

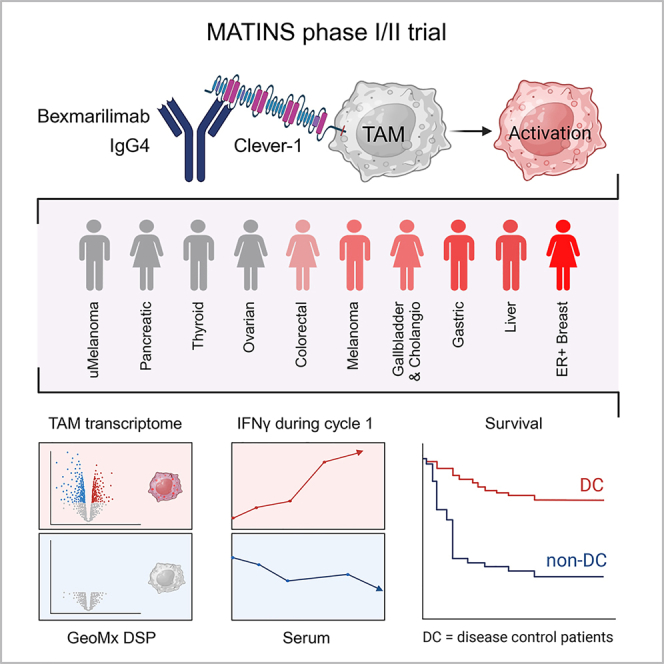
Summary
Macrophage Clever-1 contributes to impaired antigen presentation and suppression of anti-tumor immunity. This first-in-human trial investigates the safety and tolerability of Clever-1 blockade with bexmarilimab in patients with treatment-refractory solid tumors and assesses preliminary anti-tumor efficacy, pharmacodynamics, and immunologic correlates. Bexmarilimab shows no dose-limiting toxicities in part I (n = 30) and no additional safety signals in part II (n = 108). Disease control (DC) rates of 25%–40% are observed in cutaneous melanoma, gastric, hepatocellular, estrogen receptor-positive breast, and biliary tract cancers. DC associates with improved survival in a landmark analysis and correlates with high pre-treatment intratumoral Clever-1 positivity and increasing on-treatment serum interferon γ (IFNγ) levels. Spatial transcriptomics profiling of DC and non-DC tumors demonstrates bexmarilimab-induced macrophage activation and stimulation of IFNγ and T cell receptor signaling selectively in DC patients. These data suggest that bexmarilimab therapy is well tolerated and show that macrophage targeting can promote immune activation and tumor control in late-stage cancer.
Keywords: cancer, first-in-man, Clever-1, Stabilin-1, immune activation, GeoMx
Graphical abstract
Highlights
-
•
Targeting Clever-1 with bexmarilimab is well tolerated
-
•
Disease control in late-stage cancer can be achieved with bexmarilimab monotherapy
-
•
Low baseline immune activation associates with bexmarilimab benefit
-
•
Bexmarilimab converts intratumoral macrophages to support adaptive immune responses
Rannikko et al. report safety and preliminary anti-tumor efficacy of a macrophage-targeting immunotherapeutic antibody bexmarilimab in late-stage cancer. They demonstrate macrophage activation and increased IFNγ signaling in patients who achieved disease control during bexmarilimab therapy.
Introduction
In recent years, immune checkpoint blockade has become a standard of care approach in numerous malignancies. Anti-programmed death (ligand)-1 (anti-PD-(L)1) agents inhibit T cell checkpoints resulting in T cell activation and cancer cell killing. However, primary and secondary resistance are common and attempts have been made to overcome this with patient selection, combinatory approaches, and new immunotherapeutic treatment modalities.
Mounting evidence implicates the tumor microenvironment (TME) in driving resistance to immune checkpoint therapy.1 The accumulation of myeloid cells in the TME has been shown to promote tumor growth in several pre-clinical models.2 Clever-1 is a multifunctional scavenger and adhesion receptor expressed by human monocytes, subsets of immunosuppressive macrophages (M2 type), lymphatic endothelial cells, and sinusoidal endothelial cells.3,4 In macrophages, Clever-1 is involved in receptor-mediated endocytosis and recycling, intracellular sorting, and transcytosis of altered and normal self-components. In several cancers, high Clever-1 expression is associated with a poor prognosis, T cell exclusion, impaired antigen presentation, and resistance to immune checkpoint inhibitors.5,6,7 Targeting Clever-1 in various in vivo models delays tumor growth by activating cytotoxic CD8+ T cells and improves responsiveness to anti-PD-1 therapy in refractory models.8,9
Bexmarilimab (FP-1305) is a humanized monoclonal IgG4 antibody specific for human Clever-1. It contains IgG4 (S241P) heavy-chain and kappa light-chain constant regions and has been further optimized by introducing the L248E mutation to avoid Fc receptor binding.10 Thus, bexmarilimab has very low antibody-dependent cellular cytotoxicity and complement-mediated effector functions. Hence, primate toxicology studies with up to 100 mg/kg of bexmarilimab show no toxicologically relevant changes in central biological functions or histologic pathology examinations. Functionally, bexmarilimab inhibits Clever-1-mediated scavenging of modified low-density lipoproteins,11 which reduces activation of nuclear lipid receptors and potentiates tumor necrosis factor α (TNFα) release after LPS stimulus.10 Bexmarilimab is rapidly internalized with Clever-1 in endosomes where it can impair multiprotein vacuolar ATPase-mediated acidification of phago-lysosomes. This activity enhances the ability of macrophages to cross-present antigens to CD8+ T cells.11
To investigate the potential of bexmarilimab in inducing anti-tumor immune responses, a phase I/II first-in-human clinical trial (MATINS; NCT03733990) was designed to study the safety, tolerability, and early efficacy of bexmarilimab in patients with selected advanced or metastatic solid tumors (Figure S1). This basket trial approach enabled us to identify responding cancer types and biomarkers related to response. Indeed, initial results from part I of the MATINS trial shows evidence that targeting Clever-1 promotes peripheral T cell activation in a subset of patients. Here, we report part I and part II of the MATINS trial and show that patients responding favorably to bexmarilimab have low baseline systemic cytokine levels. With spatial transcriptomics profiling of pre- and post-treatment biopsy samples we show that the response coincided with intratumoral macrophage conversion and induction of adaptive immune responses.
Results
Patients and treatments in part I
Between December 13, 2018, and January 16, 2020, 30 patients with advanced colon, pancreatic, biliary tract, ovarian, or hepatocellular carcinomas or checkpoint-inhibitor-refractory cutaneous melanomas commenced intravenous bexmarilimab in five escalating dose cohorts of 0.1, 0.3, 1, 3, and 10 mg/kg every 3 weeks with five to seven patients in each dose level. The median age of included patients was 65 years (range, 30–81 years) and all had exhausted standard-of-care treatment options, with a median number of four therapy lines for advanced disease (Table 1) (range, 1–8). The median number of doses for bexmarilimab was three to five across the different dose cohorts and the median time on treatment was 2.2 months (range, 0.8–13.7).
Table 1.
Patient characteristics
| Characteristic | Number (%) Part I, n = 30 |
Number (%) Part II, n = 108 |
|---|---|---|
| Age, median | 65 | 60 |
| Gender | ||
| Male | 8 (27) | 59 (54) |
| Female | 22 (73) | 51 (46) |
| ECOG PS | ||
| 0 | 11 (37) | 45 (41) |
| 1 | 19 (63) | 65 (59) |
| Cancer type | ||
| Colorectal cancer | 15 (50) | 28∗ (26) |
| Pancreatic cancer | 8 (27) | 10 (9) |
| Ovarian cancer | 2 (7) | 10 (9) |
| Cutaneous melanoma | 2 (7) | 10 (9) |
| Biliary tract cancer | 2 (7) | 10 (9) |
| Hepatocellular carcinoma | 1 (3) | 10 (9) |
| Gastric adenocarcinoma | – | 10 (9) |
| Uveal melanoma | – | 10 (9) |
| ER+ breast cancer | – | 10 (9) |
| No. of previous lines of therapies, median | 4 | 3 |
| PD-1/PD-L1 antibodies as prior therapy | ||
| Cutaneous melanoma | 2 (100) | 10 (100) |
| Hepatocellular carcinoma | 0 (0) | 4 (40) |
| Colorectal cancer | 0 (0) | 2 (7) |
| Ovarian cancer | 1 (50) | 2 (20) |
| Uveal melanoma | – | 1 (10) |
ECOG PS, Eastern Cooperative Oncology Group performance status.
∗Two part I patients were included in part II.
Pharmacokinetics, pharmacodynamics, and immunogenicity in part I
The collected blood samples for determination of bexmarilimab showed rapid elimination of the investigational medicinal product with a terminal half-life of 13.9 h and a dose proportional elimination (Figure S2A). More than 50% receptor occupancy in circulating monocytes was observed at day 2 with dose levels higher than 1 mg/kg, but this level of occupancy did not remain throughout the entire treatment cycle and was markedly reduced by day 8 (Figure S2B). Target engagement up to 70% from baseline as measured by bexmarilimab binding on circulating soluble Clever-1 was observed at higher doses (3–10 mg/kg) and remained for 8–15 days (Figure S2C), implicating that longer target engagement is achieved with higher doses. Anti-drug antibodies were observed in three patients (12%) at cycle 2. As reported earlier, all tested dose levels resulted in increased circulating natural killer (NK) cells, CD8+ T cells and B cells, and a decreased number of circulating FoxP3+ regulatory T cells. Furthermore, circulating CD8+ and CD4+ T cells showed upregulation of CD25, CXCR3, and CD69, suggesting activation of adaptive immune responses.11
Patients and treatments in part II
Based on the clinical efficacy (disease control rates) and circulating biomarker (immune cells and cytokines) data seen in part I of the study,11 the data monitoring committee of the trial recommended a dose selection of 1.0 mg/kg every 3 weeks for part II and investigation of additional dose cohorts of 0.3 mg/kg and 3 mg/kg every 3 weeks in colorectal cancer to fully validate the optimal dosing regimen for bexmarilimab. For part II, the patients commenced treatment between February 22, 2020, and April 16, 2021. At the data cut-off, 108 patients dosed in 11 cohorts (10 patients per cohort) of specific cancer types were fully enrolled, while the anaplastic thyroid cancer cohort was still recruiting patients. Patient characteristics are presented in Table 1. In brief, the patients had received a median of three previous therapy lines in the advanced disease setting (range, 0–8). Previous PD-(L)1 targeted therapy was used in all cutaneous melanoma (100%), four hepatocellular cancer (40%), two ovarian cancer (20%), and two colorectal cancer patients (7%).
