A6-estimated lung cancer risk was observed in lighter smokers, it was marginal. These findings together suggest 1) that heavy smoking could overwhelm the effects of variation of CYP2A6 activity on lung cancer risk, and 2) that the risk varies with both CYP2A6 activity and cigarette consumption [44].
Faster CYP2A6 activity may increase levels of toxins that induce oxidative stress, increase reactive oxygen species and can injure the lungs [57, 63]. For example, faster CYP2A6 activity was associated with increased levels of polycyclic aromatic hydrocarbons and other volatile compounds found in cigarettes [64, 65]. The Nrf2 transcription factor (increased by oxidative stress) binds to the antioxidant response element 1 in CYP2A6 inducing mRNA levels which could increase production of reactive oxygen species, DNA mutations, promote more oxidative stress, lung injury, and oncogenicity ([66–68]. reviewed in [69]). CYP2A6 also mediates the activation of carcinogens like 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone [70]. In a mouse model, methoxsalen, a potent CYP2A6 inhibitor, was found to reduce the incidence and tumor multiplicity of NNK-induced adenocarcinomas [71]. Thus, there may be a role for CYP2A6 in the progression of COPD and lung cancer related to oxidative stress and/or nitrosamines.
The influence of CYP2A6 activity on lung cancer is apparent in some studies even when controlling for pack-years [44]. Consistently, five out of six PWS signals remained PWS after controlling for pack-years or CPD (Supplementary Tables 5 and 6). A decline in estimates, however, was more notable for the obstructive respiratory diseases (~20%) versus lung cancer signals (~10%), suggesting a potentially larger impact of smoking on the CYP2A6 effects on obstructive respiratory diseases.
Early onset lung cancer is associated with a worse prognosis and survival, especially for squamous cell carcinomas (i.e. the smoking-related lung cancer) [72]; this is less well understood for obstructive respiratory diseases [73]. Faster CYP2A6 metabolizers, who had an overall higher risk of lung cancer and obstructive respiratory disease, were also at greater risk of earlier diagnosis than slower CYP2A6 metabolizers (Fig. 7, Supplementary Fig. 7); the UK Biobank is an older cohort which may have limited detection of even earlier separation between CYP2A6 metabolizer groups. Our findings suggest that faster CYP2A6 metabolizers who currently smoke are not only at higher risk for acquiring respiratory disease, but do so at a younger age, furthering our understanding of the risk that faster CYP2A6 activity has on these diseases.
Tobacco smoking increases the risk of iron deficiency anemia [74]. In mice, CYP2A metabolizes bilirubin to biliverdin and CYP2A is induced by excess heme [75]. Thus CYP2A6 plays a complex, though understudied, role in heme homeostasis and catabolism. Here we showed that genetically faster CYP2A6 activity appears to be causally associated with iron deficiency anemia. We speculate that this association is unlikely to be via CYP2A6’s impact on smoking, as 1) the signal was observed in the total group and not in current smokers, and 2) we found no smoking-by-wGRS interaction effect.
We did not detect a significant association with hearing loss, as previously discovered in a PheWAS of the CYP2A6 rs113288603 variant in older, nicotine-exposed women [26]. The hearing loss PheCode in the UK Biobank phenome was divided into conductive and sensorineural. The direction of effect on conductive, but not sensorineural, hearing loss was consistent with the previous study’s finding that faster CYP2A6 activity was protective [26]; the effect was very weak in this study, and observed only among current male smokers (Supplementary Table 11).
