

Abstract
A combined physical and genetic map of the Pseudomonas putida KT2440 genome was constructed from data obtained by pulsed-field gel electrophoresis techniques (PFGE) and Southern hybridization. Circular genome size was estimated at 6.0 Mb by adding the sizes of 19 SwaI, 9 PmeI, 6 PacI, and 6 I-CeuI fragments. A complete physical map was achieved by combining the results of (i) analysis of PFGE of the DNA fragments resulting from digestion of the whole genome with PmeI, SwaI, I-CeuI, and PacI as well as double digestion with combinations of these enzymes and (ii) Southern hybridization analysis of the whole wild-type genome digested with different enzymes and hybridized against a series of probes obtained as cloned genes from different pseudomonads of rRNA group I and Escherichia coli, as P. putida DNA obtained by PCR amplification based on sequences deposited at the GenBank database, and by labeling of macrorestriction fragments of the P. putida genome eluted from agarose gels. As an alternative, 10 random mini-Tn5-Km mutants of P. putida KT2440 were used as a source of DNA, and the band carrying the mini-Tn5 in each mutant was identified after PFGE of a series of complete chromosomal digestions and hybridization with the kanamycin resistance gene of the mini-Tn5 as a probe. We established a circular genome map with an average resolution of 160 kb. Among the 63 genes located on the genetic map were key markers such as oriC, 6 rrn loci (rnnA to -F), recA, ftsZ, rpoS, rpoD, rpoN, and gyrB; auxotrophic markers; and catabolic genes for the metabolism of aromatic compounds. The genetic map of P. putida KT2440 was compared to those of Pseudomonas aeruginosa PAO1 and Pseudomonas fluorescens SBW25. The chromosomal backbone revealed some similarity in gene clustering among the three pseudomonads but differences in physical organization, probably as a result of intraspecific rearrangements.
Pseudomonas putida is a member of rRNA group I of the genus Pseudomonas. This species is able to colonize many different niches, including soil, freshwater, and the surfaces of living organisms (e.g., the roots of agriculturally important plants) (9, 37, 42, 49, 60). A number of different strains have been isolated from these niches, and a relevant property of all of them is the ability to metabolize a wide range of biogenic and xenobiotic compounds. P. putida mt-2 was isolated from soils by virtue of its ability to use 3-methylbenzoate as the sole C source (41), a property later shown to be associated with the presence of the TOL plasmid pWW0 (76). P. putida KT2440 is a cured, restriction-deficient derivative of P. putida mt-2 (13) which has been widely used in physiological and genetic studies (see references 48 and 51 for reviews). The nonpathogenic P. putida KT2440 has been shown to be an ideal host for expanding the range of substrates that it can degrade through the recruitment of genes from other microorganisms (52, 53). This strain has also been used as a vehicle for gene cloning and expression (36) and for the biotransformation of several chemicals in added-value products (8). This strain also colonizes the plant rhizosphere, which makes it potentially useful for phytorhizoremediation and for the development of biopesticides. These features make P. putida KT2440 a key strain within this genus (37, 49, 60). An international consortium is now considering sequencing its genome (73).
For efficient exploitation of this strain, thorough knowledge of its genome organization is essential. The development of pulsed-field gel electrophoresis (PFGE) and concomitant technology for the manipulation of large fragments of DNA (68) have revolutionized the analysis of bacterial chromosomes. More than 100 physical maps have been constructed (5, 12). For three members of the genus Pseudomonas—Pseudomonas aeruginosa PAO1, Pseudomonas fluorescens SBW25, and Pseudomonas syringae pv. phaseolicola—complete physical maps and detailed genetic maps have been constructed (7, 19, 46, 57, 65). In the case of P. aeruginosa, a sequencing project is providing in-depth knowledge about the species.
Unlike the situation for P. aeruginosa, little is known about the P. putida genome, and only an approximate chromosome map for one of the strains of this species has been generated, by conjugation and transduction analysis (38, 71). We present here a macrorestriction map of P. putida KT2440 developed from data obtained by PFGE and Southern blot analyses. The size of the circular chromosome was estimated to be 6.0 Mb, based on the size of the fragments generated from the digestion of the whole chromosome with I-CeuI, PacI, SwaI, and PmeI.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
P. putida EEZ15, a phosphinothricin-resistant derivative of the prototroph P. putida KT2440, was described in an earlier publication (56). P. putida EEZ15K-1 through -34 are kanamycin-resistant (Kmr) mini-Tn5 derivatives of P. putida EEZ15 (56). Escherichia coli JM109 was used to maintain different plasmids (66). Bacterial cells were grown at 30°C on Luria-Bertani (LB) culture medium. When necessary, ampicillin, chloramphenicol, kanamycin, and tetracycline were added to final concentrations of 100, 180, 25, and 15 μg/ml, respectively. Plasmids used for preparation of gene probes were extracted from E. coli host strains by the alkaline lysis method (66).
PFGE. (i) DNA preparation.
Unsheared DNA was prepared by embedding whole cells in agarose blocks. P. putida cells were grown in LB to late exponential phase; to align the origins of chromosomal replication, the culture was supplemented with chloramphenicol (180 μg/ml) and maintained for another hour. Cells were harvested by centrifugation for 10 min at 1,400 × g and washed twice with PettIV buffer (10 mM Tris-HCl [pH 7.6], 1 M NaCl) (1). Cell suspensions of different cell densities were prepared in the same buffer and mixed with an equal volume of molten 1.6% (wt/vol) low-melting-point preparative-grade agarose (Bio-Rad) to obtain agarose plugs with 0.5 × 109, 1 × 109, and 2 × 109 cells/ml. Cells were lysed by submerging the plugs in EC-lysis solution (6 mM Tris-HCl [pH 7.6], 1 M NaCl, 100 mM EDTA [pH 7.5], 0.5% [wt/vol] Brij 58, 0.2% [wt/vol] deoxycholate, 0.5% [wt/vol] sarcosyl, 1 mg of hen egg white lysozyme per ml, 20 μg of bovine pancreatic RNase per ml) for 24 h at 37°C. The EC-lysis solution was replaced with ESP solution (0.5 M EDTA [pH 9.5], 1% [wt/vol] lauryl sarcosine, 1 mg of proteinase K per ml), and incubation continued for a further 48 h at 50°C. The agarose blocks were washed several times with TE buffer (10 mM Tris-HCl, 10 mM EDTA [pH 8]) and stored at 4°C until use.
(ii) Restriction endonuclease digestion and end labeling.
Endonucleases were purchased from New England Biolabs (PmeI, PacI, and I-CeuI) and Boehringer Mannheim (SwaI). Before restriction, one-third of an agarose block was equilibrated three times with 1 ml of the recommended restriction buffer, replaced with 60 μl of fresh restriction buffer supplemented with 20 μg of bovine serum albumin per ml, 7 mM dithiothreitol, and the appropriate amount of enzyme (4, 1, 2, and 5 U for PmeI, I-CeuI, PacI, and SwaI, respectively), incubated overnight at 4°C, and then incubated at 37°C for 2 h. Double digestion was done sequentially. End labeling of PmeI- or SwaI-digested DNA was achieved by incubating each plug with 5 μCi of [α-32P]dTTP in 20 μl of Klenow buffer (10 mM MgCl2, 50 mM NaCl, 10 mM Tris-HCl [pH 7.5]) with 1 U of Klenow enzyme for 30 min at room temperature. 32P-labeled fragments were separated by conventional gel electrophoresis and transferred to nylon membranes as described below. 32P-labeled fragments were detected by autoradiography.
(iii) CHEF electrophoresis.
Contour-clamped homogeneous electric field (CHEF) electrophoresis was done in a Pharmacia-LKB Gene Navigator. Agarose gels (1.2% [wt/vol]) were run in 0.5× TBE buffer (45 mM Tris-borate, 45 mM boric acid, 1 mM EDTA [pH 8]) at 10°C unless otherwise stated. Voltage, pulse time, and total running time varied according to the size range of fragments to be separated. Specific conditions are provided in the legends for the corresponding figures. Lambda DNA concatemers (Pharmacia), laboratory-made lambda HindIII digest fragments, and chromosomes of Saccharomyces cerevisiae S-13 and Hansenula wingei (Bio-Rad) were used as molecular size DNA standards.
Southern hybridization and DNA labeling.