Safety in part I and part II
In part I of the study, no dose-limiting toxicities (DLTs) were observed, and a maximum tolerated dose (MTD) could not be defined. In part I and II, of the 138 patients who had received at least one dose of bexmarilimab, 50% reported treatment-related adverse events (AEs), of which 8.7% were grade 3–4 events, and no treatment-related deaths were observed. Of the treatment-related AEs, fatigue, pyrexia, and anemia were the most common and predominantly presented as low-grade events (Table 2). No association between the dose of bexmarilimab and treatment-related AE was observed (Table S1). Of the grade ≥3 treatment-emergent AEs, anemia (5.8%) and ascites (5.8%) were the most common (Table S2).
Table 2.
Treatment-related AEs in part I and part II
| Treatment-related AEa | All grades (n = 138) Number (%) |
Grade 3–4 (n = 138) Number (%) |
|---|---|---|
| Any | 69 (50.0) | 12 (8.7) |
| Blood and lymphatic system disorders | ||
| Anemia | 9 (6.5) | 2 (1.4) |
| Gastrointestinal disorders | ||
| Nausea | 7 (5.1) | 0 |
| Vomiting | 5 (3.6) | 0 |
| General disorders | ||
| Fatigue | 27 (19.6) | 1 (0.7) |
| Pyrexia | 12 (8.7) | 0 |
| Investigations | ||
| Aspartate aminotransferase increased | 7 (5.1) | 1 (0.7) |
| Alanine aminotransferase increased | 6 (4.3) | 0 |
| Blood alkaline phosphatase increased | 8 (5.8) | 0 |
| Metabolism and nutrition disorders | ||
| Decreased appetite | 5 (3.6) | 0 |
Since the proposed main mode-of-action of bexmarilimab is immune activation, immune-related AEs (irAEs) were of special interest. In part I, irAEs leading to treatment discontinuation were recorded in two patients (irAE class: pneumonitis of grade 1, dermatitis, myositis, thyroiditis, of grade 2, and hepatitis of grade 4), at cycles 8 and 11. The observed irAEs responded to bexmarilimab discontinuation and administration of systemic corticosteroids. In part II, eight patients developed potential irAEs (irAE class: hepatitis, colitis, pancreatitis, parathyroiditis, thyroiditis, and dermatitis), all of which resolved without sequelae (Table S3).
Efficacy in part I and II
In part I of the study, two patients—one with colorectal cancer (pretreated with five previous lines of systemic therapy) and one with cutaneous melanoma (pretreated with four previous lines of systemic therapy including anti-PD-1 and anti-CTLA-4)—had tumor shrinkage in response to bexmarilimab at dose levels of 0.3 and 1 mg/kg, respectively. The microsatellite stable, K-RAS wild type colorectal cancer patient had a long-lasting partial response (PR) according to RECIST 1.1, and while the treatment was stopped after cycle 8 due to immune-related toxicity, the response lasted until the end of follow-up (>100 days from last dose of bexmarilimab) (Figure S3). The melanoma patient who had PR in target lesions simultaneously developed symptomatic brain metastasis and was taken off study.
Of the patients in part I and II, RECIST 1.1-defined PR rate at cycle 4 was observed in one patient (1%) and stable disease (SD) in 18 patients (13%), corresponding with a disease control rate (DCR = PR + SD) of 14%. DCR was the highest in cutaneous melanoma (25%), biliary tract cancer (25%), gastric adenocarcinoma (30%), hepatocellular cancer (40%), and estrogen receptor positive (ER+) breast cancer (40%) (Table 3), translating to prolonged treatment durations (Figure 1A). Progression-free survival (PFS) and overall survival (OS) were analyzed in all patients treated in part I and II of the study (n = 138). The median PFS was 59 days (95% confidence interval [CI], 58–61 days), and OS 151 days (95% CI, 118–190 days) (Figure 1B). To define the relationship between disease control (DC) and OS, an exploratory landmark analysis according to response was conducted. The results showed improved survival (HR, 0.139; 95% CI, 0.034–0.575) for patients with DC compared with non-DC patients at cycle 4 (Figure 1C), while the duration of the previous line of therapy prior to entering the trial was similar for patients with or without DC (Figure 1D). The survival difference between DC and non-DC patients was the most drastic in cutaneous melanoma and biliary tract cancer, where no deaths were observed in the DC patients (Figure S4). The PFS2/PFS1 ratio (defined as the PFS on bexmarilimab/duration of previous treatment line) was >1.3 in 16% of patients. The PFS2/PFS1 ratio was positive in 42% of DC patients and in 12% of non-DC patients (p = 0.0031) (Table S4).
Table 3.
ORR and DC rates in parts I and II at cycle 4
| Number (%) (n = 138) | |
|---|---|
| ORR | 1 (1) |
| CR | 0 (0) |
| PR | 1 (1) |
| SD | 18 (13) |
| PD | 119 (86) |
| DCR | 19 (14) |
| Colorectal cancer (n = 43) | 2 (5) |
| Pancreatic cancer (n = 18) | 0 (0) |
| Ovarian cancer (n = 12) | 0 (0) |
| Cutaneous melanoma (n = 12) | 3 (25) |
| Biliary tract cancer (n = 12) | 3 (25) |
| Hepatocellular carcinoma (n = 11) | 4 (36) |
| Gastric adenocarcinoma (n = 10) | 3 (30) |
| Uveal melanoma (n = 10) | 0 (0) |
| ER+ breast cancer (n = 10) | 4 (40) |
ORR, objective response rate; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease; DCR, disease control rate. See also Figure S3.
Figure 1.

Bexmarilimab responses in patients showing DC
(A) Swimmer plot analysis for treatment durations and tumor responses in melanoma, gastric, and hepatocellular cancer patients of part II. The y axis shows individual patients, x axis time in days from the first bexmarilimab dose. C, cycle number; FU, follow-up visit; SURV, survival visit. Green circle, SD response; red star, PD response; blue asterisk, PD reported as AE; X: death of subject.
(B) Kaplan-Meier analysis for PFS (red) and OS (blue).
(C) Landmark Kaplan-Meier analysis for OS from cycle four according to PR/SD (red) or PD (blue).
(D) Kaplan-Meier analysis for previous therapy line treatment duration (prior entering the trial) according to PR/SD (red) or PD (blue). Circles indicate censored events. y axis, time in days.
(E) Baseline levels of IFNγ and TNFα according to DC status.
(F) Change of IFNγ levels during the first cycle of treatment according to DC. Error bars represent SE. Blue, non-DC patients; red, DC patients; SE, standard error; ∗∗p = 0.0056 for difference between DC and non-DC in the change from day 1, repeated measures ANOVA. See also Figures S2, S4, and S6, and Table S4.
Biomarker analysis of MATINS patients
Pre-treatment tumor samples were stained for Clever-1 and PD-L1 in the tumor cohorts with one identified PR or high DC (colorectal cancer, cutaneous melanoma, gastric, biliary tract and hepatocellular carcinoma). There were 78 (100%) and 43 patients (55%) who had pre-treatment tumor biopsy staining results available for Clever-1 and PD-L1, respectively. The percentage of Clever-1 positive cells and PD-L1 combined positive score (CPS) are presented in Table 4. Higher intratumoral Clever-1 positivity correlated significantly with DC (median, 15% vs. 3%; p = 0.038). Lower PD-L1 CPS was observed in DC patients (median, 1%; range, 0%–2%) in comparison to non-DC patients (median, 5%; range, 0%–100%), but this was not statistically significant. Combined PD-L1/intratumoral Clever-1 assessment showed a significantly lower PD-L1 to Clever-1 ratio in DC patients (median ratio, 0.087 vs. 1.67; p = 0.0093) (Table 4 and Figure S5).
Table 4.
Clever-1 and PD-L1 expression in pre-treatment tumors
| No. (%) | Median % (range) | p value∗ | |
|---|---|---|---|
| Whole tumor Clever-1 | 78 (100) | 15 (1–55) | |
| Non-DC | 71 (91) | 15 (1–55) | 0.064 |
| DC | 7 (9) | 20 (13–35) | |
| Stromal Clever-1 | 78 (100) | 20 (0–75) | |
| Non-DC | 71 (91) | 20 (0–75) | 0.746 |
| DC | 7 (9) | 20 (5–40) | |
| Intratumoral Clever-1 | 78 (100) | 5 (0–85) | |
| Non-DC | 71 (91) | 3 (0–85) | 0.038 |
| DC | 7 (9) | 15 (0–25) | |
| PD-L1 CPS | 39 (100) | 2 (0–100) | |
| Non-DC | 35 (90) | 5 (0–100) | 0.289 |
| DC | 4 (10) | 1 (0–2) | |
| PD-L1/intratumoral Clever-1 | 36 (100) | ||
| Non-DC | 32 (90) | 1.67 (0–30) | 0.0093 |
| DC | 4 (10) | 0.087 (0–0.3) | |
DC, disease control; CPS, combined positive score; ∗Wilcoxon two-sample test. See also Figures S5 and S6.
