

Abstract
SAR443820 (DNL788) is a selective, orally bioavailable, brain penetrant inhibitor of receptor‐interacting serine/threonine protein kinase 1 (RIPK1). This phase I first‐in‐human healthy participant study (NCT05795907) was comprised of three parts: randomized, double‐blind, placebo‐controlled single ascending dose (SAD; part 1a); 14‐day multiple ascending dose (MAD; part 2) parts that evaluated safety, tolerability, pharmacokinetics (PK), and pharmacodynamics of SAR443820; and a separate open‐label, single‐dose part 1b (PK‐cerebrospinal fluid [CSF]) to assess SAR443820 levels in CSF. SAR443820 was well‐tolerated in healthy participants, and no treatment discontinuation related to an adverse event (AE) occurred. Most common AEs were dizziness and headache. No clinically meaningful changes were noted in laboratory values, vital signs, or electrocardiogram parameters. SAR443820 had a favorable PK profile, with plasma half‐lives (geometric mean) ranged between 5.7–8.0 h and 7.2–8.9 h after single and repeated doses, respectively. There were no major deviations from dose proportionality for maximum concentration and area under the curve across SAR443820 doses. Mean CSF‐to‐unbound plasma concentration ratio ranged from 0.8 to 1.3 over time (assessed up to 10 h postdose), indicating high brain penetrance. High levels of inhibition of activated RIPK1, as measured by decrease in pS166‐RIPK1, were achieved in both SAD and MAD parts, with a maximum median inhibition from baseline close to 90% at predose (Ctrough) after multiple dosing in MAD, reflecting a marked RIPK1 target engagement at the peripheral level. These results support further development of SAR443820 in phase II trials in amyotrophic lateral sclerosis (NCT05237284) and multiple sclerosis (NCT05630547).
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Receptor‐interacting serine/threonine protein kinase 1 (RIPK1) is an intracellular protein critically involved in inflammatory signaling and multiple cell death pathways, including apoptosis and necroptosis. Increased RIPK1 activity is implicated in several neurodegenerative and neuroinflammatory diseases. Thus, a brain penetrant inhibitor of RIPK1 could be beneficial in treatment of chronic central nervous system diseases, such as amyotrophic lateral sclerosis and multiple sclerosis.
WHAT QUESTION DID THIS STUDY ADDRESS?
SAR443820 (DNL788) is a selective, orally bioavailable, brain penetrant, reversible inhibitor of RIPK1. This first‐in‐human phase I study evaluated safety, tolerability, pharmacokinetics (PK), and target engagement of SAR443820 in healthy adult participants.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
SAR443820 was generally safe and well‐tolerated following single (up to 40 mg) and repeated oral doses (up to 20 mg b.i.d.), with favorable PK, high brain penetrance, and robust peripheral RIPK1 target engagement.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The results of the phase I study support further clinical development of SAR443820 in phase II trials in patients with amyotrophic lateral sclerosis and multiple sclerosis.
INTRODUCTION
Receptor‐interacting serine/threonine protein kinase 1 (RIPK1) is an intracellular protein critically involved in multiple inflammatory pathways and cell death pathways, including apoptosis and necroptosis. RIPK1 is widely expressed in a variety of tissues in the human body, particularly in all major cell types, including astrocytes, microglia, oligodendrocytes, and neurons of the central nervous system (CNS) and peripheral immune cells, such as T‐ and B‐cells, dendritic cells, and macrophages, making it an attractive therapeutic target in neurodegenerative and neuroinflammatory diseases. 1 , 2 , 3 The kinase activity of RIPK1 plays a central role in mediating several deleterious inflammatory cell death mechanisms upon activation via tumor necrosis factor (TNF) alpha signaling through TNF receptor 1. 3 , 4 Upon activation, RIPK1 regulates both caspase‐independent necroptosis via RIPK3, and mixed lineage kinase‐like protein (MLKL) and RIPK1‐dependent apoptosis via caspase‐8. 3 , 5 , 6 Emerging evidence suggests that RIPK1 can also regulate pro‐inflammatory cytokine and chemokine production. 1 , 6 , 7 These multiple consequences of RIPK1 signaling have raised the possibility that a brain penetrant inhibitor of RIPK1 could be beneficial in preventing inflammation and necroptosis in chronic CNS diseases in humans. 1 , 3 , 8 , 9
