

Summary
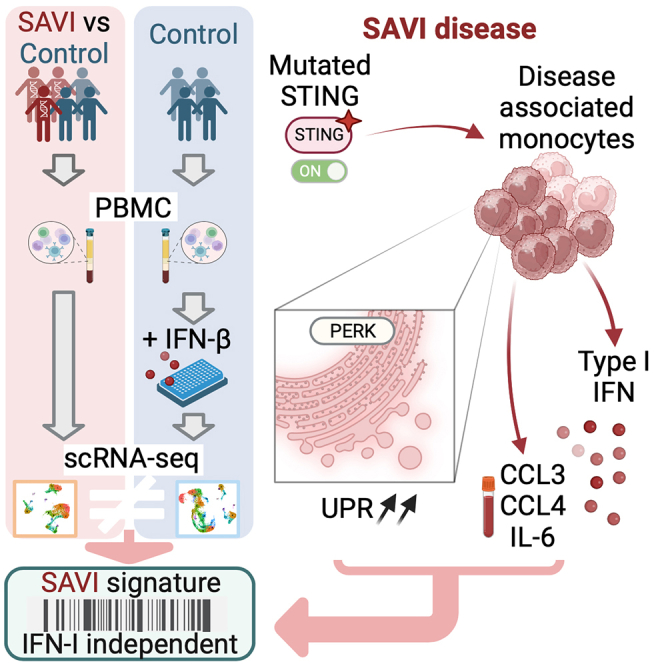
Gain-of-function mutations in stimulator of interferon gene 1 (STING1) result in STING-associated vasculopathy with onset in infancy (SAVI), a severe autoinflammatory disease. Although elevated type I interferon (IFN) production is thought to be the leading cause of the symptoms observed in patients, STING can induce a set of pathways, which have roles in the onset and severity of SAVI and remain to be elucidated. To this end, we performed a multi-omics comparative analysis of peripheral blood mononuclear cells (PBMCs) and plasma from SAVI patients and healthy controls, combined with a dataset of healthy PBMCs treated with IFN-β. Our data reveal a subset of disease-associated monocyte, expressing elevated CCL3, CCL4, and IL-6, as well as a strong integrated stress response, which we suggest is the result of direct PERK activation by STING. Cell-to-cell communication inference indicates that these monocytes lead to T cell early activation, resulting in their senescence and apoptosis. Last, we propose a transcriptomic signature of STING activation, independent of type I IFN response.
Keywords: type I interferon, SAVI, STING, unfolded protein response, integrated stress response, scRNA-seq
Graphical abstract
Highlights
-
•
SAVI patients’ monocytes are characterized by high integrated stress response
-
•
These monocytes could induce early activation, senescence, and apoptosis in T cells
-
•
Authors propose a 21-gene STING-activation signature, independent of IFN response
-
•
Authors also identify CCL3, CCL4, and IL-6 as potential biomarkers for SAVI patients
Cevins et al. explore the transcriptome of SAVI patients with gain-of-function mutations in STING1 and observe highly inflammatory monocytes characterized by ISR, potentially contributing to the loss of effector T cells. They also identify a transcriptomic signature specific to STING activation and independent of type I IFN response.
Introduction
Stimulator of interferon genes (STING)-associated vasculopathy with onset in infancy (SAVI) is a rare monogenic disease characterized by uncontrolled production of type I interferons (IFNs).1,2 Type I IFNs are the first line of defense against pathogenic infection. They lead to robust activation and increased effector functions of immune cells. However, type I IFNs are highly potent cytokines that can become detrimental when uncontrolled. SAVI is a type I interferonopathy that usually occurs during the first few years of life and is characterized by severe pulmonary and cutaneous manifestations.3 SAVI is caused by gain-of-function mutations in STING1, which codes for STING, a key component of innate immunity. To date, approximately 100 cases of SAVI have been reported.4
The STING pathway acts as a sensor of cytoplasmic nucleic acids, a potential danger signal. Upon sensing of cytoplasmic nucleic acids, by the sensor cyclic guanosine monophosphate-AMP synthase, STING is activated and changes conformation to traffic from the endoplasmic reticulum (ER) to the Golgi apparatus.5,6 There, STING triggers the phosphorylation of IRF3 and nuclear factor κB (NF-κB), leading to the expression of type I IFNs and other pro-inflammatory cytokines.7,8,9 Other molecular pathways can also be triggered by STING, such as ER stress and senescence.10,11 Moreover, STING can induce cell type-specific programs, such as apoptosis, which is induced in T cells but not in macrophages or dendritic cells.12
In SAVI, STING is constitutively activated and present at the Golgi apparatus.1,13 SAVI is associated with T cell lymphopenia and decreased percentages of effector and memory T cells.3,14 A low number of circulating natural killer (NK) cells has also been reported in some patients.15 At the molecular levels, IFN-α/β and IFN-stimulated genes (ISGs) are consistently upregulated in the plasma of all patients.1,2,3 Patients have elevated inflammatory cytokines plasma concentrations, including tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6),16 suggesting the involvement of the NF-κB pathway. Patients’ vascular endothelial cells also have increased expression of genes involved in apoptosis, cell adhesion, and coagulation.2 Many pathways are thought to be triggered by STING activation. Some of them, such as type I IFN production and NF-κB activation, are upregulated in SAVI. T cell activation or increased ER stress have also been demonstrated in murine models but have yet to be confirmed in SAVI patients.11
As uncontrolled production of type I IFNs is a hallmark of SAVI, treatment with Janus kinase (JAK) inhibitors has been proposed to patients. These drugs block the signal transduction upon IFN-α/β binding to the IFN-α/β receptor (IFNAR), by inhibiting either JAK1 and JAK2 (ruxolitinib and baricitinab) or JAK1 and JAK3 (tofacitinib). These treatments lead to partial improvements of the symptoms.3,17,18 However, in cases where the interstitial lung disease is too advanced, JAK inhibitor treatment has proved inefficient,3,17 suggesting, in SAVI, an alternative role of STING activation, independent of type I IFN signaling. A deeper understanding of the pathways modulated in patients seems to be crucial to propose alternative therapeutic strategies. Here, we aim to identify pathways modulated in a cell-type-specific manner and address their dependency to primary type I IFN production.
The functional impairment of STING gain of function has mostly been assessed using in vitro cell lines or mouse models.11,19,20 Others have used patients’ cells but evaluated individual cell types.14 Here, we propose to evaluate peripheral blood mononuclear cells (PBMCs) at the single cell level to gain a systemic overview of the transcriptomic dysregulations of SAVI, identify disease-associated cells, infer cell-cell communication, and compare the pathways specifically modulated in different immune cell types. Additionally, to decipher type I IFN-mediated signaling from other dysregulated pathways driven by STING constitutive activation, we compare the primary response to IFN-β in PBMCs coming from healthy donors to the pathways modulated in SAVI patients.
Results
Circulating effector lymphocytes are decreased in SAVI
To better understand the molecular physiopathology of SAVI, we profiled at the single-cell transcriptomic level a dataset of PBMCs from five SAVI patients, previously clinically described and carrying different gain-of-function mutations in STING1, as well as seven healthy donors (CTRLs) (SAVI dataset) (Figures 1A and S1A).1,3,15,18,21
Figure 1.

A scRNA-seq cohort of PBMCs from five SAVI patients shows loss of effector cells not replicated by challenging healthy PBMCs with IFN-β
(A) Description of the SAVI dataset: five SAVI patients, with three different STING mutations sampled before (SAVI) and under JAK inhibitor treatment (SAVI_treated), and seven healthy donors (CTRLs). Ruxo, ruxolitinib; Tofa, tofacitinib; yo, years old.
(B) Description of the IFN-β dataset.
(C) UMAP and cell type assignment of all 112,060 cells from the SAVI dataset (top) dataset separated by group (bottom).
(D) UMAP and cell type assignment of all 115,503 cells from the IFN-β dataset (top) and separated by time of IFN-β stimulation (bottom).
(E) Boxplot of the proportion of PBMCs found in several clusters of the SAVI dataset. p values are calculated by the Kruskal-Wallis test for multiple comparisons, followed by a post hoc Dunn’s test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(F) Evolution of the proportion of PBMCs found in several clusters in the IFN-β dataset.
(C and D) nCD4, naive CD4; eCD4, effector CD4; nCD8, naive CD8; eCD8, effector CD8.
In parallel, to better determine the contribution of type I IFN secretion to the transcriptomic profile of SAVI patients, we performed single cell RNA sequencing (scRNA-seq) on PBMCs from three CTRLs, stimulated with human recombinant IFN-β at different timepoints (IFN-β dataset) (Figure 1B). Response to IFN-β stimulation was assessed by following the kinetics of expression at the mRNA level of six ISGs commonly used to evaluate type I IFN response in patients1 (Figure S1B).
After quality control, integration, and unsupervised clustering, we obtained 23 cell populations in the SAVI dataset and 29 in the IFN-β dataset (Figures 1C, 1D, and S1C‒S1F). We compared cell population proportions in SAVI patients to CTRLs (Figures 1C, 1E, and S1G) and confirmed the previously described increase of naive CD4+ and CD8+ T cells and decrease of their effector/memory counterpart,14 as well as NK cells15 (particularly NK CD56bright cells). We also observed a drastic reduction in mucosal associated invariant T cells and γδ T cells in SAVI patients (Figures 1C and 1E). Significant differences in cell type proportions before and under JAK inhibitor treatment were not observed. These observations were validated by flow cytometry or cytometry by time of flight for the SAVI dataset and the IFN-β dataset, respectively (Figures S1I‒S1J). Of note, no overt decrease of effector T cells was observed in healthy PBMCs challenged in vitro with type I IFN for 36 h (Figures 1D, 1F, S1H, and S1J).
SAVI patients’ cells display type I IFN response, NF-κB activation, cell stress, and death
STING activation triggers NF-κB signaling and type I IFN production.22 Here, response to type I IFNs was increased in all PBMC populations in SAVI patients, with the strongest induction observed in monocytes and dendritic cells (DCs) (Figures 2A and S2A). This type I IFN response was only partially corrected by JAK inhibitor treatment (Figure S2A). After in vitro stimulation with IFN-β, a robust type I IFN response is measured, peaking after 2 h (Figures 2B and S2B), with the strongest type I IFN response persisting at later time points in monocytes and DCs. In the SAVI dataset, the transcriptomic signature of NF-κB activation follows the pattern of type I IFN response (Figure 2C). Under JAK inhibitor treatment, NF-κB activation was back to CTRL levels. In the IFN-β dataset, NF-κB activation was induced as soon as 1 h post-IFN-β stimulation and continued to increase in monocytes and DCs until 36 h (Figure 2D).
Figure 2.