Circulating cytokines were analyzed at baseline and over the treatment in the tumor cohorts with the highest DC rates (n = 65). Low baseline TNFα was associated with DC (p = 0.0011) and there was a tendency for lower baseline interferon γ (IFNγ) levels in DC patients (p = 0.067) (Figure 1E). A statistically significant increase of IFNγ during cycle 1 was selectively observed in DC patients (p = 0.0056; day 29) (Figure 1F) and was also associated with high tumor Clever-1 positivity (cut-off 3%; p = 0.018) (Figure S6). Altogether, these data suggest that low baseline immune activation is associated with bexmarilimab benefit.
Profiling of DC and non-DC patient tumor gene expression by spatial transcriptomics
To more comprehensively study bexmarilimab activity in the TME, we performed GeoMx digital spatial profiling combined with next-generation sequencing on pre- and post-treatment tumor biopsies. For the analysis, we selected two cancer types with high DC, namely, biliary tract cancer and ER+ breast cancer, and included patients with both pre- and post-treatment biopsies available (DC, n = 3; non-DC, n = 3). The biopsies were stained for CD68, CD31, and pan-cytokeratin to analyze gene expression separately in macrophages (CD68+), vessels (CD31+), and the remaining tumor area (CD68−CD31−) (Figures 2A and S7A, and STAR methods). Both CD68+ and CD31+ subsets are characterized to be Clever-1 positive in various tumors.5
Figure 2.

GeoMx spatial profiling reveals pro-inflammatory conversion of TAMs in DC patients
(A) Schematic for GeoMx spatial transcriptomics profiling of pre- and post-treatment biopsies from non-DC (n = 3 patients, 30 ROIs) and DC (n = 3 patients, 33 ROIs) patients. Representative images of morphology marker staining and corresponding segmentation are displayed for a single ROI.
(B) Volcano plot showing differentially expressed genes in DC patient CD68+ areas after bexmarilimab therapy.
(C) Bubble plots of top up- and downregulated pathways in CD68+ tumor areas of DC patients after bexmarilimab therapy (gene set enrichment analysis).
(D) Volcano plot showing differentially expressed genes in non-DC patient CD68+ areas after bexmarilimab therapy.
(E) Bubble plots of top up- and downregulated pathways in CD68+ tumor areas of non-DC patients after bexmarilimab therapy (gene set enrichment analysis).
(F) Heatmap of M1 and M2 macrophage gene scores on CD68+ area. Red color indicates higher M1 or M2 gene expression than overall gene expression, blue color lower. Each tile represents median of patient’s ROIs.
(G) Bar graph of M2/M1 score ratios corresponding to scores shown in (F). Each point represents patient median across ROIs and each bar patient group median.
(H) Volcano plot showing differentially activated pathways between CD68+ areas of non-DC and DC patients’ pre-treatment biopsies (gene set enrichment analysis, n = 16 ROIs [non-DC], n = 10 ROIs [DC]). Pre, pre-treatment biopsy; Post, post-treatment biopsy; ROI, region of interest; Padj, Benjamini-Hochberg-adjusted p value; ∗∗Padj < 0.01; ∗Padj < 0.05; ns, not significant. In (C) and (E), red color denotes pathway activation and blue downregulation. See also Figures S7–S9, and Supplementary Data S1 and S2.
Gene expression profiles of 63 biopsy regions, corresponding to a total of 180 segments (63 CD68+ and CD68−CD31− segments, 54 CD31+ segments) were obtained after excluding segments with a low signal-to-noise ratio (Figure S7B). The overall gene expression profiles of the segments were compared by unsupervised hierarchical clustering (Figure S7C) and principal component analysis (Figure S7D), which showed that the segment type and cancer type primarily drove differences in gene expression, as expected after successful segmentation. To further evaluate the purity of the transcriptomes in CD68+ and CD31+ areas, we performed cell type deconvolution. The resulting cell type abundancy scores validated the CD68+ segments to mainly contain macrophages and CD31+ segments to contain endothelial cells (Figure S8). Expectedly, the expression of Clever-1 (STAB1) was most abundant in the CD68+ and CD31+ segments (Figure S9A).
Bexmarilimab activates IFN signaling and M1-like gene expression in tumor-associated macrophages of MATINS DC patients
By investigating the gene expression changes in tumor-associated macrophages (TAMs) (CD68+) between pre-and post-treatment biopsies we observed upregulation of IFN-inducible genes (ICAM1, SLAMF7, GBP4, GBP5, and CD40) as well as chemokines (CCL4L2, CCL5, and CCL2) (Figure 2B and Data S1) selectively in DC patients. For instance, SLAMF7 drives strong activation and pro-inflammatory cytokine secretion in macrophages under inflammatory conditions.12 Moreover, significant upregulation of IFN signaling, antigen presentation on class I major histocompatibility complex (MHC) and adaptive immune response pathways was observed in DC patients’ TAMs after bexmarilimab therapy (Figures 2C and S9B and Data S2). In strong contrast, non-DC patients’ TAMs showed only few differentially expressed genes and downregulation of pathways related to immune system activation, such as IFNγ signaling, antigen processing and interleukin signaling (Figures 2D and 2E).
To further examine the changes in TAM phenotypes after bexmarilimab therapy, we calculated M1 and M2 macrophage scores in each CD68+ segment based on the M1 and M2 macrophage genes published by Martinez et al.13 (Figures S9C–S9E). The M1 scores increased and the M2/M1 score ratios decreased in all DC patients after bexmarilimab therapy, while similar changes were not observed in non-DC patients (Figures 2F and 2G), suggesting a beneficial pro-inflammatory conversion of TAMs specifically in DC patient tumors.
Unlike in the CD68+ tumor area, the CD31+ vessels showed alterations in pathways not directly related to immune system function rather than translation and extracellular matrix (ECM) modulation (Figures S10A–S10D). Notably, the integrin cell surface interaction pathway was downregulated during the treatment in both DC and non-DC patients’ vessels indicating decreased leukocyte binding (Figures S10B and S10D). Indeed, the parent antibody (3-372) of bexmarilimab has been shown to decrease leukocyte binding and transmigration via vascular endothelium.14,15
While investigating whether pre-existing differences in TAM phenotypes could explain subsequent bexmarilimab responses, we found only a few significantly altered pathways between DC and non-DC patients. Essentially, DC patients’ TAMs expressed higher levels of genes modulating the ECM (Figure 2H). In summary, these data demonstrate the ability of bexmarilimab to activate TAMs within human tumors. Whereas clearly different responses were observed in DC and non-DC patients’ TAMs, the TAMs were not widely different prior to therapy.
TAM activation is reflected to adjacent immune cells
We next evaluated how the CD68−CD31− tumor region was altered in DC patients to reflect the observed pro-inflammatory activation of TAMs. Significantly increased transcription was observed for IFN signaling genes (IFI16, IFI44, GBP1, GBP5, and CD40), immune cell attracting chemokines (CCL4L2, CCL5, CXCL9, CCL2, and CXCL16), lymphocyte and NK cell markers (CD3E, CD8A, CD2, NKG7, and FCGR3A), and MHC class I and II proteins (e.g., HLA-B, HLA-DRA, and HLA-DPA1) (Figure 3A and Data S1). These signs of strong immune activation were confirmed by pathway analysis that showed significant upregulation of IFNγ signaling and phosphorylation of CD3 and T cell receptor (TCR) zeta chains after bexmarilimab therapy (Figure 3B and Data S2). As with TAMs, similar changes were not observed in non-DC patients’ TME (Figures S10E and S10F). Using the SpatialDecon algorithm, we quantified the immune cell types in the CD68−CD31− segments (Figure S10G), which showed bexmarilimab-induced increase in CD4+ and CD8+ T cell abundancies in both DC and non-DC patients, while NK cell, B cell, and macrophage abundancies were increased exclusively in DC patients (Figure 3C).
Figure 3.

Tumor-infiltrating leukocyte infiltration and activation following bexmarilimab therapy in DC patients
(A) Volcano plot showing differentially expressed genes in DC patient CD68−CD31− areas (n = 3 patients, n = 10 ROIs [Pre] and 23 ROIs [Post]) after bexmarilimab therapy.
(B) Bubble plots of top up- and downregulated pathways in CD68−CD31− areas of DC patients after bexmarilimab therapy (gene set enrichment analysis). Red color denotes pathway activation and blue downregulation.
(C) Bar plots of cell abundancy scores calculated for the indicated immune cell types based on gene expression on CD68−CD31− area. Median ± interquartile range, points represent individual ROIs, n = 3 patients per group.
(D) Enrichment map of significantly altered pathways (Padj < 0.05) in CD68+ areas of DC patients after bexmarilimab therapy. The map illustrates how these pathways altered upon bexmarilimab therapy on CD68+, CD31+, and CD68−CD31− areas. Red indicates significant upregulation, blue significant downregulation and gray non-significant changes. Circle size represents pathway size and connecting line width represents the proportion of shared dataset genes between two pathways (n = 3 patients).
(E) Principal component analysis of pre-treatment biopsy ROIs based on CD68−CD31− area gene expression (n = 11,591 genes).