Increased RIPK1 activity is implicated in amyotrophic lateral sclerosis (ALS), as evidenced by increased expression and phosphorylation of RIPK1 and increased levels of its downstream signaling partners RIPK3 and MLKL in postmortem spinal cord tissue samples from patients with ALS. 10 Similar increases were observed in the spinal cord of SOD1G93A transgenic ALS mouse models, and treatment with an RIPK1 inhibitor resulted in improved survival and reduced axonal pathology in these models. 11 Furthermore, inhibition of RIPK1 activity has been found to protect against necroptotic cell death in animal models of human neurodegenerative diseases, such as multiple sclerosis, Alzheimer's disease, and ALS. 1 , 2 , 10 , 12 , 13 , 14 , 15 , 16
Few peripherally restricted (non‐brain penetrant) RIPK1 inhibitors are being developed for several chronic inflammatory diseases. GSK2982772 is in clinical development for rheumatoid arthritis, ulcerative colitis, and psoriasis, and it has been shown to be safe and well‐tolerated. 15 , 17 , 18 , 19 , 20 SAR443122 (DNL758) is being developed for ulcerative colitis (NCT05588843) and cutaneous lupus erythematosus (NCT04781816). Furthermore, previously developed brain penetrant RIPK1 inhibitors, such as DNL104 and SAR443060 (DNL747), have been tested in clinical settings. 15 In healthy volunteers, DNL104 demonstrated inhibition of RIPK1 activation as measured by a decrease in RIPK1 phosphorylation (pS166‐RIPK1) in peripheral blood mononuclear cells (PBMCs), used as a pharmacodynamic (PD) marker of peripheral drug target engagement; however, its development was discontinued due to liver function abnormalities observed during the first‐in‐human study. 8 SAR443060 was well‐tolerated in healthy participants as well as in Alzheimer's disease (NCT03757325) and ALS (NCT03757351) populations. Pharmacokinetics (PK) were similar in all three populations, and target engagement was demonstrated by dose‐dependent inhibition of pS166‐RIPK1 in PBMCs. 9
Overall, targeting RIPK1 represents an attractive opportunity for chronic inflammatory CNS diseases, and thus, it is critical to investigate the safety, tolerability, PK, and PD profiles of an inhibitor that can penetrate the brain at effective concentrations. Reported here are the results from the phase I first‐in‐human study of SAR443820 (DNL788), a selective, small‐molecule, orally bioavailable, brain penetrant, reversible inhibitor of RIPK1 with potent in vitro inhibition of RIPK1 activity, with a 50% inhibitory concentration of 3.16 nM in human PBMCs and 1.6 nM in induced pluripotent stem cell (iPSC)‐derived microglia (data not published). The results of this phase I study (NCT05795907) include assessment of safety, tolerability, PK, and target engagement after single ascending doses (SAD) and multiple ascending doses (MAD) of SAR443820 in healthy participants, and a separate single‐dose part for assessing concentration of SAR443820 in cerebrospinal fluid (CSF) to evaluate its brain penetrance.
METHODS
Study design
This single‐center, first‐in‐human, healthy participant study conducted in the United States was comprised of three parts: part 1a (SAD), part 2 (MAD), and part 1b (PK‐CSF; Figure S1). Eligible participants were healthy men or women aged 18–55 years, with body weight between 50 and 100 kg for men and between 40 and 90 kg for women, and a body mass index between 18 and 30 kg/m2. Both SAD and MAD were randomized, double‐blind, placebo‐controlled parts that evaluated SAR443820 administered under fasting conditions. Participants were fasted overnight for at least 8 h before the morning dose and for 2 h after dosing. SAR443820 was administered by oral route using 10 mg and 5 mg strength capsules or matching placebo. In SAD, four sequential ascending single doses were evaluated: 10 mg, 20 mg, 30 mg, and 40 mg, each dose per cohort. In MAD, repeated ascending doses administered as once daily (q.d.) or twice daily (b.i.d.) regimen for 14 days were evaluated: 10 mg q.d., 20 mg q.d., 15 mg b.i.d., and 20 mg b.i.d., each dose per cohort. To ensure the best conditions of safety, all SAD and MAD cohorts included a sentinel cohort with two participants dosed (1 with SAR443820 and 1 with placebo) on the first day. The remaining participants from each cohort were dosed on the following days keeping a safety observational time between each consecutive administration. In the open‐label PK‐CSF part, two single doses of SAR443820 (10 mg and 40 mg, each dose per cohort), previously tested for safety and tolerability in SAD cohorts, were sequentially administered ~30 min after breakfast.