DNA fragments separated in PFGE gels were irradiated with UV light (254 nm) for 2 min. DNA was transferred onto nylon membranes by capillary blotting for 48 h (70). Specific probes for hybridization were recovered from agarose gels with an agarose gel DNA extraction kit (Boehringer Mannheim). P. putida macrorestriction fragments used as DNA probes were obtained from a 1.2% (wt/vol) low-melting-point agarose PFGE gel by diluting the agarose with TE buffer to a final concentration of 0.3% (wt/vol) agarose, melting it at 68°C, and performing phenol, phenol-chloroform, and chloroform extractions. Finally, DNA was ethanol precipitated according to standard procedures (66). All probes were digoxigenin labeled by Klenow random primer extension according to the recommended procedure (DIG-DNA labeling and detection kit; Boehringer Mannheim). Blotted filters were prehybridized, hybridized, washed, and immunologically developed according to the supplier’s instructions. High-stringency conditions (50% [vol/vol] formamide and 42°C) were used for P. putida gene probes. For heterologous gene probes, the conditions were less stringent: the concentration of formamide in the hybridization solution was decreased to 30% (vol/vol), or the hybridization temperature was reduced to 28°C. Blots were stored at −20°C and reused several times.
Amplification of DNA by PCR.
Several probe templates were prepared by PCR amplification of genomic DNA. Genomic DNA from P. putida was prepared as described before (63). P. putida sequences were obtained from the EMBL database and used to design the following primers. For the cell division ftsZ gene, the primers 5′-GGCCCCAGTGCTTGAACGCT-3′ and 5′-TTAATCAGCCTGACGACGCA-3′ were used; amplification yielded a 1.3-kb fragment. The gene pyrB, encoding aspartate carbamoyltransferase, was obtained after PCR amplification with the primers 5′-TACTGATGGGCGGTCGCACC-3′ and 5′-CCCGCTCATGGCCATGGACA-3′; a 1.2-kb fragment was obtained. The lipoamide dehydrogenase (lpdG) gene was obtained after PCR amplification with primers 5′-GCCAGGTCGTGATTCGCCCG-3′ and 5′-CCCGCCGTGGTTTCTTATAA-3′; it yielded a 1.7-kb fragment. To obtain the gene encoding muconate-lactonizing enzyme (catB), primers 5′-GACAAGCGCGCTGATTGAAC-3′ and 5′-ACAGCGACGGGCGAAGCGCG-3′ were chosen. All PCR amplifications were performed as recommended by the manufacturer in a Perkin-Elmer DNA thermal cycler under the following conditions: 1 min at 92°C, 1 min at 60°C, and 1 min at 72°C, except for lpdG, for which the annealing temperature was 45°C.
RESULTS
Choice of restriction enzymes, separation of fragments, and estimation of P. putida KT2440 genome size.
To construct the physical map, the restriction enzymes chosen should be able to generate a manageable number of fragments. Our selection of appropriate rare-cutting endonucleases was based on the high G+C content (62.5 mol%) of P. putida (42). Three enzymes with rich A+T recognition sequences, DraI, SspI, and XbaI, were assayed. These 6-bp recognition sequence enzymes generated more than 50 fragments each (data not shown) and were considered inappropriate. SpeI, which recognizes the rare tetranucleotide CTAG (35) within its 6-bp recognition sequence, was also tested. It produced more than 40 fragments, many less than 50 kb in size, and thus was not suitable for this study. PacI, SwaI, and PmeI, which recognize an A+T-rich 8-bp target sequence, generated 6, 19, and 9 fragments respectively (Fig. 1; Table 1). These enzymes were considered suitable, as was I-CeuI, which recognizes a specific 26-bp sequence within the gene encoding the 23S rRNA (30) and which cut the P. putida KT2440 genome in six fragments (Fig. 1; Table 1).
FIG. 1.

CHEF electrophoresis of fragments of P. putida KT2440 genomic DNA predigested with different restriction enzymes. All gels were 1.2% (wt/vol) agarose and were run in 0.5× TBE buffer with the exception of gel A, which was a 0.8% (wt/vol) agarose gel run in 1× TBE buffer. (A) Running conditions: 50 V for 216 h; pulse times, 1,000 s for 24 h, followed by 1,000- to -4,000-s linearly ramped pulse for 192 h. Running conditions: (B and C) 150 V for 70 h; pulse times, 200 s for 24 h, 120 s for 24 h, and 80 s for 22 h. In gel C, partial I-CeuI fragments are indicated by arrows. (D and E) Running conditions: 145 V for 66 h; pulse times, 120 s for 24 h, 70 s for 22 h, and 60 s for 20 h. (F) Running conditions: 100 V for 72 h; 5- to 100-s linearly ramped pulse. (G) Running conditions: 450 V for 4 h, pulse times, 0.5 s. Lanes: S, SwaI; P, PmeI; Pa, PacI; C, I-CeuI; Sp, SpeI; 6, SwaI plus PmeI; 7, PacI plus PmeI; 8, SwaI plus PacI; 9, I-CeuI plus PmeI. DNA size markers were phage λ DNA concatemers (lane 1), intact phage λ DNA plus phage λ DNA digested with HindIII (lanes 2), and chromosomes of S. cerevisiae (lanes 3) and H. wingei (lanes 4). Lane 5 contains total P. putida DOT-OX3 DNA digested with SwaI. Sizes are indicated in kilobases.
TABLE 1.
Fragment sizes of all single restriction enzyme digests of SwaI, PmeI, I-CeuI, and PacI of the P. putida KT2440 chromosome
| Fragment | Fragment sizea (kb) obtained with:
|
|||
|---|---|---|---|---|
| SwaI | PmeI | I-CeuI | PacI | |
| A | 920 | 2,506 | 2,850 | 1,875 |
| B | 702 | 1,025 | 1,230 | 1,750 |
| C | 620 | 778 | 946 | 1,428 |
| D | 584 | 729 | 610 | 678 |
| E | 533 | 674 | 330 | 155 |
| F | 470 | 170 | 126 | 132 |
| G | 395b | 78 | ||
| H | 352 | 19 | ||
| I | 325 | 18 | ||
| J | 180 | |||
| K | 115 | |||
| K′ | 105 | |||
| L | 65b | |||
| M | 55 | |||
| N | 30 | |||
| N′ | 29 | |||
| O | 20 | |||
| Estimated total genome size (kb) | 5,960 | 5,997 | 6,092 | 6,018 |
Average of at least 10 independent gels. Standard deviations were in the range of 1 to 5% of the given value.
The band is a doublet.
All DNA fragments generated by restriction were separated by CHEF electrophoresis. Different running conditions were required for optimal resolution in each size range (Fig. 1A through E). For example, a period of 9 days was needed to resolve the largest fragment detected in these analyses, i.e., the 2,850-kb I-CeuI fragment A (Fig. 1A). Fragments in the size range between 1,000 and 65 kb were resolved under the running conditions specified in the legend to Fig. 1, so that the size of each fragment could be accurately determined (Fig. 1B through F). The smallest SwaI and PmeI single-digest fragments, which were smaller than 50 kb, could be detected by ethidium bromide staining (Fig. 1G). 32P-end labeling of P. putida DNA digested previously with SwaI or PmeI did not resolve any additional fragments upon autoradiographic development (Fig. 2). Table 1 summarizes the sizes of the restriction fragments obtained with the endonucleases used in this work, averaged from more than 10 separate gels. The genome size of P. putida KT2440 was estimated by adding the sizes of the fragments generated by each of the endonucleases used; this yielded an average genome size of 6.0 Mb (Table 1) with an error of less than 2%.
FIG. 2.

Identification of genomic fragments of P. putida KT2440 smaller than 15 kb after digestion with SwaI, PmeI, and SwaI plus PmeI and 32P end labeling. Total DNA of P. putida KT2440 genomic DNA was digested with SwaI (lane S), PmeI (lane P), and SwaI plus PmeI (lane S/P) and then end labeled with 32P as described in Materials and Methods. Fragments were separated in a conventional 0.8% (wt/vol) agarose gel run in 1× TAE buffer for 3 h at 5 V cm−1. The gel was exposed to Kodak photographic film and developed. Size fragments are indicated in kilobases. Bands of interest and their sizes are indicated by arrows.
The size of the genome was further confirmed when the sizes of the fragments resulting from double digestion with SwaI-PmeI and PmeI-PacI (Table 2) were added. 32P-end labeling of P. putida DNA digested with SwaI and PmeI revealed two additional fragments of 8 and 1 kb which were not detectable by ethidium bromide staining (Fig. 2).
TABLE 2.