(F) Volcano plot showing differentially activated pathways between CD68−CD31− areas of non-DC and DC patients’ pre-treatment biopsies (gene set enrichment analysis, n = 16 ROIs [non-DC], n = 10 ROIs [DC]). Top five pathways (lowest Padj) were annotated. BTC, biliary tract cancer; ER+ BRCA, estrogen receptor-positive breast cancer; mDC, myeloid dendritic cell; pDC, plasmacytoid dendritic cell; Treg, regulatory T cell; Pre, pre-treatment biopsy; Post, post-treatment biopsy; ROI, region of interest; Padj, Benjamini-Hochberg-adjusted p value; ∗∗Padj < 0.01; ∗Padj <0.05; ns, not significant. See also Figure S10, and Supplementary Data S1 and S2.
The overall pathway changes in DC patients visualized as an enrichment map showed that integrin and ECM interaction related pathways were downregulated specifically in the Clever-1+ tumor areas (CD68+ and CD31+), but not in the remaining CD68−CD31− area, which reflects the aforementioned role of Clever-1 as an adhesion molecule and successful segmentation of these three areas (Figure 3D). Furthermore, upregulation of IFN signaling and lymphocyte activation as well as downregulation of translation and respiratory electron transport were mutually observed in the CD68+ and CD68−CD31− tumor areas, but not in the CD31+ vasculature (Figure 3D).
To find common features of bexmarilimab-responsive tumors, we compared pre-treatment biopsy gene expression between DC and non-DC patients. While the CD68+ biopsy areas were relatively similar between DC and non-DC patients (Figure 2H), the CD68−CD31− areas clustered based on the response rather than the cancer type and had a high number of differentially activated pathways between DC and non-DC patients (Figures 3E and 3F). Higher expression of genes related to respiratory electron transport and lower expression of genes related to interleukin signaling in DC patient pre-treatment biopsies were observed (Figure 3F and Data S2). These observations suggest that the surrounding TME has a greater influence on bexmarilimab therapy outcome than existing TAM phenotypes. Collectively, these data illustrate that bexmarilimab therapy upregulates antigen presentation, T cell activation, and IFN signaling in tumors of DC patients, which results in enhanced chemokine production and immune cell recruitment into the TME.
Discussion
Targeting macrophages to break the immune tolerance of tumors and help to activate host immune defenses is the next cutting edge in cancer immunotherapy. Macrophage-targeted therapies could be used along with various other treatments and open entirely new therapeutic options.16,17,18 Here we report the first-in-human results of the phase I/II MATINS study, evaluating bexmarilimab, a Clever-1-targeting antibody, in patients with advanced cancer, who have exhausted standard therapeutic options. Clever-1, a scavenger receptor, is highly expressed by immunosuppressive macrophages and targeting of the receptor with therapeutic antibodies has previously been shown to result in re-programming of macrophages, reversion of macrophage-mediated immunosuppression, revitalization of antigen presentation, and T cell-mediated anti-cancer immunity.8,9,11 Thus, targeting Clever-1 represents a mechanism of action not previously investigated as anti-cancer therapy.
Bexmarilimab administration was associated with favorable tolerability with no observed DLTs in part I, and an MTD could not be defined. In part II, no additional safety signals were seen, and registered treatment-related AEs were typically low grade. The observed excellent safety profile of the drug makes it feasible to test bexmarilimab in combination with other anti-cancer agents. It is worth noting that several study patients developed irAEs after relatively long exposure to bexmarilimab, which were manageable with corticosteroids. Some of these patients also seemed to benefit from the treatment with PR or prolonged tumor stabilization. Similarly, irAEs have been linked to higher frequency of tumor responses in advanced cancer patients receiving PD-1 targeting antibodies.19
While efficacy was not the primary endpoint of the study, we found interesting preliminary evidence of anti-tumor activity for bexmarilimab. Although the RECIST 1.1-defined objective response rate was low in the entire study population, promising DC and long bexmarilimab treatment durations were observed in several patients, especially in cutaneous melanoma, gastric cancer, biliary tract cancer, hepatocellular, and ER+ breast cancer. Durable disease stabilization was associated with immune activation, as seen in the TME as well as a systemic increase in circulating IFNγ levels. Treatment benefit measured by DC was also seen in anti-PD-(L)1 pretreated patients suggesting that single-agent bexmarilimab has activity also in immunotherapy-refractory patients. No other clear associations between the traditional baseline factors and DC were observed in the study. Tumor biomarker analysis showed an association between high baseline tumor Clever-1 expression and DC. Since some non-DC patients also showed high Clever-1 expression, the data presented here are hypothesis generating. Thus, Clever-1 expression and the optimal cut-off levels as a patient enrichment strategy for bexmarilimab therapy needs further optimization and validation. As the current MATINS data consist of many tumor types, the enrichment strategy will most likely need to be validated for each tumor type separately in a prospective clinical trial. We also observed low baseline circulating levels of TNFα to be associated with DC, and IFNγ increases were observed in DC and tumor Clever-1 high patients. Low baseline circulating cytokine levels and tumor PD-L1 CPS score along with observed IFNγ increases in DC patients, including those previously treated with immune checkpoint inhibitors, could indicate that bexmarilimab is able to rewire the macrophage-mediated immunosuppression and promote T cell-mediated anti-tumor activity.
Indeed, when looking at macrophage activation signals by spatial transcriptomics, there was a clear re-programming of TAMs in DC patients reflecting the findings observed in the peripheral biomarker analysis. The observed pro-inflammatory phenotype conversion, TFRC upregulation, and downregulation of plasma lipoprotein remodeling was in line with the described changes in circulating monocytes after bexmarilimab therapy,11 indicating that monocytes can retain at least some of the properties induced by bexmarilimab in circulation while differentiating into TAMs. The immune activation was not restricted to TAMs, as the neighboring cells showed robust signs of phosphorylation of CD3 and TCR zeta chains, and co-stimulation by the CD28 family suggesting that macrophages in these areas were in close contact with T cells to facilitate their activation. These signs of TAM and lymphocyte activation were observed specifically in DC patients, but not in non-DC patients. The changes in CD31+ vessels, however, were much more similar between DC and non-DC patients, suggesting that bexmarilimab treatment benefit is associated with Clever-1 blockade in macrophages rather than endothelial cells.
It seemed that bexmarilimab increased immune cell abundance in tumors, which is contrary to published research describing impaired immune cell trafficking after Clever-1 blockade. While there is some selectivity in Clever-1-regulated trafficking,20 a recent report by Steele and colleagues21 showed that tumor lymphatics control the egress of T cells after antigen exposure by ACKR3 upregulation, thus reducing CXCL12 sensitivity and promoting their retention. The same principle may occur with Clever-1 targeting as we have previously shown that lymph node lymphatics in Clever-1 knock-out mice upregulate Ackr2 after ovalbumin-CFA administration.22 While the cell type deconvolution was performed for CD68−CD31− area, some macrophage signal inevitably remained on this segment, facilitating macrophage abundancy estimation. Unexpectedly, we detected increased macrophage abundance after bexmarilimab therapy in DC patients (Figure 3C). Inspection of representative CD68+ staining images confirmed this finding and we also observed upregulation of CCL2 in CD68+ and CD68−CD31− areas of DC patient biopsies (Figure S7A and Data S1). Typically, an increased number of TAMs would indicate a poorer outcome. However, due to the possible conversion of macrophage phenotype with bexmarilimab the recruited monocytes were no longer immunosuppressive in nature and could differentiate into immunostimulatory macrophages to support anti-tumor responses.
Curiously, some of the most upregulated genes in the CD68+ area after treatment were immunoglobulin heavy chains that may result from increased B cell numbers, for example, during the formation of tertiary lymphoid structures. Previous findings in Clever-1 knock-out mice show that they have abnormally high antibody levels under resting conditions and enhanced humoral immune responses after immunization with protein and carbohydrate antigens.23 In the MATINS patients, an increase in circulating B cells is detected up to four cycles after treatment,11 which coincides with autoantibody production against targets, such as cancer testis antigen (GAGE2), typical autoimmune disease antigens (SNRPC, TPO, and TOP1), and antigens related to the induction of innate immunity and IFN responses, e.g., TRIM21.24 Alternatively, based on a recent discovery of myeloid cells being capable of genetically recombining and expressing antibody genes,25 it is possible that the observed increase in immunoglobulins relates to bexmarilimab-induced antigen-specific tissue maintenance of macrophages.
It remains unclear what factors characterize patients who benefit from bexmarilimab therapy. The small spatial transcriptomics cohort did not allow us to make conclusive interpretations but gives insight into existing differences between the TME in DC and non-DC patients prior to therapy. The biomarker and profiling data, however, suggest that bexmarilimab would have better efficacy in tumors with low pre-existing circulating IFNγ and interleukin signaling, respectively. Since disease stabilization and prolonged survival was observed with the study drug in conjunction with macrophage re-programming and immune activation, additional analysis was carried out to characterize whether SD responses would be associated with signs of clinically meaningful anti-tumor activity. Interestingly, we observed a prolongation of survival in patients with PR/SD without this being associated with longer previous line of treatment duration, a surrogate for PFS. Furthermore, PFS2/PFS1 ratio of >1.3 has often been proposed to indicate therapeutic benefit,26 and this was observed significantly more frequently in PR/SD than PD patients. This is suggestive for the disease-stabilizing effect of bexmarilimab and, even more important, a prolonged survival effect. A randomized trial is required to fully characterize the survival benefit seen with immune activation achieved with bexmarilimab.