Limitations of this study include a potential healthy volunteer selection bias, since the UK Biobank is composed of mostly healthy participants. This may also underpower the ability to detect other a priori associations, such as smoking-related cardiovascular diseases. Although we report smoking-adjusted, unadjusted, interaction, and stratified data, there may be an unaccounted for collider bias that may bias the reported estimates. Testing mediation effects of smoking in the CYP2A6-disease relationships in the future will aid in disentangling indirect (via smoking) and direct effects of CYP2A6 on disease risk. While logistic regression models are inherently heteroscedastic [48], potential heteroscedasticity in our PheWAS models may have biased the estimates; thus, feature selection and engineering is needed before these models are used for disease prediction as opposed to discovery. Additionally, the PheCode system was built from ICD codes only, limiting the examination of phenotypes not captured by this system (e.g. cancer histological subtypes). Although the wGRS captures the largest NMR CYP2A6 activity phenotype to date, it does not account for all variation in CYP2A6 activity. We did not look at the influence of medications taken by the UK Biobank participants in this study; however, we speculate a negligible influence on our findings considering the low reported counts of medications known to interact with CYP2A6 (e.g. Letrozole). The censoring assumption in survival analysis may introduce bias if one group did not receive a diagnosis due to factors unaccounted for (e.g. access to healthcare) or if there are other time-varying covariates (e.g. other lifestyle factors or comorbidities). While it is difficult to eliminate bias, survival analysis is informative as it captures the dynamic nature of the risk factor better than traditional regression models (e.g. logistic regression). Lastly, we were unable to examine non-European ancestry groups due to low case numbers (and thus power), especially in smokers, in the UK Biobank.
In summary, this is the first study to demonstrate, using a hypothesis-free approach in a large biobank, an association of CYP2A6 wGRS with lung cancers and obstructive respiratory diseases and to provide evidence supporting causation. Faster metabolizers were also at greater risk for a disease at a younger age. Additionally, this is the first study to identify potential novel associations between CYP2A6 and iron deficiency anemias, constipation, appendicitis, and ureteral calculus. Our findings suggest there may be an opportunity to incorporate CYP2A6 genotyping into early lung cancer screening programs to enhance identification of those at greater risk.
Funding
We thank the CAMH Specialized Computing Cluster which is funded by the Canada Foundation for Innovation, Research Hospital Fund, the UK Biobank Resource (under Application Number 55371), the PNAT team members, the UK Biobank access management team, the SCC support team, the developers and maintainers of the open-sourced packages and software used, and PNAT and Biobank participants for making this study possible, funding from the Canada Research Chairs program (R.F.T.), CIHR grants FDN-154294 (R.F.T.) and PJY-159710 (R.F.T., J.K., and M.J.C.), NIH grant DA020830 (R.F.T. and C.L.), the Centre for Addiction and Mental Health and the CAMH Foundation, and a University of Toronto Fellowship (H.G.).
Conflict of interest statement: None declared.
Data availability
The UK biobank and clinical trial datasets included in this study are restricted to approved collaborators. The analysis code is available upon request.
Supplementary Material
Contributor Information
Haidy Giratallah, Department of Pharmacology and Toxicology, University of Toronto, 1 King’s College Circle, Toronto, ON M5S 1A8, Canada; Campbell Family Mental Health Research Institute, CAMH, 250 College St, Toronto, ON M5T 1R8, Canada.
Meghan J Chenoweth, Department of Pharmacology and Toxicology, University of Toronto, 1 King’s College Circle, Toronto, ON M5S 1A8, Canada; Campbell Family Mental Health Research Institute, CAMH, 250 College St, Toronto, ON M5T 1R8, Canada; Department of Psychiatry, University of Toronto, 1 King’s College Circle, Toronto, ON M5S 1A8, Canada.
Jennie G Pouget, Campbell Family Mental Health Research Institute, CAMH, 250 College St, Toronto, ON M5T 1R8, Canada; Department of Psychiatry, University of Toronto, 1 King’s College Circle, Toronto, ON M5S 1A8, Canada.
Ahmed El-Boraie, Department of Pharmacology and Toxicology, University of Toronto, 1 King’s College Circle, Toronto, ON M5S 1A8, Canada; Campbell Family Mental Health Research Institute, CAMH, 250 College St, Toronto, ON M5T 1R8, Canada.
Alaa Alsaafin, Department of Pharmacology and Toxicology, University of Toronto, 1 King’s College Circle, Toronto, ON M5S 1A8, Canada; Campbell Family Mental Health Research Institute, CAMH, 250 College St, Toronto, ON M5T 1R8, Canada.