Fragment sizes resulting from double digestion of P. putida KT2440 DNAa
| Fragment | Fragment sizeb (kb) obtained with SwaI + PmeI | Name | Fragment size (kb) obtained with PmeI + PacI | |
|---|---|---|---|---|
| SwaI-A | 920 | PP1 | 1,390 | |
| SP1 | 680 | PP2 | 1,115 | |
| SP2 | 560 | PmeI-E | 678 | |
| SwaI-E | 533 | PP3 | 548 | |
| SP3 | 450 | PP4 | 510 | |
| SwaI-G | 395 | PP5 | 446 | |
| SP4 | 390 | PP6 | 384 | |
| SwaI-H | 352 | PP7 | 337 | |
| SwaI-I | 325 | PmeI-F | 180 | |
| SP5 | 320 | PacI-E | 155 | |
| SP6 | 172 | PacI-F | 132 | |
| SP7 | 165 | PacI-G/PP8 | 78c | |
| SP8 | 130 | PmeI-H | 19 | |
| SwaI-K′ | 105 | PmeI-I | 19 | |
| SwaI-L* | 65c | |||
| SP9 | 60 | |||
| SwaI-M | 55 | |||
| SP10 | 45 | |||
| SP11/SwN | 30* | |||
| SwaI-N′ | 29 | |||
| SP12 | 21 | |||
| SwaI-O | 20 | |||
| PmeI-H | 18 | |||
| PmeI-I | 18 | |||
| SP13 | 8 | |||
| SP14 | 1 | |||
| Estimated total genome size (kb) | 5,977 | 6,068 |
The fragments were designated SwaI, PmeI, or PacI to indicate that the corresponding SwaI, PmeI, or PacI fragment was not cut by SwaI, PmeI, or PacI, respectively. The fragments resulting from the cut of a SwaI or PmeI fragment by the other enzyme are designated SP followed by a number (1 to 14) such that the higher the number, the smaller the DNA fragment. Accordingly, fragments were designated PP followed by a number when they resulted from the digestion of a PmeI fragment by PacI and vice versa.
Average of at least three independent gels. Standard deviations are in the range of 1 to 5% of the given values.
The band is a doublet.
In the course of mapping, the identity of the fragment patterns between successive plug preparations was checked. No change in fragment pattern was observed during the period of study.
Construction of the physical map.
Two approaches were combined to organize the restriction fragments into a map: (i) physical methods and (ii) hybridization analysis.
(i) Physical methods.
In addition to total digestion of the chromosomal DNA of P. putida with SwaI, PacI, PmeI, and I-CeuI, we tried to obtain partial digestions with these enzymes by reducing either the amount of enzyme or the incubation time. For I-CeuI, two partial fragments (Fig. 1C) of 750 and 470 kb were observed. These fragments can correspond only to the combinations I-CeuI-D (610 kb)–I-CeuI-F (126 kb) and I-CeuI-E (330 kb)–I-CeuI-F (126 kb), respectively. Therefore, these results unequivocally link the fragments in the order I-CeuI-D–I-CeuI-F–I-CeuI-E).
Partial digestion with SwaI generated a wide range of fragments, but only two of them (420 and 220 kb) could be unequivocally assigned to the combinations SwaI-H (352 kb)–SwaI-L (65 kb) and SwaI-J (180 kb)–SwaI-N (30 kb) (not shown).
To extend the limited information derived from partial digestion of chromosomal DNA, we analyzed the SwaI-PmeI double digests of the P. putida KT2440 chromosome. This was expected to provide information about linkage of the fragments generated by SwaI and PmeI (Fig. 1B, D, and F). Fragment SwaI-A remained uncut by PmeI and was assumed to be contained within PmeI-A or PmeI-B. Fragment SwaI-B included a PmeI restriction site yielding a smaller fragment of 680 kb (Table 1). Fragments SwaI-C and SwaI-D were both cut by PmeI; one of them generated a 560-kb fragment (Fig. 1B), but the other fragment generated could not be identified unequivocally. SwaI-E and the double SwaI-G fragments, SwaI-H, SwaI-I, SwaI-K′, SwaI-L, and SwaI-M remained uncut by PmeI (Table 2; Fig. 1D and F).
To further exploit this information, we used a modified version of two-dimensional restriction fragment analysis as described by Bautsch et al. (2) and Römling et al. (65). Total chromosomal DNA was digested with PmeI, and the resulting fragments (PmeI-A through PmeI-G) were initially separated on a CHEF electrophoresis gel and identified (Fig. 3A). An agarose plug with each of the DNA bands was then removed from the gel and further digested with SwaI, and the new fragments were separated in a second electrophoresis (Fig. 3B). As a control, total chromosomal DNA of P. putida KT2440 was digested with SwaI. These analyses revealed that fragments SwaI-A, SwaI-H, SwaI-I, SwaI-L, SwaI-M, and SwaI-N were internal fragments of PmeI-A, and as expected, two new fragments were generated by SwaI within PmeI-A. These were called SP3 (450 kb) and SP12 (21 kb) (Table 2). PmeI-B contained SwaI-E, SwaI-K′, and SwaI-G fragments and two new fragments, designated SP9 (60 kb) and SP13 (8 kb) (Table 2). PmeI-C generated fragments SP1 (680 kb) and SP8 (130 kb). PmeI-D contained the SwaI-N fragment and yielded two new fragments called SP2 (560 kb) and SP6 (172 kb). PmeI-E yielded SP4 (390 kb) and SP5 (330 kb) upon digestion with SwaI, whereas PmeI-F contained SwaI-O and yielded fragments SP7 (165 kb) and SP14 (1 kb). Digestion of PmeI-G with SwaI resulted in fragments SP10 (45 kb) and SP11 (30 kb). This approach allowed us to locate all of the SwaI PmeI-site-free fragments within a PmeI fragment. Based on these data, we linked some of the PmeI fragments to each other, on the assumption that the SP fragments generated in the SwaI/PmeI double digestion would equal the size of a previously determined SwaI fragment. For example, fragments SP3 (450 kb) and SP8 (130 kb) were assumed to come from SwaI-D (584 kb) and therefore to establish the linkage between PmeI-A and PmeI-C. Similar examples linked fragments PmeI-F with PmeI-E and PmeI-E with PmeI-B.
FIG. 3.

Locations of SwaI restriction sites within PmeI fragments. (A) Separation of P. putida KT2440 PmeI fragments in the first electrophoresis; (B) separation of SwaI fragments generated upon digestion of different PmeI fragments. In gel A, two different lanes of total DNA digested with PmeI were run side by side. After ethidium bromide staining of one of them (A), the agarose plugs containing the PmeI fragment of interest were excised and digested with SwaI. Finally DNA fragments were separated in the second electrophoresis (B). Lanes: 1, phage lambda DNA undigested and digested with HindIII; 9, phage lambda DNA concatemers: S, P. putida KT2440 genomic DNA digested with SwaI. Lanes 2, 3, 4, 5, 6, 7, and 8 correspond to fragments PmeI-A, PmeI-B, PmeI-C, PmeI-D, PmeI-E, PmeI-F, and PmeI-G, respectively, digested with SwaI. Sizes are indicated in kilobases.
The order of the SwaI fragments included in PmeI-A could not be unequivocally assigned on the basis of the available information. Useful information was derived from strain DOT-OX3, a P. putida KT2440 mutant unable to synthesize the O antigen of the lipopolysaccharide that had been generated after mutagenesis with mini-Tn5′ luxAB (Fig. 1D, lane 4). When we tried to locate the position of the mini-Tn5 on the chromosome of this strain, we found that fragments SwaI-A and SwaI-H had disappeared and two new fragments, of 750 and 480 kb, had appeared. The sum of the sizes of these two new fragments is similar to the sum of the sizes of SwaI-A and SwaI-H (Fig. 1D). This finding unequivocally linked these two fragments and suggested that this mutant must contain an inversion of at least 150 kb.
In summary, the above series of analyses allowed us to establish that SwaI-A, SwaI-H, and SwaI-L were linked, as were SwaI-J and SwaI-N. We also found that fragments E, F, and D resulting from I-CeuI digestion were also linked. PmeI-A was linked to PmeI-C, whereas PmeI-E was linked to PmeI-F and PmeI-B. The order of fragments in the intact chromosome was PmeI-F,E,B. We identified a number of SwaI fragments within each of the PmeI fragments. In all, the above pattern of fragment linkage provided a low-resolution map in which approximately two-thirds of the chromosome backbones have been established.
(ii) Hybridization analysis.
To enhance the physical map, we used Southern blot analysis of a series of P. putida KT2440 derivatives labeled with mini-Tn5-Km. This series of mutants is called P. putida EEZ15K-x where x is 3, 8, 12, 14, 16, 17, 22, 23, 24, or 30 (56). As a probe, the Kmr determinant gene was used and the position of the Kmr gene in P. putida EEZ15K-x was established. Table 3 summarizes the hybridization data obtained for the DNA of each mutant cut with SwaI, PmeI, and I-CeuI. This analysis confirmed the location of the different SwaI fragments within the PmeI fragments. In addition, it revealed that fragments I-CeuI-E, I-CeuI-F, and I-CeuI-D were located within PmeI-A. We also found that I-CeuI-C was contained within PmeI-A, as surmised from evidence that the Kmr cassette in P. putida EEZ15K-30 was located in SwaI-A, PmeI-A, and I-CeuI-C. The same was true for EEZ15K-16 except that in this mutant the mini-Tn5 lies within the SwaI-H fragment. The finding that the Kmr cassette in mutant EEZ15K-23 was located in SwaI-L and I-CeuI-E and that the mini-Tn5 in mutant EEZ15K-14 was in SwaI-I and in I-CeuI-E linked SwaI-I with the set of fragments SwaI-A–SwaI-H–SwaI-L.