Type I IFN and NF-κB signature scores are elevated in SAVI patients
(A and B) Signature score of the type I IFN response in (A) the SAVI dataset and (B) the IFN-β dataset (nCD4, naive CD4; eCD4, effector CD4; nCD8, naive CD8; eCD8, effector CD8; γδ, γδ T cells).
(C and D) Violin plot of the signature score of NF-κB activation in (C) the SAVI dataset and (D) the IFN-β dataset (dark lines indicate medians).
(E and F) Heatmap of the pathway enrichment analysis (E) between SAVI and CTRL and (F) between healthy PBMCs stimulated with IFN-β for 36 h and unstimulated PBMCs. Dots indicate non-significant pathways (Bonferroni-Hochberg corrected p values >0.05). Side color bar indicate groups of pathways based on broader functions.
To explore the dysregulated pathways in SAVI patients, we performed differential expression and pathway analysis between untreated SAVI and CTRLs in each major cell population. We observed, in most cell types, an enrichment in pathways involved in response to IFNs and/or linked to inflammation (Figure 2E; Table S1) that were downregulated by JAK inhibitors but remained higher than in CTRLs (Figures S2A and S2C; Table S1). These pathways were also found to be activated in PBMCs treated with IFN-β for 36 h (Figure 2F; Table S2). Cell death pathways were enriched in both SAVI patients and in PBMCs stimulated with IFN-β for 36 h. JAK inhibition also seemed to partially correct the induction of these cell death-related pathways (Figure S2C). Interestingly, whereas actin/integrin-related pathways are predicted to be downregulated in all PBMCs in SAVI patients, in healthy PBMCs 36 h after IFN-β stimulation, these pathways are predicted to be upregulated in T and B cells while inhibited in monocytes and NK (Figures 2E and 2F). Additionally, in both PBMCs from SAVI patients and in vitro-stimulated PBMCs, we observed a robust modulation of pathways linked to the ISR, with downregulation of EIF2 signaling and oxidative phosphorylation as well as an upregulation of the unfolded protein response (UPR). Of note, EIF2 signaling is predicted to be strongly inhibited in most cell types with the exception of T cells. In patients’ CD8+ T cells, EIF2 signaling is predicted to be activated both prior and during treatment, indicating JAK-STAT pathway-independent mechanism. In the IFN-β dataset, UPR was induced as soon as 4 h with increasing levels of induction throughout the time course. Meanwhile, ISR and EIF2 responses were inhibited in all populations in a time-dependent manner (Figure S2D; Table S3).
T lymphocytes of SAVI patients are hyperactivated, senescent, and apoptotic with increased IFN-β signaling
Imbalance between naive and effector T cells, along with proliferation defects, has previously been described in SAVI.14 To better characterize transcriptomic dysregulations in patients’ circulating T cells, we performed differential analysis between untreated SAVI patients and CTRLs in each cluster of the T cell compartment (Table S1). Pathways analyses revealed a strong enrichment of inflammatory response genes (Figure 3A). It also pointed to actin and integrin-related pathways and particularly the leukocyte extravasation signaling (Figure 3A). Several other associated pathways, including RAC signaling and integrin signaling, were decreased in effector cells. We hypothesized that this may reflect an impaired lymphocyte trafficking machinery, which was supported by the decrease of a lymphocyte trafficking signature score particularly in effector T cells (Figure 3B). This signature was also decreased after IFN-β challenge of PBMCs from CTRLs (Figure 3C), suggesting that type I IFNs may cause impaired T cell trafficking in SAVI patients.
Figure 3.

T cells of SAVI have impaired signature score of trafficking machinery, and naive cells have increased activation markers and senescence signature score
(A) Heatmap of the pathway enrichment analysis between SAVI and CTRL. Dots indicate non-significant pathways (Bonferroni-Hochberg corrected p values > 0.05). Side color bar indicate groups of pathways based on broader functions.
(B and C) Heatmaps of a lymphocyte trafficking signature in T cells of (B) the SAVI dataset and (C) the IFN-β dataset.
(D and E) Feature plots of CD69 mRNA expression level in T cells in (D) the SAVI dataset and (E) the IFN-β dataset. nCD4, naive CD4; eCD4, effector CD4; nCD8, naive CD8; eCD8, effector CD8.
(F and G) T cell activation signature score in T cells of (F) the SAVI dataset and (G) the IFN-β dataset over the time course of IFN-β stimulation.
(H and I) Senescence signature score in T cells of (H) the SAVI dataset and (I) the IFN-β dataset over the time course of IFN-β stimulation.
(J and K) Apoptosis signature score in (J) the SAVI dataset and (K) the IFN-β dataset over the time course of IFN-β stimulation. (J and F) Dark lines indicate medians. (G–I and K) Each dot is the average score of the signature for a sample.
As T cells of StingN153S/+ mice were shown to undergo spontaneous activation,11 we examined the expression of the early activation marker CD69. Patients displayed increased CD69 mRNA levels (Figures 3D and S3A), mostly in naive T cells (Figures 1C and S1E). CD69 expression was also induced by IFN-β challenge at early timepoints but returned to basal levels between 4 and 12 h after stimulation (Figures 3E and S3B). A global T cell activation signature score was increased in both SAVI patients and IFN-β-challenged cells, mostly in naive cells of SAVI patients, and in all T cell clusters in IFN-β-challenged PBMCs (Figures 3F and 3G).
We then evaluated a senescence signature and the proportion of cells in the growth and mitotic phases (G2/M) of the cell cycle (Figures 3H, 3I, S3C, and S3D). Patients’ T cells had increased senescence signature score compared with CTRLs, reduced by JAK inhibitor treatment in naive T cells (Figure 3H). We also observed significantly decreased proportion of naive T cells from SAVI patients in the G2/M phase, partially rescued by the treatment, suggesting reduced proliferation capacity (Figure S3C). Similarly, in the IFN-β dataset, IFN-β induced increased senescence and decreased proportion of cells in G2/M phase, even though the proportion of cell in the G2/M phase is back to normal after 36 h (Figures 3I and S3D). Finally, we revealed increased transcriptomic signature score of apoptosis, particularly in patients’ naive T cells, restored by JAK inhibitor treatment (Figure 3J). The IFN-β-treated cells showed a sharp increase in apoptosis signature score after 2 h of treatment, while the signature score is decreased afterward (Figure 3K).
Taken together, these data suggest that naive T cells are highly activated in SAVI but have reduced ability to proliferate and differentiate into effector cells, ultimately leading to cell death.
Identification of disease-associated monocytes in SAVI patients, presenting hyperinflammation and transcriptomic signature of an ISR
As type I IFN response and NF-κB signaling signature scores were predominantly detected in monocytes and DCs, we examined these populations in greater details. We observed a shift in the distribution of classical monocytes, without changes of global monocytes proportions (Figures 1G, S1G, S1I, 4A, 4B, and S4A). Cluster 17, named “disease-associated cluster,” is composed at 85% of cells from patients, with 66.7% of cells from patients before JAK inhibitor treatment, and 18.3% of cells from patients under treatment (Figures 4B and S4A). In this disease-associated cluster (Figure S1E), both type I IFN response score and NF-κB activation score were elevated. Other monocytes (clusters 5 and 12) from SAVI patients also had an elevated score for both signatures, although to a lesser extent (Figure 4C). Expression of type I and type III IFNs were increased in SAVI patients compared with CTRLs and partially decreased upon JAK inhibitor treatment (Figure S4B). In SAVI patients, we observed that disease-associated monocytes were the main source of IFN-β mRNA (Figures 4D and S4C). We then performed differential expression analysis between disease-associated monocytes of SAVI patients (cluster 17) and their classical monocytes (cluster 5). We observed 984 differentially expressed genes (Figures 4E and S4D; Table S4), with the upregulation of key players involved in the ISR, cell activation, and transcripts of pro-inflammatory cytokines including IL1B and several type I IFNs (such as IFNB1, IFNA1, and IFNA2). Pathway analysis revealed a strong enrichment of genes negatively regulating the EIF2 signaling pathway, which can be associated to increased ISR23 (Figures 4F and S4D; Table S5). To validate the upregulation of the ISR, we evaluated PPP1R15A, which encodes the ISR marker GADD34. PPP1R15A expression reached the highest level in the disease-associated monocytes of the SAVI group (Figures 4E and 4G). An UPR signature,24 known to contribute to the ISR, was similarly upregulated in patients’ disease-associated monocytes. Notably, patients’ non-classical monocytes expressed increased PPP1R15A and UPR signature score. JAK inhibitor treatments seem to partially reduce these pathways in both non-classical and disease-associated clusters (Figure 4G). In the stimulated monocytes of the IFN-β dataset, both PPP1R15A and the UPR signature score were increased after 36 h of stimulation (Figure S4E). Among the other pathways upregulated in the disease-associated cluster, we report pathways linked to response to IFN, production of inflammatory cytokines, and cell death along with an enrichment of genes involved in oxidative phosphorylation (Figure S4D). Overall, our data suggest that this disease-associated monocytic cluster corresponds with classical monocytes presenting a high level of inflammation and ISR at the transcriptomic level and likely related to the constitutive STING activation.
Figure 4.

SAVI patients present a disease-associated cluster of monocytes characterized by elevated type I IFN response, NF-κB activation, and ISR
(A) UMAP of the monocytes and DCs in the SAVI dataset, separated by groups. Black circle indicates cluster 17 (disease-associated monocytes).
(B) Composition of monocyte and DC clusters in the SAVI dataset.
(C) Violin plots of the signature score of type I IFN response (top) and of NF-κB activation (bottom) in the SAVI dataset.
(D) Dot plot of the expression levels of all type I and type III IFNs, in each monocyte and DC clusters, in the SAVI group.
(E) Volcano plot of the differentially expressed gene (DEGs) between the cells from SAVI patients in cluster 17 (disease-associated monocytes) and cluster 5 (classical monocytes).
(F) Pathway enrichment analysis between cells from SAVI patients in cluster 17 (disease-associated monocytes) and cluster 5 (classical monocytes). Side color bar indicate groups of pathways based on broader functions.
(G) Violin plot of the expression of PPP1R15A which codes for GADD34 (top) and a UPR signature of 85 genes (bottom) in the SAVI dataset. (C and G) Dark lines indicate medians.