Caryn Lerman, Norris Comprehensive Cancer Center, University of Southern California, 1441 Eastlake Ave, Los Angeles, CA 90033, United States.
Jo Knight, Department of Psychiatry, University of Toronto, 1 King’s College Circle, Toronto, ON M5S 1A8, Canada; Data Science Institute, Lancaster University Medical School, Lancaster LA1 4YE, United Kingdom.
Rachel F Tyndale, Department of Pharmacology and Toxicology, University of Toronto, 1 King’s College Circle, Toronto, ON M5S 1A8, Canada; Campbell Family Mental Health Research Institute, CAMH, 250 College St, Toronto, ON M5T 1R8, Canada; Department of Psychiatry, University of Toronto, 1 King’s College Circle, Toronto, ON M5S 1A8, Canada.
References
- 1. Nakajima M, Yamamoto T, Nunoya KI. et al. Characterization of CYP2A6 involved in 3′-hydroxylation of cotinine in human liver microsomes. J Pharmacol Exp Ther 1996;277:1010–5. [PubMed] [Google Scholar]
- 2. Tiano HF, Wang RL, Hosokawa M. et al. Human CYP2A6 activation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)—mutational specificity in the GPT gene of AS52 cells. Carcinogenesis 1994;15:2859–66. [DOI] [PubMed] [Google Scholar]
- 3. El-Boraie A, Tyndale RF. The role of pharmacogenetics in smoking. Clin Pharmacol Ther 2021;110:599–606. [DOI] [PubMed] [Google Scholar]
- 4. Tanner JA, Tyndale RF. Variation in CYP2A6 activity and personalized medicine. J Pers Med 2017;7:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Inoue K, Yamazaki H, Shimada T. CYP2A6 genetic polymorphisms and liver microsomal coumarin and nicotine oxidation activities in Japanese and Caucasians. Arch Toxicol 2000;73:532–9. [DOI] [PubMed] [Google Scholar]
- 6. Tanner JA, Zhu AZ, Claw KG. et al. Novel CYP2A6 diplotypes identified through next-generation sequencing are associated with in-vitro and in-vivo nicotine metabolism. Pharmacogenet Genomics 2018;28:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nunoya K, Yokoi T, Kimura K. et al. A new deleted allele in the human cytochrome P450 2A6 (CYP2A6) gene found in individuals showing poor metabolic capacity to coumarin and (+)-cis-3,5-dimethyl-2-(3-pyridyl)thiazolidin-4-one hydrochloride (SM-12502). Pharmacogenetics 1998;8:239–50. [DOI] [PubMed] [Google Scholar]
- 8. Loukola A, Buchwald J, Gupta R. et al. A genome-wide association study of a biomarker of nicotine metabolism. PLoS Genet 2015;11:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buchwald J, Chenoweth MJ, Palviainen T. et al. Genome-wide association meta-analysis of nicotine metabolism and cigarette consumption measures in smokers of European descent. Mol Psychiatry 2021;26:2212–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bloom J, Hinrichs AL, Wang JC. et al. The contribution of common CYP2A6 alleles to variation in nicotine metabolism among European-Americans. Pharmacogenet Genomics 2011;21:403–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dempsey D, Tutka P, Jacob P. et al. Nicotine metabolite ratio as an index of cytochrome P450 2A6 metabolic activity. Clin Pharmacol Ther 2004;76:64–72. [DOI] [PubMed] [Google Scholar]
- 12. Lea RA, Dickson S, Benowitz NL. Within-subject variation of the salivary 3HC/COT ratio in regular daily smokers: prospects for estimating CYP2A6 enzyme activity in large-scale surveys of nicotine metabolic rate. J Anal Toxicol 2006;30:386–9. [DOI] [PubMed] [Google Scholar]
- 13. Giratallah HK, Chenoweth MJ, Addo N. et al. Nicotine metabolite ratio: comparison of the three urinary versions to the plasma version and nicotine clearance in three clinical studies. Drug Alcohol Depend 2021;223:108708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Benowitz NL, Pomerleau OF, Pomerleau CS. et al. Nicotine metabolite ratio as a predictor of cigarette consumption. Nicotine Tob Res 2003;5:621–4. [DOI] [PubMed] [Google Scholar]
- 15. Kubota T, Nakajima-Taniguchi C, Fukuda T. et al. CYP2A6 polymorphisms are associated with nicotine dependence and influence withdrawal symptoms in smoking cessation. Pharmacogenomics J 2006;6:115–9. [DOI] [PubMed] [Google Scholar]
- 16. Gu DF, Hinks LJ, Morton NE. et al. The use of long PCR to confirm three common alleles at the CYP2A6 locus and the relationship between genotype and smoking habit. Ann Hum Genet 2000;64:383–90. [DOI] [PubMed] [Google Scholar]