TABLE 3.
Localization of the Kmr determinant in the chromosomes of Kmr derivatives of P. putida EEZ15K-xa
| Kmr mutant | Fragment
|
||
|---|---|---|---|
| SwaI | PmeI | I-CeuI | |
| EEZ15K-3 | I | A | F |
| EEZ15K-8 | K′ | B | A |
| EEZ15K-12 | D | A | D |
| EEZ15K-14 | I | A | E |
| EEZ15K-16 | H | A | C |
| EEZ15K-17 | C | D | A |
| EEZ15K-22 | B | C | B |
| EEZ15K-23 | L | A | E |
| EEZ15K-24 | G | E | A |
| EEZ15K-30 | A | A | C |
Conditions for digestion and separation of fragments were as described in the legend for Fig. 1A, B, and D.
Given that SwaI-D established the linkage between PmeI-A and PmeI-C, the position of the Kmr cassette in mutant EEZ15K-22 within fragments SwaI-B, PmeI-C, and I-CeuI-B confirmed the connection between fragments SwaI-D and SwaI-B as well as that between fragments I-CeuI-D and I-CeuI-B.
We deduced that the largest I-CeuI-A fragment included PmeI-B, PmeI-D, and PmeI-E, because mutants EEZ15K-8, EEZ15K-17, and EEZ15K-24 were located within these fragments (Table 3). From the information thus obtained, the linkage of all I-CeuI fragments was established as I-CeuI-A,C,E,F,D,B.
In addition, the 51 gene probes listed in Table 4 were used against P. putida KT2440 chromosomal DNA digested with SwaI, PmeI, I-CeuI, PacI, SwaI-PmeI, etc. (Tables 5 and 6). This allowed us to further define the map. As an example, the hybridization gel with the npt gene probe and the dnaJ-dnaB-carAB set of genes is shown in Fig. 4.
TABLE 4.
Gene probes used for hybridization in this study
| Gene | Function | Species of origin | Reference(s) |
|---|---|---|---|
| arg | Arginine requirement | Pseudomonas fluorescens | 46 |
| bkdR | Positive transcriptional activator of the bkd operon | P. putida | 31 |
| catB | Muconate-lactonizing enzyme | P. putida | 21 |
| dnaJ, dapB, carA,B | P. fluorescens | 45 | |
| dctA | C4-dicarboxylate transport | P. fluorescens | 4 |
| fabF | Fatty acid synthesis | Escherichia coli | 32 |
| fliA, cheY | Flagellin synthesis, chemotaxis factors | P. aeruginosa | 34 |
| fliP,R | Flagellar biosynthetic genes | P. putida | 69 |
| ftsZ | Cell division protein | P. putida | 79 |
| gltB | Major subunit of glutamate dehydrogenase | P. putida | 10 |
| gyrB | DNA gyrase subunit B | P. putida | 77 |
| hemA, prf1M | Glutamyl-tRNA reductase, protein release factor 1 | P. aeruginosa | 24 |
| hemB | 5-Aminolevulonic acid dehydratase | P. aeruginosa | 23 |
| hemL | Glutamate-1-semialdehyde-1,2-aminomutase | P. aeruginosa | 23 |
| hemN | Oxygen-independent copro-porphyrinogen III-dehydrogenase | P. aeruginosa | 23 |
| qor, hemF, aroE | Quinone oxidoreductase, Oxygen-dependent copro-porphyrinogen III-oxidase, shikimate 5-dehydrogenase | P. aeruginosa | 72 |
| IS1246 | Insertion sequence | P. putida | 59 |
| hip | Integration host factor subunit | P. putida | 33 |
| him | Integration host factor subunit | P. putida | 33 |
| leu | Leucine requirement | P. fluorescens | 46 |
| lexA | Sodium dodecyl sulfate | P. putida | 3 |
| lpdG | Lipoamide dehydrogenase | P. putida | 43 |
| lps | Lipopolysaccharide biosynthesis | P. putida | 54 |
| modABCD | Molybdate transport operon | E. coli | 58 |
| msrA | Methionine sulfoxide reductase | E. coli | 39 |
| npt | Assimilatory nitrate reductase gene | P. putida | 10 |
| oriC | Chromosomal replication origin | P. aeruginosa | 78 |
| osmE | Osmotically inducible envelope protein | E. coli | 15 |
| tolQRAB, orpL, orf2 | Peptidoglycan-associated lipoprotein, cell envelope Tol complex periplasmic proteins | P. putida | 64 |
| panB,C | α-Ketopantoate hydroxymethyl transferase, pantothenate synthetase | P. fluorescens | 46 |
| pca, qui, pob | Protocatechuate p-hydroxybenzoate catabolism | Acinetobacter calcoaceticus | 11 |
| pcaG,H | Protocatechuate metabolism | P. putida | 40 |
| pcaK,F | 4-Hydroxybenzoate transport protein, β-ketoadipyl coenzyme A thiolase | P. putida | 17 |
| pilK | Pilin biosynthesis | E. coli | 6 |
| putC | Regulator region of the proline operon | P. putida | 47 |
| pyrB | Aspartate carbamoyltransferase | P. putida | 67 |
| recA | RecA protein | P. aeruginosa | 20 |
| rpoD | RpoD ς factor | P. putida | 16 |
| rpoE | RpoE ς factor | E. coli | 18 |
| rpoN | RpoN ς factor | P. putida | 26 |
| rpoS | RpoS ς factor | P. putida | 55 |
| rrnA–F | rRNA operons | P. aeruginosa | 27 |
| thr | Threonine requirement | P. fluorescens | 46 |
| thrS, infC rpmL, rplT | P. syringae | 25 | |
| trkA | Putative potassium uptake system | P. putida | 69 |
| ttgA,B, oprD | Toluene-tolerant genes | P. putida | 50, 69 |
| uvrA | UV resistance | P. aeruginosa | 62 |
| uvrB | UV resistance | P. aeruginosa | 61 |
TABLE 5.
Fragments restricted with SwaI, PmeI I-CeuI, and PacI which gave a hybridization signal with several gene probes
| Probe(s) | Restriction fragments hybridizing toa:
|
|||
|---|---|---|---|---|
| SwaI | PmeI | I-CeuI | PacI | |
| arg | H (H + L) | A | C | |
| catB | C | D | ||
| dnaJ, dapB, carA,B | A | A | ||
| dctA | B | |||
| fliA, cheYZ | J | D | A | B |
| fliP,R | J | D | ||
| ftsZ | B | C | B | |
| gltB | A | A | ||
| gyrB | H (H + L) | A | C | C |
| hemA,M, pRf1 | D | A | D | C |
| hemL | A | A | C | B |
| hemN | J (J + N) | D | A | |
| qor, hemF, aroE | H | A | C | |
| IS1246 | G | B | A | A |
| hip | B | C | B | |
| him | G | E | A | |
| leu | A | A | C | |
| lexA | F | E | B | |
| lpdG | D | A | ||
| lps | B | C | A | |
| npt | B | C | ||
| oriC | H | A | C | |
| pal, tolB | B | C | B | |
| panB | A | A | C | |
| pcaG | A | A | C | |
| pcaK | B | C | B | |
| pilK | Unspecific signal | |||
| putC | A | |||
| pyrB | A | C | ||
| recA | B | C | B | A |
| rpoD | I | A | E | C |
| rpoN | D | A | D | C |
| rpoS | B | C | B | A |
| rrs | A, D, F, I, L, M | A, C, E | ||
| thr | B | C | B | |
| thrS, infC, rpmL, rplT | G | E | A | |
| trkA | A | A | ||
| ttgA,B, oprD, rpmL | B | C | ||
| rplT | ||||
| uvrA | I | A | F | |
| uvrB | F | F | B | |
Fragments in parentheses indicate that a hybridization signal with the indicated partial digestion fragment was obtained.
TABLE 6.