The pharmacokinetic (PK) and pharmacodynamic profile of bexmarilimab was characterized by rapid elimination, receptor occupancy decay in circulating monocytes, and recovery of circulating soluble Clever-1 levels. Because of the faster than anticipated elimination of bexmarilimab, sampling carried out in part I did not enable us to define a comprehensive PK profile of the drug. We speculate that rapid elimination of bexmarilimab might be related to rapid internalization of bexmarilimab and its target as well as the short half-life of circulating monocytes (∼1 day) and replenishment of new ones from the bone marrow. The short elimination of bexmarilimab did not, however, affect the immune-activating pharmacodynamic effects. The further expansion cohorts of the MATINS trial will investigate more frequent dosing intervals and escalating dose levels to fully optimize the dosing and efficacy of bexmarilimab.
In conclusion, we present the results of part I and II of the MATINS trial, investigating immunotherapeutic targeting of Clever-1 with bexmarilimab. Bexmarilimab demonstrated an excellent safety and tolerability profile with immune activation and disease-stabilizing effect as monotherapy in several late-stage, treatment-refractory metastatic solid tumors. Further trials will investigate bexmarilimab for the treatment of acute myeloid leukemia and myelodysplastic syndrome, since blast cells commonly express high levels of Clever-1, and in earlier lines of therapy for the treatment of solid tumors in combination with other anti-cancer agents such as PD-(L)1 inhibitors.
Limitations of the study
We acknowledge this study to have the following limitations that affect the interpretation of the presented results. First, bexmarilimab therapy efficacy was evaluated here based on observed DC, which can be achieved more easily in cancer types that progress more slowly. To evaluate whether DC and non-DC patient groups were comparable prior to bexmarilimab therapy, we analyzed the duration of the previous line of therapy in these groups and observed no difference. In the survival comparisons, landmark analysis excluding early progression/deaths was used to control for selection bias. Furthermore, we also show with the pronounced positivity of the PFS2/PFS1 ratio in DC patients that bexmarilimab treatment leads to prolonged DC and clinically meaningful stabilization in these patients. If bexmarilimab had no effect on these DC patients’ clinical disease behavior, their PFS2 should be shorter than PFS1 leading to similar PFS2/PFS1 ratio among DC and non-DC patients. Second, the overall low number of patients with DC diminishes the strength of our conclusions. However, while the overall DC rate in the study is low, in certain cancers, such as gastric cancer, biliary tract cancer, hepatocellular carcinoma, melanoma, and ER+ breast cancer, the DC was as high as 30%–40%. Finally, the low number of available paired pre- and post-treatment biopsies (n = 12 biopsies, n = 3 DC patients, n = 3 non-DC patients, n = 63 analyzed biopsy regions) limits the generalization of the spatial transcriptomics results. Yet, observing macrophage and lymphocyte activation specifically in DC patients, and not in non-DC patients, suggests these effects to be related to bexmarilimab treatment rather than unrelated processes.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bexmarilimab (FP-1305) | Abzena | Batch: P81901A, P81902A, P81903A |
| Anti-bexmarilimab Fab fragment AbD30055 | Bio-Rad | N/A |
| Mouse anti-human CD14 (clone M5E2, Pacific Blue) | BD Pharmingen | Cat# 558121; RRID: AB_397041 |
| Human IgG4 (S241/L248E) isotype control | Abzena | N/A |
| Rat anti-human Clever-1 (clone AB FUMM 9–11) | InVivo BioTech | AK1013 |
| Mouse anti-human Stabilin-1 (clone 4G9) | Santa Cruz | Cat# sc-293254 |
| Mouse anti-human pan-cytokeratin (clone AE-1/AE-3, AF488) | Novus Biologicals | Cat# NBP2-33200AF488 |
| Mouse anti-human CD68 (clone KP1, AF594) | Santa Cruz | Cat# sc-20060AF594 |
| Mouse anti-human CD31 (clone JC/70A, AF647) | Abcam | Cat# ab215912; RRID: AB_2890260 |
| Chemicals, peptides, and recombinant proteins | ||
| SYTO 83 orange fluorescent nucleic acid stain | ThermoFisher Scientific | Cat# S11364 |
| Critical commercial assays | ||
| PD-L1 IHC 22C3 pharmDx assay | Agilent Technologies | Code SK006 |
| V-PLEX Proinflammatory Panel 1 Human | Meso Scale Discovery | Cat# K15049D |
| GeoMx human whole transcriptome atlas | NanoString | Cat# GMX-RNA-NGS-HuWTA |
| Deposited data | ||
| GeoMx spatial transcriptomics data | This paper | GEO: GSE240138 |
| Software and algorithms | ||
| FlowJo v10.7.1 | BD | https://www.flowjo.com |
| GeoMx DSP Control Center v2.4 | NanoString | https://nanostring.com/products/geomx-digital-spatial-profiler/geomx-dsp-overview/ |
| ImageJ v1.53q | National Institute of Health | https://fiji.sc |
| R v4.0.4 | R Core Team27 | https://www.r-project.org |
| RStudio v1.4.1106 | RStudio, PBC | http://www.rstudio.com/ide |
| ComplexHeatmap v2.6.2 | Gu et al.28 | https://github.com/jokergoo/ComplexHeatmap |
| Cytoscape v3.9.1 | Shannon et al.29 | https://cytoscape.org |
| SpatialDecon v1.0.0 | Danaher et al.30 | https://github.com/Nanostring-Biostats/SpatialDecon |
Resource availability
Lead contact
Any further information or requests should be directed to, and will be fulfilled by the lead contact, Dr. Maija Hollmén (maijal@utu.fi).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Participant-level clinical data is deposited to MATINS trial master database (managed by the trial sponsor) and can be accessed via the corresponding authors by reasonable request. The GeoMx spatial transcriptomics data have been deposited at the GEO database under the accession number GSE240138.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study Participant details
Patients
This international, first-in-human, open-label, non-randomized phase I/II, dose escalation study (Figure S1) was run according to Good Clinical Practice and the Declaration of Helsinki. The study was approved by local institutional review boards and national medicinal agencies (MATINS; NCT03733990, EudraCT 2018-002732-24). All participants provided written, informed consent before any trial-related investigations or treatment took place. Eligible patients had advanced (inoperable or metastatic), treatment-refractory, histologically confirmed hepatocellular carcinoma, gallbladder or intra- or extrahepatic biliary tract carcinomas, colorectal cancer, serous poorly differentiated ovarian cancer, pancreatic ductal adenocarcinoma, or immune checkpoint inhibitor refractory cutaneous melanoma without standard treatment options available. In addition, part II included patients with uveal melanoma, gastric adenocarcinoma, ER + breast cancer, and anaplastic thyroid cancer. Patients were ≥18 years old with a life expectancy of ≥12 weeks, an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, adequate organ function, and no ongoing systemic infections, brain metastasis, history of autoimmune disease (except type 1 diabetes, celiac disease, hypothyroidism requiring only hormone replacement, vitiligo, psoriasis, or alopecia), use of concurrent antineoplastic therapies, or systemic steroids, and measurable disease (part II only). Detailed eligibility criteria can be found at https://www.clinicaltrials.gov/study/NCT03733990.
Method details
Procedures
The selection of the initial dose of 0.3 mg/kg was based on the nonclinical safety findings and affinity, binding, and receptor occupancy of bexmarilimab to Clever-1 and monocytes in vitro. No relevant toxicity was observed with 100 mg/kg dosing in non-human primates and 0.3 mg/kg dose should result in Clever-1 receptor occupancy in monocytes. Dose escalation for 0.1, 0.3, 1, 3, and 10 mg/kg (every 3 weeks intravenously) was characterized by two-stage time-to-event continual reassessment method (TITE-CRM) which allowed for more accurate determination of the MTD and staggered patient accrual without the need for complete DLT follow-up of previously treated patients, compared to conventional dose-finding designs.31 AEs were collected according to the National Cancer Institute-Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 5.0.
Outcomes
The primary objectives of part I of the study were to characterize safety, tolerability, MTD, and define the recommended dose of bexmarilimab for parts II and III of the trial. Secondary objectives included pharmacokinetic (PK) profiling, evaluation of immunogenicity of bexmarilimab, and preliminary efficacy according to the objective response rate (ORR) and immune-related ORR. DLTs were defined as a treatment-related grade ≥3 AE or laboratory abnormality occurring ≤63 days following the first dose of bexmarilimab with exceptions of grade 3 infusion reactions, nausea/vomiting, thrombocytopenia, or neutropenia. Furthermore, grade 2 AST/ALT accompanied with 2x ULN elevation of bilirubin (with normal AST/ALT at baseline), or dose delay of second cycle of bexmarilimab due to drug-related toxicity by ≥ 14 days were considered as DLTs. In part II, the primary objectives were safety, tolerability and preliminary efficacy of bexmarilimab monotherapy with ORR, DCR, and immune-related ORR in distinct cancer type specific expansion groups. Secondary objectives included Clever-1 expression in each tumor type, PK profile and immunogenicity of bexmarilimab, predictive biomarkers of efficacy, and duration of response.
Pharmacokinetics and receptor occupancy
The PK profile of a single dose (during cycle 1) and repeated doses (during cycles 1–5) of bexmarilimab were determined by repeated measurements of the drug concentration in the circulation. Peak concentration (Cmax), through concentration (Cmin), area under the plasma concentration versus time curve (AUC), clearance, volume of distribution, terminal half-life (t1/2), receptor occupancy (RO) on circulating monocytes and circulating soluble Clever-1 levels at each dose level were determined.
PK analysis
Anti-bexmarilimab Fab Fragment AbD30055 (Bio-Rad) was coated on 96-well plates and blocked. Dilution series of bexmarilimab was used for standard curve preparation. The standard and the diluted samples were added to the wells. After washing, the assay was visualized by the subsequent additions of HRP-labelled mouse anti-human IgG4 (Fc) antibody and a chromogenic substrate (TMB). The concentration of bexmarilimab in samples was back calculated from a calibration curve.