- 17. Strasser AA, Benowitz NL, Pinto AG. et al. Nicotine metabolite ratio predicts smoking topography and carcinogen biomarker level. Cancer Epidemiol Biomark Prev 2011;20:234–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schnoll RA, George TP, Hawk L. et al. The relationship between the nicotine metabolite ratio and three self-report measures of nicotine dependence across sex and race. Psychopharmacology 2014;231:2515–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park SL, Murphy SE, Wilkens LR. et al. Association of CYP2A6 activity with lung cancer incidence in smokers: the multiethnic cohort study. PLoS One 2017;12:e0178435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McKay JD, Hung RJ, Han Y. et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet 2017;49:1126–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yadav VK, Katiyar T, Ruwali M. et al. Polymorphism in cytochrome P4502A6 reduces the risk to head and neck cancer and modifies the treatment outcome. Environ Mol Mutagen 2021;62:502–11. [DOI] [PubMed] [Google Scholar]
- 22. El-Boraie A, Taghavi T, Chenoweth MJ. et al. Evaluation of a weighted genetic risk score for the prediction of biomarkers of CYP2A6 activity. Addict Biol 2020;25:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. El-Boraie A, Chenoweth MJ, Pouget JG. et al. Transferability of ancestry-specific and cross-ancestry CYP2A6 activity genetic risk scores in African and European populations. Clin Pharmacol Ther 2021;110:975–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Denny JC, Ritchie MD, Basford MA. et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 2010;26:1205–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smith GD, Ebrahim S. Mendelian randomization: prospects, potentials, and limitations. Int J Epidemiol 2004;33:30–42. [DOI] [PubMed] [Google Scholar]
- 26. Polimanti R, Jensen KP, Gelernter J. Phenome-wide association study for CYP2A6 alleles: rs113288603 is associated with hearing loss symptoms in elderly smokers. Sci Rep 2017;7:1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bycroft C, Freeman C, Petkova D. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018;562:203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chenoweth MJ, Novalen M, Hawk LW Jr. et al. Known and novel sources of variability in the nicotine metabolite ratio in a large sample of treatment-seeking smokers. Cancer Epidemiol Biomark Prev 2014;23:1773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lerman C, Schnoll RA, Hawk LW Jr. et al. Use of the nicotine metabolite ratio as a genetically informed biomarker of response to nicotine patch or varenicline for smoking cessation: a randomised, double-blind placebo-controlled trial. Lancet Respir Med 2015;3:131–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Crews KR, Gaedigk A, Dunnenberger HM. et al. Clinical pharmacogenetics implementation consortium guidelines for cytochrome P450 2D6 genotype and codeine therapy: 2014 update. Clin Pharmacol Ther 2014;95:376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. The UK Biobank . Genotyping and quality control of UK Biobank, a large-scale, extensively phenotyped prospective resource. Biology 2015. https://biobank.ctsu.ox.ac.uk/crystal/crystal/docs/genotyping_qc.pdf. [Google Scholar]
- 32. Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015;31:3555–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wei WQ, Bastarache LA, Carroll RJ. et al. Evaluating phecodes, clinical classification software, and ICD-9-CM codes for phenome-wide association studies in the electronic health record. PLoS One 2017;12:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bastarache L. Using phecodes for research with the electronic health record: from PheWAS to PheRS. Annual Review of Biomedical Data Science 2021;4:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Team, R.C . R Foundation for Statistical Computing. Vienna, Austria, 2021. https://www.R-project.org/. [Google Scholar]
- 36. Piekos JA, Hellwege JN, Zhang YF. et al. Uterine fibroid polygenic risk score (PRS) associates and predicts risk for uterine fibroid. Hum Genet 2022;141:1739–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc, B: Stat 1995;57:289–300. [Google Scholar]
