

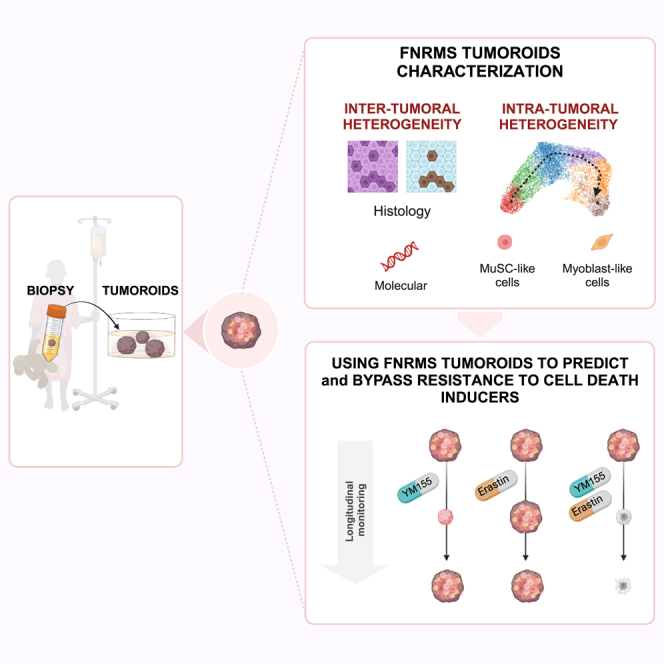
Summary
Rhabdomyosarcoma (RMS) is the main form of pediatric soft-tissue sarcoma. Its cure rate has not notably improved in the last 20 years following relapse, and the lack of reliable preclinical models has hampered the design of new therapies. This is particularly true for highly heterogeneous fusion-negative RMS (FNRMS). Although methods have been proposed to establish FNRMS organoids, their efficiency remains limited to date, both in terms of derivation rate and ability to accurately mimic the original tumor. Here, we present the development of a next-generation 3D organoid model derived from relapsed adult and pediatric FNRMS. This model preserves the molecular features of the patients’ tumors and is expandable for several months in 3D, reinforcing its interest to drug combination screening with longitudinal efficacy monitoring. As a proof-of-concept, we demonstrate its preclinical relevance by reevaluating the therapeutic opportunities of targeting apoptosis in FNRMS from a streamlined approach based on transcriptomic data exploitation.
Keywords: rhabdomyosarcoma, tumor-derived organoids, tumoroid, pediatric oncology, resistance, heterogeneity, child, apoptosis, cell death
Graphical abstract
Highlights
-
•
Relapsed rhabdomyosarcoma (RMS) 3D organoids can be derived from needle biopsies
-
•
RMS 3D organoids finely preserve inter- and intra-tumor heterogeneity
-
•
Transcriptomic approach reveals Survivin as a key apoptosis blockage point in RMS
-
•
RMS 3D organoids are powerful tools to design and/or reexplore drug combinations
Savary et al. develop finely tuned next-generation RMS 3D organoids amenable for high-content assays to contribute to the design of efficient therapeutic combinations needed to improve the survival of patients with rhabdomyosarcoma.
Introduction
Rhabdomyosarcomas (RMS) gather a group of heterogeneous cancers representing notably 5% of all pediatric solid tumors.1,2 They share similarities to embryonic muscle tissue,3 and two main subclasses have been defined based on histological features in the pediatric population, namely the embryonal (ERMS; 70% of all RMS) and the alveolar (ARMS; 20% of all RMS) subclasses.4 Molecular advances resulted in the identification of pathognomonic PAX3/7-FOXO1 chromosomal translocations as being associated with 85% ARMS.5,6,7 ERMS and ARMS lacking this translocation, i.e., fusion-negative RMS (FNRMS), are an even more heterogeneous and complex subgroup, still defined by default.8,9,10 Their 5-year survival rate ranges from 60% to 80% for patients with localized tumors but drops to 20% for those who relapsed or had metastases at diagnosis, with only a slight improvement in prognosis over the last 20 years.11 Innovative therapeutic strategies are thus required notably for these patients with poor prognosis.
To meet this challenge, major efforts have been made to design in vitro models mimicking RMS tumors in their diversity and complexity to (1) better understand RMS biology and (2) facilitate high-throughput drug screenings. Protocols have notably been proposed to establish tumor-derived organoids (i.e., tumoroids).12,13,14 However, approaches to develop FNRMS in vitro models, although promising, have until yet been hampered by (1) derivation efficiency rate below 20% from patients’ samples, (2) lack of preservation of original tumors’ molecular specificities beyond genetics, and (3) difficulties to expand them in 3D.12,13,14 These pitfalls complicate both the modeling of inter-tumoral heterogeneity and the long-term evaluation of new therapeutic strategies. Here, we present the establishment of an original relapse FNRMS-derived organoid model, which has overcome all these limitations and allows the highly efficient derivation of 3D cultures directly from patients’ tumors in a unique optimized culture medium. These tumoroids recapitulate precisely the histological and molecular characteristics of aggressive FNRMS tumors, and they accurately preserve their stem cell hierarchy and intra-tumoral heterogeneity even after several passages and cryopreservation as 3D cultures. By combining bioinformatics analyses of bulk and single-cell transcriptomic data, we demonstrate their usefulness to design and evaluate new drug combinations from a streamlined proof-of-concept approach based on targeting apoptosis.
Results
Design and characterization of an original FNRMS-derived organoid model that finely preserves tumor of origin’s features
By extensively deciphering active signaling cascades that could support tumor cell growth based on transcriptomic datasets (Figure S1), we designed an M3 culture medium and established a protocol to rapidly generate and expand over a long term (>6 months) adult and pediatric relapse FNRMS-derived organoids (designated as RMS_Os). RMS_Os were successfully established directly in 3D from patient or patient-derived xenograft tumor specimens (100% efficiency, 5/5 samples; Figure 1A; Table S1). We also improved significantly the derivation of primary 2D FNRMS models directly from patients’ samples at diagnosis by using a supplemented M3 medium (M5) with 100% growth (n = 6/6) versus, respectively, 17% in classically used DMEM-FBS 10% (n = 1/6 samples) or 16% growth in the recently described BM1∗ medium (n = 4/25).13 Tumorigenicity of RMS_Os was demonstrated by their ability to give rise to tumors when orthotopically (tibialis anterior) xenografted in immunocompromised mice (RMS_XG; Figures 1B and S2A). Contrary to the methods available until now,13,14 RMS_Os were derived successfully from tumors located in diverse regions, with varying histological etiologies and at different ages, without necessarily requiring a prior grafting step in patient-derived xenograft (PDX) models (Table S1). Moreover, RMS_Os precisely recapitulate the histological features of their tumor of origin (Figures 1B, S2A, and S2B), which is also a major improvement to obtain tumors’ phenocopies. For example, RMS1_O preserves the histology of its primary tumor (RMS1_T), with cells of variable sizes, some with small nuclei and often reduced cytoplasm and some rhabdomyoblast cells, whereas RMS2_O, like RMS2_T, displays a more undifferentiated state (Figure 1B). RMS_Os also preserve the expression pattern of Desmin and Myogenin diagnostic markers and the proliferation rates of their tumor of origin (Figures 1B, S2A, and S2B).
Figure 1.

Design and characterization of an original FNRMS-derived organoid model that finely preserves tumor of origin’s features
(A) Pipeline of organoids (RMS_Os) derivation from fresh FNRMS tumors. Samples were obtained from patients undergoing biopsy/surgery (Table S1). Five RMS_Os have been established and expanded using the protocol described in the STAR Methods. RMS1_O and RMS2_O, derived from pediatric patients 1 and 2, respectively, are shown. From days 3 to 15 post-seeding, RMS_Os expand to 1,000 (RMS1_O) and 1,500 μm (RMS2_O) diameters. White scale bar: 1 cm. Black scale bar: 200 μm. Expansion over 8–10 weeks allows the full characterization (histologic and multi-omic), the biobanking, and the preclinical use (e.g., middle-throughput drug screening) of the organoid lines.
(B) Representative hematoxylin phloxine saffron (HPS) and immunohistochemistry (IHC) characterization of RMS_Os using clinical markers routinely used for RMS diagnosis. RMS_Os were matched blind by anatomopathologists to their tumors of origin in all cases. _T, tumor; _O, organoid _XG, xenograft derived from RMS_O line. Scale bar: 50 μm.
(C) Heatmap of pairwise Pearson correlation coefficients based on global transcriptomic expression profile showing the clustering of RMS_Os with their paired tissues of origin (RMS_Ts). Beside RMS_Os and RMS_Ts, samples are designed according to (1) the medium in which they were derived, (2) their 2D or 3D culture method, and (3) their passage at time of collection (P), as well as their cryopreservation/reanimation when applicable (∗), and then labeled as follows: Culture Medium_Culture method _Passage (∗ when applicable). M3, optimized tumoroid medium; M2; incomplete medium; DMEM, DMEM +10% FBS. Two biological replicates per condition. Patient 1-derived models and tumor (RMS1): pink; patient 2-derived models and tumor (RMS2): blue.
(D) PCA of RNA-seq data from tumors and models plotted in 2D, using their projections onto the first two PCs (Dim1 and Dim2). Each data point represents one sample. The names and color code correspond to those described in (C). Two biological replicates per condition.
(E) Functional enrichment of differentially expressed (DE) genes between 2D cell lines and 3D RMS_Os from patient 1. Left scatterplots represent the top 6 enriched Gene Ontology (GO) pathways in 2D models, and right scatterplots correspond, reciprocally, to the top 6 enriched GO pathways in 3D RMS_Os. Dots are colored according to their adjusted statistical probabilities with a blue (lower significance) to yellow (higher significance) gradient and sized by the counts of genes matching the biological process.
(F) Hierarchical clustering analysis based on the centered-normalized expression values of RMS tumor and differentiation markers highlights the high level of similarities between RMS_Os and their corresponding tumor samples. Top left column indicates whether the indicated genes are markers of stem (progenitors/satellite cells [SCs]), committed muscle (muscle differentiation), or cancer cells (RMS). Each sample is designed as above (see C). Two biological replicates per condition.
(G and H) Preservation of tumor mutational (G) and methylation (H) profiles in RMS_Os.
(G) Heatmap of pairwise Pearson correlation coefficients between all samples. The correlation coefficients were calculated on the variant allele frequencies, and the hierarchical clustering was based on the Euclidean distance. Two biological replicates per condition except for tissue sample (n = 1).
(H) PCA plot of normalized methylation values of tissue samples (top) and in vitro samples (2D or 3D) cultured in 2D (DMEM or M5 media) or 3D (_O) (bottom) for RMS1, RMS2, RMS7, and RMS9 patients. Two biological replicates per condition except for RMS2_T (n = 1).
Previously established RMS-derived organoids only partially reproduced the transcriptional characteristics of their original tumors, allowing us to only cluster them according to their fusion status.13 Pearson’s correlation heatmap and principal-component analysis (PCA) established from transcriptomic profiling unveil that RMS_O models are clearly grouped with their respective tumors of origin, mostly through the first dimension (Dim1), which largely reflects inter-patient heterogeneity in PCA (Figures 1C and 1D). RMS_Os, but also their equivalent 2D models cultured in M3 medium (M3_2D), cluster with their corresponding tumors in an unsupervised analysis even after several passages (Figure 1C), contrary to 2D and 3D models cultured, respectively, in DMEM-FBS 10% (DMEM_2D) or an incomplete organoid medium (M2_3D). However, functional gene set enrichment highlights that cultures propagated in 2D are characterized by unspecific processes, including those related to cell adhesion regulation, whereas RMS_Os display the expected developmental and muscle identity (Figure 1E). Hierarchical clustering analysis on RMS markers confirms the high level of similarities between RMS_Os and their corresponding tumor samples, even after cryopreservation, while unveiling notable differences with 2D cultures, even those grown in M3 (M3_2D) (Figure 1F). For example, the expression patterns of cancer (GPC3, FGFR4, IGF2, IGF1R, DUSP6, and MYCN) and muscle (PAX7, MYOD1, TTN, MYL1, and MYH3) pediatric markers are comparable in RMS1_O and its tumor of origin, while they are downregulated in M3_2D cultures (Figure 1F). These markers are barely expressed in pediatric RMS2_T and its matched RMS2_O, which reciprocally both present a strong expression of MEOX2, a specific marker of early paraxial mesoderm.15 Furthermore, DNA sequencing (DNA-seq) analysis validates the preservation of the tumor of origin’s variant allelic fraction (VAF) by RMS_Os, indicating that they retain major clones and subclonal populations even after cryopreservation, unlike DMEM_2D cultures (Figures 1G and S2D). Of note, DMEM_2D cultures acquire in vitro, before any cryopreservation, two genetic alterations predicted by automatic tools (PolyPhen-2 and SIFT) to be deleterious and recently described as oncogenic.16 Finally, methylome analysis indicates that only RMS_O models allow us to discriminate patients like the original tumors (Figure 1H), whereas in vitro 2D propagation leads to a loss of inter-patient heterogeneity regardless of the medium (Figure 1H, bottom).