Fragments restricted with SwaI/I-CeuI, SwaI/PmeI, I-CeuI/PmeI, PacI/PmeI, and PacI/SwaI which gave a hybridization signal with several gene probes
| Probe | Restriction fragments hybridizing toa:
|
||||
|---|---|---|---|---|---|
| SwaI/ I-CeuI | SwaI/PmeI | I-CeuI/PmeI | PacI/ PmeI | PacI/SwaI | |
| arg | SwaI-H | SwaI-H | CeuI-C | ||
| catB | SP2 | ||||
| gyrB | SwaI-H | CeuI-C | |||
| him | SwaI-G | SwaI-G | 420 kb | ||
| leu | SwaI-A | CeuI-C | |||
| lexA | SP5 | 320 kb | |||
| lpdG | SP3 | ||||
| lps | PP5 | 475 kb | |||
| npt | SP1 | ||||
| panB,C | SwaI-A | CeuI-C | |||
| putC | 640 kb | ||||
| recA | SP1 | PP5 | 475 kb | ||
| rpoD | SwaI-I | CeuI-E | SwaI-I | ||
| rpoN | SwaI-D | SP3 | 494 kb | ||
| rpoS | SP1 | 739 kb | 475 kb | ||
| thr | 700 kb | SP1 | 660 kb | ||
| thrS, infC, rpmL | SwaI-G | 420 kb | |||
| trkA | SwaI-A | ||||
| urvB | SP7 | PmeI-F | |||
The fragment to which the probe hybridized is shown. The fragment is designated by a name when it was unequivocally identified in single or double digests of the whole genome; otherwise it is designated by fragment size.
FIG. 4.

Hybridization analysis with several gene probes. P. putida KT2440 DNA was digested with SwaI (lanes S), PmeI (lanes P), and SwaI plus PmeI (lanes 1), and DNA fragments were separated by CHEF electrophoresis. The three lanes on the left were separated from the two other lanes, and each set of DNA was transferred to a nylon membrane. In panel B, the three lanes on the left correspond to hybridization with the npt gene probe, while the other two lanes correspond to hybridization with the dnaJ-dapB-carAB set of genes. Sizes are indicated in kilobases.
Analysis of all hybridization assays unequivocally located all I-CeuI and PacI fragments except fragments PacI-E and PacI-F, which, because they showed the same location within SwaI-C, PmeI-D, and I-CeuI-A, could be exchanged with each other. We were able to locate most but not all SwaI and PmeI fragments. The positions of the smaller SwaI (SwaI-K through -O) and PmeI (PmeI-G through -I) fragments remained undetermined. These fragments were separated with PFGE, and fragments smaller than 130 kb were extracted and purified from agarose gels (Table 6). These fragments were used directly as probes against blots of chromosomal DNA digested with SwaI, PmeI, and I-CeuI. A single SwaI or PmeI fragment used as a probe should produce a single band when hybridized against a filter carrying fragments of a digest produced by the same enzyme and should produce one or more bands when hybridized to a digest produced by one of the other enzymes. The data from these experiments are summarized in Table 7. Figure 5 shows an example in which the SwaI-J fragment was used as a probe. It hybridized with itself (180 kb) and with the PmeI-D fragment (729 kb) in single-digest assays and with the SP6 (172 kb) fragment in the PmeI-SwaI double digest (Fig. 5). It also hybridized with the I-CeuI fragment A (not shown). Similar assays allowed us to unequivocally locate the SwaI-G, SwaI-J, SwaI-K′, and SwaI-O fragments, as well as the PmeI-F, PmeI-G, PmeI-H, and PmeI-I fragments (Table 7).
TABLE 7.
Hybridization with genomic restriction fragments isolated from agarose gels
| Probe | Restriction fragment(s) hybridizing toa:
|
|||
|---|---|---|---|---|
| SwaI | SwaI + PmeI | PmeI | I-CeuI | |
| PmeI-F | F | ND | F | B |
| PmeI-G | C | ND | G | ND |
| PmeI-H | C | ND | H | ND |
| PmeI-I | B | ND | I | ND |
| SwaI-G | G | G | A/E | ND |
| SwaI-J | J | SP6 | D | A |
| SwaI-K | K (L) (M) | SP10 | G | ND |
| SwaI-K′ | K′ | K′ | B | ND |
| SwaI-L | MB | ND | MB | MB |
| SwaI-M | MB | ND | MB | MB |
| SwaI-N,N′ | N, N′ (A) | ND | A/D (B) (C) | ND |
| SwaI-O | O | ND | F | B |
Fragments in parentheses gave weaker hybridization signals and were not necessarily adjacent. These weak signals were thought to arise from the presence of repeated sequences contained in the genomic restriction fragments. ND, not determined; MB, multiple bands.
FIG. 5.
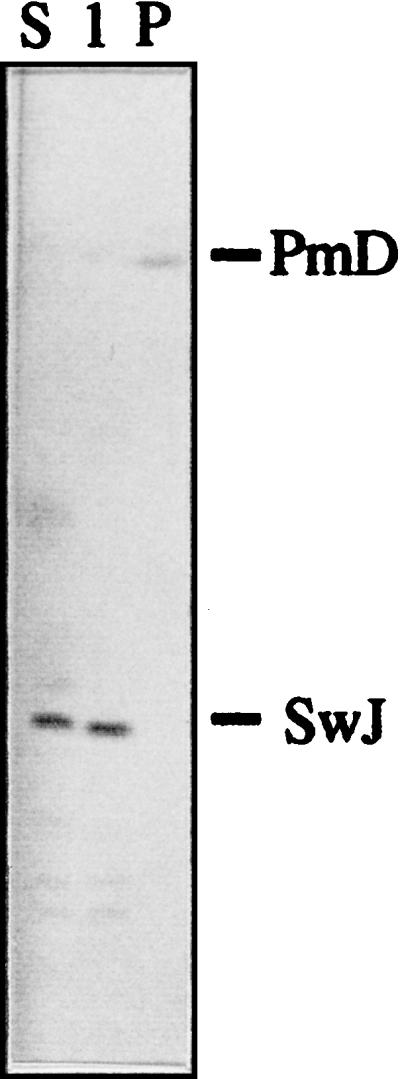
Hybridization analysis with small chromosomal fragments. Fragment SwaI-J was extracted from a gel and used as a probe against P. putida KT2440 DNA digested with SwaI (lane S), PmeI (lane P), or SwaI plus PmeI (lane 1).
Hybridization with fragments SwaI-L and SwaI-M produced multiple bands with all of the enzymes tested; these bands represent rrn operons within these fragments, as shown by hybridization with the rrn gene probe (Fig. 6). Fragments SwaI-L and SwaI-M were located on the basis of the positions of the I-CeuI restriction sites and the hybridization data.
FIG. 6.

Hybridization analysis with the rrs gene as a probe. P. putida KT2440 genomic DNA was digested with SwaI (lanes S), PmeI (lanes P), I-CeuI (lanes C), SwaI plus PmeI (lanes 1), or I-CeuI plus PmeI (lanes 2). The sizes (in kilobases) indicated on the right correspond to S. cerevisiae chromosomes used as a size marker.
Construction of the gene map.
Forty-five previously identified genes or gene clusters were located on the P. putida KT2440 physical map (Tables 5 to 7; Fig. 7) by probing chromosomal digests with available cloned genes and PCR fragments. The gene probes used for this purpose are listed in Table 4.
FIG. 7.

Physical and genetic map of P. putida KT2440. The circular chromosome is represented as a series of overlapping fragments for the enzymes SwaI (Sw), PmeI (Pm), Pac (Pa), and I-CeuI (Ce). Genetic loci were assigned to restriction fragments by Southern hybridization analysis; groups of markers that hybridize at the same restriction fragments are underlined. The exact positions of the six rDNA operons are indicated.
The backbone of the chromosome is defined by the position of the rrn loci rrs (16S), rrl (23S), and rrf (5S) and their distribution with respect to the origin of replication (5). I-CeuI recognizes a specific 26-bp sequence within the rrl gene. The mapped I-CeuI restriction sites localized six rrn operons, which we have designated rrnA through -F, in the physical map of P. putida KT2440 (Fig. 7). A specific rrs probe was used to validate the existence of an rrn operon within the SwaI and PmeI fragments that contain the I-CeuI restriction sites (Fig. 6). Hybridization of the rrs probe to I-CeuI chromosomal digests gave no signal with fragment I-CeuI-B but gave a signal with the rest of the I-CeuI fragments (Fig. 6).
Probes consisting of more than a single gene, such as hemA-hemM-prf1, dnaJ-dapB-carAB, and pcaG-pcaH, hybridized to a single band only, suggesting that these genes are probably contiguous in P. putida KT2440. The positions of the genes necessary for leucine, threonine, and arginine biosynthesis were mapped by Southern hybridization with cosmid clones from a P. fluorescens SBW25 genomic library (46). Each clone hybridized to a single band under high-stringency conditions, showing that these species are similar in gene organization within these cosmid areas.