- 38. Yavorska OO, Burgess S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol 2017;46:1734–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hemani G, Zhengn J, Elsworth B. et al. The MR-base platform supports systematic causal inference across the human phenome. elife 2018;7:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hemani G, Tilling K, Smith GD. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet 2017;13:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Conway JR, Lex A, Gehlenborg N. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 2017;33:2938–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wilkinson L. venneuler: Venn and Euler Diagrams. 2011. R package version 1.1-0. https://CRAN.R-project.org/package=venneuler
- 43. Therneau TM. A Package for Survival Analysis in R. 2020. R package version 3.2-7. https://CRAN.R-project.org/package=survival
- 44. Wassenaar CA, Dong Q, Wei QY. et al. Relationship between CYP2A6 and CHRNA5-CHRNA3-CHRNB4 variation and smoking behaviors and lung cancer risk. J Natl Cancer Inst 2011;103:1342–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Burgess S, Thompson SG. Use of allele scores as instrumental variables for Mendelian randomization. Int J Epidemiol 2013;42:1134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Palmer TM, Lawlor DA, Harbord RM. et al. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat Methods Med Res 2012;21:223–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miyamoto M, Umetsu Y, Dosaka-Akita H. et al. CYP2A6 gene deletion reduces susceptibility to lung cancer. Biochem Biophys Res Commun 1999;261:658–60. [DOI] [PubMed] [Google Scholar]
- 48. Yuan JM, Nelson HH, Carmella SG. et al. CYP2A6 genetic polymorphisms and biomarkers of tobacco smoke constituents in relation to risk of lung cancer in the Singapore Chinese health study. Carcinogenesis 2017;38:411–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wassenaar CA, Ye YQ, Cai QY. et al. CYP2A6 reduced activity gene variants confer reduction in lung cancer risk in African American smokers-findings from two independent populations. Carcinogenesis 2015;36:99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Minematsu N, Nakamura H, Iwata M. et al. Association of CYP2A6 deletion polymorphism with smoking habit and development of pulmonary emphysema. Thorax 2003;58:623–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chenoweth MJ, Ware JJ, Zhu AZX. et al. Genome-wide association study of a nicotine metabolism biomarker in African American smokers: impact of chromosome 19 genetic influences. Addiction 2018;113:509–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dai JC, Zhu M, Wang YZ. et al. Identification of risk loci and a polygenic risk score for lung cancer: a large-scale prospective cohort study in Chinese populations. Lancet Respir Med 2019;7:881–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cho MH, Castaldi PJ, Wan ES. et al. A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Hum Mol Genet 2012;21:947–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sin S, Choi HM, Lim J. et al. A genome-wide association study of quantitative computed tomographic emphysema in Korean populations. Sci Rep 2021;11:16692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Thorgeirsson TE, Gudbjartsson DF, Surakka I. et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet 2010;42:448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tindle HA, Duncan MS, Greevy RA. et al. Lifetime smoking history and risk of lung cancer: results from the Framingham heart study. J Natl Cancer Inst 2018;110:1201–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Terzikhan N, Verhamme KMC, Hofman A. et al. Prevalence and incidence of COPD in smokers and non-smokers: the Rotterdam study. Eur J Epidemiol 2016;31:785–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. King C, Mulugeta A, Nabi F. et al. Mendelian randomization case-control PheWAS in UK Biobank shows evidence of causality for smoking intensity in 28 distinct clinical conditions. EClinicalMedicine 2020;26:100488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Eisner MD, Jacob P, Benowitz NL. et al. Longer term exposure to secondhand smoke and health outcomes in COPD: impact of urine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Nicotine Tob Res 2009;11:945–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Oelsner EC, Balte PP, Bhatt SP. et al. Lung function decline in former smokers and low-intensity current smokers: a secondary data analysis of the NHLBI pooled cohorts study. Lancet Respir Med 2020;8:34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health . 2020. https://www.cdc.gov/tobacco/sgr/2020-smoking-cessation/index.html.