Then, these FNRMS-derived organoids (1) are established and propagated in 3D directly from pediatric and adult patients’ samples with high efficiency and (2) precisely preserve the genetic, epigenetic, and gene expression profiles’ characteristics of their original tumors, which is a prerequisite to mimic inter-patient heterogeneity in the context of personalized medicine approaches.
FNRMS-derived organoids preserve functional intra-tumor heterogeneity
Intra-tumoral heterogeneity is widely considered a key driver of resistance to treatment. Preservation of the recently described RMS tumor hierarchy14,17 is then a prerequisite in a preclinical perspective. To define the relevance of our RMS_Os, we performed droplet-based single-cell RNA-seq (scRNA-seq). Using an unsupervised Leiden algorithm, we identified 6 clusters on uniform manifold approximation and projection (UMAP), each expressing a specific subset of biomarker genes (Figures 2A, S2E, and S2F; Table S2). Trajectory inference analyses unveiled the expected myogenic-like differentiation sequence (Figures 2B, 2C, and S2G), with features of muscle satellite cells in clusters 4–3, whereas clusters 5-2-6 resemble myoblasts at different cell-cycle stages and are subsequently grouped into a single myoblast-like proliferative cluster (Figures 2C, 2D, and S2G). Gene set enrichment analyses confirmed that clusters 4–3 encompass quiescent satellite-like cells enriched in a hypoxic gene signature (Figures 2E and 2F), whereas cycling myoblast-like cells (clusters 5-2-6) are reciprocally enriched in genes involved in quiescence exit and in the previously reported pediatric cancer signature18 (Figures 2E and 2F). As expected from the expression of key muscle marker genes described in the literature,19,20,21 clusters 4-3 express CRYAB, GLUL, and MTX1, three genes robustly defining satellite cells in human muscle tissue scRNA-seq analyses,19,21 and the mesenchymal marker CD44, recently described to be expressed by RMS satellite-like cells (Figure 2F).17,22 Cells in cluster 1 specifically express PAX7, suggesting a commitment toward myogenic differentiation and satellite cell activation (Figure 2F).23 This differentiation ends at clusters 5-2-6, which express EZH2, a gene specifically induced in activated satellite cells during myogenesis,23 and early myoblastic differentiation markers, such as MSTN and TPM424 (Figure 2F). Importantly, these myoblast-like tumor cells fail to express late markers of muscular differentiation (Table S2). Thus, proliferating myoblast-like cells at the end of the myogenic continuum described in this RMS_O model seem to be “halted” in an early stage of differentiation, confirming recent studies on RMS tumor hierarchy.14,17 We then wondered whether precise preservation of the tumor hierarchy allows us to model drug response and relapse. As a proof of concept, we showed that RMS1_O, RMS2_O, and RMS3_O reproduce the vincristine resistance profiles observed in patients 1, 2, and 3 (Figures 2G–2K and S2H–S2J), with a resistant subpopulation remaining viable even under high doses of treatment. Importantly, the ability to expand RMS_Os over time in 3D allows us to mimic the regrowth of tumor cells post-treatment washout (Figures 2H–2K, S2I, and S2J).
Figure 2.

FNRMS-derived organoids preserve functional intra-tumor heterogeneity
(A and B) scRNA-seq UMAP of RMS1_O showing cluster identities (A) and unsupervised trajectory inference analysis using scVelo (B). Data from 2 biological replicates at passages P13 and P14.
(C) Module scores of quiescent muscle satellite cell (top) and myoblast (bottom) expression programs displayed on scRNA-seq UMAP of RMS1_O.
(D) Module scores of cycling progenitors and G1/S (top) and G2/M (bottom) cell-cycle phases.
(E) Functional enrichment between quiescent muscle satellite-like cells (clusters 4-3) and myoblast proliferative cells (clusters 5-2-6) (see STAR Methods). Dots are colored according to their adjusted statistical probabilities with a yellow (lower significance) to blue (higher significance) gradient and sized by the count number of genes matching the biological process. MuSC, muscle satellite cells; SkM, skeletal muscle.
(F) Dot plot representing gene expressions of specific myogenic differentiation markers between cluster groups. Dots are sized according to the percentage of cells in each cluster group that express the gene (transcript level > 0) and color coded by average gene expression levels across cells.
(G–K) Efficiency of vincristine on RMS_Os.
(G) Vincristine dose-response curves performed on RMS_Os. Viability is expressed as a percentage of the value in untreated cells (CellTiter-Glo). Means ± SD are represented (n = 3).
(H–K) RMS_Os were treated or not with vincristine (5 nM) for 3 days. Treatments were then stopped, and regrowth of structures was evaluated within 50 days in each case.
(H and I) RMS1_O (H) and RMS2_O (I) regrowth rate within 30 days after treatment washout.
(J and K) Representative bright-field images of RMS1_O (J) and RMS2_O (K) at t = 0 and 3 days, with or without (vehicle) vincristine treatment. When regrowth of tumoroids was observed and tumoroids reached a size of 4–6 × 105 μm2, they were split to ensure that their renewal properties were preserved. WO, washout. Scale bar: 400 μm.
Thus, the RMS_O model preserves the cell states previously described in patients’ tumors, allowing us to model the impact of intra-tumor heterogeneity on drugs’ response.
FNRMS-derived organoids can be used for drug screenings: Proof of concept on targeting apoptosis blockage points
Therapies targeting apoptotic pathways are the prototypical example of promising strategies, which have so far fallen short of expectations in patients.25 We reasoned that by combining a rationalized approach based on the targeting of appropriate apoptotic blocking points and the use of FNRMS tumoroid models that faithfully reproduce their tumor of origin, we could optimize the design of apoptotic inducer therapeutic combinations.
Separated clusters for FNRMS and fusion-positive RMS (FPRMS) were identified on UMAP in two independent cohorts, solely based on the expression of an 86-gene apoptotic signature (Figures 3A and S3A; Table S3). Interestingly, causal inference approaches by Ingenuity Pathway Analysis26 indicate that cell death transcriptional regulatory networks are significantly activated in FNRMS compared to FPRMS in cohorts 1 (p < 0.0001, Z score = 1.94; Figure 3B) and 2 (p < 0.0001, Z score = 1.41; Figure S3B). Using a cross-validation strategy, we generated an apoptotic gene metascore based on a two-gene signature using BNIP3 (p < 0.05 in 186/250 iterations) and FASLG (p < 0.05 in 170/250 iterations) (Figure S3C) that efficiently discriminates patients with metascore-high (BNIP3/FASL-high) FNRMS as those with a significantly poorer outcome compared to metascore-low patients (Figure 3C and S3D). Considering the clinical relevance of the apoptotic cascade’s status, we sought to identify apoptotic effectors that block its execution in FNRMS. We unveiled that 47% pro-apoptotic and 54% anti-apoptotic genes are, respectively, expressed at significantly higher and lower levels in tumors than in their healthy skeletal muscle’s counterparts (Figure 3D; Table S4). Interestingly, pro-apoptotic effectors overexpressed in tumors are mostly upstream in the apoptotic cascade (Figure 3E). Conversely, BIRC5, which encodes the downstream IAP (inhibitor of apoptosis protein) Survivin, is significantly overexpressed in FNRMS (log 2 fold change [log2(FC)] = 2.9; p < 0.0001) in two independent cohorts (Figures 3D, 3E, S3E, and S3F). To define whether FNRMS may be dependent on BIRC5 expression for survival, we built a custom-made library of 20 drugs where compounds were chosen based on their ability to directly (6 apoptotic targeting agents) or indirectly (5 DNA repair inhibitors) activate the apoptosis mechanisms. As conventional chemotherapies are the gold standard of RMS treatment, we also added 9 conventional chemotherapies as controls. Using this drug library, we performed a medium-scale drug screening on 2D RMS cell lines (Figure 3F). As expected, and has already been shown by others,12,13,27 FNRMS cells are highly susceptible to the Survivin-expression inhibitor YM-155, with 10 nM of this compound being sufficient to drive massive cell death in all four tested 2D FNRMS cell lines (Figure 3F). In contrast, FNRMS cells are resistant to other compounds antagonizing IAP as SMAC mimetics, as well as to BCL-2 and DNA repair inhibitors, confirming the central role of Survivin in blocking apoptosis execution (Figure 3F).
Figure 3.

FNRMS-derived organoids can be used for drug screenings: Proof of concept on targeting apoptosis blockage points
(A) UMAP of FNRMS and FPRMS samples (cohort 1) based on the expression of apoptotic effectors (see Table S3).
(B) Activation state of apoptotic cascades using Ingenuity Pathway Analysis (IPA) in FNRMS versus FPRMS samples (cohort 1). Significant difference corresponds to Z score >0 and p < 0.0001.
(C) Survival analyses on the apoptotic metascore in the training (cohort 1) and test (cohort 3) FNRMS datasets. Kaplan-Meier curves were generated using its dichotomized form defined by a cross-validated optimal cut-point procedure in a cohort-dependent manner. Differences of overall survival (cohort 1) and event-free survival (cohort 3) probabilities between both groups were tested using log-rank tests, and associated statistical probabilities are displayed on the graph. Numbers of patients at risk are indicated in the tables below the curves. Time-dependent receiver operating characteristic (ROC) curves and hazard ratios were generated using continuous apoptotic metascore.
(D) Heatmap of apoptotic genes expression levels significantly DE (FDR < 0.05) between FNRMS and healthy muscle samples (cohort 4; Table S4). Samples in columns are clustered using Ward’s method on the inverse Spearman’s correlation coefficient matrix. Pro-apoptotic genes are displayed with bold text.
(E) Mapping of main pro-apoptotic (rectangle) and anti-apoptotic (circle) effectors significantly DE (FDR < 0.05; UP = FC > 1.5; DN [down] = FC < 1.5) between FNRMS and healthy muscle samples. Genes with altered expression compared to healthy tissue are colored in red or green when alterations are potentially associated with apoptotic blockage or induction, respectively. Drug-target genes are indicated with an orange label.
(F–H) Drug screenings on 2D and 3D RMS models. We used a customized library composed of a panel of 20 drugs, including IAP (pink), BCL-2 (green), and DNA repair (orange) inhibitors, as well as conventional chemotherapy agents (blue). For each drug of our screens, IC50, defined as the half maximal inhibitory concentration values, were established based on the Selleckchem online database. Drugs were distributed at 4 (3D RMS_Os) to 6 (2D RMS cell lines) different concentrations chosen to cover at least 3 log of concentrations and to include the aforementioned IC50 (see STAR Methods). Data were normalized to negative control wells (DMSO only). The pIC50 was calculated by using 3 technical replicates for each compound (see STAR Methods).
(F) Heatmap representing the sensitivity (pIC50) of four 2D FNRMS and FPRMS cell lines. Color gradient: blue (low sensitivity) to yellow (high sensitivity).
(G) Heatmap representing the sensitivity (pIC50) of two 3D FNRMS organoid lines. Color gradient: blue (low sensitivity) to yellow (high sensitivity). Black boxes indicate a resistant organoid model to the identified drug (IC50 not reached).
(H) High-content drug screening performed on FNRMS-derived organoids. Top: boxplots depicting the cytotoxicity of apoptosis inducers and chemotherapeutic agents on RMS1_Os as measured by the log of dead/live intensity ratio after treatment. Boxes represent the 25%, 50%, and 75% quartiles, and whiskers represent the minimum and maximum values. Each circle represents a test realized on one RMS1_O sphere, with an average of 46 spheres per condition. Dashed horizontal line represents the DMSO control (gray) and the gambogic acid (GA, brown) median baselines, and full horizontal line and greyed area represent the 3× standard deviation of DMSO. Dose 100 nM, sole common dose between all drugs was chosen for this representation. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, Dunnett’s test. Bottom: fluorescence microscopy images of RMS1_Os exposed to DMSO (negative control), 10 μM GA, 100 nM vincristine (positive controls), and 100 nM YM-155 (bottom). Images show Calcein AM (viable, green) and EthD1 (dead, red). Each image depicts 55 organoids, with corresponding outline of segmentation and microcavities shown in white. Scale bars: 400 μm (white) and 200 μm (red).