No homology was detected to several E. coli probes (fabF, asmE, mobABCD, msrA, and rpoE) or to pca-qui-pob from Acinetobacter calcoaceticus, to bkdR from P. putida PpG2, or to hemB from P. aeruginosa, even under low-stringency conditions.
DISCUSSION
We have generated the first complete physical map for the enzymes I-CeuI, PacI, PmeI, and SwaI in P. putida KT2440. A single chromosome was shown to be circular and to have an estimated size of 6.0 Mb. Two independent mapping approaches, (i) analysis of fragments resulting from digestion of the whole genome with rare-cutting restriction enzymes and (ii) Southern hybridization, were used to minimize possible errors and to validate the data obtained separately with each approach. A similar method was used to construct the maps of Haemophilus influenzae (29) and Mycoplasma mycoides (44). In the P. putida KT2440 map, a total of 40 restriction sites (6 I-CeuI, 6 PacI, 9 PmeI, and 19 SwaI) were positioned on the map, achieving an average resolution of approximately 160 kb. Our procedure ensures that DNA fragments larger than 18 kb in size were detected by ethidium bromide staining of whole genome DNA and that the use of 32P-labeled fragments detected fragments of up to 1 kb. It is unlikely that we overlooked fragments smaller than 1 kb because we avoided overrunning the gels when fragments were labeled with 32P. The genome of this P. putida strain is only 100 kb larger than the 5.9-Mb P. aeruginosa PAO1 genome (65), 1.78 Mb larger than the average Pseudomonas stutzeri genome (14), about 400 kb larger than the P. syringae pv. phaseolica genome (7), and about 600 kb smaller than the 6.63-Mb P. fluorescens SWB25 genome (46).
A partial genetic map of the P. putida KT2440 genome was obtained by Southern hybridization with 51 probes (Table 4), of which 38 gave positive signals locating a total of 63 genes (Fig. 7), including key markers such as oriC, recA, gyrB, rpoS, rpoN, rpoD, and the rDNA operons. Figure 7 summarizes the results obtained for more than 100 gels resulting from single or double digestion with rare-cutting enzymes in a large series of hybridization experiments. Several auxotrophic markers (leu, thr, and arg) were positioned, as were genes involved in pilus biosynthesis and motility, lipopolysaccharide production, and inorganic nitrogen assimilation. In addition, we located several catabolic operons (genes) for the metabolism of aromatic compounds. P. putida KT2440 is a nonaggressive root colonizer (37). The genes involved in C4-dicarboxylate transport (dctA, -B, and -D) are important for rhizosphere colonization (37). In this study, we located the dctA gene.
The distribution of signals allowed us to identify a genetically dense region around oriC; the rrn operons seem to be grouped in this region with five of the six operons occupying one-third of the genetic map (Fig. 7). P. putida contains six rRNA loci, designated rrnA through -F. Localization of the I-CeuI sites precisely positioned the rrn operons (30). Assuming the 5′-16S-23S-3′ orientation, the hybridization data with a 16S rRNA probe suggested that the rDNA genes are organized in a typical eubacterial manner, with the rrn operons transcribed divergently. The six copies of rrn operons in P. putida KT2440 contrast with the five rrn operons in P. fluorescens SBW25 (46) and P. syringae pv. phaseolica (7) and the four described for P. aeruginosa PAO1 (65). Among eubacteria, the number of rRNA operons varies between 14 in Clostridium beijerinkii (75) and 1 in some species of the genus Mycobacterium (28). This finding suggests different patterns of gene rearrangements not only among different genera but also within the same genus.
This information provides a basis for comparing different members of the genus Pseudomonas. Figure 8 shows the locations of the common genetic markers in P. putida KT2440, P. fluorescens SBW25, and P. aeruginosa PAO1. It is accepted that translocatable elements allow bacterial chromosomes to acquire new genes by lateral transfer from other species, so that a different map location for genes performing the same function in species derived from a common ancestor does not necessarily reflect a chromosomal rearrangement since the time of divergence, but may mean that the two species have independently acquired that gene (27). However, the presumed ancestor of P. aeruginosa, P. putida, and P. fluorescens probably already possessed all or many of the genes required for DNA recombination, metabolism of simple metabolites, and uptake of compounds abundant in the environment (such as dicarboxylic acids), and so these markers should be reliable indicators of chromosomal rearrangements. The limited number of hybridization experiments with cosmids derived from P. fluorescens suggests that at least in the cases we studied, clusters of genes were conserved, although our limited information does not allow us to draw definitive conclusions. A detailed comparison of the genomic organization of different pseudomonads thus awaits further experimental analysis.
FIG. 8.

Comparison of the maps of P. fluorescens SBW25, P. putida KT2440, and P. aeruginosa PAO1. To facilitate comparisons, the circular maps were opened at an arbitrarily chosen point and positioned with respect to the origin of replication. Only the genes found in both P. putida and one of the other bacteria are shown, connected by a dashed line. The position of each gene is expressed as a ratio of the position of that gene relative to oriC.
Determination of the physical and partial genetic map of the P. putida genome constitutes a significant step forward in terms of comparative genome analysis and will aid the genome sequencing project. The map provides a sound framework for studies of the taxonomy of Pseudomonas species as well as for studies of the colonization of different niches and the utilization of different nutrients by these bacteria.
ACKNOWLEDGMENTS
P. fluorescens SBW25 cosmids were kindly provided by P. Rainey. We thank J. Sokatch, C. W. Ronson, J. Cronan, H. Otnake, A. Segura, L. Eberl, S. Harayama, D. Jahn, P. A. Williams, V. de Lorenzo, J. Barbé, M. I. Ramos-González, R. Gunsalus, N. Brot, B. Holloway, C. Gutierrez, J. J. Rodríguez-Herva, L. N. Ornston, G. Mosqueda, C. S. Harwood, C. Ramos, J. Horn, K. Makino, and D. Hill for strains and/or plasmids. We acknowledge the comments on this work offered by B. Tümmler and P. Rainey. We thank K. Shashok for language improvements and M. M. Fandila for typing the manuscript.
This work was supported by grants from the European Commission (BIO4-CT97-2183) and CICYT (BIO 97-0641).
REFERENCES
- 1.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: John Wiley & Sons; 1991. [Google Scholar]
- 2.Bautsch W, Grothues D, Tümmler B. Genome fingerprinting of Pseudomonas aeruginosa by two dimensional field inversion gel electrophoresis. FEMS Microbiol Lett. 1988;52:255–258. [Google Scholar]
- 3.Calero S, Garriga X, Barbé J. Analysis of the DNA damage-mediated induction of Pseudomonas putida and Pseudomonas aeruginosa lexA gene. FEMS Microbiol Lett. 1993;110:65–70. doi: 10.1111/j.1574-6968.1993.tb06296.x. [DOI] [PubMed] [Google Scholar]
- 4.Challis B C. Molecular characterization of the C4-dicarboxylate transport system of P. fluorescens and its role in root colonization. M.Sc. thesis. Dunedin, New Zealand: University of Otago; 1994. [Google Scholar]
- 5.Cole S T, Saint Girons I. Bacterial genomics. FEMS Microbiol Rev. 1994;14:139–160. doi: 10.1111/j.1574-6976.1994.tb00084.x. [DOI] [PubMed] [Google Scholar]
- 6.Darzins A. The pilG gene product, required for Pseudomonas aeruginosa pilus production and twitching motility, is homologous to the enteric, single-domain response regulator CheY. J Bacteriol. 1993;175:5934–5944. doi: 10.1128/jb.175.18.5934-5944.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Ita M E, Marsch-Moreno R, Guzmán P, Alvarez-Morales A. Physical map of the chromosome of the phytopathogenic bacterium Pseudomonas syringae pv. phaseolica. Microbiology. 1998;144:493–501. doi: 10.1099/00221287-144-2-493. [DOI] [PubMed] [Google Scholar]
- 8.Delgado A, Wubbolts M G, Abril M A, Ramos J L. Nitroaromatics are substrates for the TOL plasmid upper-pathway enzymes. Appl Environ Microbiol. 1992;58:415–417. doi: 10.1128/aem.58.1.415-417.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Weger L A, Bakker P A H M, Schippers B, van Loosdrecht M C M, Lugtenberg B J J. Pseudomonas spp. with mutational changes in the O-antigenic side chain of their lipopolysaccharide are affected in their ability to colonize potato roots. NATO ASI (Adv Sci Inst) Ser. 1989;H36:197–202. [Google Scholar]
- 10.Eberl, L. 1998. Personal communication.