The study was conducted in accordance with Good Clinical Practice as required by the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines and the ethical principles of the Declaration of Helsinki. The study protocol complied with the recommendations of the 18th World Health Congress (Helsinki, 1964) and all applicable amendments, and was approved by the Institutional Review Board. All participants provided written informed consent prior to study enrollment.
Safety assessments
Safety of SAR443820 was assessed by physical examination, neurological examination, monitoring adverse events (AEs), serious AEs (SAEs), AEs of special interest (AESIs), and potentially clinically significant abnormalities (PCSAs) in clinical laboratory tests (hematology, clinical chemistry, and urinalysis), vital signs (body temperature, blood pressure, and heart rate [HR]), and electrocardiogram (ECG) parameters (HR, respiratory rate, pulse rate [PR], QRS duration, QT, QTcF, and QTcB intervals) measured in the standard 12‐lead ECG or ECGs extracted from 24‐h Holter ECG data. The AESIs were based on the standard practice in phase I protocol and/or preclinical toxicology findings of SAR443820 and AEs reported in other RIPK1 inhibitors. These AESIs included seizure, alanine aminotransferase (ALT) increase greater than or equal to two‐fold the upper limit of normal (ULN), QTc greater than or equal to 500 ms, pregnancy in women exposed to SAR443820, and symptomatic overdose with SAR443820.
SAR443820 concentration‐ECG analysis
A thorough time‐matched ECG parameter analysis in relation with corresponding SAR443820 concentrations on day 1 (SAD) and day 14 (MAD) over 24 h compared to the baseline prior to the first dosing was performed on ECG extracted from 24‐h Holter ECG data as per the ICH guidance. 21 , 22 , 23 The concentration‐ECG analysis data were collected only for data building.
Pharmacokinetic analysis
A bioanalytical method was developed and validated for the quantification of SAR443820 in human plasma and CSF using the high‐performance liquid chromatography mass spectrometry method with a lower limit of quantification of 1 ng/mL.
In SAD, plasma samples were collected before dosing and at 10 postdose timepoints on day 1, and one sample each on days 2 and 3. In MAD, plasma samples were collected at predose and at 10–11 postdose timepoints on days 1 and 14; in addition, predose samples were collected on days 2, 3, 5, 7, and 11, and one sample each on days 15, 16, and 17.
The following plasma PK parameters were calculated from plasma SAR443820 concentrations obtained after single (SAD) or repeated (MAD) oral doses: maximum plasma concentration observed (C max); time to reach C max (T max); area under the plasma concentration versus time curve from time zero to the real time T last(AUClast); AUC extrapolated to infinity (AUC); AUC calculated using the trapezoidal method during a dosage interval (tau), with tau = 12 h for b.i.d. regimen and tau = 24 h for q.d. regimen (AUCtau); half‐life associated with the terminal slope (T 1/2z); apparent total body plasma clearance after single dose (CL/F); apparent total body plasma clearance doses at steady‐state (CLss/F); the apparent volume of distribution (Vz/F); and concentration observed just before treatment administration during repeated dosing (C trough).
In the PK‐CSF part, plasma sampling was conducted at predose and at 10 postdose timepoints, including one plasma sampling at the same time of CSF sampling. SAR443820 concentrations in CSF were determined following lumbar puncture after single oral dosing of 10 mg and 40 mg in two dedicated cohorts. Only one CSF sample was collected per participant on day 1 within the following time intervals: 1–3 h or 4–6 h or 8–10 h postdose, matching with plasma sampling timepoints. SAR443820 concentrations in CSF were reported and concentration ratio between CSF and unbound plasma was calculated using the in vitro unbound fraction in human plasma.
The plasma 4β‐hydroxycholesterol on day 14 versus day 1 predose concentration ratios were calculated to evaluate the potential for CYP3A4 enzyme induction by SAR443820 after repeated doses in MAD. The plasma 4β‐hydroxycholesterol data were collected only for data building.