RO analysis
Peripheral blood mononuclear cells were plated at 0.2×106 cells/well in round-bottom 96-well plates. All wells were stained with anti-human CD14-Pacific Blue (clone M5E2, BD Pharmingen) together with 10 ng/μL in-house conjugated (AF647) anti-Clever-1 antibody bexmarilimab (competitor). In-house conjugated Irrelevant isotype control human IgG4 (S241/L248E) was used for signal normalization. Flow cytometry was run on the LSRFortessa (BD) and analyzed with FlowJo software v10.7.1 (BD).
Target engagement on circulating Clever-1
Serum samples were analyzed for bexmarilimab target engagement on soluble Clever-1 using a sandwich-ELISA (enzyme-linked immunosorbent assay) method, in which capture antibody (9–11), blocking agents, samples, detecting antibodies (biotinylated bexmarilimab), and finally detection agents (Eu-Labeled Streptavidin) were introduced to the wells in successive incubation periods followed by a washing step in between. The baseline sample value was set as 1 and the proceeding samples in cycle 1 are shown as percent decrease from baseline indicating the level of bexmarilimab binding on soluble Clever-1.
Tumor biomarker analysis
Pre-treatment tumor samples were immunohistochemically (IHC) stained for Clever-1 and PD-L1, and circulating cytokines were analyzed using a panel approach.
IHC analysis for Clever-1 and PD-L1
FFPE samples with 4–5 μm sections were stained with Ventana Benchmark Ultra (Roche Diagnostics, Basel, Switzerland). For Clever-1 staining, UltraView Universal DAB Detection Kit (Roche Diagnostics) combined with Clever-1 primary antibody (clone 4G9, Santa Cruz, cat. sc-293254) at 1:100 dilution was used. PD-L1 staining was performed using 22C3 pharmDx assay (Agilent Technologies) according to manufacturer’s instructions. Interpretation and scoring were performed by board-certified pathologists using bright-field microscopy.
Percentage of Clever-1 positive viable cells (the number of all Clever-1 positive cells divided by the total number of cells, multiplied by 100) were scored irrespectively of location, intratumorally, and in stroma. PD-L1 was scored as combined positive score (CPS) by calculating the number of PD-L1 staining cells (tumor cells, lymphocytes, macrophages) divided by the total number of viable tumor cells, multiplied by 100.
Circulating cytokine analysis
Samples for circulating cytokines were collected on cycles 1–4. On the cycles 1,2, and 4, analysis was done pre-dose, day 2, day 8, and day 15, and on cycle 3, pre-dose. Cytokines were measured from serum with V-PLEX Proinflammatory Panel 1 Human from Meso Scale Discovery (MSD, Rockville, Maryland, US) by Translational Biomarker Solutions, Labcorp Drug Development, UK.
GeoMx digital spatial profiling of tumor biopsies
The spatial gene expression profiles of pre- and post-treatment tumor biopsies were analyzed using NanoString’s GeoMx Digital Spatial Profiler (DSP). The method uses immunofluorescence staining to annotate tissue morphology and RNA probes coupled to photocleavable oligonucleotide tags to measure gene expression. Oligonucleotide tags are released from selected tissue regions using UV exposure (area of illumination, AOI) and quantitated by next-generation sequencing (NGS). Tissue sections (5 μm) from tumor samples were mounted on Superfrost slides and baked at 60°C in a drying oven for 1 h. After deparaffinization and rehydration, the protein targets were retrieved by dipping the slides into DEPC-treated water at 100°C for 10 s, followed by incubating the slides in 1x Tris-EDTA (pH 9.0) at 100°C for 20 min without further heating. The slides were then washed with phosphate-buffered saline (PBS) at room temperature for 5 min. To expose the RNA targets, the slides were next incubated in preheated Proteinase K solution (0.1 μg/mL) at 37°C for 20 min and washed with PBS for 5 min, followed by incubation in 10% neutral buffered formalin (NBF) for 5 min, NBF-stop buffer (2 × 5 min), and washing with PBS for 5 min, all at room temperature.
Hybridization was performed using the GeoMx Human Whole Transcriptome Atlas (cat. GMX-RNA-NGS-HuWTA) kit following the manufacturer’s instructions. Briefly, Atlas probe mix was diluted in Buffer R (pre-heated to 37°C) and applied to the slides. The sections were then covered with a Grace Bio-Labs HybriSlip for the hybridization reaction and incubated at 37°C overnight (16 h) in a hybridization oven. The following day, the slides were washed with 2x saline sodium citrate (SSC), 2x SSC-T (20x SSC +0.7mL 10% Tween 20 + 62.3 mL DEPC-treated water), and 2x SSC-50% formamide to remove the HybriSlip and reduce the binding of off-target probes, and moved back into 2x SSC buffer.
For the morphology staining, sections were blocked with Buffer W at room temperature for 30 min (protected from light) and stained with SYTO 83 (Invitrogen, cat. S11364, 1:35000), PanCK (Novus Biologicals, cat. NBP2-33200AF488, 1:200), CD68 (Santa Cruz, cat. sc-20060AF594, 1:40), and CD31 (Abcam, cat. ab215912, 1:110) morphology markers diluted in buffer W for 2 h in a humid chamber at room temperature. After staining, the sections were washed with 2x SSC for 2 times and immediately loaded on the GeoMx DSP.
The regions of interest (ROIs) were selected based on the abundance of CD68 and CD31 staining in pan-cytokeratin rich areas to achieve at least 50 cells in each segment. The segmentation of ROIs into the three areas of illumination (AOI) was performed in following order: 1.) selection of CD68+pan-cytokeratin- area, 2.) selection of CD31+ area and 3.) selection of the remaining DNA+ area. These three non-overlapping areas are referred as CD68+, CD31+ and CD68−CD31− throughout the text.
Image scan and probe tag collection were performed using the GeoMx DSP instrument. The resulting libraries were sequenced on Illumina NovaSeq 6000 system with following read length configuration: R1: 27, i7: 8, i5: 8 and R2: 27. The sequencing data was demultiplexed and converted into FASTQ files with bcl2fastq2 Conversion Software (Illumina, v2.2.0). Raw FASTQ files were subjected to adapter removal, merging of paired-end reads, read alignment to barcode IDs and PCR duplicate removal with GeoMx NGS Pipeline software (NanoString, v2.3.3.10), and resulting DCC files were uploaded into GeoMx DSP Control Center software (Nanostring, v.2.4). High-resolution TIFF images of ROIs were exported from GeoMx DSP Control Center to ImageJ (NIH, USA, v1.53q) and ROIs with representative quantity of CD68+ cells were displayed by selecting 400 μm × 400 μm regions (limited by smallest ROI) from the ROI center.
GeoMx NGS readout data analysis
GeoMx analysis workflow consisting of quality control, normalization, differential gene expression analysis and pathway analysis was run in GeoMx DSP Control Center according to NanoString’s recommendations (GeoMx-NGS Data Analysis User Manual, SEV_00090-05 for software v2.4). Subsequent analyses and visualizations were performed with R (v4.0.4, RStudio v1.4.1106, tidyverse v1.3.1).27,32 For quality control, negative probes identified as outliers by Grubbs test were excluded in each segment. Signal-to-noise ratio [Q3/geoMean(Negative probes)] was calculated for each segment and segments with signal-to-noise ratio ≤1 were excluded from further analyses. Genes exceeding both the limit of quantitation calculated based on negative probe counts and value 2 in at least 15% of segments were defined as confidently expressed and used in further analyses. For analyses not directly comparing different segment types (differential gene expression and pathway analyses), this gene filtering was performed separately for CD68+, CD31+ and CD68−CD31− segments. Filtered data were normalized by Q3 values calculated based on all genes passing the aforementioned filters, and normalized counts used in all further analyses and visualizations.
Unsupervised hierarchical clustering and principal component analyses were performed with R (functions hclust and prcomp, respectively) using log2-transformed normalized counts. Hierarchical clustering was visualized as a heatmap using ComplexHeatmap package (v2.6.2)28 and first three principal components as a 3D scatterplot using scatterplot3d package (v0.3-41).33 Differentially expressed genes (DEGs) were identified using linear mixed models to account for multiple observations (ROIs) from the same biopsy. DEGs between post- and pre-biopsies were identified separately for DC and non-DC patient group using a linear mixed model with biopsy type as a fixed effect and patient ID as a random effect. DEGs between DC and non-DC patients’ pre-biopsies were identified similarly by using a linear mixed model with response group as a fixed effect and patient ID as a random effect. Obtained p values and log2Fold Changes were plotted as volcano plots and genes passing Benjamini-Hochberg-adjusted p value (Padj) threshold 0.05 were indicated by point color. Significantly altered pathways between the indicated conditions were identified with GeoMx DSP Control Center that performs gene set enrichment analyses using fGSEA package34 and Reactome pathway database (v78). Pathways with <20% coverage (% of pathway genes expressed in the dataset) were excluded from the analysis, resulting in 76.3% median coverage. Obtained normalized enrichment scores and Benjamini-Hochberg-adjusted p values (Padj) were plotted as volcano plots or as bubble plots with dot area corresponding to -log10Padj value. Pathways with Padj value lower than 0.05 were visualized as an enrichment map using Cytoscape’s (v3.9.1)29 Enrichment Map app (v3.3.4)35 with yFiles Organic Layout. The created network was clustered by applying Markov clustering algorithm with default settings and resulting clusters were manually annotated based on annotations created with Cytoscape’s AutoAnnotate app (v1.4.0).36
To evaluate TAM phenotypes, M1 and M2 macrophage scores were calculated for each CD68+ segment based on genes differentially expressed in M1 and M2 macrophages13 (Table 1 in Martinez et al.) and passing our aforementioned gene filtering criteria. The expression levels of these M1 and M2 genes were plotted as a heatmap after log2-transformation and z-scoring. M1 and M2 scores were calculated separately for each CD68+ segment by dividing average M1 (or M2) gene expression level by average overall gene expression level. For evaluating cell type abundancies within the CD68−CD31−, CD68+ and CD31+ biopsy areas, we used SpatialDecon package (v1.0.0)30 and its safeTME tumor-immune deconvolution cell profile matrix with default settings, except for providing safeTME cell type matches for major cell type calculation and raw count matrix for data-point weighting.