- 11.Elsemore D A, Ornston L N. The pca-pob supraoperonic cluster of Acinetobacter calcoaceticus contains quiA, the structural gene for quinate-shikimate dehydrogenase. J Bacteriol. 1994;176:7659–7666. doi: 10.1128/jb.176.24.7659-7666.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fonstein M, Haselkorn R. Physical mapping of bacterial genomes. J Bacteriol. 1995;177:3361–3369. doi: 10.1128/jb.177.12.3361-3369.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franklin F C H, Bagdasarian M, Bagdasarian M M, Timmis K N. Molecular and functional analysis of the TOL plasmid pWW0 from Pseudomonas putida and cloning of genes for the entire regulated aromatic ring meta-cleavage pathway. Proc Natl Acad Sci USA. 1981;78:7458–7462. doi: 10.1073/pnas.78.12.7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ginard M, Lalucat J, Tummler B, Römling U. Genome organization of Pseudomonas stutzeri and resulting taxonomic and evolutionary considerations. Int J Syst Bacteriol. 1997;47:132–143. doi: 10.1099/00207713-47-1-132. [DOI] [PubMed] [Google Scholar]
- 15.Gutierrez C, Gordia S, Bonnassie S. Characterization of the osmotically inducible gene osmE of Escherichia coli K-12. Mol Microbiol. 1995;16:553–563. doi: 10.1111/j.1365-2958.1995.tb02418.x. [DOI] [PubMed] [Google Scholar]
- 16.Harayama, S. Personal communication.
- 17.Harwood C S, Nichols N N, Kim M K, Ditty J L, Parales R E. Identification of the pcaRKF gene cluster from Pseudomonas putida: involved in chemotaxis, biodegradation, and transport of 4-hydroxybenzoate. J Bacteriol. 1994;176:6479–6488. doi: 10.1128/jb.176.21.6479-6488.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiratsu K, Amemura M, Nashimoto H, Shinagawa H, Makino K. The rpoE gene of Escherichia coli, which encodes ςE, is essential for bacterial growth at high temperature. J Bacteriol. 1995;177:2918–2922. doi: 10.1128/jb.177.10.2918-2922.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holloway B W, Römling U, Tümmler B. Genomic mapping of Pseudomonas aeruginosa PAO. Microbiology. 1994;140:2907–2929. doi: 10.1099/13500872-140-11-2907. [DOI] [PubMed] [Google Scholar]
- 20.Horn J M, Ohman D E. Transcriptional and translational analyses of recA mutant alleles in Pseudomonas aeruginosa. J Bacteriol. 1988;170:1637–1650. doi: 10.1128/jb.170.4.1637-1650.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Houghton J E, Brown T M, Appel A J, Hughes E J, Ornston L N. Discontinuities in the evolution of Pseudomonas putida cat genes. J Bacteriol. 1995;177:401–412. doi: 10.1128/jb.177.2.401-412.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Housiaux P J, Hill D F, Peterson G B. Nucleotide sequence of a gene for 5S ribosomal RNA from Pseudomonas aeruginosa. Nucleic Acids Res. 1988;16:2721. doi: 10.1093/nar/16.6.2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hungerer C, Troup B, Römling U, Jahn D. Cloning, mapping and characterization of the Pseudomonas aeruginosa hemL gene. Mol Gen Genet. 1995;248:375–380. doi: 10.1007/BF02191605. [DOI] [PubMed] [Google Scholar]
- 24.Hungerer C, Weiss D S, Thauer R K, Jahn D. The hemA gene encoding glutamyl-tRNA reductase from the archaeon Methanobacterium thermoautotrophicum strain Marburg. Bioorg Med Chem. 1996;4:1089–1095. doi: 10.1016/0968-0896(96)00098-3. [DOI] [PubMed] [Google Scholar]
- 25.Kitten T, Willis D K. Suppression of a sensor kinase-dependent phenotype in Pseudomonas syringae by ribosomal proteins L35 and L20. J Bacteriol. 1996;178:1548–1555. doi: 10.1128/jb.178.6.1548-1555.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Köhler T, Harayama S, Ramos J L, Timmis K N. Involvement of Pseudomonas putida RpoN ς factor in regulation of various metabolic functions. J Bacteriol. 1989;171:4326–4333. doi: 10.1128/jb.171.8.4326-4333.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kolstø A-B. Dynamic bacterial genome organization. Mol Microbiol. 1997;24:241–248. doi: 10.1046/j.1365-2958.1997.3501715.x. [DOI] [PubMed] [Google Scholar]
- 28.Krawiec S, Riley M. Organization of the bacterial chromosome. Microbiol Rev. 1990;54:502–539. doi: 10.1128/mr.54.4.502-539.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J J, Smith H O, Redfield R J. Organization of the Haemophilus influenzae Rd genome. J Bacteriol. 1989;171:3016–3024. doi: 10.1128/jb.171.6.3016-3024.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu S, Hessel A, Sanderson K E. Genomic mapping with I-CeuI, an intron-encoded endonuclease specific for genes for ribosomal RNA, in Salmonella spp., Escherichia coli, and other bacteria. Proc Natl Acad Sci USA. 1993;90:6874–6878. doi: 10.1073/pnas.90.14.6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madhusudhan K T, Huang N, Sokatch J R. Characterization of BkdR-DNA binding in the expression of the bkd operon of Pseudomonas putida. J Bacteriol. 1995;177:636–641. doi: 10.1128/jb.177.3.636-641.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Magnuson K, Carey M R, Cronan J E., Jr The putative fabJ gene of Escherichia coli fatty acid synthesis is the fabF gene. J Bacteriol. 1995;177:3593–3595. doi: 10.1128/jb.177.12.3593-3595.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marqués S, Gallegos M T, Manzanera M, Holtel A, Timmis K N, Ramos J L. Activation and repression of transcription at the double tandem divergent promoters for xylR and xylS genes of the TOL plasmid of Pseudomonas putida. J Bacteriol. 1998;180:2889–2894. doi: 10.1128/jb.180.11.2889-2894.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masduki A, Nakamura J, Ohga T, Umezaki R, Kato J, Ohtake H. Isolation and characterization of chemotaxis mutants and genes of Pseudomonas aeruginosa. J Bacteriol. 1995;177:948–952. doi: 10.1128/jb.177.4.948-952.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McClelland M, Jones R, Patel Y, Nelson M. Restriction endonucleases for pulsed-field mapping of bacterial genomes. Nucleic Acids Res. 1987;15:5985–6005. doi: 10.1093/nar/15.15.5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mermod N, Lehrbach P R, Reineke W, Timmis K N. Transcription of the TOL plasmid toluate catabolic pathway operon of Pseudomonas putida is determined by a pair of co-ordinately and positively regulated overlapping promoters. EMBO J. 1984;11:2461–2466. doi: 10.1002/j.1460-2075.1984.tb02156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Molina L, Ramos C, Ronchel M C, Molin S, Ramos J L. Field release of biologically contained Pseudomonas putida strains with biodegradative potential. Appl Environ Microbiol. 1998;64:2073–2078. doi: 10.1128/aem.64.6.2072-2078.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morgan A F, Dean H F. Chromosomal map of Pseudomonas putida PPN, and a comparison of gene order with the Pseudomonas aeruginosa PAO chromosomal map. J Gen Microbiol. 1985;131:885–896. doi: 10.1099/00221287-131-4-885. [DOI] [PubMed] [Google Scholar]
- 39.Moskovitz J, Rahman M A, Strassman J, Yancey S O, Kushner S R, Brot N, Weissbach H. Escherichia coli peptide methionine sulfoxide reductase gene: regulation of expression and role in protecting against oxidative damage. J Bacteriol. 1995;177:502–507. doi: 10.1128/jb.177.3.502-507.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mosqueda, G. 1998. Personal communication.
- 41.Nakazawa T, Yokota T. Benzoate metabolism in Pseudomonas putida (arvilla) mt-2: demonstration of two benzoate pathways. J Bacteriol. 1973;15:262–267. doi: 10.1128/jb.115.1.262-267.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palleroni N J. Section 4, family I, Pseudomonadaceae. In: Sneath P H A, Mair N S, Sharpe E M, Holt J G, editors. Bergey’s manual of systematic bacteriology. Vol. 1. Baltimore, Md: The Williams & Wilkins Co.; 1986. pp. 141–219. [Google Scholar]
- 43.Palmer J A, Hatter K, Sokatch J R. Cloning and sequence analysis of the LPD-glc structural gene of Pseudomonas putida. J Bacteriol. 1991;173:3109–3116. doi: 10.1128/jb.173.10.3109-3116.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pyle L E, Finch L R. A physical map of the genome of Mycoplasma mycoides subspecies mycoides Y with some functional loci. Nucleic Acids Res. 1988;16:6027–6039. doi: 10.1093/nar/16.13.6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rainey, P. B. 1998. Personal communication.