Pharmacodynamic analysis
Activity of RIPK1 when stimulated can be detected by an increase in autophosphorylation at serine 166 in the RIPK1 protein. 9 Human PBMCs are a primary cell type that expresses endogenous RIPK1. Therefore, the ability of SAR443820 to inhibit RIPK1 activity, as measured by pS166‐RIPK1 inhibition in PBMC lysates isolated from blood samples, was investigated to assess the peripheral target engagement. The pS166‐RIPK1 levels in human PBMC lysates and percentage change from baseline were calculated. In SAD, six blood samples (at predose; at 1, 3, 5, 8 h postdose on day 1; and at 24 h postdose on day 2) were collected. In MAD, blood samples were collected on day ‐1 or day 1 (at predose), on day 7 (one sample), on day 14 (at predose; at 1, 3, 5, 8 h postdose), and on days 15 and 16 (one sample each).
Statistical analysis
Statistical analyses were performed separately per study part (part 1a, part 1b, and part 2), with participants under placebo were pooled from different cohorts for part 1a and part 2. Safety population included all participants exposed to investigational medicinal product (IMP), regardless of the duration of the treatment administered. All safety assessments (AEs, laboratory parameters, and vital signs) were summarized using descriptive statistics. Safety analysis was focused on treatment‐emergent period – the time from the first IMP administration up to the end‐of‐study visit (included). AEs were coded according to the Medical Dictionary for Regulatory Activities versions 23.1 (part 1) and 24.0 (part 2). Severity was graded (grades 1–4) according to the US Food and Drug Administration Guidance for Industry. 24 Number (%) of participants experiencing treatment‐emergent AEs (TEAEs) and PCSAs in clinical chemistry, vital signs, and ECGs were summarized by dose level group.
Concentration‐ECG population included all randomized participants treated by SAR443820 with at least one ECG assessment time‐matched with a PK concentration (ECGs centrally read coming from 24‐h Holter ECG). Participants randomized in the placebo group were also included if they had at least one missing change from baseline in ECG parameter; their SAR443820 concentrations were set to 0 by convention. Exposure–response analyses between ECG parameters (change from time‐matched baseline in 24‐h Holter ECG data) and corresponding SAR443820 concentrations were performed using graphical tools and regression methods over 24 h on day 1 (SAD) or day 14 (MAD) postdose. Predictions (point estimates with 90% confidence interval [CI]) at selected SAR443820 concentrations (geometric mean, C max for each dose) were computed using a linear mixed effect model.
PK population included all participants exposed to SAR443820 with no major or critical deviations related to SAR443820 intake and for whom PK data were considered sufficient and interpretable. PK parameters were summarized by descriptive statistics for each dose level group. In SAD, dose proportionality was assessed using a linear model on C max, AUClast and AUC. In MAD, all PK analyses were performed separately for q.d. and b.i.d. regimens. Steady‐state was assessed on C trough using a nonlinear model. Accumulation was assessed using a linear model on log‐transformed accumulation ratio. Dose proportionality was assessed using a power model on C max and AUCtau on day 1 and day 14, separately. In the PK‐CSF part of the study, CSF concentration was described by dose level group and by timepoint and CSF‐to‐unbound plasma concentration ratios were calculated by dose and by timepoint. Given there was no evidence of an effect of dose on the CSF‐to‐unbound plasma concentration ratios, the latter were described by pooling the data from the two dose level groups and were presented by time intervals 1–3 h or 4–6 h or 8–10 h. In MAD, 4β‐hydroxycholesterol on day 14 versus day 1 predose concentration ratios were summarized using descriptive statistics by dose level group and point estimates with 90% CI for the geometric mean of the log‐transformed day 14/day 1 ratio were provided by dose level group and overall, using an analysis of covariance.
PD population included all participants in the safety population with no major or critical deviations related to IMP and/or PD measurements, for whom the PD data were considered sufficient and interpretable. Levels of pS166‐RIPK1 and percentage of change from baseline were summarized descriptively by dose level group and timepoints. Line plots of median (Q1–Q3) were provided for percent change from baseline in pS166‐RIPK1 at multiple timepoints up to 24 h in SAD and up to 48 h in MAD.
RESULTS
Study participants
The SAD part of the study was conducted between November 30, 2020, and April 28, 2021. The MAD part of the study was conducted between February 26, 2021, and July 20, 2021. A total of 84 participants were enrolled in the study. In the SAD part, 32 participants were randomized: six participants in each cohort received SAR443820 (single dose of 10 mg, 20 mg, 30 mg, or 40 mg) and two participants in each cohort received placebo. In part 1b, 12 participants were enrolled: six into each of the two cohorts of SAR443820 (10 mg or 40 mg). In the MAD part, 40 participants were randomized: eight participants in each cohort received SAR443820 10 mg q.d., 20 mg q.d., 15 mg b.i.d., or 20 mg b.i.d., and two participants in each cohort received placebo (Figure 1). The demographics and baseline characteristics are summarized in Table 1.