Since FNRMS-derived organoids are stable and expandable, we performed the same drug screening on two of these models to demonstrate their potential for preclinical use. We used classical cell viability assay (Figure 3G) and a robust and sensitive image-based high-content assay that allowed us to miniaturize the drug screening and perform it simultaneously on 55 spheres per RMS organoid line and per dose of each therapeutic compound (Figure 3H). Consistently with transcriptomic data extrapolation, both RMS_O models were sensitive to the BIRC5 inhibitor YM-155 at a level comparable to that of conventional chemotherapies but not to other apoptosis inducers (Figure 3G, 3H, S3G, and S3H). Of note, the response of RMS organoids to overall drugs, and notably YM-155, is weaker than the one observed for 2D RMS cell lines (Figures 3F–3G). Interestingly, we observed that some cells remained viable, even at a dose of YM-155 corresponding to 100 times its IC50, suggesting that not all RMS cell states present the same sensitivity to this compound (Figure 3H, bottom).
Thus, FNRMS-derived organoids are robust tools for drug screening and can be used to define therapeutic potential but also to anticipate resistance to compounds such as YM-155, pinpointed from a transcriptional mapping approach of apoptotic pathways.
FNRMS-derived organoids are innovative tools to design new drug combinations targeting all tumor cell populations
To identify YM-155 RMS resistant cell states, we assessed the expression profile of apoptosis effectors in RMS_Os, which accurately maintain the original tumor pattern of apoptotic gene expression (Figure S3F). We observed that BIRC5 is specifically expressed by the MKI67-positive proliferative myoblast population (Figure 4A and 4B). Consistently, although YM-155 induced a massive wave of cell death and a major destruction of tumoroid structures (Figures 4C and S4A), 61% tumoroids treated grew back from cells that had not been eliminated within 1 month post-washout (Figures 4D and S4B). To eliminate those cells, we therefore looked for another target gene more specifically expressed in the BIRC5-negative quiescent satellite cell-like population. We identified the voltage-dependent anion-selective channel protein-2 encoding gene (VDAC2), which we selected as a putative candidate because it is both involved in cell death regulation and pharmacologically targetable28,29 (Figure 4E). A therapeutic combination of YM-155 and Erastin, a known inhibitor of VDAC2 activity notably leading to ferroptosis,29 is sufficient to induce a massive destruction of tumoroid structures (Figures 4F and 4G) at doses largely ineffective in monotherapy (Figure S4C). Most importantly, while both monotherapies are largely insufficient to block RMS1_O regrowth, their combination is highly effective even after several weeks (Figures 4F, 4G, S4D, and S4E). Importantly, renewal properties of RMS1_O treated with YM-155 are preserved, as shown by their ability to be split over time (Figures 4G and S4E).
Figure 4.

FNRMS-derived organoids are innovative tools to design new drug combinations targeting all tumor cell populations
(A) MKI67 (top) and BIRC5 (bottom) expressions in RMS1_O scRNA-seq UMAP. Data from 2 biological replicates at passages P13 and P14.
(B) Immunofluorescence (top) showing heterogeneous expression of BIRC5 (in green) among tumor cells on a tumoroid section (RMS1_O). Overlap between the BIRC5-encoded protein Survivin and Ki67 (in red) in proliferative cells is shown in enlarged boxes (middle). Nuclei are stained in blue. White scale bar: 50 μm. Orange scale bar: 10 μm. Bottom: quantification of unstained (blue), Ki67+ (red), Survivin+ (green), or Ki67+/Survivin+ (yellow) cells (n = 3 spheres).
(C and D) Survivin inhibitor YM-155 shows only transient efficiency on tumoroids. Representative images of live/dead immunofluorescence staining of RMS1_O treated for 2 days with vehicle (control) or YM-155 condition, 1 (C) or 20 days (D) post-washout of the treatment. Quantification was performed by measuring the ratio of surface area of live (green) to dead cells (red) (right). Scale bar: 200 μm. n = 3 spheres at least per condition.
(E) VDAC2 expression in RMS1_O visualized on scRNA-seq UMAP.
(F and G) Efficiency of Erastin/YM-155 combination on RMS1_O. RMS1_Os were treated with YM-155, Erastin, or a combination of both for 2 days. Treatments were then stopped, and regrowth of structures was evaluated within 80 days in each case (curves and images are from one representative experiment out of 3; control n = 5; Erastin n = 6; YM-155 n = 12; Erastin+YM-155 n = 16).
(F) Growth curves of RMS1_O after treatment washout in the different conditions tested. Each curve corresponds to the area of one tumoroid over time.
(G) Representative bright-field images of RMS1_O in the different conditions tested. When regrowth of RMS1_O was observed and tumoroids reached a size of 4–6 × 105 μm2, they were split to ensure that their renewal properties were preserved (red lines). Scale bar: 800 μm.
Because they accurately reproduce tumor cell states while maintaining their self-renewal and 3D expansion properties, these last-generation FNRMS-derived organoids are crucial tools to define effective therapeutic strategies in longitudinal preclinical approaches.
Discussion
RMS remain a clinical challenge due to the insufficient effectiveness of current treatments.30 Although elegant neurosphere-like rhabdosphere models have been developed from 2D cell lines to enable the study and targeting of the tumor stem compartment thought to be at the origin of chemoresistance, one of the major obstacles to improving their management is the lack of preclinical tools that accurately and precisely reproduce the characteristics of their tumors of origin in their heterogeneous and dynamic component. Organoid models derived from patient tumors (i.e., tumoroids) have been demonstrated to efficiently bridge the gap between in vitro and xenograft models to predict the actual human response in the field of adult epithelial cancers.31 However, so far, the derivation of FNRMS in vitro cultures has only been possible with rather low efficiency or from PDX models.12,13,14 Moreover, beyond genetics, an important challenge remained to faithfully reproduce the epigenetic/transcriptional profiles of the original FNRMS tumors and their heterogeneous cell states. Finally, the design of new therapeutic combinations, targeting all tumor populations, relies on the possibility of cultivating and expanding tumoroids over the long term in 3D, to preserve as much as possible the characteristics of their original tumors.
Here, we show that it is feasible to derive high-efficiency tumoroids from aggressive FNRMS that reproduce the original tumor’s features, including its genetic, epigenetic, and transcriptional profiles, and that can be expanded in 3D over a long period of time, thereby meeting the tumor-derived organoid definition.31 Preservation of the 3D structure is a key issue to reproduce cell-cell contacts, as well as physical constraints existing in malignancies, and thus to precisely reproduce tumor behavior. Indeed, although the 2D lines that we and others12,13 established recapitulate partial tumor characteristics, they also present significant caveats as exemplified here by the artifactual enrichment in cell adhesion signatures or the loss of the original DNA methylation pattern. Moreover, our protocol meets the challenge to allow the systematic derivation of all RMS_Os included so far, which is a prerequisite from a personalized medicine perspective considering both the heterogeneity and scarcity of these tumors. Last, the direct derivation of in vitro FNRMS organoids from patients’ tumors is crucial in a preclinical setting: although in vivo models remain of interest for evaluating new therapeutic combinations from a clinical perspective, patient-derived FNRMS organoid models offer a unique opportunity (1) to avoid time loss, operational complexity, and additional costs associated with in vivo drug screening and (2) to precisely dissect the mechanisms associated with resistance, including distinguishing tumor-autonomous from microenvironment-driven factors.12 The creation of biocollection of RMS_Os is highly relevant considering that (1) this subgroup is the most prevalent in children and adolescents, (2) a collection of such models, which we initiated here, is crucial to provide proxies of the high level of complexity resulting from the inter-patient heterogeneity in the FNRMS group, and (3) the survival rate of patients at relapse is about 20%, highlighting the need for models to establish new, efficient therapeutic approaches.
Beyond simple assessment of drug vulnerability, we also established the proof of concept of the efficiency of these FNRMS-derived organoid models in optimized drug screening by showing that they provide leads to design and/or reexplore the therapeutic potential of drug combinations while explaining their limits and the potential origins of resistance observed in patients. Targeting BIRC5 has already been proposed as a putative therapeutic approach in several cancers including RMS,12,13,27,32 but no combination including a Survivin inhibitor has shown clinical efficacy. Although we do not yet foresee a therapeutic application, the use of our FNRMS-derived organoid models sheds new light on these contradictory data and underline the possible interest in reevaluating YM-155 therapeutic potential in the context of optimized combinations targeting all tumor cell states. Indeed, BIRC5 expression seems to be restricted to the myoblast proliferative clusters—consistent with the dual role of Survivin in both apoptosis inhibition and cell-cycle promotion33—thereby explaining the transient effect of YM-155 on this model. Because they finely reproduce intra-tumor heterogeneity and are expandable over a long-term period in 3D, which was not the case with the tumor-derived models proposed so far, these FNRMS-derived organoids then offer a sizable opportunity to reexplore the specific vulnerabilities of each tumor cell state, to target them therapeutically using relevant drug combinations, and to model long-term therapeutic response. We have focused here on the evaluation of a few drugs in this proof of concept, but the stability and amplifiability of these models mean that larger drug screening can also be performed. In other words, the use of these FNRMS-derived organoid models could reconcile the promising data obtained in vitro and the failures observed in the clinic to rapidly provide new effective therapeutic opportunities to prevent and anticipate resistance and relapse.