- 46.Rainey P B, Bailey M J. Physical and genetic map of the Pseudomonas fluorescens SBW25 chromosome. Mol Microbiol. 1996;19:521–533. doi: 10.1046/j.1365-2958.1996.391926.x. [DOI] [PubMed] [Google Scholar]
- 47.Ramos, C., L. Molina, and S. Vílchez. 1998. Personal communication.
- 48.Ramos J L, Díaz E, Dowling D, de Lorenzo V, Molin S, O’Gara F, Ramos C, Timmis K N. The behavior of bacteria designed for biodegradation. Bio/Technology. 1994;12:1349–1356. doi: 10.1038/nbt1294-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramos J L, Duque E, Huertas M J, Haïdour A. Isolation and expansion of the catabolic potential of a Pseudomonas putida strain able to grow in the presence of high concentrations of aromatic hydrocarbons. J Bacteriol. 1995;177:3911–3916. doi: 10.1128/jb.177.14.3911-3916.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramos J L, Duque E, Segura A. Efflux pumps involved in toluene tolerance in Pseudomonas putida DOT-T1E. J Bacteriol. 1998;180:3323–3329. doi: 10.1128/jb.180.13.3323-3329.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramos J L, Marqués S, Timmis K N. Transcriptional control of the Pseudomonas TOL plasmid catabolic operons is achieved through an interplay of host factors and plasmid-encoded regulators. Annu Rev Microbiol. 1997;51:341–373. doi: 10.1146/annurev.micro.51.1.341. [DOI] [PubMed] [Google Scholar]
- 52.Ramos J L, Timmis K N. Experimental evolution of catabolic pathways of bacteria. Microbiol Sci. 1987;4:228–237. [PubMed] [Google Scholar]
- 53.Ramos J L, Waserfallen A, Rose K, Timmis K N. Redesigning metabolic routes: manipulation of TOL plasmid pathway for catabolism of alkylbenzoates. Science. 1987;235:593–596. doi: 10.1126/science.3468623. [DOI] [PubMed] [Google Scholar]
- 54.Ramos-González M I. Liberación al medio ambiente de microorganismos manipulados genéticamente. Ph.D thesis. Granada, Spain: University of Granada; 1993. [Google Scholar]
- 55.Ramos-González M I, Molin S. Cloning, sequencing and phenotypic characterization of an rpoS gene from Pseudomonas putida KT2440. J Bacteriol. 1998;180:3421–3431. doi: 10.1128/jb.180.13.3421-3431.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramos-González M I, Ramos-Díaz M A, Ramos J L. Chromosomal gene capture mediated by the Pseudomonas putida TOL catabolic plasmid. J Bacteriol. 1994;176:4635–4641. doi: 10.1128/jb.176.15.4635-4641.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ratnaningsih E, Dharmsthiti S, Krishnapillai Y, Morgan A, Sinclair K, Holloway B W. A combined physical and genetic map of Pseudomonas aeruginosa PAO. J Gen Microbiol. 1990;136:2351–2356. doi: 10.1099/00221287-136-12-2351. [DOI] [PubMed] [Google Scholar]
- 58.Rech S, Deppenmeier U, Gunsalus R P. Regulation of the molybdate transport operon, modABCD, of Escherichia coli in response to molybdate availability. J Bacteriol. 1995;177:1023–1029. doi: 10.1128/jb.177.4.1023-1029.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reddy B R, Shaw L E, Sayers J R, Williams P A. Two identical copies of IS1246, a 1275 base pair sequence related to other bacterial insertion sequences, enclose the xyl genes on TOL plasmid pWW0. Microbiology. 1994;140:2305–2307. doi: 10.1099/13500872-140-9-2305. [DOI] [PubMed] [Google Scholar]
- 60.Reniero, D., J. J. Rodríguez-Herva, L. Molina, E. Galli, J. L. Ramos, and E. Duque. Colonization of the corn root system by wild-type Pseudomonas putida KT2440 and mutants with altered surfaces. Submitted for publication.
- 61.Rivera E, Vila L, Barbé J. The uvrB gene of Pseudomonas aeruginosa is not DNA damage inducible. J Bacteriol. 1996;178:5550–5554. doi: 10.1128/jb.178.18.5550-5554.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rivera E, Vila L, Barbé J. Expression of the Pseudomonas aeruginosa uvrA gene is constitutive. Mutat Res. 1997;377:149–155. doi: 10.1016/s0027-5107(97)00061-4. [DOI] [PubMed] [Google Scholar]
- 63.Robson R L, Chesshyre J, Wheeler C, Jones R, Woodley P, Postgate J R. Genome size and complexity in Azotobacter chroococcum. J Gen Microbiol. 1984;130:1603–1612. doi: 10.1099/00221287-130-7-1603. [DOI] [PubMed] [Google Scholar]
- 64.Rodríguez-Herva J J, Ramos-González M I, Ramos J L. The Pseudomonas putida peptidoglycan-associated outer membrane lipoprotein is involved in maintenance of the integrity of the cell envelope. J Bacteriol. 1996;178:1699–1706. doi: 10.1128/jb.178.6.1699-1706.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Römling U, Grothues D, Bautsch W, Tümmler B. A physical genome map of Pseudomonas aeruginosa PAO. EMBO J. 1989;13:4081–4089. doi: 10.1002/j.1460-2075.1989.tb08592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sambrook J, Fritsch E F, Maniatis E. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 67.Schurr M J, Vickrey J F, Kumar A P, Campbell A L, Cunin R, Benjamin R C, Shanley M S, O’Donovan G A. Aspartate transcarbamoylase genes of Pseudomonas putida: requirement for an inactive dihydroorotase for assembly into the dodecameric holoenzyme. J Bacteriol. 1995;177:1751–1759. doi: 10.1128/jb.177.7.1751-1759.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schwartz D C, Saffran W, Welsh J, Haas R, Goldleuberg M, Canter C R. New techniques for purifying large DNAs and studying their properties and packaging. Cold Spring Harbor Symp Quant Biol. 1982;47:189–195. doi: 10.1101/sqb.1983.047.01.024. [DOI] [PubMed] [Google Scholar]
- 69.Segura, A. 1998. Personal communication.
- 70.Smith C L, Klco S R, Cantor C R. Pulsed-field gel electrophoresis and the technology of large DNA molecules. In: Davies K E, editor. Genome analysis. Oxford, England: IRL Press; 1988. pp. 41–72. [Google Scholar]
- 71.Strom A D, Hirst R, Petering J, Morgan A. Isolation of high frequency of recombination donors from Tn5 chromosomal mutants of Pseudomonas putida PPN and recalibration of the genetic map. Genetics. 1990;126:497–503. doi: 10.1093/genetics/126.3.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Troup B, Jahn M, Hungerer C, Jahn D. Isolation of the hemF operon containing the gene for the Escherichia coli aerobic coproporphyrinogen III oxidase by in vivo complementation of a yeast HEM13 mutant. J Bacteriol. 1994;176:673–680. doi: 10.1128/jb.176.3.673-680.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tümmler, B. 1998. Personal communication.
- 74.Watson R J. Analysis of the C-4-dicarboxylate transport gene of Rhizobium meliloti: nucleotide sequence and deduced products of dctA, dctB and dctD. Mol Plant-Microbe Interact. 1990;3:174–184. doi: 10.1094/mpmi-3-174. [DOI] [PubMed] [Google Scholar]
- 75.Wilkinson S R, Young D I, Morris J G, Young M. Molecular genetics and the initiation of solvent to genesis in Clostridium beijerinckii (formerly Clostridium acetobutylicum) NCIMB 8052. FEMS Microbiol Rev. 1995;17:275–285. doi: 10.1111/j.1574-6976.1995.tb00211.x. [DOI] [PubMed] [Google Scholar]
- 76.Worsey M J, Williams P A. Metabolism of toluene and xylenes by Pseudomonas putida (arvilla) mt-2: evidence for a new function of the TOL plasmid. J Bacteriol. 1975;124:7–13. doi: 10.1128/jb.124.1.7-13.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamamoto S, Harayama S. PCR amplification and direct sequencing of gyrB genes with universal primers and their application to the detection and taxonomic analysis of Pseudomonas putida strains. Appl Environ Microbiol. 1995;61:1104–1109. doi: 10.1128/aem.61.3.1104-1109.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yee T W, Smith D W. Pseudomonas chromosomal replication origins: a bacterial class distinct from Escherichia coli-type origins. Proc Natl Acad Sci USA. 1990;87:1278–1282. doi: 10.1073/pnas.87.4.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yim, L., and M. Vicente. 1995. GenBank accession no. U29400.