Cell-to-cell communication inference predicts that hyperinflammatory monocytes in SAVI could drive hyperactivation and death of effector T cells
To better understand how monocytes from SAVI patients may influence T cells (Figures 3 and 4), we inferred monocyte-to-T cell communications. Thus, we used ICELLNET,25 which computes a score of cell-to-cell communication based on the transcriptomic level of ligands expressed by a sender cell type (here, both clusters of classical monocytes: cluster 5 and cluster 17) and the transcriptomic level of their receptors expressed by a receiving cell type (here, T cells) (Figure 5A). We applied this method to the SAVI dataset to infer communications between classical monocytes and each T cell cluster in the SAVI group and the CTRL group separately (Figure 5B). We reproduced a similar analysis on the IFN-β dataset for each time point (Figure 5C), allowing us to decipher IFN-dependent from IFN-independent predicted interactions. Hierarchical clustering of T cells based on their communication score with monocytes showed a clear separation between clusters from untreated SAVI patients and CTRLs (Figure 5D). We observed a predicted increase of type I IFN-IFNAR interactions in SAVI patients, which probably reflects the drastic upregulation of IFN transcripts in the monocytes of SAVI patients (Figure 4D), although this interaction is not predicted in the IFN-β dataset (Figures 5D and 5E). Other chemokines and pro-inflammatory cytokines, including CXCL9, CXCL10, and CXCL11 with their common receptor CXCR3, as well as the pair IL-6/IL-6R, are predicted to have increased interactions in a type I IFN-dependent manner (Figures 5D and 5E). In contrast, increased interactions of IL-12 and IL-27 with their receptors is likely type I IFNs-independent, as absent from the predictions in the IFN-β dataset.
Figure 5.

Cell-to-cell communication inference indicate that hyperinflammatory monocytes of SAVI drive activation and death of T cells through cytokine secretion
(A–C) (A) Cell-to-cell communications are inferred from the expression of ligands (secreted or presented at the membrane) expressed by a sender cell (here monocytes) and receptors expressed by a receiver cell (here, T cells). UMAP of the clusters involved in the inference in (B) the SAVI dataset and (C) the IFN-β dataset.
(D) Heatmap of the score of each ligand/receptor pair between each T cell cluster and the monocytes, either in the SAVI or the CTRL group. Hierarchical clustering based on Pearson correlation. Colored arrows indicate association to specific pathways.
(E) Heatmap of the score of the same ligand/receptor pair as observed in the SAVI dataset, between each T cell cluster and the monocytes, in each time point of the IFN-β dataset.
(F) Volcano plot of the differential protein secretion between SAVI patients and associated CTRL for 96 proteins measured from the plasma. Genes written in red are in agreement with cell-to-cell interaction predictions.
(G) Volcano plot of the differential protein secretion between PBMCs stimulated with IFN-β for 36 h and unstimulated for 96 proteins measured from the supernatant using the Olink inflammation panel. Genes written in red are also increased in the plasma of SAVI.
(H and I) Heatmaps of the scaled mRNA levels of the 12 proteins found upregulated in the blood of SAVI patients in (F), in (H) the SAVI patients, and (I) each cell type in cell treated with IFN-β for 36 h.
Strikingly, the strongest predicted interaction in SAVI patients was reported for the CCL19/CCR7 ligand/receptor couple, which plays an essential role in T cell activation.26 This communication was not inferred in healthy PBMCs treated with IFN-β or in SAVI patients under JAK inhibitor treatment (Figures 5E and S5A), arguing for a mechanism independent of type I IFN production. Checkpoint interactions, such as CD80/CD28, Nectin2/CD226, Nectin2/TIGIT, and Nectin2/CD96, involved in T cells stimulations27 were also increased in both SAVI patients and after IFN-β stimulation but should be interpreted with caution as monocytes are not professional antigen-presenting cells. Moreover, our analyses predicted enhanced crosstalk between LAG3, involved in immune exhaustion and regulation of effector function,28 and its ligands LGALS3 and MHC class II molecules in SAVI, corresponding to the strongest induction predicted in the IFN-β dataset (Figure S5B). We also observed in SAVI patients and IFN-β stimulated cells increased communications involved in apoptosis induction with TNFSF10/TNFRSF10B and LGALS1/CD69 (Figures 5D and 5E).29,30 In CD8+ T cell interactions with monocytes, the pair MICA/KLRK1 was predicted to be increased both in SAVI patients and IFN-β stimulated cells, an interaction leading to cell lysis31 (Figure 5D). Overall, these data point toward a role of the disease-associated monocytes in favoring T cells chronic activation and death.
Circulating cytokines in the plasma of SAVI patients as biomarkers of the disease
As several cytokines/chemokines were upregulated in SAVI patients at the transcriptomic level, we evaluated 96 inflammation-related proteins in both datasets using the proteomic proximity extension assays. We confirmed the previously known increase of IL-616 and observed increased plasmatic levels of CXCL10 and CCL19 (Figure 5F), which were predicted to play a role in monocyte-to-T cell communications (Figure 5D). Other pro-inflammatory cytokines, such as CCL3, CCL4, CCL7, and CCL8, were also increased in the plasma of SAVI patients compared with CTRLs (Figure 5F). The secretory profile of PBMCs stimulated with IFN-β for 36 h was characterized by induction of CXCL10, VEGFA, and CCL19 (Figure 5G). To assess the correspondence between the protein and the mRNA levels of the cytokines measured in the plasma of SAVI patients, we evaluated the expression of the 12 cytokines found to be increased in the plasma of SAVI patients in the scRNA-seq data (Figure 5H). Our results suggest a strong role of the disease-associated monocytes in the secretion of several of these cytokines and, therefore, validate our cell-to-cell communication predictions (Figures 5H and S5C). Of note, CCL3, CCL4, and IL-6 were among the most upregulated genes in the disease-associated monocytes (Figure 4E). IFN-β-stimulated PBMCs had a more homogeneous expression of inflammatory cytokines across cell types, in particular monocytes, DCs, and NK cells (Figures 5I and S5D). The specific elevation of type I IFN-independent cytokines, in particular CCL3, CCL4, and IL-6, suggests that they may be used as blood biomarkers of SAVI to aid the differential diagnosis with other type I interferonopathies.
A transcriptomic signature specific to STING activation and independent of response to IFN-β
We next sought to identify a transcriptomic signature of STING activation, independent of response to IFN-β stimulation (Figure 6A; Table S6). The first step consisted in extracting the 95 genes most specific to the disease-associated monocytes cluster (step 1, Figure 6B). One gene, G0S2, was filtered out from the list as its expression was not increased in SAVI compared with CTRLs (step 2, Figure 6C). To obtain a type I IFN-independent signature, another step of the workflow was removing all genes related to type I IFNs and type I IFN response. We first removed type I IFN transcripts that were found in the list (step 3, Figure 6C) and genes upregulated by IFN-β at any time point of the IFN-β dataset, in monocytes and DCs (step 4, Figure 6D). Consequently, 21 genes remained in the STING-activation signature (Figure S6A). These genes were mostly associated with TNF-α signaling via NF-κB (Figure 6E). We confirmed that this signature was specific to the SAVI patients, as compared with the CTRL group, and partially reduced in JAK inhibitor-treated patients (Figure 6F). We also confirmed in the IFN-β dataset that the resulting signature score was not increased by type I IFN at any time point (Figure S6B).
Figure 6.

Design of a STING-activation signature, independent of type I IFN response, based on the transcriptome of the disease-associated monocytes
(A) Workflow used for the extraction of an IFN-independent STING-activation signature.
(B) Volcano plot of the differentially expressed genes between cluster 17 and cluster 5, in SAVI. The 95 genes highlighted in pink have log2FC > 1 and were selected as the basis of the STING activation signature.
(C) Heatmap of the 95 genes of selected in (B), in the monocyte and DCs of CTRL, SAVI, and SAVI treated. Blue writing represents the genes whose expression is higher in CTRL than SAVI and are filtered out in step 2. Red writing represents the IFN transcripts that are filtered out in step 3.
(D) Upset plot of the genes of the STING-activation signature. Red bar represents the genes uniquely found in the signature and not upregulated by IFN-β at any time point. These 21 genes are kept in the final STING activation signature.
(E) Bar chart of pathways significantly enriched for the 21 genes of the STING activation signature in MSigDB_Hallmark_2020. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗ p < 0.0001. Numbers indicate genes detected/total genes in the pathway. Color scale is proportional to adjusted p value.
(F) Feature plot of the signature score of the STING activation signature in monocytes and DCs in the SAVI dataset.
(G) Violin plot of the signature score of the STING activation signature in the monocytes and DCs in the ADUS100 dataset. Dark lines indicate medians.
To further evaluate the relevance of this signature in the context of STING activation, independent of type I IFN response, we designed an in vitro model of STING activation, in which PBMCs from a CTRL were stimulated with ADUS100 (a STING agonist), with or without JAK inhibitor. After scRNA-seq, type I-IFN response and NF-κB activation signature score were both increased by ADUS100 and dampened by JAK inhibitor (Figure S6C). We observed a strong increase of the STING-activation signature score in ADUS100-stimulated monocytes and DCs, which remained elevated under JAK inhibitor (Figure 6G). The STING activation signature score was restricted to monocyte and DCs in both the SAVI patients (Figure S6D) and the ADUS100-stimulated PBMCs (Figure S6E).
Overall, this list of 21 genes may represent a signature specific to STING activation and independent of type I IFN response.
Identification of a second genetic variant in PERK in a SAVI patient with a low UPR
While analyzing the transcriptome of SAVI patients, we identified several pathways that were differentially modulated in P1. We noted, in this patient, a marked reduction of the UPR, which was largely increased in other patients (Figure 7A). Of note, IFN-β stimulation induced a slight increase of the UPR (Figure S7A). This observation is accompanied by low expression of the ISR marker PPP1R15A in monocytes and DCs (Figure 7B). Further exploration showed differences in induction of the apoptosis signature and NF-κB signaling, although less drastic than the UPR (Figures 7C and 7D). These differences are even observable when compared with P2, P1’s father’s homozygotic twin (Figure S7E). In contrast, type I IFN response is found similarly increased in P1 as compared with other SAVI patients (Figures S7B–S7D). Of note, P1 is the only patient in this study to present with a systemic lupus erythematosus (SLE)-like phenotype,1 in addition to the SAVI phenotype.
Figure 7.

Exome analysis of a patient with low UPR reveals another SNP in EIF2AK3
(A) Heatmap of a UPR signature in the SAVI dataset.
(B) Violin plot of PPP1R15A expression (encoding GADD34) in monocytes and DCs of the SAVI dataset. Dark lines indicate medians.
(C) Heatmap of an apoptosis signature in the SAVI dataset.
(D) Heatmap of an NF-κB activation signature in the SAVI dataset.
(E) Workflow of the exome analysis based on the 85 genes of the UPR signature. MAF, major allele frequency.
(F) Table of the four SNPs found in P1.
(A, C, and D) Gray squares indicate clusters not found in a sample.