Quantification and statistical analysis
The data cut-off date for analysis was Jan 31st, 2022. ORR and DCR were evaluated at cycle 4 according to RECIST 1.1. PFS was evaluated from the date of the first dose of bexmarilimab until documented disease progression, death, or end of follow-up, the former two counted as events. OS was calculated from the date of the first dose of bexmarilimab until death or end of follow-up, the former was counted as an event. Previous line of therapy duration was calculated from the first treatment date until therapy discontinuation. Landmark analysis for OS was analyzed from the cycle 4 treatment date until death or end of follow-up, the former was counted as an event. PFS and OS were analyzed using the Kaplan-Meier method and Cox-regression with 95% confidence interval. For the IHC biomarker analysis, Wilcoxon two-sample test was applied. Changes in cytokine levels over time were analyzed with repeated measures ANOVA model. For the PFS2/PFS1 ratio, percentage of patients with ratio of >1.3 were scored positive.26 The descriptive parameters summarizing the data center and spread are indicated in the Y axes or figure legends. The data were analyzed by a statistician employed by the sponsor and by the senior academic authors.
Analysis populations
DLT population (n = 30) included all the patients treated in part I dose escalation cohorts who had received at least one dose of bexmarilimab and had at least three-week follow-up period after the 1st dose. Safety population and efficacy analysis populations (ORR, PFS, OS, duration of response) comprised of all the patients who had received at least one dose of bexmarilimab (n = 138). Waterfall plot analysis (n = 24) was performed for all the patients who had received at least one dose of bexmarilimab and had data from the follow-up RECIST 1.1 evaluation available. For the landmark analysis (n = 91), all the study subjects who were alive at cycle 4 (9 weeks after the 1st dose of bexmarilimab) were included. Previous line of therapy duration population (n = 134) comprised of all the study subjects who had received at least one dose of bexmarilimab, had received a previous line of therapy for advanced disease and had accurate dates available for the beginning and end of the treatment. Bexmarilimab pharmacokinetics were analyzed from part I patients who had repeated pharmacokinetic samples available (n = 30). Clever-1 receptor occupancy (n = 28) and soluble Clever-1 (n = 28) populations included all the study subjects treated in part I of the trial who had samples available for RO or soluble Clever-1 analysis, respectively. Tumor Clever-1 (n = 78) and PD-L1 (n = 43) IHC was performed for all the study subjects who had received at least one dose of bexmarilimab, belonged to the specific cohorts with one identified PR or high DC rates ≥25% (colorectal cancer, cutaneous melanoma, gastric, biliary tract cancer and hepatocellular cancer), who had adequate pre-treatment tumor samples available for analysis, and had successful IHC staining and positive cell scoring results available. GeoMx DSP set (n = 6 patients; n = 180 spatial transcriptomics profiles) included all the patients who had paired pre- and post-treatment biopsies available, and had successful GeoMx DSP morphology staining and hybridization probe quantification.
Additional resources
Clinical trial details for the MATINS trial: https://www.clinicaltrials.gov/study/NCT03733990.
Acknowledgments
We thank the study participants, the investigators, and the research team who contributed to the study. We also want to thank Mari Parsama, Teija Kanasuo, Riikka Sjöroos, Sari Mäki, and Maritta Pohjansalo for excellent technical assistance. GeoMx Digital Spatial Profiling was performed at FIMM Single-Cell Analytics and Sequencing units supported by HiLIFE and Biocenter Finland. This study was funded by Faron Pharmaceuticals (Turku, Finland), European Union’s Horizon 2020 research and innovation program (ID: 960914), the Academy of Finland (M.H.), Business Finland (M.H.), Cancer Society of Finland (M.H. and J.H.R.), the Sigrid Jusélius Foundation (M.H.), Orion Research Foundation (J.H.R.), and the Paulo Foundation (J.H.R.). The graphical abstract was created with BioRender.
Authors contributions
J.H.R., S.S., C.Y., J.M., M.K.K., J.J., J.P.K., M.H., and P.B. designed the study; L.V., M.D.M., A.P., D.R., T.S., S.I., Y.T.M., D.G., S.P.A., P.J., and A.M. recruited and treated the patients, and collected the data; J.H.R., Y.X., S.K., and M.H. optimized and performed GeoMx; J.H.R., C.Y., J.M., M.K.K., J.J., J.P.K., M.H., and P.B. analyzed and interpreted the data; J.H.R., J.P.K., M.H., and P.B. wrote the first draft of the manuscript; all authors reviewed the manuscript and approved the final version.
Declaration of interests
L.V. declares consulting fees from Adaptherapy; stock or stock options for Resolved. M.D.M. declares payment or honoraria for lectures, presentations, speakers bureaus, and manuscript writing or educational events from Janssen and MSD. A.P. declares payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from Incyte and Roche; support for attending meetings and/or travel from Gilead; participation on a Data Safety Monitoring Board or Advisory Board for Incyte, Novartis, Gilead, and BeiGen. D.B. declares consulting fees from Merck AG, Pfizer, Bayer, Cantargia AB, Faron Pharmaceuticals, and Servier. T.S. declares payment or honoraria for lectures, presentations, speakers bureaus, and manuscript writing or educational events from BMS; participation on a Data Safety Monitoring Board or Advisory Board for Faron Pharmaceuticals, Merck, and Novartis; leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid for Finnish Oncology Society. S.I. declares grants or contracts from any entity from Roche and AstraZeneca; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from BMS, Roche, Astra Zeneca, Takeda, Novartis, MSD, Pierre Fabre, and Boehringer-Ingelheim. S.S. declares consulting fees from Faron Pharmaceuticals, participation on a Data Safety Monitoring Board or Advisory Board for Faron Pharmaceuticals. Y.T.M. declares consulting fees from Eisai, Roche, AstraZeneca, and Ipsen; payment or honoraria for lectures, presentations, speakers bureaus, and manuscript writing or educational events for Bayer and Boston Scientific; and participation on a Data Safety Monitoring Board or Advisory Board for Faron Pharmaceuticals. D.G. declares consulting fees from Clinigen and McCann Health; payment or honoraria for lectures, presentations, speakers bureaus, and manuscript writing or educational events from Cancer Drug Development Fund and Pfizer. S.P.A. declares payment or honoraria for lectures, presentations, speakers bureaus, and manuscript writing or educational events from Bayer, BMS, ASCO, and Exelixis; participation on a Data Safety Monitoring Board or Advisory Board for AstraZeneca, Seagen, and QED Therapeutics/Helsinn; leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid for Board-Support New India. P.J. declares participation on a Data Safety Monitoring Board or Advisory Board for Faron Pharmaceuticals. C.Y. declares consulting fees from Faron Pharmaceuticals; and payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from Bayer. J.M. declares all support for the present manuscript from Faron Pharmaceuticals (employment); patents planned, issued or pending for Faron Pharmaceuticals; participation on a Data Safety Monitoring Board or Advisory Board for Faron Pharmaceuticals; and stock or stock options for Faron Pharmaceuticals. M.K.K. declares all support for the present manuscript from Faron Pharmaceuticals (employment); patents planned, issued or pending for Faron Pharmaceuticals; and stock or stock options for Faron Pharmaceuticals. J.J. declares all support for the present manuscript from Faron Pharmaceuticals (employment); grants or contracts from any entity from European Innovation Council; and stock or stock options for Faron Pharmaceuticals. J.P.K. declares all support for the present manuscript from Faron Pharmaceuticals; consulting fees from MSD, BMS, Roche, Pfizer, and AstraZeneca; payment or honoraria for lectures, presentations, speakers bureaus, and manuscript writing or educational events from MSD, BMS, Roche, AstraZeneca, and Sanofi; payment for expert testimony from Sanofi; support for attending meetings and/or travel from BMS; participation on a Data Safety Monitoring Board or Advisory Board for Faron Pharmaceuticals; and stock or stock options from Faron Pharmaceuticals. A.M. declares payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from Janssen, Chugai, Novartis, and Bayer; support for attending meetings and/or travel from Amgen; and participation on a Data Safety Monitoring Board or Advisory Board for Janssen, Takeda, Genmab, Merck, and Faron Pharmaceuticals. M.H. declares all support for the present manuscript from Faron Pharmaceuticals; patents issued or pending for Faron Pharmaceuticals; and stock or stock options from Faron Pharmaceuticals. P.B. declares consulting fees from Faron Pharmaceuticals, Herantis Pharma, MSD Oncology, Ipsen, Oncorena, and TILT biotherapeutics; participation on a Data Safety Monitoring Board or Advisory Board for Faron Pharmaceuticals, TILT biotherapeutics, and Oncorena; leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid for Terveystalo (employment); stock or stock options for Terveystalo, TILT biotherapeutics; and other financial or non-financial interests for Faron pharmaceuticals, stock ownership (spouse).
Published: December 5, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101307.
Contributor Information
Maija Hollmén, Email: maijal@utu.fi.