FIGURE 1.
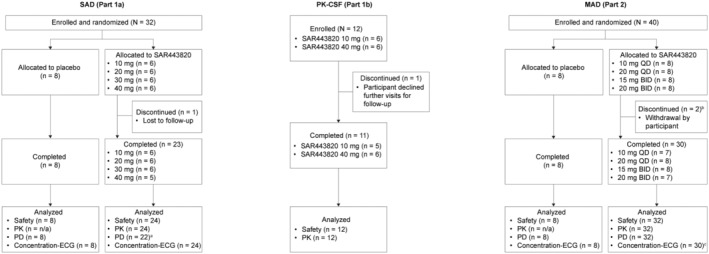
Disposition of trial participants. aTwo participants excluded due to error in sample processing. bTwo participants withdrew for personal reasons without any related safety/tolerability concerns. c Two participants prematurely withdrew before day 14. BID, twice daily; CSF, cerebrospinal fluid; ECG, electrocardiogram; n/a, not applicable; MAD, multiple ascending dose; QD, once daily; PD, pharmacodynamic; PK, pharmacokinetic; SAD, single ascending dose.
TABLE 1.
Demographics and baseline characteristics (Safety population).
| Characteristic | SAD (part 1a) | PK‐CSF (part 1b) | MAD (part 2) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo (n = 8) | SAR443820 | SAR443820 | Placebo (n = 8) | SAR443820 | ||||||||
| 10 mg (n = 6) | 20 mg (n = 6) | 30 mg (n = 6) | 40 mg (n = 6) | 10 mg (n = 6) | 40 mg (n = 6) | 10 mg q.d. (n = 8) | 20 mg q.d. (n = 8) | 15 mg b.i.d. (n = 8) | 20 mg b.i.d. (n = 8) | |||
| Age, years, mean (SD) | 34.5 (11.0) | 29.0 (5.9) | 36.2 (12.8) | 32.8 (10.7) | 25.3 (8.2) | 29.3 (11.9) | 29.5 (12.9) | 35.0 (13.2) | 31.0 (5.1) | 28.9 (8.1) | 37.5 (10.8) | 28.3 (6.4) |
| Sex, n (%), Male | 2 (25.0) | 3 (50.0) | 2 (33.3) | 4 (66.7) | 0 | 4 (66.7) | 6 (100.0) | 5 (62.5) | 3 (37.5) | 5 (62.5) | 6 (75.0) | 6 (75.0) |
| Race, n (%), White | 6 (75.0) | 4 (66.7) | 5 (83.3) | 4 (66.7) | 6 (100.0) | 5 (83.3) | 6 (100.0) | 6 (75.0) | 4 (50.0) | 8 (100) | 6 (75.0) | 5 (62.5) |
| Weight, kg, mean (SD) | 70.4 (13.0) | 71.5 (8.5) | 69.1 (7.0) | 76.1 (11.8) | 58.4 (9.5) | 72.3 (14.0) | 81.8 (8.1) | 80.8 (11.8) | 74.1 (10.0) | 82.5 (10.5) | 76.9 (11.1) | 81.7 (9.1) |
| BMI, kg/m2, n (%) | ||||||||||||
| 18.5 to <25 | 6 (75.0) | 5 (83.3) | 3 (50.0) | 3 (50.0) | 5 (83.3) | 4 (66.7) | 3 (50.0) | 4 (50.0) | 2 (25.0) | 1 (12.5) | 5 (62.5) | 3 (37.5) |
| 25 to <30 | 2 (25.0) | 1 (16.7) | 3 (50.0) | 3 (50.0) | 1 (16.7) | 2 (33.3) | 3 (50.0) | 4 (50.0) | 5 (62.5) | 6 (75.0) | 3 (37.5) | 5 (62.5) |
| ≥30 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (12.5) | 1 (12.5) | 0 | 0 |
Abbreviations: b.i.d., twice daily; BMI, body mass index; CSF, cerebrospinal fluid; MAD, multiple ascending dose; PK, pharmacokinetics; q.d., once daily; SAD, single ascending dose; SD, standard deviation.