These observations prompted us to reanalyze the exome sequencing previously performed for this patient, including her mother and her paternal uncle (P2).1 We specifically explored the 85 genes of the UPR signature (Figure 7E). Exome analysis revealed SNPs in four genes: CTH, VEGFA, ZCCHC8, and EIF2AK3 (Figure 7F). Variants in CTH and VEGFA are intronic or in untranslated regions and, therefore, may not induce a change in protein function. In contrast, variants in ZCCHC8 and EIF2AK3 induced core changes in amino acid chain and may be of importance. While the STING p.V155M mutation was inherited from the father, both variants were inherited from the healthy mother, suggesting they could play a role in the transcriptomic differences observed between P1 and her uncle P2, regarding the UPR signature score (Figure S7E). However, the variant in ZCCHC8 had an allele count in the population higher than expected for a pathogenic variant (it was seen 110 times in gnomAD, giving an allele frequency of 0.000405), and ZCCHC8 seemed to be a downstream target of the UPR pathway. Therefore, we focused on the variant in EIF2AK3. This private missense variant (p.P649T) in EIF2AK3, which encodes PERK, a key activator of the UPR, was confirmed by Sanger sequencing (Figure S7F). PERK was recently shown to be activated upon STING activation through direct interaction between its kinase domain and the C-terminal tail of STING.20 Once activated, PERK induces the phosphorylation of key components of the ISR. The p.P649T variant carried is in the kinase domain of PERK, which carries the ISR-inducing function. Importantly, the UPR signature score of the mother who carries the variant (C9) appeared in T and B lymphocytes slightly lower than the score observed in the healthy paternal aunt (C8), who is not carrying the EIF2AK3variant (Figure 7A). Overall, these findings support a potential causal effect of the p.P649T variant of PERK in the reduction of the UPR signature in P1.
Discussion
Here, we combined scRNA-seq on PBMCs from five SAVI patients with the analysis of in vitro IFN-β-stimulated PBMCs from CTRLs (1–36 h). We highlighted the main pathways dysregulated in the immune cells of SAVI patients, including inflammation, exacerbated ISR and cell death, and extracted, from disease-associated monocytes, 21 genes specific to STING activation but independent of type I IFN response. We also reported in the plasma of SAVI patients a specific elevation of CCL3, CCL4, and IL-6 independent of a primary stimulation by type I IFN. Together, our findings provide a deeper understanding of the SAVI pathogenesis.
Effector T cell lymphopenia is a clinical hallmark of SAVI,1,14 confirmed in our cohort of SAVI patients. We report that naive T cells are maintained in an early activation state, which concords with previous reports suggesting that T cells carrying a gain-of-function STING1 mutation are constitutively activated.11,32 While activation should prompt T cell proliferation, we highlight increased activation of the senescence pathway in patients’ naive T cells, associated with decreased proportions of cells reaching the G2M phase of the cell cycle. This particular phenotype may be linked to the increased EIF2 signaling observed in patients’ T cells. Indeed, a STING-PERK-eIF2a axis has been shown to induce senescence in a human cell line.20 Our data have shown that EIF2 signaling is activated, particularly in CD8+ T cells, both prior and during JAK inhibitor treatment, reinforcing the link between STING activation, EIF2 signaling, and senescence, independently of type I IFN signaling. In addition, several studies have highlighted the ability of chronic IFN-β to trigger senescence33,34 and our observations from the IFN-β dataset support these studies, although the decrease in cell cycle is only transiently observed. Moreover, we have observed an increase in apoptosis transcriptomic signature, suggesting that activated senescent T cells may head toward programmed cell death. These results concur with the antiproliferative role of STING,14 as well as its pro-senescence10,35 and pro-apoptotic1,12,14 roles. Altogether, our results could suggest that the decrease of effector T cells may result from the altered naive T cell compartment. We propose that naive T cells could lose their ability to transition into an effector state due to an hyperactivation state combined with premature senescence and apoptosis.
Clustering of the SAVI dataset revealed a disease-associated cluster composed of 85% of patients’ cells. This cluster was of classical monocyte origin and presented an increased expression of activation and inflammatory markers. It had elevated GADD34 gene expression, an increased UPR signature score, and decreased ribosomal genes expression, which altogether implies activation of the ISR. While our results did not allow us to conclude on the exact mechanism driving the ISR, the recent discovery of the direct activation by STING of an ISR mediator, PERK, suggests that this could be mediated through PERK.20 PERK is an ER stress sensor that, like inactivated STING, is found on the ER membrane. Upon unfolded protein sensing, PERK phosphorylates eIF2α, thereby triggering the ISR. Its direct activation by STING, independent of unfolded proteins, has recently been demonstrated in human cell lines and in mice, and was shown to be responsible for organ fibrosis as well as cellular senescence, both hallmarks of SAVI.20 These observations suggest that elevated UPR in this cluster reflects a STING-induced activation of PERK. However, as PERK is not the only trigger for UPR, we cannot exclude that other mechanisms are involved in these disease-associated monocytes.
Intriguingly, we observed in the patient P1, as opposed to other patients, a low UPR signature score. This observation helped us to reveal a missense p.P649T variant in EIF2AK3, encoding PERK, inherited from her healthy mother. This residue lies in the kinase domain of PERK, which is responsible for eIF2α phosphorylation, a key event for the initiation of the ISR. This never-described variant was predicted to be highly deleterious, suggesting that it might be highly detrimental in combination with STING-activating mutations. As the kinase domain of PERK is responsible for eIF2α phosphorylation, it strengthens the importance of PERK-mediated ISR in disease-associated monocytes of SAVI patients.
By inferring monocyte-to-T cells interactions,25 we provided clues that hyperinflammatory monocytes could promote activation of T cells, as well as cell death. We revealed the establishment of known T cell-activating signaling in SAVI patients, with increase of CCL19/CCR7 and LGALS1/CD69, which play a role in T cell activation.26,29 Additionally, we note several communications that trigger T cell death, such as TRAIL(TNFSF10)/TNFRSF10B36 or MIKA/KLRK1,31 or involved in immune exhaustion such as LAG3 and its ligands.28 These data point toward a potent role of the monocytes to prime T cells toward activation and cell death. Although these interactions were predicted at the transcriptomic level, several of the cytokines involved in the predicted monocyte-to-T cells communications were also elevated at the protein level in the plasma of SAVI patients.
To further evaluate the relevance of the disease-associated cells in the pathogenesis of SAVI, we suggest that their presence should be investigated in the lungs of SAVI patients, as the role of monocytes and monocyte-derived macrophages is increasingly recognized in pulmonary fibrosis.37,38,39,40 The ISR has also been proposed as an important driver of pulmonary fibrosis,41 which further supports the hypothesis that the disease-associated monocytes may drive the pulmonary damage in SAVI patients and could constitute an interesting indication for treatment monitoring. Other potential targets of interest in SAVI are PERK and the ISR, which are the targets of compounds under development for other diseases.42
While JAK inhibitors are beneficial to SAVI patients, they do not allow for complete remission, and patients with advanced lung disease prior to treatment show little to no amelioration.3 Here, we have assessed the transcriptomic impact of JAK inhibition, supporting an incomplete impact of the treatment at the molecular level. In naive T cells, while treatments seemed to correct the senescence and apoptosis, they did not correct the activation of EIF2 signaling, nor restore the proportion of T cells to normal levels.
Current diagnosis of type I interferonopathies, including SAVI, relies on clinical evaluation followed by measurement of ISGs in PBMCs or whole blood of suspected patients. This approach is common to all type I interferonopathies, and subsequent sequencing (either Sanger or exome sequencing) is required to pinpoint a specific gene.1 There is therefore an unmet need for an easy-to-use, differential diagnosis strategy between the different type I interferonopathies. We noticed elevated CCL3, CCL4, and IL-6 at the transcriptomic level in the disease-associated monocytes and at the protein level in the plasma, and we propose that they be added to the panel of six ISGs routinely measured at the transcriptomic level. Elevated expression of these cytokines associated with high ISG score could lead to a suspicion of STING1 gain-of-function mutation. In addition, we identified a STING-activation signature and have tested if it could serve as another tool to discriminate SAVI patients from other interferonopathies. We have observed that patients with SAVI had a uniquely high level of the signature compared with patients with Aicardi-Goutières syndrome or COPA syndrome (data not shown), reinforcing the specificity of that signature. Interestingly, IL1B, encoding inflammasome activation marker IL-1β, is part of the STING activation signature, consistent with STING’s ability to induce NLRP3 inflammasome activation.43 Even though the inflammasome activates IL-1β at the protein level, IL-1β activation may lead to a positive feedback loop enhancing IL1B gene expression.44 Therefore, the presence of IL1B in the STING activation signature may reflect the activation of NLRP3. Furthermore, the SAVI signature may be relevant in the stratification of SLE patients. Indeed, SLE has been shown the represent not one disease but up to seven group of “sub-diseases,”45 and STING may be activated in some patients.46,47 Thus, we think that our signature may bring new insights into SLE patient stratification.
Limitations of the study
Our study has several limitations, including a low number of samples and the exclusive assessment of PBMCs, both caused by the rareness of SAVI patients and the consequent freezing of any blood sample. However, PBMCs exclude several cell types that may have been interesting to study in SAVI.4 In endothelial cells, STING activation was shown to control T cells transendothelial migration,48 potentially into the lungs.49 Endothelial cells in patients’ lungs also display elevated immune functions,19 further emphasizing their critical role in the SAVI pathogenesis. In addition, fibroblasts have also been implicated in lung fibrosis of SAVI patients.50 Nonetheless, the view of the immune system provided by PBMCs, even partial, has allowed to draw several meaningful conclusions regarding the pathogenesis of SAVI.
To reinforce the analysis, we compared the results driven from these patients to a dataset of PBMC stimulated with IFN-β. The system that we have set up only represents the consequence of an acute IFN-β stimulation while SAVI is a chronic disease with an onset in the first weeks of life, making comparison of the molecular pathways challenging to interpret. However, comparing the patients’ PBMCs with the IFN-β stimulation PBMCs has led to several insights into disease pathogenesis.
Finally, in an attempt to overcome the limitation of studying proteomic pathways at the transcriptomic level, we have used functional signatures composed of targets of a pathway, as opposed to components of said pathways. Nevertheless, some observations made on the transcriptomics data were validated in the plasma at the protein level. In addition, other papers have already reported at the protein level what we have observed at the transcriptomic level,14 further reinforcing the strength of the transcriptomic analysis.