Limitations of the study
To fully investigate RMS organoids’ potential for predicting clinical treatment responses, a comprehensive study involving multiple patients and tumor-derived organoids would be needed. In this report, this was hampered by the small sample size of patients. Thus, the next challenge will be to establish a biocollection of tumoroid models sufficient to mimic RMS inter-patient heterogeneity at diagnosis and relapse. It will also be necessary to optimize the tumoroids’ derivation pipeline (1) to integrate a stromal component to reconstitute the tumor niche in its complexity and (2) to make it compatible in terms of time/cost with use in personalized and/or precise medicine approaches, aiming at predicting the best treatment for each patient.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Omni map DAB kit | Roche Diagnostics | cat. no. 760-158 |
| Mouse monoclonal anti-desmin | Dako | cat. no. M0760; RRID:AB_2335684 |
| Mouse monoclonal anti-myogenin | Dako | cat. no. M3559; RRID:AB2250893 |
| Mouse monoclonal anti-ki67 | Dako | cat. no. M7240; RRID:AB_2142367 |
| Rabbit monoclonal anti-survivin | Cell Signaling | cat. no. 2808S, RRID:AB_2063948 |
| Biological samples | ||
| Rhabdomyosarcoma patients | BRCs of CLB, HCL-HFME (Tissu-Tumorothèque Est) and Institut Curie. | N/A |
| Patient-derived xenografts (PDX) | St Jude Hospital | SJRHB013758_X2 |
| Critical commercial assays | ||
| Chromium Single Cell 5′ v3.1 assay | 10X Genomics |
https://www.10xgenomics.com/ |
| NovaSeq 6000 platform | Illumina | https://www.illumina.com/ |
| CellTiter-Glo® 2.0 Cell Viability Assay | Promega | cat. no. G9243 |
| Live/DEAD™ Viability/Cytotoxicity Kit | Thermo Fisher Scientific | cat. no. 10237012 |
| Deposited data | ||
| Raw and normalized microarray data | R2 platform34; GEO database; Array Express; Shern et al.10 | http://r2.amc.nl; GEO: GSE28511; ArrayExpress: E-TABM-1202; N/A |
| -omics data generated for this paper | GEO database | GEO: GSE248183 |
| Normalized RNA-seq data | St. Jude Cloud35 | https://www.stjude.cloud |
| Human reference genome NCBI build 38, GRCh38.p13 | Genome Reference Consortium36 | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| GENCODE gene annotation (homo sapiens, v37) | GENCODE37 | https://www.gencodegenes.org/ |
| Resource website for apoptotic genes (human) | Diez et al.38 | http://deathbase.org/ |
| Resource for biological pathways (human) | This paper; MSigDB39 | https://www.gsea-msigdb.org/gsea/msigdb |
| Homo_sapiens_assembly38.fasta, Homo_sapiens_assembly38.fasta.fai, Homo_sapiens_assembly38.dict, Homo_sapiens_assembly38.dbsnp138.vcf, hapmap_3.3.hg38.vcf.gz, Mills_and_1000G_gold_standard.indels.hg38.vcf.gz, 1000G_omni2.5.hg38.vcf.gz, 1000G.phase3.integrated.sites_only.no_MATCHED_REV.hg38.vcf | Resource bundle Google bucket | https://storage.googleapis.com/genomics-public-data/resources/broad/hg38/v0/ |
| af-only-gnomad.hg38.vcf.gz, 1000g_pon.hg38.vcf.gz | GATK Best Practices Google bucket | https://storage.googleapis.com/gatk-best-practices/somatic-hg38/ |
| homo_sapiens_vep_108_GRCh38.tar.gz | Ensembl | https://ftp.ensembl.org/pub/release-108/variation/indexed_vep_cache/ |
| Experimental models: Cell lines | ||
| Rhabdomyosarcoma cell line (RD) | ATCC | cat. no. 85111502 |
| Rhabdomyosarcoma cell line (Rh30) | ATCC | cat. no. crl-2061 |
| Rhabdomyosarcoma cell line (RDAbl) | N/A | N/A; RD subclone |
| Rhabdomyosarcoma cell line (Rh36) | N/A | (RRID:CVCL_M599) |
| Experimental models: Organisms/strains | ||
| Mouse xenografts: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ |
Charles River | RRID:IMSR_JAX:005557 |
| Software and algorithms | ||
| ImageJ | internet | https://imagej.nih.gov/ij/ |
| Phenochart 1.1.0 | internet | Akoya Biosciences |
| CaseViewer | internet | 3D Histech |
| HALO | Indica Labs | |
| Oligo | Carvalho BS, Irizarry RA40 | https://www.bioconductor.org/packages/release/bioc/html/oligo.html |
| DESeq2 | Love et al.41 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| limma | Ritchie et al.42 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| ComplexHeatmap | Gu et al.43 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| ggpubr | Kassambara44 | https://rpkgs.datanovia.com/ggpubr/ |
| rstatix | Kassambara45 | https://CRAN.R-project.org/web/packages/rstatix/index.html |
| QIAGEN Ingenuity Pathway Analysis (IPA) | Krämer et al.46 | https://digitalinsights.qiagen.com/IPA |
| progeny | Schubert et al.47 | https://www.bioconductor.org/packages/release/bioc/html/progeny.html |
| survival | Therneau48 | https://CRAN.R-project.org/package=survival |
| survminer | Kassambara et al.49 | https://CRAN.R-project.org/package=survminer |
| maxstat | Hothorn50 | https://CRAN.R-project.org/package=maxstat |
| timeROC | Blanche et al.51 | https://cran.r-project.org/web/packages/timeROC/index.html |
| survivalROC | Heagerty and Saha-Chaudhuri52 | https://CRAN.R-project.org/package=survivalROC |
| FastQC | Andrews53 | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Cutadapt | Martin54 | https://github.com/marcelm/cutadapt |
| STAR | Dobin et al.55 | https://github.com/alexdobin/STAR |
| HTseq-count | Anders et al.56 | https://github.com/simon-anders/htseq |
| org.Hs.e.g.,.db | Carlson57 | https://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html |
| FactoMineR | Lê et al.58 | https://cran.r-project.org/web/packages/FactoMineR/index.html |
| factoextra | Kassambara and Mundt59 | https://CRAN.R-project.org/package=factoextra |
| RcolorBrewer | Neuwirth60 | https://CRAN.R-project.org/package=RColorBrewer |
| Cell Ranger | Zheng et al.61 | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| Seurat | Stuart et al.62 | https://cran.r-project.org/web/packages/Seurat/index.html |
| scDblFinder | Germain et al.63 | https://bioconductor.org/packages/release/bioc/html/scDblFinder.html |
| scran | Lun et al.64 | https://bioconductor.org/packages/release/bioc/html/scran.html |
| scanpy | Wolf et al.65 | https://github.com/scverse/scanpy |
| slingshot | Street et al.66 | https://www.bioconductor.org/packages/release/bioc/html/slingshot.html |
| scVelo | Bergen et al.67 | https://github.com/theislab/scvelo/blob/master/docs/source/index.rst |
| loom | Linnarsson Lab68 | https://github.com/linnarsson-lab/loompy |
| fgsea | Korotkevich et al.69 | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| R | R Core Team (2022). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. | https://www.R-project.org/ |
| python | Van Rossum, G. & Drake, F.L., 2009. Python 3 Reference Manual, Scotts Valley, CA: CreateSpace. | https://www.python.org/ |
| presto | Korsunsky et al.70 | https://github.com/immunogenomics/presto |
| bwa-mem2 | Vasimuddin et al.71 | https://anaconda.org/bioconda/bwa-mem2 |
| Picard | Broad Institute72 | https://anaconda.org/bioconda/picard |
| Mosdepth | Pedersen et al.73 | https://anaconda.org/bioconda/mosdepth |
| Ensembl Variant Effect Predictor | McLaren et al.74 | https://anaconda.org/bioconda/ensembl-vep |
| Fiji | Schindelin et al.75 | https://doi.org/10.1038/nmeth.2019 |
| CellProfiler | Lamprecht MR et al.76 | https://cellprofiler.org/ |
| R - Tidyverse | Ritz C et al.77 | https://tidyverse.tidyverse.org. |
| DOPPLatform | N/A | https://www.doppl.ch/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Laura Broutier (laura.broutier@lyon.unicancer.fr).
Materials availability
This study did not generate new unique reagent.
Data and code availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Laura Broutier (laura.broutier@lyon.unicancer.fr). All -omic datasets generated during this study are avalaible via GEO repository with the accession number GSE248183. All data analyses’ codes will be made available upon request. This paper also analyzes existing, publicly available data. The accession numbers for these datasets are listed in the key resources table. There are restrictions to the availability of biobanked RMS_O due to the lack of an external centralized repository for their distribution and our need to maintain the stock.
Experimental model and study participant details
Patient samples
Leftovers from RMS samples (n = 14) were obtained through biopsies/resections performed at the Pediatric Hematology and Oncology Institute (iHOPE, Lyon) or Hôpital Femme Mère Enfant (HFME, Lyon) or Institut Curie (Paris). Tumor pieces were put in a sterile saline solution (0.9%), while confirmed to be RMS by anatomopathologists. For each RMS sample, tissues were split into four parts and processed for histology, RNA and DNA isolation, or dissociated and processed for RMS derivation in 2D cell lines and organoids (i.e., RMS_O). Samples were used in the context of patient diagnosis/clinical care. Indeed, non-used parts of the samples might be employed for research if the patient is not opposed to it (information notice transmitted to each patient). This study was approved by the ethical review board of the BRC of the Center Léon Bérard (n°BB-0033-00050, N° 2020-02). This BRC quality is certified according to AFNOR NFS96900 (N° 2009/35884.2) and ISO 9001 (Certification N° 2013/56348.2). Biological material collection and retention activity are declared to the Ministry of Research (DC-2008-99 and AC-2019-3426). The study had all necessary regulatory approvals and informed consents are available for all patients. Fourteen patients were diagnosed with fusion-negative RMS (FNRMS) according to FISH assays, including 11 embryonal RMS (ERMS, 3 relapsed pediatric tumors, 1 relapsed adult tumor and 7 pediatric primary tumors), 1 relapsed pleiomorphic RMS and 2 adult spindle cell RMS (primary tumors).
Animal studies
Female NSG-NOD SCID mice (6 weeks old) were obtained from Charles River animal facility. The mice were housed in sterilized filter-topped cages and maintained in the P-PAC pathogen-free animal facility (D 69 388 0202). All animal studies were performed in strict compliance with relevant guidelines validated by the local Animal Ethic Evaluation Committee (C2EA-15) and authorized by the French Ministry of Education and Research (Authorization APAFIS#28836).
Method details
Derivation and culture of tumor-derived organoids and 2D cell lines
RMS tissues (∼5–125 mm3) were minced into small pieces, digested in a solution containing collagenase D (0.125 mg/mL Roche, cat. no. 1108866001) diluted in HBSS (Gibco, cat. no. 14025050) and washed using Advanced DMEM/F-12 medium (Gibco, cat. no. 12634010) supplemented with HEPES (1X, Gibco, cat. no. 15630106), GlutaMAX (1X, Gibco, cat. no. 35050038) and Penicillin-Streptomycin (1X, Gibco, cat. no. 15140122). After centrifugation, tumor cell suspensions were plated in 96-well to 6-well plates (2D, Corning, cat. no. 353046, 3D, Corning, cat. no. 3471) to ensure a sufficient density (10,000 viable cells per 1 cm2) in DMEM supplemented with 10% FBS (Gibco, cat. no. 26140079) and Penicillin-Streptomycin (1X, Gibco, cat. no. 15140122) or in a low-serum (Promocell, cat. no. C-23260) optimized M3 and M5 media, notably supplemented with Penicillin-Streptomycin (1X, Gibco, cat. no. 15140122) and EGF (10 ng/mL) and bFGF (1 ng/mL). Culture media were changed twice a week, and RMS_O and 2D models were split every 2 weeks using TrypLE Express Enzyme (Thermo Fisher Scientific, cat. no. 12605010). All cultures were tested every month for mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza, cat. no. LT07-318), in accordance with the manufacturer’s instructions. To prepare frozen vials, all RMS models were dissociated using TrypLE Express Enzyme (Thermo Fisher Scientific, cat. no. 12605010) and resuspended in Recovery Cell Culture Freezing medium (Gibco, cat. no. 12648010).
Histological analyses
The histological match between the tumoroid and its tumor-of-origin was a major criterion for selecting the derivation protocol. In brief, RMS_Os were fixed and processed as described before.78 Immunohistochemistry (IHC) was performed on an automated immunostainer (Ventana discoveryXT, Roche) using rabbit Omni map DAB kit. Organoids’ slides were stained with HPS (Hematoxylin Phloxine Saffron), or the following antibodies: anti-Desmin (1/50, Dako, cat. no. M0760), anti-Myogenin (1/100, Dako, cat. no. M3559), and anti-Ki67 (1/100, Dako, cat. no. M7240). Then, slides were incubated in relevant antibody-HRP conjugate for 1 h at room temperature (RT) and finally revealed with 3,3′-diaminobenzidine (DAB) for 5 min, and counterstained with Gill’s-hematoxylin. Following IHC, slides were mounted using Pertex (Histolab, cat. no. 00801-EX). Co-immunofluorescence (IF) was performed on Bond RX automated immunostainer (Leica biosystems) using OPAL detection kits (ref NEL871001KT, AKOYA bioscience). Primary antibodies specific to Survivin (1/400, Cell Signaling, cat. no. 2808S) and Ki67 (1/100, Dako, cat. no. M7240) were applied 30 min at RT. Sequential immunofluorescence was performed using OPAL 520 (Survivin, green), OPAL 690 (Ki-67, red), and cells were counterstained with DAPI. Slides were then mounted in Prolong Gold Antifad Reagent (Invitrogen, cat. no. P36930). Sections were scanned with panoramic scan II (3D Histech, Hungary) at 40× for IHC and using the Vectra POLARIS device (Akoya bioscience) for multiplexed IF.
Molecular profiling by multiome sequencing
RNA and genomic DNA (gDNA) from both RMS tissues and matched in vitro models cultured in 2D or 3D conditions with DMEM, M3 and M5 media were extracted using the Allprep DNA/RNA/miRNA universal kit (Qiagen, cat. no. 80224) and Arcturus PicoPure RNA Isolation Kit (Thermo Fisher Scientific, cat. no. KIT0204) only for small tumor pieces, following manufacturer’s instructions. Samples were then characterized at the molecular levels by RNA-seq (n = 29), DNA-seq (n = 11) and methylation array (n = 25).
For RNA-seq library construction, 100 to 1 000 ng of total RNAs were used. Libraries were prepared with Illumina Stranded mRNA Prep (Illumina, cat. no. 20040534) following recommendations. Quality was further assessed using the TapeStation 4200 automated electrophoresis system (Agilent) with High Sensitivity D1000 ScreenTape (Agilent). All libraries were sequenced (2 × 75 bp) using NovaSeq 6000 (Illumina) according to the standard Illumina protocol.