Petri Bono, Email: petri.bono@terveystalo.com.
Supplemental information
References
- 1.Petitprez F., Meylan M., de Reyniès A., Sautès-Fridman C., Fridman W.H. The Tumor Microenvironment in the Response to Immune Checkpoint Blockade Therapies. Front. Immunol. 2020;11:784. doi: 10.3389/fimmu.2020.00784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noy R., Pollard J.W. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kzhyshkowska J. Multifunctional receptor stabilin-1 in homeostasis and disease. Sci. World J. 2010;10:2039–2053. doi: 10.1100/tsw.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palani S., Maksimow M., Miiluniemi M., Auvinen K., Jalkanen S., Salmi M. Stabilin-1/CLEVER-1, a type 2 macrophage marker, is an adhesion and scavenging molecule on human placental macrophages. Eur. J. Immunol. 2011;41:2052–2063. doi: 10.1002/eji.201041376. [DOI] [PubMed] [Google Scholar]
- 5.Hollmén M., Figueiredo C.R., Jalkanen S. New tools to prevent cancer growth and spread: a ‘Clever’ approach. Br. J. Cancer. 2020;123:501-509 doi: 10.1038/s41416-020-0953-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tervahartiala M., Taimen P., Mirtti T., Koskinen I., Ecke T., Jalkanen S., Boström P.J. Immunological tumor status may predict response to neoadjuvant chemotherapy and outcome after radical cystectomy in bladder cancer. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-12892-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin S.Y., Hu F.F., Miao Y.R., Hu H., Lei Q., Zhang Q., Li Q., Wang H., Chen Z., Guo A.Y. Identification of STAB1 in Multiple Datasets as a Prognostic Factor for Cytogenetically Normal AML: Mechanism and Drug Indications. Mol. Ther. Nucleic Acids. 2019;18:476–484. doi: 10.1016/j.omtn.2019.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karikoski M., Marttila-Ichihara F., Elima K., Rantakari P., Hollmén M., Kelkka T., Gerke H., Huovinen V., Irjala H., Holmdahl R., et al. Clever-1/stabilin-1 controls cancer growth and metastasis. Clin. Cancer Res. 2014;20:6452–6464. doi: 10.1158/1078-0432.CCR-14-1236. [DOI] [PubMed] [Google Scholar]
- 9.Viitala M., Virtakoivu R., Tadayon S., Rannikko J., Jalkanen S., Hollmén M. Immunotherapeutic Blockade of Macrophage Clever-1 Reactivates the CD8+ T-cell Response against Immunosuppressive Tumors. Clin. Cancer Res. 2019;25:3289–3303. doi: 10.1158/1078-0432.CCR-18-3016. [DOI] [PubMed] [Google Scholar]
- 10.Hollmén M., Maksimow M., Rannikko J.H., Karvonen M.K., Vainio M., Jalkanen S., Jalkanen M., Mandelin J. Nonclinical characterization of bexmarilimab, a Clever-1-targeting antibody for supporting immune defense against cancers. Mol Cancer Ther. 2022;21:1207-1218 doi: 10.1158/1535-7163.MCT-21-0840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Virtakoivu R., Rannikko J.H., Viitala M., Vaura F., Takeda A., Lönnberg T., Koivunen J., Jaakkola P., Pasanen A., Shetty S., et al. Systemic blockade of Clever-1 elicits lymphocyte activation alongside checkpoint molecule downregulation in patients with solid tumors: Results from a phase I/II clinical trial. Clin. Cancer Res. 2021;27:4205–4220. doi: 10.1158/1078-0432.CCR-20-4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simmons D.P., Nguyen H.N., Gomez-Rivas E., Jeong Y., Jonsson A.H., Chen A.F., Lange J.K., Dyer G.S., Blazar P., Earp B.E., et al. SLAMF7 engagement superactivates macrophages in acute and chronic inflammation. Sci. Immunol. 2022;7 doi: 10.1126/sciimmunol.abf2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez F.O., Gordon S., Locati M., Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J. Immunol. 2006;177:7303–7311. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 14.Irjala H., Elima K., Johansson E.L., Merinen M., Kontula K., Alanen K., Grenman R., Salmi M., Jalkanen S. The same endothelial receptor controls lymphocyte traffic both in vascular and lymphatic vessels. Eur. J. Immunol. 2003;33:815–824. doi: 10.1002/eji.200323859. [DOI] [PubMed] [Google Scholar]
- 15.Salmi M., Koskinen K., Henttinen T., Elima K., Jalkanen S. CLEVER-1 mediates lymphocyte transmigration through vascular and lymphatic endothelium. Blood. 2004;104:3849–3857. doi: 10.1182/blood-2004-01-0222. [DOI] [PubMed] [Google Scholar]
- 16.Mantovani A., Allavena P., Marchesi F., Garlanda C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022;21:799–820. doi: 10.1038/s41573-022-00520-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goswami S., Anandhan S., Raychaudhuri D., Sharma P. Myeloid cell-targeted therapies for solid tumours. Nat. Rev. Immunol. 2023;23:106–120. doi: 10.1038/s41577-022-00737-w. [DOI] [PubMed] [Google Scholar]
- 18.Pittet M.J., Michielin O., Migliorini D. Clinical relevance of tumour-associated macrophages. Nat. Rev. Clin. Oncol. 2022;19:402–421. doi: 10.1038/s41571-022-00620-6. [DOI] [PubMed] [Google Scholar]
- 19.Das S., Johnson D.B. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer. 2019;7:306. doi: 10.1186/s40425-019-0805-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shetty S., Weston C.J., Oo Y.H., Westerlund N., Stamataki Z., Youster J., Hubscher S.G., Salmi M., Jalkanen S., Lalor P.F., Adams D.H. Common lymphatic endothelial and vascular endothelial receptor-1 mediates the transmigration of regulatory T cells across human hepatic sinusoidal endothelium. J. Immunol. 2011;186:4147–4155. doi: 10.4049/jimmunol.1002961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steele M.M., Jaiswal A., Delclaux I., Dryg I.D., Murugan D., Femel J., Son S., du Bois H., Hill C., Leachman S.A., et al. T cell egress via lymphatic vessels is tuned by antigen encounter and limits tumor control. Nat. Immunol. 2023;24:664–675. doi: 10.1038/s41590-023-01443-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tadayon S., Dunkel J., Takeda A., Eichin D., Virtakoivu R., Elima K., Jalkanen S., Hollmén M. Lymphatic Endothelial Cell Activation and Dendritic Cell Transmigration is Modified by Genetic Deletion of Clever-1. Front. Immunol. 2021;12:602122. doi: 10.3389/fimmu.2021.602122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dunkel J., Viitala M., Karikoski M., Rantakari P., Virtakoivu R., Elima K., Hollmén M., Jalkanen S., Salmi M. Enhanced Antibody Production in Clever-1/Stabilin-1-Deficient Mice. Front. Immunol. 2018;9:2257. doi: 10.3389/fimmu.2018.02257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vuorinen E., Björkman M.L., Virtakoivu R., Jalkanen J., Aakko S., Takeda A., Budde P., Zucht H.-D., Brautigam M., Ahangarianabhari B., et al. Abstract 2269: Bexmarilimab Induces B-Cell Activation and Autoantibody Production. AACR. Cancer Research. 2023;83:2269. [Google Scholar]
- 25.Neumaier M., Giesler S., Ast V., Roemer M., Voß T.D., Reinz E., Costina V., Schmelz M., Nürnberg E., Nittka S., et al. Opsonization-independent antigen-specific recognition by myeloid phagocytes expressing monoclonal antibodies. Sci. Adv. 2023;9 doi: 10.1126/sciadv.adg1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Massard C., Michiels S., Ferté C., Le Deley M.C., Lacroix L., Hollebecque A., Verlingue L., Ileana E., Rosellini S., Ammari S., et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017;7:586–595. doi: 10.1158/2159-8290.CD-16-1396. [DOI] [PubMed] [Google Scholar]
- 27.R Core Team . R Foundation for Statistical Computing; Vienna, Austria: 2021. R: A Language and Environment for Statistical Computing.https://www.R-project.org/ [Google Scholar]
- 28.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 29.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Danaher P., Kim Y., Nelson B., Griswold M., Yang Z., Piazza E., Beechem J.M. Advances in mixed cell deconvolution enable quantification of cell types in spatial transcriptomic data. Nat. Commun. 2022;13:385. doi: 10.1038/s41467-022-28020-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheung Y.K., Chappell R. Sequential designs for phase I clinical trials with late-onset toxicities. Biometrics. 2000;56:1177–1182. doi: 10.1111/j.0006-341x.2000.01177.x. [DOI] [PubMed] [Google Scholar]
- 32.Wickham H., Averick M., Bryan J., Chang W., McGowan L.D.A., François R., Grolemun G., Hayes A., Henry L., Hester J., et al. Welcome to Tidyverse. [DOI]
- 33.Ligges U., Mächler M. scatterplot3d - An R Package for Visualizing Multivariate Data. J. Stat. Software. 2003;8 [Google Scholar]
- 34.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast gene set enrichment analysis. [DOI]
- 35.Merico D., Isserlin R., Stueker O., Emili A., Bader G.D. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kucera M., Isserlin R., Arkhangorodsky A., Bader G.D. AutoAnnotate: A Cytoscape app for summarizing networks with semantic annotations. F1000Res. 2016;5:1717. doi: 10.12688/f1000research.9090.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Participant-level clinical data is deposited to MATINS trial master database (managed by the trial sponsor) and can be accessed via the corresponding authors by reasonable request. The GeoMx spatial transcriptomics data have been deposited at the GEO database under the accession number GSE240138.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.