Despite these limitations, our results provide a deeper understanding of the dysregulated responses in patient immune cells and how monocytes and their downstream effect on T cells can reflect SAVI pathogenesis, as well as proposing biomarker candidates for the disease.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD8a, BV711, clone RPA-T8 | Biolegend | 301044; RRID:AB_11218793 |
| CD11c, PE/Dazzle 594, clone 3.9 | Biolegend | 301642; RRID:AB_2564083 |
| CD16, BV570, clone 3G8 | Biolegend | 302036; RRID:AB_2632790 |
| CD19, AF488, clone HIB19 | Biolegend | 302219; RRID:AB_389313 |
| CD25, PerCP/Cy5.5, clone BC96 | Biolegend | 302626; RRID:AB_2125479 |
| CD45RA, AF488, clone HI100 | Biolegend | 304114; RRID:AB_528816 |
| CD123, BV711, clone 6H6 | Biolegend | 306030; RRID:AB_2566353 |
| HLA-DR, PE/Cy5, clone L243 | Biolegend | 307608; RRID:AB_314686 |
| HLA-DR, BV570, clone L243 | Biolegend | 307638; RRID:AB_2650882 |
| CD24, PerCP/Cy5.5, clone ML5 | Biolegend | 311116; RRID:AB_10962689 |
| IgM, BV421, clone MHM-88 | Biolegend | 314516; RRID:AB_2561443 |
| CD3, BV510, clone OKT3 | Biolegend | 317332; RRID:AB_2561943 |
| CD4, BV605, clone OKT4 | Biolegend | 317438; RRID:AB_11204077 |
| CD279 (PD1), PE, clone EH12.2H7 | Biolegend | 329906; RRID:AB_940481 |
| CD1c, AF647, clone L161 | Biolegend | 331510; RRID:AB_1186032 |
| CD161, PE, clone HP-3G10 | Biolegend | 339904; RRID:AB_1501086 |
| TCR Vα24-Jα18, BV421, clone 6B11 | Biolegend | 342916; RRID:AB_2564004 |
| CD141, PE/Cy7, clone M80 | Biolegend | 344110; RRID:AB_256122 |
| IgD, PE, clone IA6-2 | Biolegend | 348204; RRID:AB_10553900 |
| CD127, BV650, clone A019D5 | Biolegend | 351326; RRID:AB_2562095 |
| TCR Vα7.2, PE/Cy7, clone 3C10 | Biolegend | 351712; RRID:AB_2561993 |
| CCR7, BV421, clone G043H7 | Biolegend | 353208; RRID:AB_10915137 |
| CCR6, BV650, clone G034E3 | Biolegend | 353426; RRID:AB_2563869 |
| CXCR3, PE/Dazzle 594, clone G025H7 | Biolegend | 353736; RRID:AB_2564287 |
| CD38, PE/Cy7, clone HB-7 | Biolegend | 356608; RRID:AB_2561903 |
| CCR4, PE/Dazzle 594, clone L291H4 | Biolegend | 359420; RRID:AB_2564094 |
| CD57, PerCP/Cy5.5, clone HNK-1 | Biolegend | 359622; RRID:AB_2565930 |
| CD56, Pacific Blue, clone 5.1H11 | Biolegend | 362520; RRID:AB_2564096 |
| CD56, BV510, clone 5.1H11 | Biolegend | 362534; RRID:AB_2565633 |
| CD14, Pacific Blue, clone 63D3 | Biolegend | 367122; RRID:AB_2687385 |
| CD45, PerCP, clone 2D1 | Biolegend | 368506; RRID:AB_2566358 |
| CD45, BV605, clone 2D1 | Biolegend | 368524; RRID:AB_2715826 |
| Zombie NIR | Biolegend | 423105 |
| CD4, PE-AF610, clone S3.5 | Thermofisher | MHCD0422; RRID:AB_10371763 |
| Biological samples | ||
| Human blood samples | Patients and healthy donors | This paper |
| Human blood samples | Etablissement Francais du Sang | This paper |
| Human PBMC | PromoCell | 434Z036 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant human IFN-β | PBL assay | 11410 |
| ADUS100 | Sanofi compound | RA14447001A |
| JAK1/2-inhibitor (baricitinib) | Sanofi compound | RA14836171 |
| Ficoll | Eurobio Scientific | Cat#CMSMSL01-01 |
| Fetal Bovine Serum | Gibco, Thermo Fisher Scientific | Cat#10270106 |
| DMSO | Sigma Aldrich | Cat#D2650 |
| Fc Block | Miltenyi Biotec | 130-059-901 |
| TruStain FcX | Biolegend | 4422302 |
| PierceTM 16% Formaldehyde (w/v), Methanolfree | Thermo Fisher Scientific | Cat# 28906 (10∗1 mL) |
| Cell ID Intercalator | Standard BioTools | 201325 |
| pBSSVD2005 | Addgene | 21826 |
| EQ Beads | Standard BioTools | Cat# 201078 |
| Critical commercial assays | ||
| Maxpar Direct Immune Profiling Assay | Standard BioTools | Cat# 201325 |
| Chromium Single Cell 3′ Library & Gel Bead Kit v2 | 10X Genomics | Not available anymore |
| Chromium Single Cell 3′ Library & Gel Bead Kit v3 | 10X Genomics | 1000075 |
| Chromium Single Cell 3′ Library & Gel Bead Kit v3.1 | 10X Genomics | 1000268 |
| Proximity extension assay Inflammation | Olink | Targeted 96 inflammation |
| Oligonucleotides | ||
| EIF2AK3 primers: Forward TCAGATATGAACAGCCTTCAGTGT Reverse AACCAAAATTTCACAAGTGGCT | Eurofins Genomics | N/A |
| Deposited data | ||
| Single-Cell RNA-sequencing data – SAVI dataset | This paper | GSE226598 |
| Single-Cell RNA-sequencing data – IFN dataset | This paper | GSE226572 |
| Software and algorithms | ||
| CellRanger V6 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| R V3.6.1 | R-project | https://www.r-project.org/ |
| Seurat v4 | Hao et al.51 | https://satijalab.org/seurat/ |
| FlowJo v10 | BD | https://www.flowjo.com/solutions/flowjo |
| EnrichR | Chen et al.52 | https://maayanlab.cloud/Enrichr/ |
| Spotfire® v.10.3.2.7 | TIBCO® | https://www.tibco.com/products/tibco-spotfire |
| CyTOF software version 8.0.14050 | Standard BioTools | https://go.fluidigm.com/cytofsw/v8 |
| MaxPar Pathsetter V.2.0.45.4 | Standard BioTools | https://www.standardbio.com/products/software/maxpar-pathsetter |
| Ingenuity pathway analysis v57662101 | Qiagen | https://www.qiagen.com/us/products/discovery-and-translational-research/next-generation-sequencing/informatics-and-data/interpretation-content-databases/ingenuity-pathway-analysis |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mickaël Ménager (mickael.menager@institutimagine.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
The scRNA-seq data from the SAVI and the IFN-β datasets have been deposited in GEO and are publicly available as of the date of publication, under the superseries number GSE226601, with respective series number GSE226598 and GSE226572 for the SAVI and IFN-β datasets respectively. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant detail
We performed analysis on 5 SAVI patients previously described clinically, as mentioned in the first paragraph of the results section, and selected for their gain-of-function mutations in STING1. Patients are female or male between 4 years old and 34 years old (P1, P2, P4, P5, P6), P2 being P1’s uncle. 7 healthy controls in the SAVI dataset were selected for their absence of STING1 mutation and for matching age range of SAVI patients. Control donors are male and female under 18 years old (for C7, C10, C11) or adults (C1, C2, C8, C9) among which two samples are relatives, C8 being P1’s paternal aunt and C9 being P1’s mother. Healthy donors for generation of the IFN datasets are male of 19–22 years old and were obtained from the Etablissement Français du Sang. Primary PBMCs ex vivo culture is described in method details part of the STAR Methods text.
Method details
Sample collection of the SAVI dataset
Peripheral blood samples were collected on lithium heparin. Blood samples were centrifuged at 2,300 rpm (1,065g) for 10 min to collect supernatant (plasma), which was stored at −20°C. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation (2,200 rpm or 974g without break for 30 min) using Ficoll (Eurobio Scientific, Les Ulis, France). After centrifugation, the pellet was resuspended in Phosphate-Buffered Saline (PBS) (Thermo Fisher scientific, Illkirch, France) and cells were centrifuged at 1500 rpm (453g) for 5 min. Finally, the pellet was frozen in a medium containing 90% of Fetal Bovine Serum (FBS) (GIBCO, Thermo Fisher scientific, Illkirch, France) and 10% of Dimethyl Sulfoxide (DMSO) (Sigma Aldrich, St. Quentin Fallavier, France), and stored in liquid nitrogen.
Cell phenotyping by flow cytometry
For each sample of the SAVI dataset, 500,000 cells were stained. After an initial wash and 350g centrifugation, Zombie NIR Fixable Viability kit was used to stain dead cells according to manufacturer instructions. Cells were washed and resuspended in 200μL of wash buffer (PBS, 5% BSA, 1mM EDTA), before staining with 90μL of antibody mix with antibodies diluted 1/50. The antibodies used in each one of the three panels are listed in Table S7. Fc Block (Biolegend Europ, Netherland) and wash buffer were used to avoid unspecific bindings. For each panel, extracellular staining has been performed for 30 min at 4°C protected from light. Samples were analyzed using the Sony SP6800 spectral flow cytometers.
IFN-β stimulation of healthy PBMC
Buffy coats from three healthy young male (19–22 years old) were obtained from the Etablissement Français du Sang. PBMCs were isolated using Ficoll (Eurobio Scientific, Les Ulis, France) density gradient and centrifugation at 800g without brakes for 25 min. Following centrifugation, cells were washed in wash buffer and centrifuged at 160g for 9 min at 4°C and the pellet was resuspended in 10mL of wash buffer. Cells were plated in 96-wells tissue culture plates, with 150,000 cells per wells in an initial volume of 125μL of RPMI (Gibco), 10% FBS (heat inactivated, Sigma), 10 mM HEPES, 55 μM β-mercaptoethanol, 6 mM L-glutamine, 50 μg/mL Gentamicin and 100 U/mL Penicilin/Streptomycin (Gibco). One plate was made for each donor for each timepoint to plate a total of 15 million cells per condition.
Cells were challenged with 25μL of recombinant IFN-β (Human interferon beta 1a, mammalian, Catalog No 11410-2, PBL assay Science, USA) at 6,000 IU/mL for a final concentration of 1000 U/mL per well, at one of several timepoints: 1h, 2h, 4h, 8h, 12h, 24h or 36h. Stimulations were done so that all cells, even unstimulated, would be kept a total of 36h in culture. After 36h of culture at 37°C, 5% of CO2, cells were collected and counted. 10,000 cells were sent to scRNA-seq and 2 million to cell phenotyping by CyTOF. Supernatant was frozen at −20°C and kept for analysis of secreted proteins.