For DNA-seq library construction, a total of 200 ng input gDNA per sample was used for SureSelect XT low input library preparation (Agilent). A next-generation sequencing (NGS) Custom Hybridization capture-based product (Agilent, Santa Clara, USA) has been designed to detect single nucleotide variants, insertions, and deletions on a 740 gene-target panel (2.7Mbp size). Library preparations were performed according to the SureSelect XT low input Target Enrichment System for Illumina Paired-End Multiplexed Sequencing Library protocol (Version C2, July 2019). DNA samples were first sheared by enzymatic DNA fragmentation, using Agilent’s SureSelect XT low input Enzymatic Fragmentation Kits, and adaptors were ligated to end repaired DNA. Adapter-ligated libraries were purified using AMPure XP beads (Beckman Coulter, Inc., Brea, CA, USA), amplified, and then purified. Quality and quantity of libraries were determined by TapeStation using a High Sensitivity D1000 ScreenTape (Agilent). Next, 500 ng of each library was hybridized with the SureSelect capture library. Hybridized libraries were purified with Dynabeads MyOne Streptavidin T1 magnetic beads (Thermo Fisher Scientific, Waltham, USA). Beads with captured DNA were then washed once with wash buffer to remove non-specific binding. After all wash steps, the beads were suspended in 25 μL of nuclease free water. DNA bound to streptavidin beads was amplified by PCR using SureSelect post capture primer mix and Herculase II Fusion DNA polymerase. The cycling conditions were as follows: 98°C for 2 min; followed by 9 cycles of 98°C for 30 s, 60°C for 30 s, and 72°C for 1 min; and a final extension at 72°C for 5 min. After PCR, streptavidin beads were removed using a magnet stand, and the PCR products were further purified with AMPure XP beads. High quality libraries were identified with an Agilent TapeStation using High Sensitivity D1000 ScreenTape and then pooled for sequencing. Sequencing of SureSelect enriched libraries was performed on an Illumina NovaSeq 6000 platform (Illumina) on an S1 200-cycle cartridge (2 × 100bp).
For methylation array construction, 100 to 500 ng of gDNA was hybridized to Illumina Infinium HumanMethylationEPIC BeadChip (850K) arrays according to the manufacturer’s protocol recommendations.
Single-cell RNA sequencing of FNRMS-derived organoids
For single-cell suspension preparation, FNRMS-derived organoids were dissociated using TrypLE Express Enzyme (Thermo Fisher Scientific, cat. no. 12605010). Cells were then filtered through a 30-μm strainer (Miltenyi Biotec, cat. no. 130-098-458), centrifuged at 500 × g for 5 min, resuspended in complete culture medium and sorted using a FACSAria (BD Biosciences). Cells were centrifuged again at 500 × g for 8 min and resuspended in PBS (Gibco, cat. no. 14190-094) with 0.04% BSA (Sigma-Aldrich, cat. no. A7030) for a final cell concentration of 1 000 cells/μL. Approximately 20 μL of isolated cells were loaded on a 10X Genomic chip and run on the Chromium Controller system (10X Genomics) to target 10 000 cells per sample. Gene expression data were generated with the Chromium Single Cell 5′ v3.1 assay (10X Genomics) and sequenced on the NovaSeq 6000 platform (S1 flow cell, Illumina).
Drug screening and assays on RMS in vitro models
For each drug of our screen, IC50, defined as half maximal inhibitory concentration values, was established based on Selleckchem online database. Drugs were distributed at 4 (3D-RMS_O) to 6 (2D-RMS cell lines) different doses chosen to cover at least 3 log of concentrations and to include the aforementioned IC50. Thus, depending on the IC50 identified, 3 different ranges were used (1) from 10 p.m. to 100 nM for YM-155 (2) from 1 nM to 1 μM for Vincristine, Vinorelbine, SN38, Doxorubicine, Topotecan and Paclitaxel and (3) from 100 nM to 100 μM for Etoposide, Melphalan, Gemcitabine, Olaparib, Berzosertib, KU-60019, Prima-1, Nutlin-3, Navitoclax, Venetoclax, LCL-161, Birinapant et GDC-0152.
Drug screening on RMS 2D-cell lines
Living cells from RDAbl (2x103 cells/well) and RD, Rh36 FNRMS or RH30 FPRMS (4x103cells/well) RMS cell lines were seeded in 384-well plates (Corning, cat. no. 3830) and incubated in the presence of a selection of 20 drugs. Briefly, cells were grown in DMEM medium supplemented with 10% Fetal Bovine Serum (Gibco, cat. no. 26140079), 1% Penicillin-Streptomycin (Gibco, cat. no. 15140122), 1% GlutaMAX (Gibco, cat. no. 35050038), and 1% Non-Essential Amino Acids (Gibco, cat. no. 11140035). Drugs were carefully distributed with the Echo 550 liquid dispenser (Labcyte). Cell viability was measured using CellTiter-Glo 2.0 Cell Viability Assay (Promega, cat. no. G9243) after 72 h of drug incubation and luminescence was read using a Pherastar plate reader (BMG Labtech).
Drug screenings and assays on 3D tumor-organoids
Phenotypic drug screening
U-bottom microwell hydrogel-based arrays (Gri3D, SUN bioscience) were fabricated and conditioned as previously described.79 Gri3D 96 well-plates with 55 microwells of 600 μm in diameter per well were used to perform high-throughput drug screening on RMS_O. Hydrogel arrays were equilibrated with 150 μL of medium for at least 30 min at 37°C. Tumoroids were dissociated to single cell with TrypLE Express (Thermo Fisher Scientific, cat. no. 12605036). Cells were then centrifuged at 500 × g for 5 min and counted with NucleoCounter NC-3000 and Trypan blue exclusion assay. Cells were seeded at a density of 2x105 cells/mL. Plates were kept at 37°C in 5% CO2 before further processing. RMS_Os were treated after 3 days of culture for 72 h, and their growth was monitored with CELLCYTE X. Tumoroids were labeled with calcein-AM and ethidium homodimer-1 using the Live/DEAD Viability/Cytotoxicity Kit (Thermo Fisher Scientific, cat. no. 10237012) for mammalian cells according to the manufacturer’s protocol and were imaged using an Opera Phenix Plus High-Content Screening System.
CellTiter-Glo drug screening
Tumoroids were dissociated to single cell with TrypLE Express (Thermo Fisher Scientific, cat. no. 12605036). Cells were then centrifuged at 500 × g for 5 min and counted with NucleoCounter NC-3000 and Trypan blue exclusion assay. Cells were seeded at a concentration of 5000 cells/well in 96-well microplates (Corning, cat. no. 4515). Plates were kept at 37°C in 5% CO2. RMS_Os were treated after 3 days of culture for 72 h. Cell viability was measured by adding a volume of CellTiter-Glo reagent (Promega, cat. no. G9683) equal to the volume of cell culture medium present in each well and gently shaking the plate for 5 min at RT to induce cell lysis. All acquisitions of luminescence were performed on a Spark microplate reader (Tecan) with a 400 ms exposition and auto-attenuation. Relative luminescence units (RLU) of each well were normalized to the mean RLU from the DMSO negative control wells as 100% viability. Three technical replicates per condition were performed for each experiment.
Drug assays
IC50 determination
Tumoroids were dissociated and plated at 5x103 cells/well in 96-well plates (Corning, cat. no. 4515). RMS_Os were allowed to form for 3 to 4 days, and then treated with serial dilutions of YM-155 (Selleckchem, cat. no. S1130), Erastin (Selleckchem, cat. no. S7242), or Vincristine (Selleckchem, cat. no. S1241). Impact of treatments on intracellular ATP content was measured using the CellTiter-Glo 3D Cell Viability Assay (Promega, cat. no. G9681) after 2 (Erastin/YM-155) or 3 days (Vincristine). Relative luminescence units (RLU) of each well were normalized to the mean RLU from the DMSO negative control wells as 100% viability. Gambogic acid (10 μM, Cayman Chemical, cat. no. 14761) was used as a positive control. All acquisitions of luminescence were performed on a Spark microplate reader (Tecan) with a 400 ms exposition and auto-attenuation. Three technical replicates per condition were performed for each experiment.
Regrowth experiments
Tumoroids were seeded at 5x103 cells/well in 96-well plates (Corning, cat. no. 4515). After 3 days, RMS_Os were treated either with DMSO (negative control), Vincristine (at 5 nM for RMS1_O, 10 nM for RMS2_O and 40 nM for RMS3_O according to their respective dose-response curves), 0.25 μM Erastin (according to RMS1_O dose-response curve), 25 nM YM-155 (according to RMS1_Os dose-response curve), or a combination of both compounds (0.25 μM Erastin; 25 nM YM-155). In brief, we selected systematically and specifically (for each RMS-organoid line) the first concentration inducing the maximum toxicity for each agent of interest. Two (Erastin, YM-155 and combo) or three (Vincristine) days after, RMS_Os were washed to remove the drugs. Due to phenotypic differences, RMS1_O and RMS3_O were collected and washed twice in 1 mL of fresh medium and replaced in new wells with complete culture medium, while RMS2_O were progressively washed by successively removing and adding fresh medium until the residual vincristine concentration was diluted below 0.01 nM. Culture medium was renewed twice a week and regrowth was carefully monitored with CELLCYTE X and a classical inverted tissue culture microscope equipped with a digital camera (Zeiss, Axiovision). When reaching the growth plateau, FNRMS-derived organoids were split and reseeded at 5x103 cells/well. Cell viability was assessed at different time points during the regrowth experiment by using the LIVE/DEAD Viability/Cytotoxicity Kit (Invitrogen, cat. no. L3224). Acquisition images were captured using EVOS M7000 microscope.
Xenograft models
For orthotopic grafts, cells from RMS1_O (n = 3x105) and RMS2_O (n = 5x105) were prepared in 50% culture medium-50% Matrigel Low Growth Factor (Corning, cat. no. 356231) and were injected orthotopically into the tibialis anterior muscle of mice. Visible tumors developed in approximately 2–3 months (RMS1_O) and 3–4 weeks (RMS2_O). Mice were culled when the tumor reached the limit endpoint (600 mm3).
Quantification and statistical analysis
Immunohistochemistry quantifications
Fast digital quantitative analysis was performed using HALO software. In brief, masks were manually set up allowing negative, weak, and strong detection of all expression markers. Data were plotted using Prism 9.3.1 GraphPad; Mann–Whitney U tests were applied on at least n = 3 areas (for tissues) or n = 2 biological replicates (RMS organoids) to assess statistical significance.
RNA sequencing analysis
Raw FASTQ files were processed using the following steps. Quality control was performed using FastQC (v.0.11.9), followed by trimming of adapter sequences with Cutadapt (v.3.4) using -a CTGTCTCTTATACACATCT and -A CTGTCTCTTATACACATCT parameters. Reads were mapped using STAR (v.2.7.9) to the human reference genome assembly GRCh38.p13 with --seedSearchStartLmax 38 --outFilterMatchNminOverLread 0.66 --outReadsUnmapped Fastx --outSAMmultNmax −1 --outMultimapperOrder Random --outFilterScoreMinOverLread 0.66 --quantMode TranscriptomeSAM --outSAMstrandField intronMotif --twopassMode Basic --limitSjdbInsertNsj 1324910 parameters. Gene expression data were generated with HTseq-count (v.0.13.5) using --order pos --stranded reverse parameter and symbols were annotated with their respective Ensembl gene IDs using the package org.Hs.e.g.,.db v3.14.0 based on Gencode v37 (Ensembl v103). To assess the concordance of FNRMS tumors with their matched organoids and models, raw HTseq counts for all tissues and derived-models were loaded using DESeq2 R library with the “design” parameter combining sample conditions (tissue/culture, 2D/3D culture). Genes with low counts, i.e., less than 10 reads across samples, were then filtered. Gene expressions were normalized using the vst function of DESeq2 R library with parameter blind = FALSE and only protein coding genes were kept for further analysis. DESeq-normalized data were extracted using the DESeq function (DESeq2 R library). Principal Component Analysis (PCA) and Hierarchical Clustering on Principal Components (HCPC) were performed using FactoMineR (v.2.4) and factoextra (v1.0.7) R libraries. Heatmaps were generated using ComplexHeatmap R library (v2.10.0) with Euclidean distance as the clustering method and color palettes of RcolorBrewer R library (v.1.1–3). All analyses were performed in an R statistical environment (v.4.1.2) using DESeq2 (v1.34.0) library.