- 62. Crispo A, Brennan P, Jöckel KH. et al. The cumulative risk of lung cancer among current, ex- and never-smokers in European men. Br J Cancer 2004;91:1280–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brucker N, Moro AM, Charao MF. et al. Biomarkers of occupational exposure to air pollution, inflammation and oxidative damage in taxi drivers. Sci Total Environ 2013;463-464:884–93. [DOI] [PubMed] [Google Scholar]
- 64. Pezzuto A, Lionetto L, Ricci A. et al. Inter-individual variation in CYP2A6 activity and chronic obstructive pulmonary disease in smokers: perspectives for an early predictive marker. Biochim Biophys Acta Mol basis Dis 2021;1867:165990. [DOI] [PubMed] [Google Scholar]
- 65. Carroll DM, Murphy SE, Benowitz NL. et al. Relationships between the nicotine metabolite ratio and a panel of exposure and effect biomarkers: findings from two studies of US commercial cigarette smokers. Cancer Epidemiol Biomark Prev 2020;29:871–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ande A, Earla R, Jin MY. et al. An LC-MS/MS method for concurrent determination of nicotine metabolites and the role of CYP2A6 in nicotine metabolite-mediated oxidative stress in SVGA astrocytes. Drug Alcohol Depend 2012;125:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chen X, Owoseni E, Salamat J. et al. Nicotine enhances alcoholic fatty liver in mice: role of CYP2A5. Arch Biochem Biophys 2018;657:65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yokota S, Higashi E, Fukami T. et al. Human CYP2A6 is regulated by nuclear factor-erythroid 2 related factor 2. Biochem Pharmacol 2011;81:289–94. [DOI] [PubMed] [Google Scholar]
- 69. Nakamura H, Takada K. Reactive oxygen species in cancer: current findings and future directions. Cancer Sci 2021;112:3945–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jalas JR, Hecht SS, Murphy SE. Cytochrome p450 enzymes as catalysts of metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco specific carcinogen. Chem Res Toxicol 2005;18:95–110. [DOI] [PubMed] [Google Scholar]
- 71. Maenpaa J, Juvonen R, Raunio H. et al. Metabolic interactions of methoxsalen and coumarin in humans and mice. Biochem Pharmacol 1994;48:1363–9. [DOI] [PubMed] [Google Scholar]
- 72. Etzel CJ, Lu M, Merriman K. et al. An epidemiologic study of early onset lung cancer. Lung Cancer 2006;52:129–34. [DOI] [PubMed] [Google Scholar]
- 73. Soriano JB, Polverino F, Cosio BG. What is early COPD and why is it important? Eur Respir J 2018;52:1801448. [DOI] [PubMed] [Google Scholar]
- 74. Vivek A, Kaushik RM, Kaushik R. Tobacco smoking-related risk for iron deficiency anemia: a case-control study. J Addict Dis 2023;41:128–36. [DOI] [PubMed] [Google Scholar]
- 75. Lamsa V, Levonen AL, Sormunen R. et al. Heme and Heme biosynthesis intermediates induce Heme Oxygenase-1 and cytochrome P450 2A5, enzymes with putative sequential roles in Heme and bilirubin metabolism: different requirement for transcription factor nuclear factor erythroid-derived 2-like 2. Toxicol Sci 2012;130:132–44. [DOI] [PubMed] [Google Scholar]
- 76. Bloom AJ, Harari O, Martinez M. et al. Use of a predictive model derived from in vivo endophenotype measurements to demonstrate associations with a complex locus, CYP2A6. Hum Mol Genet 2012;21:3050–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The UK biobank and clinical trial datasets included in this study are restricted to approved collaborators. The analysis code is available upon request.