Cell phenotyping by cytometry by time-of-flight
High dimensional immune profiling of the cultured cells was obtained using the Maxpar Direct Immune Profiling System (Fluidigm, Inc France) with a 30-marker antibody panel, for CyTOF (Cytometry by Time-Of-Flight), (Table S7). Briefly, 2 million cells were washed with MaxPar Cell Staining Buffer, and the cells were transferred in polypropylene tubes and incubated at room temperature for 10 min with 5 μL of TruStain FcX (Biolegend Europ, Netherland). Cells were then transferred into the tube containing the dry antibody cocktail, vortexed and incubated for 30 min at room temperature. After washing, the cells were fixed with 1mL of 1.6% paraformaldehyde solution (SigmaAldrich, France) and incubated at room temperature for 10 min. After washing, 1mL CellID intercalator-Ir at 125nM (pentamethylcyclopentadienyl-Ir (III)- dipyridophenazine, Fluidigm, Inc France) was added onto the cells and incubated overnight. Cells were then stored at −80°C until acquisition.
Cells were thawed at room temperature then washed 3 times in Maxpar Cell Acquisition Solution Plus (CAS+), a high-ionic-strength solution, and resuspended at a concentration of 1 million cells per mL in CAS+. 10% of EQ Four Element Calibration Beads were added to the cells immediately before acquisition. Sample acquisition and data normalization were made on the CyTOF XT mass cytometer and CyTOF software version 8.0.14050 (Fluidigm, Inc Canada) at the “Plateforme de Cytométrie de la Pitié-Salpetriere (CyPS).” An average of 500,000 events were acquired per sample. Dual count calibration, noise reduction, cell length threshold between 10 and 150 pushes, and a lower convolution threshold equal to 12 were applied during acquisition. Data cleaning was performed using Maxpar Pathsetter software v2.0.45. 4 parameters (center, offset, residual and width) were used to resolve ion fusion events (doublets) from single events using the Gaussian distribution generated by each event. After data cleaning, the program produces new FCS files consisting of only intact live single cells.
Cleaned FCS files were loaded in R using the flowCore package. FCS files from all samples were concatenated into a SingleCellExperiment object. Clustering was performed using the cluster function from the CATALYST package to identify 60 initial clusters. Clusters were manually identified based on marker expression following Fluidigm Maxpar Immune Profiling manufacturer instruction. Clusters were merged into cohesive cell populations, resulting in 26 final cell types (including 3 groups of contaminants that were removed). Kinetics of cell type proportions over time was plotted using the ggplot2 package.
scRNA-seq libraries and sequencing
scRNA-seq experiments were performed at the “Labtech SingleCell@Imagine”, in 3 batches for the SAVI dataset, and 3 batches for the IFN-β dataset. 8 libraries of the STING1 batch were generated using Chromium Single Cell 3′ Library & Gel Bead Kit v.2 (10x Genomics) according to the manufacturer’s protocol, while the remaining 9 libraries (STING2 and STING3) were generated using Chromium Single Cell 3′ Library & Gel Bead Kit v.3 (10x Genomics). Briefly, cells were counted, diluted at 1,000 cells/μL in PBS+0.04% and 20,000 cells were loaded in the 10x Chromium Controller to generate single-cell gel-beads in emulsion. After reverse transcription, gel-beads in emulsion were disrupted. Barcoded complementary DNA was isolated and amplified by PCR. Following fragmentation, end repair and A-tailing, sample indexes were added during index PCR. All libraries were sequenced on a Novaseq (Illumina). The 8 purified libraries generated from the v2 kit were sequenced with 26 cycles of read 1, 8 cycles of i7 index and 98 cycles of read 2, while the 9 libraries generated with v3 were sequenced with 28 cycles of read 1, 8 cycles of i7 index and 91 cycles of read 2.
The 24 scRNA-seq libraries of the IFN-β dataset were generated using Chromium Single Cell Next GEM 3′ Library & Gel Bead Kit v.3.1 (10x Genomics) according to the manufacturer’s protocol, following the same steps as in the SAVI dataset. The purified libraries were sequenced on a Novaseq (Illumina) with 28 cycles of read 1, 8 cycles of i7 index and 91 cycles of read 2.
Bioinformatics analysis of scRNAseq data
Sequencing reads were demultiplexed and aligned to the human reference genome (hg38), and counted using the CellRanger Pipeline v6.0. Unfiltered RNA UMI counts were loaded into Seurat v4 for quality control, data integration and downstream analyses. Apoptotic cells and empty sequencing capsules were excluded by filtering out cells with low number of features (Filters were defined based on the batch: STING1: nFeature <300; STING2 and STING3 <500; IFN-stim <750) or a mitochondrial content higher than 20%. Data from each sample were normalized using sctransform, before batch correction using Seurat’s FindIntegratedAnchors, on the 3,000 most variable features. For the IFN-β dataset, for computational efficiency, integration anchors were determined with a canonical correlation analysis, using all unstimulated samples as reference. For the SAVI dataset, to avoid over-aligning samples, a reciprocal principal component analysis (PCA) was used to find integration anchors. On the resulting integrated datasets, we computed the PCA on the 3000 most variable genes, before computing a UMAP (on 20 PCs for SAVI and 30 PCs for IFN-stimulation). Community detection was performed using the graph-based modularity-optimization Louvain algorithm from Seurat’s FindClusters function with a 1.2 resolution. Cell types labels were assigned to resulting clusters based on a manually curated list of marker genes as well as previously defined signatures of the PBMC subtypes.53 Despite filtering for high quality cells, 11/34 clusters in the SAVI dataset and 7/36 in the IFN-β dataset stood out as poor quality clusters and were removed from further analysis. They represented low UMI cells, dying cells, cycling cells, doublets, or contaminants (progenitor cells, basophils, platelets or megakaryocytes). A total of 112,060 and 115,503 cells were kept in the SAVI and IFN-β datasets respectively. Transcriptomic signatures scores were calculated using Seurat’s AddModuleScore function. The signatures and their origin are described in Table S7. Differential expression testing was conducted using the FindMarkers function in Seurat, with default Wilcoxon testing. p-values were controlled using Bonferroni correction. Genes with an absolute log(fold-change) ≥0.25 and an adjusted p value ≤ 0.05 were selected as differentially expressed. Pathway analysis was performed using both the Ingenuity pathway analysis v57662101 software (IPA (QIAGEN Inc.) and EnrichR.52,54 Heatmaps were extracted from the comparison module in IPA. Pathways with an absolute Z score lower than 2 or a Bonferroni-Hochberg corrected p values higher than 0.05 were filtered out. The TRRUST transcription factors 201921 used for the transcription factors enrichment analysis was performed using Enrich R.
The cell cycle phase was defined using the CellCycleScoring function of Seurat V4, with the default gene lists and adjusting the parameters to better reflect expected proportions of CTRL cells in each phase of the cell cycle.
Cell-to-cell interactions were predicted using the ICELLNET framework.25 We filtered out filtered the gene expression matrix to only keep the genes that were expressed in at least 5% of the cells in at least one of the clusters. We combined together cluster 5 and cluster 17 into a single classical monocyte group and considered this group as the “central cells”. Each cluster of CD4+ or CD8+ T cell was considered as the “partner cells”. In the SAVI dataset, the predictions were performed separately for the CTRLs, the SAVI, and the SAVI_treated groups. In the IFN-β dataset, the predictions were performed separately for each timepoint.
Cytokine measurements
The plasma samples and conditioned media from in vitro stimulated PBMC were analyzed by proximity extension assays (PEA), a targeted, antibody-based, proteomics method. We used the PEA Olink Target96 Inflammation panel which targets 92 biomarkers. Briefly, 2 DNA-labeled antibodies bind to a targeted protein, followed by hybridization of the oligonucleotides when the antibodies come in close proximity to each other. Adding DNA polymerase to the sample generates a unique PCR target sequence, which is then detected and quantified using a quantitative PCR (qPCR) readout (BiomarkHD, Data collection and Real-Time PCR analysis software 4.7.1, Fluidigm).
The Olink PEA Quality control process consists of technical controls to monitor the performance of all 3 steps of the assays (immunoreaction, extension, and amplification/detection) as well as the individual samples. Internal controls are spiked into each sample to provide a non-human assay control, a positive extension control consisting of an antibody coupled to a unique DNA-pair and, finally, a positive detection control based on a double stranded DNA amplicon. In addition, each experimental run includes a control strip with control samples used to estimate the accuracy of protein detection and quantification (intra- and inter-coefficient of variation). A negative control (buffer) run in triplicate is used to set background levels and calculate limit of detection (LOD), a plate control (plasma pool) is run in triplicate for plate normalization, and a sample control (reference plasma) is included in duplicate to estimate CV between runs.
Data are presented as NPX (Normalized Protein eXpression) values. NPX is Olink’s relative protein quantification unit on log2 scale. Data generation of NPX (obtained using Olink NPX Manager 3.3.2.434) consists of normalization to the extension control (known standard), log2-transformation, and level adjustment using the plate control (plasma sample).
Serial dilutions of samples are performed to determine a potential Hook effect (observed when there is an antigen excess relative to the reagent antibodies, resulting in falsely lower values). Conditioned media are tested at 1:1, 1:4 and 1:10 dilutions for Target96 Inflammation. Plasma samples are not diluted.
For plasma cytokine measurement, differential protein expression was performed using Multiple comparisons parametric test (TIBCO Spotfire v.10.3.2.7) with Benjamini-Hochberg (BH) False Discovery Rate adjustment. For the IFN-β dataset, pairwise analysis by donor was performed by timepoint. For patient samples, SAVI samples were compared to healthy donors. An adjusted p value < 0.05 was considered statistically significant.
Exome analysis
The exome sequencing used in this study has been described by Jeremiah et al..1 Exome sequencing was performed by the Center National de Génotypage, Institut de Génomique, CEA. After quality control by the DNA Bank Laboratory, genomic DNA (3μg) was captured using in-solution enrichment methodology (Human All Exon v5 – 50 Mb, Agilent Technologies, CA, USA). Library preparation and exome enrichment protocol (∼20,000 targeted genes) was performed on an automated platform, using NGSx (PerkinElmer Inc, MA, USA) and Bravo (Agilent Technologies, CA, USA) robots respectively, according to manufacturer’s instructions (SureSelect, Agilent Technologies CA, USA). After normalization and quality control, exome enriched libraries were sequenced on a HiSEQ 2000 (Illumina Inc., CA, USA) as paired-end 100 base reads. Sequencing was performed in order to provide a mean cover of at least 60 to 70X for each sample. Image analysis and base calling was performed using Illumina Real Time Analysis (RTA) Pipeline. Sequence quality parameters were assessed daily throughout the 12 days sequencing run. Sequences were aligned to the reference human genome hg19 using the Burrows-Wheeler Aligner. Downstream processing was carried out with the Genome Analysis Toolkit (GATK), SAMtools, and Picard, following documented best practices (http://www.broadinstitute.org/gatk/guide/topic?name=best-practices).