DNA sequencing analysis
All tools were used with default parameters unless otherwise specified. Quality control of reads was performed using FastQC (v.0.11.9). Unique Molecular Identifiers (UMIs) were extracted and deduplicated using UMI-tools (v.1.1.2). Reads from each sequencing lane were mapped separately to the hg38 human genome using bwa-mem2 (v.2.2.1). Optical duplicates were further removed using Picard MarkDuplicates (v.2.27.4). This step is also where the separate sequencing lanes BAM files were merged into a per-sample BAM file. Mosdepth (v.0.3.3) was used to generate coverage quality control. Then, GATK (v.4.3.0.0) Best-Practices workflow Somatic short variant discovery (SNVs + Indels) (https://gatk.broadinstitute.org/hc/en-us/articles/360035894731-Somatic-short-variant-discovery-SNVs-Indels-) was used to call somatic mutations. Briefly, the following tools were used: BaseRecalibrator, ApplyBQSR, Mutect2 (with parameter ‘--callable-depth 1’) in multi-sample tumor-only mode (with sample groups as RMS1, RMS2), and variant filtering with LearnReadOrientationModel, GetPileupSummaries, CalculateContamination and FilterMutectCalls. Germline variants were filtered with vcftools (v.0.1.16)
‘vcf-isec’ (using germline variants from gnomAD and 1000 Genomes VCF files). ‘vcf-merge’ was used to merge all samples into a single VCF file. A custom Python script was used to re-annotate variants whose FILTER value was wrongly modified by vcf-merge. Remaining variants were annotated using Ensembl Variant Effect Predictor (VEP, v.108.1). Another Python custom script was used to produce Table S3B. Briefly, the script reformated the VCF output from VEP into a tabular output, keeping only certain columns and re-calculated Allelic Frequency (AF) values and determined variant presence/absence for each sample. The AF was calculated using the Allele Depth (AD) value and dividing the variant allele with the most reads by the sum of the AD values for all alleles. Presence/absence calls were made using the AD values: if the AD value for the alternative allele was non-zero, then the variant was considered “Present”. If we only had the reference allele or the variant was not called for the sample, it was considered “Absent”. Heatmaps were made using ComplexHeatmap R library (v2.10.0).
DNA methylation array analysis
Methylation data were analyzed using minfi (R library v.1.44.0). Normalization was performed for all samples using functional normalization (FunNorm) with default parameters. Filtering was performed to remove (1) poor performing probes, i.e., detection p value above 0.01 in one or more samples, (2) probes located on sex chromosomes, (3) probes known to have common SNPs at the CpG site using dropLociWithSnps function; and (4) cross-reactive probes published by Chen et al. (2013) and Pidsley et al. (2016). Normalized methylation M-values were extracted to perform principal component analysis (PCA) using factoextra (R library v.10.7).
Single cell RNA-seq data analysis
To generate gene-barcode count matrices, raw sequencing reads were processed using mkfastq and count (Cell Ranger v.3.1.0, 10x Genomics). The raw base call (BCL) files were demultiplexed into FASTQ files and aligned to the hg38 human genome as reference. Overall, 23 993 cells (RMS1_O_P13, n = 11 627; RMS1_O_P14, n = 12 366) passed the quality control criteria. Each single cell dataset was imported using Read10X function and converted into a Seurat object with CreateSeuratObject function with at least min.features = 200 and min.cells = 3. To retain only high-quality cells, we applied a joint filtration based on number of unique molecular identifier (nUMI), number of detected genes (nGene) and number of mitochondrial counts (mitoRatio) criteria (Luecken & Theis, 2019). For each sample, independently, we retained cells within a three median absolute deviation (MAD) around the population median for these metrics, combined with absolute quality thresholds. We considered low-quality cells as cells with (1) low (nGene <200 genes) and high (nGene > 3MAD) number of detected genes; (2) high mitochondrial gene content (mitoRatio > 3MAD); and (3) cells with relatively high library sizes (nUMI >4 500). We predicted doublets/multiplets, i.e., multiple cells captured within the same droplet or reaction volume, using the scDblFinder R library (v.1.10.0) but kept this variable as indicative. The single cell datasets were then merged and normalized using methods adapted from scran pipeline (scran R library v.1.24.0) comprising quickCluster, computeSumFactors with min.mean = 0.1 and logNormCounts steps. The highly variable genes (HGVs) were detected using three algorithms including scran, Seurat and a rank custom strategy. The scran method comprises: (1) a modelGeneVar function that models the variance of the log-expression profiles for each gene; (2) a metadata function to fit the mean-variance trend; and (3) a getTopHVGs function to extract the top features. The Seurat V3 algorithm was implemented in the highly_variable_genes function (scanpy python library v.1.8.2) and consists of ranking genes according to a normalized variance procedure. The custom strategy (1) ranks genes according to their expression levels for each cell; (2) measures the standard deviation of rank for each gene across overall cells; and (3) sort genes based on their ranked expression levels; and (4) select the most variable ones. For each strategy, we selected the top 2 000 most variable genes and retained a list of 1 158 genes that were detected in at least two of the three methods. Variable features included the top 484 of these most variable genes and 245 genes known to be biologically relevant in the process of myogenic differentiation (12,14,15) and were used for principal component analysis (PCA) using RunPCA function. We kept the first 9 principal components (PCs) for analysis based on the ElbowPlot method that allows a visualization of the standard deviation of each PC. The most contributing dimensions were then chosen based on two metrics: (1) the percent of variation associated with each PC (cumulative percent of variation >90% and percent of variation >5%); and (2) the percent change in variation between consecutive PCs (>0.1% to keep including PCs). Clusters were identified with the FindClusters function using a resolution set to 0.3 and the Leiden algorithm. Briefly, this strategy comprises local moving of nodes, refinement of the partition and aggregation of the network based on the refined partition, as previously described (Traag et al., 2019). Cluster identities of the cells were then mapped on a UMAP using the RunUMAP function. Specific marker genes for clusters were identified using the FindAllMarkers function with only.pos = TRUE, min.pct = 0.25 and test.use = “MAST”. Trajectory inference analyses were performed using slingshot R library (v.2.4.0) with start.clus = 4 and stretch = 0 for a supervised strategy and scVelo python library (v.0.2.4) for an unsupervised one based on RNA velocity data generated by loom python library (v.3.0.6). To assess the functional differences between the quiescent satellite (clusters 4-3) and myoblast-proliferative (clusters 5-2-6) cells, we performed fast differential expression analyses between both conditions using wilcoxauc function (R library presto v1.0.0). We then ranked all the genes based on their area under receiver operator curve (auROC) value and performed Gene Set Enrichment Analysis (GSEA) using fgsea R library (v.1.22.0) with minSize = 15, maxSize = 500 and nperm = 1000 on HALLMARK (H), Gene Ontology (subcategory: Biological Processes), curated (C2) and cell type (C8) gene signatures downloaded from MSigDB (http://www.gsea-msigdb.org/), Human Protein Atlas (HPA) (https://www.proteinatlas.org/) and literature-based gene sets. Of note, custom gene sets are lists of genes resulting from the intersection of literature-based gene sets (Myoblasts intersection: PMID: 31053169 and PMID: 32011235; MuSC intersection: PMID: 32396864 and PMID: 32234209 and PMID: 31937892). Overall, 14 818 gene sets were tested and statistical probabilities were adjusted based on the number of tested biological processes using the FDR method. Only significantly enriched pathways (FDR <0.01 and absolute Normalized Enrichment Score (NES) > 2) from the custom, skeletal muscle cells, pediatric cancers and hypoxic gene-sets were retained for Figure 4E. Analyses were performed in an R statistical environment (v.4.1.3) using Seurat R library (v.4.1.1) and python environment (v.3.9.10).
Gene expression analysis of publicly available muscle and RMS datasets
Three microarray datasets were downloaded from public databases. E-TABM-1202 (cohort 1)79 raw microarray data (.CEL files) with 101 RMS samples are accessible at the ArrayExpress platform (https://www.ebi.ac.uk/arrayexpress/) and were normalized using the Robust Multiarray Average (RMA) algorithm (oligo R library v.1.58.0). Schäfer and Welle (cohort 4, Schafer-Welle-56-MAS5.0-u133a) log2-transformed data comprising 26 healthy muscles and 30 RMS samples were downloaded from the R2 genomic platform (http://r2.amc.nl) using the gene reporter selection mode, i.e., HugoOnce algorithm that selects a single probeset to represent a gene. GSE28511 (cohort 5) quantile normalized data80 were downloaded from the GEO database (www.ncbi.nlm.nih.gov/geo/) and were then log2-transformed. After quality control, we removed the GSM706247 normal sample (tumor adjacent skeletal muscle cell) subject to high levels of tumor-in-normal contamination leading to a dataset of 5 healthy muscles and 18 RMS samples. Last, Javed Khan and colleagues kindly shared Khan collection’s log2-transformed data (cohort 3) with 86 RMS samples.10 Gene reporter selection was performed by selecting the probeset with the highest average expression levels across samples, except for the Schäfer and Welle dataset with default probeset assignment.
St. Jude RNA-seq data (cohort 2) of 60 RMS samples have been retrieved from St. Jude Cloud (https://www.stjude.cloud) and generated as described.81 Briefly, read mapping was done using STAR (v.2.7.9a) on the hg38 human genome and gene-level counts were generated using HTSeq-count based on the Gencode v31 gene annotations. We focused on transcripts with consistent annotations, i.e., protein-coding genes, and filtered those with less than 10 reads in overall samples. Gene expression data were normalized using a variance-stabilizing transformation procedure with vst function (DESeq2 R library v.1.34.0). To remove unwanted variability driven by technical and non-biological factors, we used the removeBatchEffect function implemented in the limma R library (v.3.50.3) and specified the “fusion status” as the variable to consider in the linear model.
Apoptotic genes expression profiling and pathway activation scores
We selected manually curated genes, known to encode proteins involved in apoptosis and other forms of cell death mechanisms, from the Deathbase platform (http://deathbase.org/, downloaded on March 31, 2022). Only genes characterized in the Homo sapiens organism were selected. Based on this list of 86 genes (Table S3), we performed differential expression analyses using limma R library (v.3.50.3) for microarray data and DESeq2 R library (v.1.34.0) using Shrunken log2 fold changes (LFC) for RNA-seq data. We tested gene expression differences between (1) FNRMS versus FPRMS samples in the cohort 1 (E-TABM-1202), cohort 2 (St. Jude) and cohort 3 (Khan); and (2) healthy muscles versus FNRMS samples in the cohort 4 (Schäfer and Welle) and cohort 5 (GSE28511). Statistical probabilities were adjusted using the FDR method. Only apoptotic genes with significant differences between both conditions (FDR <0.05) were then selected for visualization. Visualization plots were generated with the ComplexHeatmap R library (v.2.10.0) using ward.D2 clustering on the inverse Spearman’s correlation coefficient matrix to assess both the distance between samples and the distance between genes. Single gene expression comparison between normal and tumor samples was performed using ggboxplot (ggpubr R library v.0.4.0) for visualization and rstatix (v.0.7.0) for statistical analysis using Wilcoxon signed-rank test. Ingenuity Pathway Analysis (IPA) was performed with QIAGEN IPA (v.01-20-04, https://digitalinsights.qiagen.com/IPA) to predict downstream effects on biological functions based on the expression log fold change ratio of apoptotic genes with significant differences between conditions (FDR <0.05), i.e., normal versus tumoral or FNRMS versus FPRMS samples.