We searched for SNPs in the 85 genes of the UPR signature, to extract the SNPs found in less than 5% of the general population. No other filter was used.
We generate a HOPE report55 for the p.P649T mutation in PERK (encoded by EIF2AK3), by using PERK uniprot identifier Q9NZJ5.
Generation of the ADUS100 dataset
Human PBMC samples were obtained from Promocell (C-12907 promocell, lot 434Z036). Briefly, after thawing and a quality control on cell viability (>90%), a minimal of of 2∗106 cells cells were incubated either with or without ADUS100 (3mM final concentration), an STING agonist, during 4h in RPMI1640 medium complemented with 10% heat inactivated fetal bovine serum. In some conditions, cells were pre-incubated 15min with a JAK1-2 inhibitor (baricitinib, 10mM final concentration) prior and during ADUS100 exposure. After 4h, cells were washed with PBS without Ca/Mg containing 0.04% BSA and a minimum of 6,105 harvested for single cell analysis (10X Genomics). Libraries were generated using Chromium Single Cell 3′ Library & Gel Bead Kit v.2 (10x Genomics) according to the manufacturer’s protocol. Libraries were sequenced on a Novaseq (Illumina), with 26 cycles of read 1, 8 cycles of i7 index and 98 cycles of read 2.
Primary and SV40-fibroblasts
Patient’s primary fibroblasts were obtained by cultivating skin biopsies in RPMI 1640 supplemented with 20% FBS, 1% penicillin/streptomycin, 1X gentamicin, and 1X amphotericin B. Primary fibroblasts were transformed by lipofection of pBSSVD2005 (Addgene #21826) coding the large-T antigen of Simian virus-40. After 3 to 4 cycles of cultures, fibroblasts change of their shape and growing rate and were considered immortalized. This was later confirmed by evaluating expression of SV40 by confocal microscopy. SV40-fibroblasts were then maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin.
Sanger sequencing
Sanger sequencing was performed on SV40-fibroblast genomic DNA to confirm the next-generation sequencing results for EIF2AK3. The primers used for PCR are as follow: Forward TCAGATATGAACAGCCTTCAGTGT; Reverse AACCAAAATTTCACAAGTGGCT.
Purified PCR products were directly sequenced using BigDye Terminators (version 1.1) and a 3500xL Genetic Analyzer (Applied Biosystems).
Quantification and statistical analysis
Statistical tests for cellular composition analysis in both the CyTOF and scRNA-seq datasets were performed in R v3.6.1. Kruskal-Wallis test followed by post-hoc multiple comparison Dunn’s test was applied to assess differences in cell population proportions (∗: p ≤ 0.05; ∗∗: p ≤ 0.01; ∗∗∗: p ≤ 0.001).
Acknowledgments
The study was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM) and the Université Paris Cité, by the Atip-Avenir, by government grants managed by the Agence National de la Recherche as part of the “Investment for the Future” program (Institut Hospitalo-Universitaire Imagine, grant ANR-10-IAHU-01, Recherche Hospitalo-Universitaire, grant ANR-18-RHUS-0010), by a Sanofi iAward Europe, and by the “Emergence ville de Paris” program. F.R.L. received grants from the Centre de Référence des Déficits Immunitaires Héréditaires (CEREDIH), the Agence National de la Recherche (ANR-14-CE14-0026-01 “Lumugene” as PI, and ANR-18-CE17-0001 “Action”, ANR-19-CE15-028 « Lympho-Sting; ANR-21-CE17-0044 « PREDICT JIA »; ANR-22-CE15-0047-02 « BREAK-ITP » as Partner), and the Fondation pour la recherche Médicale (FRM: EQU202103012670).
C.C. and L.D. are recipients of CIFRE PhDs (Sanofi) and Imagine Thesis Awards. Q.R. is a recipient of an Institut Imagine MD-PhD fellowship (funded by the Fondation Bettencourt Schueller) and a Société Nationale Française de Médecine Interne (SNFMI) fellowship.
We thank the LabTech Single-Cell@Imagine, supported by the Paris Region and the “Investissement d’avenir” program through 2019 ATF funding – Sésame Filières PIA (grant 3877871), for the generation of scRNA-seq libraries. We thank Imagine genomic and bioinformatics core facilities and the Pitié-Salpêtrière Cytometry platform CyPS for advice and technical assistance. We sincerely thank the patients and their families for participating in the study.
Author contributions
Conceptualization: C.C., G.B., F. Augé, M.D., F.R.L., and M.M.M.; data curation: C.C., T.F., F.C., and L.C.; formal analysis: C.C., L.D., M.B., Q.R., C. Masson, and G.B.; investigation: C.C., L.D., M.B., Q.R., M.L., B.S., A.R., B.H., C. Meunier, M.P., M.Z., B.P.P., and V.G.P.; methodology: C.C., G.B., and M.M.M.; funding acquisition: J.C.G., F. Augé, M.D., and M.M.M.; resource: C.B., E.C., B.S., B.B.M., A.B., P.Q., M.L.F., and B. Neven; supervision: J.C.G., G.B., F. Augé, A. Fischer, M.D., F.R.L., and M.M.M.; visualization: C.C., L.D., M.B., Q.R., and M.L.; writing – original draft: C.C., and M.M.M.; writing – review and editing: C.C., M.B., L.G., B. Nal, P.P., M.L.F., B. Neven, G.B., F. Augé, A. Fischer, M.D., F.R.L., and M.M.M.
Declaration of interests
C.C., F.R.L., and M.M.M. are listed as inventors on a patent application related to this article (European Patent Application no. PCT/FR2023/050433, entitled “A gene signature for diagnosing stimulator of interferon genes (STING)-associated vasculopathy with onset in infancy (SAVI)”). F.R.L. and M.M.M. received grants from Sanofi (iAward Europe and research collaboration contract). C.C., L.D., M.D., F.A., G.B., J.C.G., and A.R. are or were employees of Sanofi and may hold shares and/or stock options in the company.
Inclusion and diversity
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. We avoided “helicopter science” practices by including the participating local contributors from the region where we conducted the research as authors on the paper.
Published: December 19, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101333.
Supplemental information
References
- 1.Jeremiah N., Neven B., Gentili M., Callebaut I., Maschalidi S., Stolzenberg M.-C., Goudin N., Frémond M.L., Nitschke P., Molina T.J., et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J. Clin. Invest. 2014;124:5516–5520. doi: 10.1172/JCI79100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu Y., Jesus A.A., Marrero B., Yang D., Ramsey S.E., Sanchez G.A.M., Tenbrock K., Wittkowski H., Jones O.Y., Kuehn H.S., et al. Activated STING in a Vascular and Pulmonary Syndrome. N. Engl. J. Med. 2014;371:507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frémond M.L., Hadchouel A., Berteloot L., Melki I., Bresson V., Barnabei L., Jeremiah N., Belot A., Bondet V., Brocq O., et al. Overview of STING-Associated Vasculopathy with Onset in Infancy (SAVI) Among 21 Patients. J. Allergy Clin. Immunol. Pract. 2021;9:803–818.e11. doi: 10.1016/j.jaip.2020.11.007. [DOI] [PubMed] [Google Scholar]
- 4.David C., Frémond M.L. Lung Inflammation in STING-Associated Vasculopathy with Onset in Infancy (SAVI) Cells. 2022;11:318. doi: 10.3390/cells11030318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dobbs N., Burnaevskiy N., Chen D., Gonugunta V.K., Alto N.M., Yan N. STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe. 2015;18:157–168. doi: 10.1016/j.chom.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishikawa H., Ma Z., Barber G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abe T., Barber G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol. 2014;88:5328–5341. doi: 10.1128/JVI.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yum S., Li M., Fang Y., Chen Z.J. TBK1 recruitment to STING activates both IRF3 and NF-κB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2100225118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanaka Y., Chen Z.J. STING Specifies IRF3 phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci. Signal. 2012;5:ra20. doi: 10.1126/scisignal.2002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dou Z., Ghosh K., Vizioli M.G., Zhu J., Sen P., Wangensteen K.J., Simithy J., Lan Y., Lin Y., Zhou Z., et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550:402–406. doi: 10.1038/nature24050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu J., Chen Y.-J., Dobbs N., Sakai T., Liou J., Miner J.J., Yan N. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J. Exp. Med. 2019;216:867–883. doi: 10.1084/jem.20182192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gulen M.F., Koch U., Haag S.M., Schuler F., Apetoh L., Villunger A., Radtke F., Ablasser A. Signalling strength determines proapoptotic functions of STING. Nat. Commun. 2017;8:427. doi: 10.1038/s41467-017-00573-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Staels F., Betrains A., Doubel P., Willemsen M., Cleemput V., Vanderschueren S., Corveleyn A., Meyts I., Sprangers B., Crow Y.J., et al. Adult-Onset ANCA-Associated Vasculitis in SAVI: Extension of the Phenotypic Spectrum, Case Report and Review of the Literature. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.575219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cerboni S., Jeremiah N., Gentili M., Gehrmann U., Conrad C., Stolzenberg M.-C., Picard C., Neven B., Fischer A., Amigorena S., et al. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J. Exp. Med. 2017;214:1769–1785. doi: 10.1084/jem.20161674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Picard C., Thouvenin G., Kannengiesser C., Dubus J.-C., Jeremiah N., Rieux-Laucat F., Crestani B., Belot A., Thivolet-Béjui F., Secq V., et al. Severe Pulmonary Fibrosis as the First Manifestation of Interferonopathy (TMEM173 Mutation) Chest. 2016;150:e65–e71. doi: 10.1016/j.chest.2016.02.682. [DOI] [PubMed] [Google Scholar]
- 16.Tang X., Xu H., Zhou C., Peng Y., Liu H., Liu J., Li H., Yang H., Zhao S. STING-Associated Vasculopathy with Onset in Infancy in Three Children with New Clinical Aspect and Unsatisfactory Therapeutic Responses to Tofacitinib. J. Clin. Immunol. 2020;40:114–122. doi: 10.1007/s10875-019-00690-9. [DOI] [PubMed] [Google Scholar]
- 17.Lin B., Berard R., Al Rasheed A., Aladba B., Kranzusch P.J., Henderlight M., Grom A., Kahle D., Torreggiani S., Aue A.G., et al. A novel STING1 variant causes a recessive form of STING-associated vasculopathy with onset in infancy (SAVI) J. Allergy Clin. Immunol. 2020;146:1204–1208.e6. doi: 10.1016/j.jaci.2020.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frémond M.L., Rodero M.P., Jeremiah N., Belot A., Jeziorski E., Duffy D., Bessis D., Cros G., Rice G.I., Charbit B., et al. Efficacy of the Janus kinase 1/2 inhibitor ruxolitinib in the treatment of vasculopathy associated with TMEM173-activating mutations in 3 children. J. Allergy Clin. Immunol. 2016;138:1752–1755. doi: 10.1016/j.jaci.2016.07.015. [DOI] [PubMed] [Google Scholar]
- 19.Gao K.M., Motwani M., Tedder T., Marshak-Rothstein A., Fitzgerald K.A. Radioresistant cells initiate lymphocyte-dependent lung inflammation and IFNγ-dependent mortality in STING gain-of-function mice. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2202327119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang D., Liu Y., Zhu Y., Zhang Q., Guan H., Liu S., Chen S., Mei C., Chen C., Liao Z., et al. A non-canonical cGAS–STING–PERK pathway facilitates the translational program critical for senescence and organ fibrosis. Nat. Cell Biol. 2022;24:766–782. doi: 10.1038/s41556-022-00894-z. [DOI] [PubMed] [Google Scholar]
- 21.Munoz J., Rodière M., Jeremiah N., Rieux-Laucat F., Oojageer A., Rice G.I., Rozenberg F., Crow Y.J., Bessis D. Stimulator of Interferon Genes–Associated Vasculopathy With Onset in Infancy: A Mimic of Childhood Granulomatosis With Polyangiitis. JAMA Dermatol. 2015;151:872–877. doi: 10.1001/jamadermatol.2015.0251. [DOI] [PubMed] [Google Scholar]
- 22.Balka K.R., Louis C., Saunders T.L., Smith A.M., Calleja D.J., D’Silva D.B., Moghaddas F., Tailler M., Lawlor K.E., Zhan Y., et al. TBK1 and IKKε Act Redundantly to Mediate STING-Induced NF-κB Responses in Myeloid Cells. Cell Rep. 2020;31 doi: 10.1016/j.celrep.2020.03.056. [DOI] [PubMed] [Google Scholar]
- 23.Pakos-Zebrucka K., Koryga I., Mnich K., Ljujic M., Samali A., Gorman A.M. The integrated stress response. EMBO Rep. 2016;17:1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reich S., Nguyen C.D.L., Has C., Steltgens S., Soni H., Coman C., Freyberg M., Bichler A., Seifert N., Conrad D., et al. A multi-omics analysis reveals the unfolded protein response regulon and stress-induced resistance to folate-based antimetabolites. Nat. Commun. 2020;11:2936. doi: 10.1038/s41467-020-16747-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noël F., Massenet-Regad L., Carmi-Levy I., Cappuccio A., Grandclaudon M., Trichot C., Kieffer Y., Mechta-Grigoriou F., Soumelis V. Dissection of intercellular communication using the transcriptome-based framework ICELLNET. Nat. Commun. 2021;12:1089. doi: 10.1038/s41467-021-21244-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan Y., Chen R., Wang X., Hu K., Huang L., Lu M., Hu Q. CCL19 and CCR7 Expression, Signaling Pathways, and Adjuvant Functions in Viral Infection and Prevention. Front. Cell Dev. Biol. 2019;7:212. doi: 10.3389/fcell.2019.00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shrager S.H., Kiel C. SnapShot: APC/T Cell Immune Checkpoints. Cell. 2020;183:1142–1142.e1. doi: 10.1016/j.cell.2020.10.007. [DOI] [PubMed] [Google Scholar]
- 28.Grebinoski S., Zhang Q., Cillo A.R., Manne S., Xiao H., Brunazzi E.A., Tabib T., Cardello C., Lian C.G., Murphy G.F., et al. Autoreactive CD8+ T cells are restrained by an exhaustion-like program that is maintained by LAG3. Nat. Immunol. 2022;23:868–877. doi: 10.1038/s41590-022-01210-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cedeno-Laurent F., Dimitroff C.J. Galectin-1 research in T cell immunity: Past, present and future. Clin. Immunol. 2012;142:107–116. doi: 10.1016/j.clim.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fulda S. Tumor-necrosis-factor-related apoptosis-inducing ligand (TRAIL) Adv. Exp. Med. Biol. 2014;818:167–180. doi: 10.1007/978-1-4471-6458-6_8. [DOI] [PubMed] [Google Scholar]
- 31.Guerra N., Lanier L.L. Editorial: Emerging Concepts on the NKG2D Receptor-Ligand Axis in Health and Diseases. Front. Immunol. 2020;11:562. doi: 10.3389/fimmu.2020.00562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kieper W.C., Jameson S.C. Homeostatic expansion and phenotypic conversion of naïve T cells in response to self peptide/MHC ligands. Proc. Natl. Acad. Sci. USA. 1999;96:13306–13311. doi: 10.1073/pnas.96.23.13306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frisch S.M., MacFawn I.P. Type I interferons and related pathways in cell senescence. Aging Cell. 2020;19 doi: 10.1111/acel.13234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu Q., Katlinskaya Y.V., Carbone C.J., Zhao B., Katlinski K.V., Zheng H., Guha M., Li N., Chen Q., Yang T., et al. DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep. 2015;11:785–797. doi: 10.1016/j.celrep.2015.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang H., Wang H., Ren J., Chen Q., Chen Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA. 2017;114:E4612–E4620. doi: 10.1073/pnas.1705499114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang S., El-Deiry W.S. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22:8628–8633. doi: 10.1038/sj.onc.1207232. [DOI] [PubMed] [Google Scholar]
- 37.Misharin A.V., Morales-Nebreda L., Reyfman P.A., Cuda C.M., Walter J.M., McQuattie-Pimentel A.C., Chen C.-I., Anekalla K.R., Joshi N., Williams K.J.N., et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017;214:2387–2404. doi: 10.1084/jem.20162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCubbrey A.L., Barthel L., Mohning M.P., Redente E.F., Mould K.J., Thomas S.M., Leach S.M., Danhorn T., Gibbings S.L., Jakubzick C.V., et al. Deletion of c-FLIP from CD11bhi Macrophages Prevents Development of Bleomycin-induced Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018;58:66–78. doi: 10.1165/rcmb.2017-0154OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fraser E., Denney L., Antanaviciute A., Blirando K., Vuppusetty C., Zheng Y., Repapi E., Iotchkova V., Taylor S., Ashley N., et al. Multi-Modal Characterization of Monocytes in Idiopathic Pulmonary Fibrosis Reveals a Primed Type I Interferon Immune Phenotype. Front. Immunol. 2021;12:623430. doi: 10.3389/fimmu.2021.623430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kessler N. 2022. Monocyte-derived Macrophages Aggravate Pulmonary Vasculitis via cGAS/STING/IFN-mediated Nucleic Acid Sensing; p. 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barabutis N. Unfolded Protein Response in Lung Health and Disease. Front. Med. 2020;7:344. doi: 10.3389/fmed.2020.00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Derisbourg M.J., Hartman M.D., Denzel M.S. Modulating the integrated stress response to slow aging and ameliorate age-related pathology. Nat. Aging. 2021;1:760–768. doi: 10.1038/s43587-021-00112-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaidt M.M., Ebert T.S., Chauhan D., Ramshorn K., Pinci F., Zuber S., O’Duill F., Schmid-Burgk J.L., Hoss F., Buhmann R., et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell. 2017;171:1110–1124.e18. doi: 10.1016/j.cell.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schindler R., Ghezzi P., Dinarello C.A. IL-1 induces IL-1. IV. IFN-gamma suppresses IL-1 but not lipopolysaccharide-induced transcription of IL-1. J. Immunol. 1990;144:2216–2222. [PubMed] [Google Scholar]
- 45.Banchereau R., Hong S., Cantarel B., Baldwin N., Baisch J., Edens M., Cepika A.-M., Acs P., Turner J., Anguiano E., et al. Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell. 2016;165:551–565. doi: 10.1016/j.cell.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.An J., Durcan L., Karr R.M., Briggs T.A., Rice G.I., Teal T.H., Woodward J.J., Elkon K.B. Expression of Cyclic GMP-AMP Synthase in Patients With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017;69:800–807. doi: 10.1002/art.40002. [DOI] [PubMed] [Google Scholar]
- 47.Thim-Uam A., Prabakaran T., Tansakul M., Makjaroen J., Wongkongkathep P., Chantaravisoot N., Saethang T., Leelahavanichkul A., Benjachat T., Paludan S., et al. STING Mediates Lupus via the Activation of Conventional Dendritic Cell Maturation and Plasmacytoid Dendritic Cell Differentiation. iScience. 2020;23 doi: 10.1016/j.isci.2020.101530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anastasiou M., Newton G.A., Kaur K., Carrillo-Salinas F.J., Smolgovsky S.A., Bayer A.L., Ilyukha V., Sharma S., Poltorak A., Luscinskas F.W., Alcaide P. Endothelial STING controls T cell transmigration in an IFNI-dependent manner. JCI Insight. 2021;6 doi: 10.1172/jci.insight.149346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao K.M., Chiang K., Korkmaz F.T., Janardhan H.P., Trivedi C.M., Quinton L.J., Gingras S., Fitzgerald K.A., Marshak-Rothstein A. Expression of a STING Gain-of-function Mutation in Endothelial Cells Initiates Lymphocytic Infiltration of the Lungs. bioRxiv. 2023 doi: 10.1016/j.celrep.2024.114114. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xie X., Wu X., Zhao D., Liu Y., Du Q., Li Y., Xu Y., Li Y., Qiu Y., Yang Y. Fluvoxamine alleviates bleomycin-induced lung fibrosis via regulating the cGAS-STING pathway. Pharmacol. Res. 2023;187 doi: 10.1016/j.phrs.2022.106577. [DOI] [PubMed] [Google Scholar]
- 51.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M. III, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021 Jun 24;184(13):3573-3587.e29. [DOI] [PMC free article] [PubMed]
- 52.Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Monaco G., Lee B., Xu W., Mustafah S., Hwang Y.Y., Carré C., Burdin N., Visan L., Ceccarelli M., Poidinger M., et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep. 2019;26:1627–1640.e7. doi: 10.1016/j.celrep.2019.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Venselaar H., Te Beek T.A.H., Kuipers R.K.P., Hekkelman M.L., Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinf. 2010;11:548. doi: 10.1186/1471-2105-11-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The scRNA-seq data from the SAVI and the IFN-β datasets have been deposited in GEO and are publicly available as of the date of publication, under the superseries number GSE226601, with respective series number GSE226598 and GSE226572 for the SAVI and IFN-β datasets respectively. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.