Establishment of a prognostic apoptotic metascore in patients with FNRMS
Only FNRMS patients with known survival time and status information were selected for analysis (cohorts 1 and 3). The cohort 1 (E-TABM-1202) was used as the training set and the cohort 3 (Khan) as the independent test set. For each apoptotic gene, univariate Cox proportional hazards models were performed to test the prognostic value of each gene. To limit optimism bias, the selection strategy was based on a leave-10-out cross-validation procedure with 250 iterations in the training set. Genes were ranked based on their statistical significance (p < 0.05) across iterations and those significantly associated with the overall survival probability of patients with FNRMS in at least 150 (60%) iterations were included in the multivariate Cox proportional hazards model. Proportional hazard hypothesis was checked using Schoenfeld residuals using cox.zph function (survival R library). To explore collinearity between predictor genes, associations were assessed with Pearson correlation coefficients using cor function (stats R library v.4.1.3) with method = “pearson”. For each sample, the apoptotic metascore was calculated as the sum of the predictor genes expression levels weighted by the regression coefficients of the training model, generated on the FNRMS samples of the cohort 1 (E-TABM-1202). For each cohort, an independent optimal risk cut point was identified to define two groups, high and low apoptotic metascore, among FNRMS. For each of the 250 iterations, a cut point of the metascore was identified using the surv_cutpoint function (survminer R library). This algorithm relies on the maxstat function (maxstat R library v.0.7–25) that performs a test of independence between a quantitative predictor X (here, the apoptotic metascore) and a censored response Y (here, the survival status) using maximally selected rank statistics. This defines which cutpoint μ in X determines two groups of observations regarding the response Y and measures the difference between both groups as the absolute value of an appropriate standardized two-sample linear rank statistic of the responses. We retained as final threshold the median of overall cutpoints (n = 250). Both groups defined by low and high apoptotic metascore have been studied in more detail from a discriminatory point of view. Kaplan-Meier survival curves were drawn using the ggsurvplot function (survminer R library). Survival curves in high and low metascore groups were compared using log rank tests in the training and test sets. Dynamic receiver operating characteristic (ROC) curves were built using timeROC R library (v.0.4). All statistical analyses were performed in the R statistical environment (v.4.1.3) using survival (v.3.3–1, Therneau 2022), survivalROC (v.1.0.3, Heagerty 2013) and survminer (v.0.4.9, Kassambara 2021) libraries.
Immunofluorescence quantifications
Co-immunoflorescence survivin and Ki67
Manual quantifications of unstained (blue = DAPI) Ki67+ cells (red), Survivin+ cells (green) or Ki67+/Survivin+ cells (yellow) in RMS1_O were performed using ImageJ Fiji software (n = 3 spheres).
Quantitative analysis methods for drug screenings/assays on RMS in vitro models
CellTiter-Glo drug screening on 2D-RMS cell lines and 3D-RMS organoids and IC50 determination on 3D-RMS organoids
Data were normalized to negative control wells (DMSO only). IC50, defined as half maximal inhibitory concentration values, were deduced from dose-response curves obtained using Prism 9.3.1 (GraphPad). For drug screening heatmaps, the negative logarithm of IC50 (pIC50 = -log(IC50)) was also calculated for each compound to compare their efficacy.
Phenotypic drug screening on 3D-RMS organoids
The image analysis was conducted using the Doppl platform, a high-throughput automated analysis platform specialized in screening assays for organoid growth on the Gri3D plate from SUN Bioscience. This technology implements a CellProfiler pipeline, allowing for the automated detection and segmentation of organoids, coupled with imaging techniques to measure specific fluorescent signals. The Calcein signal represents organoid viability, while the Ethidium signal represents cell death in organoids. The readout R of our assay for a compound C is a ratio of the mean fluorescent signals, calculated as with D the Ethidium signal, L the Calcein signal. Data generated by the Doppl platform for image analysis were subsequently used to generate the statistics and plots presented in this paper. We utilized the tidyverse package for data manipulation. The statistical test performed to assess the significance of our compound was Dunnett’s test, using the negative control (DMSO) as the baseline
Regrowth experiments
LIVE/DEAD immunofluorescence staining were manually quantified by measuring the surface area of live (green) and dead (red) cells on at least 5 organoids (n = 3 different experiment) using ImageJ Fiji software and then plotted using Prism 9.3.1 GraphPad.
Acknowledgments
We thank the patients and their families who consented to participate in this study. We also thank all facilities from the CRCL and clinical teams from IHOPe, HCL-HFME, and Institut Curie as well as the BRCs of CLB, HCL (Tissu-Tumorothèque Est), and Institut Curie for their support and contributions. We are grateful to Séverine Tabone-Eglinger, Corinne Perrin, Lesly Chambault, Deborah Sitbon, Gaelle Pierron, Loïc Sebileau, and Anne-Sophie Bonne for their help with the management of regulatory procedures and tissue collection. We also thank Dr. Xavier Morelli and Carine Derviaux of the HiTS platform of the CRCM for their help with the 2D cell line medium-throughput drug screening and Dr. Akram Ghantous from IARC for his help with methylome analysis. We thank Brigitte Manship for editorial assistance. We thank the parents’ charities that support this work (“Enfants, Cancers, Santé,” “Imagine for Margo,” “L’Etoile de Martin,” “Le sourire de Lucie,” “Aidons Marina,” “Un Horizon d’Espoir,” “Nos P’tites Etoiles,” “César Gibaud, une enfance sans cancer,” and “Cassandra contre les Leucémies”). This research was supported by the Foundation ARCECI innovation award (USA) awarded to L.B., the ANR JCJC program (CHILD-SARC; ANR-19-CE14-0001-01), INCa (PEDIAHR20-035, N2020-178), the DevWeCan2 LabEx (ANR-10-LABX-0061), the Convergence Institute Plascan (ANR-17-CONV-0002), the Ligue départementale Saône et Loire, Ain, and Haute-Savoie, and the Ligue Nationale Contre le Cancer. C.S. and A.T. received financial support respectively from the Fondation de France, the Ligue Nationale Contre le Cancer and Fondation ARC.
Author contributions
C.S., P.H., L.L., A.T., M.C., and L.B. conceived and designed the experiments and analyses. M.C. and L.B. analyzed the data with contributions from C.S., P.H., L.L., A.T., C.C., M.B., C. Deligne, A.L.G., V.B., J.-Y.B., T.D., O.N., A.R., M.C.-B., and N.B. Bioinformatic analyses were performed by C.S. and T.D. with the help of L.T., C. Degletagne, A.B., P.R., V.M., L.O., M.C., and L.B. Tumor sample processing and characterization was performed by A.L.G., L.L., C. Deligne, N.M., V.B., and L.B., with contributions from O.D., D.O., S.W., S.E.Z., J.-Y.B., I.R., C.P., F.D., C.B., and N.C. Metascore design and statistical analysis were performed by C.S., M.C., L.B., and D.M.-B. M.L.G., K.M., and E.P. performed drug screening on 2D cell lines. C.C. performed the middle-throughput drug screenings on tumoroids. L.C., M.B., P.H., and A.T. did the drug(s) experiments with longitudinal monitoring on tumoroids. M.C.-B, A.D., A.R. and N.B. provided detailed technical advice. Sequencing was performed on the CRCL platform with the help of C. Deligne, L.T., and V.A. Immunostainings and histological analyses were performed with the help of the anatomopathological platform of CRCL/HCL/Institut Curie with the help of S.L., N.G., and S.E.Z. M.C. and L.B. wrote the manuscript with contributions from C.S., P.H., L.L., A.T., and A.R. M.C. and L.B. are co-corresponding authors since they both supervised the work, designed the experiments, and organized the presentation of results in terms of text and figures. More specifically, L.B. oversaw the derivation, characterization, and use of organoid models for drug screening and identification of effective drug combinations and M.C. the rationalized approach based on the targeting of appropriate apoptotic blocking points to bypass resistance to apoptotic inducers often observed in RMS. All authors read and approved the manuscript.
Declaration of interests
The Ecole Polytechnique Fédérale de Lausanne has filed for patent protection on technology used for the phenotypic drug screening performed on 3D organoid, and N.B. is named as an inventor on those patents; N.B is also shareholder in SUN bioscience SA and DOPPL SA, which are commercializing those patents.
Published: December 19, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101339.
Contributor Information
Marie Castets, Email: marie.castets@lyon.unicancer.fr.
Laura Broutier, Email: laura.broutier@lyon.unicancer.fr.
Supplemental information
References
- 1.Siegel R., Naishadham D., Jemal A. Cancer statistics, 2012. Cancer J. Clin. 2012;62:10–29. doi: 10.3322/caac.20138. CA. [DOI] [PubMed] [Google Scholar]
- 2.Hawkins D.S., Spunt S.L., Skapek S.X. Children’s Oncology Group’s 2013 Blueprint for Research: Soft Tissue Sarcomas. Pediatr. Blood Cancer. 2013;60:1001–1008. doi: 10.1002/pbc.24435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saab R., Spunt S.L., Skapek S.X. In: Current Topics in Developmental Biology Cancer and Development. Dyer M.A., editor. Academic Press; 2011. Chapter 7 - Myogenesis and Rhabdomyosarcoma: The Jekyll and Hyde of Skeletal Muscle; pp. 197–234. [DOI] [PubMed] [Google Scholar]
- 4.Skapek S.X., Ferrari A., Gupta A.A., Lupo P.J., Butler E., Shipley J., Barr F.G., Hawkins D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Primer. 2019;5:1. doi: 10.1038/s41572-018-0051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barr F.G., Galili N., Holick J., Biegel J.A., Rovera G., Emanuel B.S. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993;3:113–117. doi: 10.1038/ng0293-113. [DOI] [PubMed] [Google Scholar]
- 6.Davis R.J., D’Cruz C.M., Lovell M.A., Biegel J.A., Barr F.G. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. 1994;54:2869–2872. [PubMed] [Google Scholar]
- 7.Sorensen P.H.B., Lynch J.C., Qualman S.J., Tirabosco R., Lim J.F., Maurer H.M., Bridge J.A., Crist W.M., Triche T.J., Barr F.G. PAX3-FKHR and PAX7-FKHR Gene Fusions Are Prognostic Indicators in Alveolar Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2002;20:2672–2679. doi: 10.1200/JCO.2002.03.137. [DOI] [PubMed] [Google Scholar]
- 8.Parham D.M., Barr F.G. Classification of rhabdomyosarcoma and its molecular basis. Adv. Anat. Pathol. 2013;20:387–397. doi: 10.1097/PAP.0b013e3182a92d0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williamson D., Missiaglia E., de Reyniès A., Pierron G., Thuille B., Palenzuela G., Thway K., Orbach D., Laé M., Fréneaux P., et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010;28:2151–2158. doi: 10.1200/JCO.2009.26.3814. [DOI] [PubMed] [Google Scholar]
- 10.Shern J.F., Chen L., Chmielecki J., Wei J.S., Patidar R., Rosenberg M., Ambrogio L., Auclair D., Wang J., Song Y.K., et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4:216–231. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arndt C.A.S., Bisogno G., Koscielniak E. Fifty years of rhabdomyosarcoma studies on both sides of the pond and lessons learned. Cancer Treat Rev. 2018;68:94–101. doi: 10.1016/j.ctrv.2018.06.013. [DOI] [PubMed] [Google Scholar]
- 12.Manzella G., Schreck L.D., Breunis W.B., Molenaar J., Merks H., Barr F.G., Sun W., Römmele M., Zhang L., Tchinda J., et al. Phenotypic profiling with a living biobank of primary rhabdomyosarcoma unravels disease heterogeneity and AKT sensitivity. Nat. Commun. 2020;11:4629. doi: 10.1038/s41467-020-18388-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meister M.T., Groot Koerkamp M.J.A., de Souza T., Breunis W.B., Frazer-Mendelewska E., Brok M., DeMartino J., Manders F., Calandrini C., Kerstens H.H.D., et al. Mesenchymal tumor organoid models recapitulate rhabdomyosarcoma subtypes. EMBO Mol. Med. 2022;14:e16001. doi: 10.15252/emmm.202216001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel A.G., Chen X., Huang X., Clay M.R., Komarova N.L., Krasin M.J., Pappo A., Tillman H., Orr B.A., McEvoy J., et al. The myogenesis program drives clonal selection and drug resistance in rhabdomyosarcoma. Dev. Cell. 2022;57:1226–1240.e8. doi: 10.1016/j.devcel.2022.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugii H., Grimaldi A., Li J., Parada C., Vu-Ho T., Feng J., Jing J., Yuan Y., Guo Y., Maeda H., et al. The Dlx5-FGF10 signaling cascade controls cranial neural crest and myoblast interaction during oropharyngeal patterning and development. Development. 2017;144:4037–4045. doi: 10.1242/dev.155176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rekhi B., Upadhyay P., Ramteke M.P., Dutt A. MYOD1 (L122R) mutations are associated with spindle cell and sclerosing rhabdomyosarcomas with aggressive clinical outcomes. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc. 2016;29:1532–1540. doi: 10.1038/modpathol.2016.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Danielli S.G., Porpiglia E., De Micheli A.J., Navarro N., Zellinger M.J., Bechtold I., Kisele S., Volken L., Marques J.G., Kasper S., et al. Single-cell profiling of alveolar rhabdomyosarcoma reveals RAS pathway inhibitors as cell-fate hijackers with therapeutic relevance. Sci. Adv. 2023;9:eade9238. doi: 10.1126/sciadv.ade9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whiteford C.C., Bilke S., Greer B.T., Chen Q., Braunschweig T.A., Cenacchi N., Wei J.S., Smith M.A., Houghton P., Morton C., et al. Credentialing preclinical pediatric xenograft models using gene expression and tissue microarray analysis. Cancer Res. 2007;67:32–40. doi: 10.1158/0008-5472.CAN-06-0610. [DOI] [PubMed] [Google Scholar]
- 19.Xi H., Langerman J., Sabri S., Chien P., Young C.S., Younesi S., Hicks M., Gonzalez K., Fujiwara W., Marzi J., et al. A Human Skeletal Muscle Atlas Identifies the Trajectories of Stem and Progenitor Cells across Development and from Human Pluripotent Stem Cells. Cell Stem Cell. 2020;27:158–176.e10. doi: 10.1016/j.stem.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Micheli A.J., Laurilliard E.J., Heinke C.L., Ravichandran H., Fraczek P., Soueid-Baumgarten S., De Vlaminck I., Elemento O., Cosgrove B.D. Single-Cell Analysis of the Muscle Stem Cell Hierarchy Identifies Heterotypic Communication Signals Involved in Skeletal Muscle Regeneration. Cell Rep. 2020;30:3583–3595.e5. doi: 10.1016/j.celrep.2020.02.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barruet E., Garcia S.M., Striedinger K., Wu J., Lee S., Byrnes L., Wong A., Xuefeng S., Tamaki S., Brack A.S., et al. Functionally heterogeneous human satellite cells identified by single cell RNA sequencing. Elife. 2020;9:e51576. doi: 10.7554/eLife.51576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei Y., Qin Q., Yan C., Hayes M.N., Garcia S.P., Xi H., Do D., Jin A.H., Eng T.C., McCarthy K.M., et al. Single-cell analysis and functional characterization uncover the stem cell hierarchies and developmental origins of rhabdomyosarcoma. Nat. Cancer. 2022 doi: 10.1038/s43018-022-00414-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Almada A.E., Wagers A.J. Molecular circuitry of stem cell fate in skeletal muscle regeneration, ageing and disease. Nat. Rev. Mol. Cell Biol. 2016;17:267–279. doi: 10.1038/nrm.2016.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terry E.E., Zhang X., Hoffmann C., Hughes L.D., Lewis S.A., Li J., Wallace M.J., Riley L.A., Douglas C.M., Gutierrez-Monreal M.A., et al. Transcriptional profiling reveals extraordinary diversity among skeletal muscle tissues. Elife. 2018;7:e34613. doi: 10.7554/eLife.34613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh P., Lim B. Targeting Apoptosis in Cancer. Curr. Oncol. Rep. 2022;24:273–284. doi: 10.1007/s11912-022-01199-y. [DOI] [PubMed] [Google Scholar]
- 26.Krämer A., Green J., Pollard J., Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ueno T., Uehara S., Nakahata K., Okuyama H. Survivin selective inhibitor YM155 promotes cisplatin-induced apoptosis in embryonal rhabdomyosarcoma. Int. J. Oncol. 2016;48:1847–1854. doi: 10.3892/ijo.2016.3438. [DOI] [PubMed] [Google Scholar]
- 28.Roy S.S., Ehrlich A.M., Craigen W.J., Hajnóczky G. VDAC2 is required for truncated BID-induced mitochondrial apoptosis by recruiting BAK to the mitochondria. EMBO Rep. 2009;10:1341–1347. doi: 10.1038/embor.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Y., Luo M., Zhang K., Zhang J., Gao T., Connell D.O., Yao F., Mu C., Cai B., Shang Y., et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat. Commun. 2020;11:433. doi: 10.1038/s41467-020-14324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yohe M.E., Heske C.M., Stewart E., Adamson P.C., Ahmed N., Antonescu C.R., Chen E., Collins N., Ehrlich A., Galindo R.L., et al. Insights into pediatric rhabdomyosarcoma research: Challenges and goals. Pediatr. Blood Cancer. 2019;66:e27869. doi: 10.1002/pbc.27869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tuveson D., Clevers H. Cancer modeling meets human organoid technology. Science. 2019;364:952–955. doi: 10.1126/science.aaw6985. [DOI] [PubMed] [Google Scholar]
- 32.Caldas H., Holloway M.P., Hall B.M., Qualman S.J., Altura R.A. Survivin-directed RNA interference cocktail is a potent suppressor of tumour growth in vivo. J. Med. Genet. 2006;43:119–128. doi: 10.1136/jmg.2005.034686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wheatley S.P., Altieri D.C. Survivin at a glance. J. Cell Sci. 2019;132:jcs223826. doi: 10.1242/jcs.223826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.R2 Genomics Analysis and Visualization Platform. https://hgserver1.amc.nl/cgi-bin/r2/main.cgi.
- 35.McLeod C., Gout A.M., Zhou X., Thrasher A., Rahbarinia D., Brady S.W., Macias M., Birch K., Finkelstein D., Sunny J., et al. St. Jude Cloud: A Pediatric Cancer Genomic Data-Sharing Ecosystem. Cancer Discov. 2021;11:1082–1099. doi: 10.1158/2159-8290.CD-20-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneider V.A., Graves-Lindsay T., Howe K., Bouk N., Chen H.-C., Kitts P.A., Murphy T.D., Pruitt K.D., Thibaud-Nissen F., Albracht D., et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017;27:849–864. doi: 10.1101/gr.213611.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frankish A., Diekhans M., Jungreis I., Lagarde J., Loveland J.E., Mudge J.M., Sisu C., Wright J.C., Armstrong J., Barnes I., et al. GENCODE 2021. Nucleic Acids Res. 2021;49:D916–D923. doi: 10.1093/nar/gkaa1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Díez J., Walter D., Muñoz-Pinedo C., Gabaldón T. DeathBase: a database on structure, evolution and function of proteins involved in apoptosis and other forms of cell death. Cell Death Differ. 2010;17:735–736. doi: 10.1038/cdd.2009.215. [DOI] [PubMed] [Google Scholar]
- 39.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carvalho B.S., Irizarry R.A. A framework for oligonucleotide microarray preprocessing. Bioinforma. Oxf. Engl. 2010;26:2363–2367. doi: 10.1093/bioinformatics/btq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinforma. Oxf. Engl. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 44.Kassambara A. 2023. Ggpubr: “Ggplot2” Based Publication Ready Plots.https://rpkgs.datanovia.com/ggpubr/ [Google Scholar]
- 45.Kassambara A. 2023. Rstatix: Pipe-Friendly Framework for Basic Statistical Tests. [Google Scholar]
- 46.Krämer A., Green J., Pollard J., Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinforma. Oxf. Engl. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schubert M., Klinger B., Klünemann M., Sieber A., Uhlitz F., Sauer S., Garnett M.J., Blüthgen N., Saez-Rodriguez J. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun. 2018;9:20. doi: 10.1038/s41467-017-02391-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Therneau, T.M., until 2009), T.L. (original S.->R port. maintainer R., Elizabeth A., Cynthia C. 2023. Survival: Survival Analysis; p. 5. [Google Scholar]
- 49.Kassambara A., Kosinski M., Biecek P., Fabian S. 2021. Survminer: Drawing Survival Curves Using “ggplot2.”. [Google Scholar]
- 50.Hothorn T. 2017. Maxstat: Maximally Selected Rank Statistics; pp. 7–25. [Google Scholar]
- 51.Blanche P. 2019. timeROC: Time-dependent ROC Curve and AUC for Censored Survival Data. Version 0.4. [Google Scholar]
- 52.Heagerty P.J., Saha-Chaudhuri, packaging by P . 2022. survivalROC: Time-dependent ROC Curve Estimation from Censored Survival Data. [Google Scholar]
- 53.Andrews S. Babraham Institute; 2010. FastQC: A Quality Control Tool for High Throughput Sequence Data (Babraham Bioinformatics. [Google Scholar]
- 54.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17:10–12. [Google Scholar]
- 55.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinforma. Oxf. Engl. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carlson M., Falcon S., Pages H., Li N. Vol. 3. R Package Version; 2019. Org. Hs. eg. db: Genome wide annotation for Human; p. 3. [Google Scholar]
- 58.Lê S., Josse J., Husson F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008;25:1–18. [Google Scholar]
- 59.Kassambara A., Mundt F. Package ‘factoextra. ’ Extr. Vis. Results Multivar. Data Anal. 2017;76 [Google Scholar]
- 60.Neuwirth E. 2022. RColorBrewer: ColorBrewer Palettes; pp. 1–3. [Google Scholar]
- 61.Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Germain P.-L., Lun A., Garcia Meixide C., Macnair W., Robinson M.D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Research. 2021;10:979. doi: 10.12688/f1000research.73600.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lun A.T.L., McCarthy D.J., Marioni J.C. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Research. 2016;5:2122. doi: 10.12688/f1000research.9501.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Street K., Risso D., Fletcher R.B., Das D., Ngai J., Yosef N., Purdom E., Dudoit S. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018;19:477. doi: 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bergen V., Lange M., Peidli S., Wolf F.A., Theis F.J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 2020;38:1408–1414. doi: 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
- 68.Linnarsson . 2022. Loompy v3.0. (Linnarsson Lab) [Google Scholar]
- 69.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2021 Preprint at. [Google Scholar]
- 70.Korsunsky I., Nathan A., Millard N., Raychaudhuri S. Presto scales Wilcoxon and auROC analyses to millions of observations. bioRxiv. 2019 Preprint at. [Google Scholar]
- 71.Vasimuddin M., Misra S., Li H., Aluru S. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS); 2019. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems; pp. 314–324. [Google Scholar]
- 72.Broad Institute Picard Toolkit. https://broadinstitute.github.io/picard/.
- 73.Pedersen B.S., Quinlan A.R. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics. 2018;34:867–868. doi: 10.1093/bioinformatics/btx699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lamprecht M.R., Sabatini D.M., Carpenter A.E. CellProfiler: free, versatile software for automated biological image analysis. Biotechniques. 2007;42:71–75. doi: 10.2144/000112257. [DOI] [PubMed] [Google Scholar]
- 77.Ritz C., Baty F., Streibig J.C., Gerhard D. Dose-Response Analysis Using R. PLoS One. 2015;10 doi: 10.1371/journal.pone.0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gonzalez A.L., Luciana L., Le Nevé C., Valantin J., Francols L., Gadot N., Vanbelle C., Davignon L., Broutier L. Staining and High-Resolution Imaging of Three-Dimensional Organoid and Spheroid Models. J. Vis. Exp. JoVE. 2021 doi: 10.3791/62280. [DOI] [PubMed] [Google Scholar]
- 79.Brandenberg N., Hoehnel S., Kuttler F., Homicsko K., Ceroni C., Ringel T., Gjorevski N., Schwank G., Coukos G., Turcatti G., et al. High-throughput automated organoid culture via stem-cell aggregation in microcavity arrays. Nat. Biomed. Eng. 2020;4:863–874. doi: 10.1038/s41551-020-0565-2. [DOI] [PubMed] [Google Scholar]
- 80.Missiaglia E., Williamson D., Chisholm J., Wirapati P., Pierron G., Petel F., Concordet J.-P., Thway K., Oberlin O., Pritchard-Jones K., et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012;30:1670–1677. doi: 10.1200/JCO.2011.38.5591. [DOI] [PubMed] [Google Scholar]
- 81.Li L., Sarver A.L., Alamgir S., Subramanian S. Downregulation of microRNAs miR-1, -206 and -29 stabilizes PAX3 and CCND2 expression in rhabdomyosarcoma. Lab. Investig. J. Tech. Methods Pathol. 2012;92:571–583. doi: 10.1038/labinvest.2012.10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Laura Broutier (laura.broutier@lyon.unicancer.fr). All -omic datasets generated during this study are avalaible via GEO repository with the accession number GSE248183. All data analyses’ codes will be made available upon request. This paper also analyzes existing, publicly available data. The accession numbers for these datasets are listed in the key resources table. There are restrictions to the availability of biobanked RMS_O due to the lack of an external centralized repository for their distribution and our need to maintain the stock.