Safety and tolerability
Overall, SAR443820 was well‐tolerated in all study parts with no AE‐related treatment discontinuation reported (Table 2). During SAD, no SAEs or severe TEAEs were observed in the SAR443820 groups; one participant in the placebo group experienced a grade 3 event of blood creatinine phosphokinase increase, which was attributed to a pre‐existing condition, and not related to IMP or study procedure. During MAD, no SAEs or severe TEAEs occurred. In the PK‐CSF part of the study, one participant in the SAR443820 40 mg group experienced a grade 3 SAE of CSF leakage on day 3 that required hospitalization. The participant also had radiculopathy (grade 3), coccydynia, back pain, and vomiting. All AEs, including the SAE, were due to the procedural complication of lumbar puncture and were considered unrelated to SAR443820. Corrective treatment for CSF leakage consisted of blood patch leading to relief.
TABLE 2.
Overall summary of TEAEs (Safety population).
| n (%) | SAD (part 1a) | PK‐CSF (part 1b) | MAD (part 2) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo (n = 8) | SAR443820 | SAR443820 | Placebo (n = 8) | SAR443820 | ||||||||
| 10 mg (n = 6) | 20 mg (n = 6) | 30 mg (n = 6) | 40 mg (n = 6) | 10 mg (n = 6) | 40 mg (n = 6) | 10 mg q.d. (n = 8) | 20 mg q.d. (n = 8) | 15 mg b.i.d. (n = 8) | 20 mg b.i.d. (n = 8) | |||
| Any TEAE | 5 (62.5) | 2 (33.3) | 1 (16.7) | 3 (50.0) | 3 (50.0) | 4 (66.7) | 6 (100.0) | 5 (62.5) | 4 (50.0) | 3 (37.5) | 6 (75.0) | 4 (50.0) |
| Any grade ≥3 TEAE | 1 (12.5) | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 0 |
| Any treatment‐emergent SAE | 0 | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 0 |
| Any treatment‐emergent AESI | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 1 (12.5) | 0 |
Abbreviations: AESI, adverse event of special interest; b.i.d., twice daily; CSF, cerebrospinal fluid; MAD, multiple ascending dose; PK, pharmacokinetics; q.d., once daily; SAD, single ascending dose; SAE, serious adverse event; TEAE, treatment‐emergent adverse event.
During SAD, no AESIs were reported. In MAD, one participant in the SAR443820 15 mg b.i.d. group reported an AESI of ALT increase (2.29 ULN) on day 9 and then increased up to 2.71 ULN on day 15 (24 h post last dosing). This ALT increase was associated with a slight concomitant increase in aspartate aminotransferase (AST; 1.43 ULN) whereas the other liver function tests (alkaline phosphatase, bilirubin, and gamma glutamyl transferase) were within the normal range. Liver transaminases returned within normal ranges on day 27. This event was considered as IMP‐related but did not lead to permanent treatment discontinuation. In the PK‐CSF part of the study, one participant in the SAR443820 10 mg group experienced an AESI of ALT increase on day 2 (1.5 ULN) and then increased up to 2.4 ULN, meeting the criterion of AESI (≥2‐fold ULN) on day 7 (end‐of‐study visit). This AESI was considered related to SAR443820 and was not resolved due to participant declining follow‐up assessments. It is important to note that none of these AESIs were reported in the cohorts receiving the highest dose of SAR443820. Moreover, these instances of ALT increase in both cases were asymptomatic and isolated (or slightly associated with AST increase <2‐fold ULN).
The most frequently reported TEAEs were the nervous system disorders, with dizziness (SAD) and headache (PK‐CSF and MAD) being the most common AEs (Tables S1 and S2). Summary of TEAEs for each participant in parts 1a, 1b, and 2 are listed in Table S3.
During this first‐in‐human study, a few PCSAs were observed for hematology, clinical chemistry, vital signs, and ECG parameters. None of the PCSAs in the SAR443820 groups were considered as clinically relevant.
SAR443820 concentration‐ECG analysis
In both SAD and MAD, a time‐matched concentration‐ECG parameters relationship analysis was conducted on QTcF, HR, PR, and QRS. The results did not indicate any signal for SAR443820 administered up to a single dose of 40 mg and 14‐day dose of 20 mg b.i.d. to cause any clinically relevant modifications of ECG parameters (Table S4).
Pharmacokinetics
Plasma PK parameters of SAR443820 in all study parts are summarized in Table 3 and mean concentration‐time profiles are illustrated in Figure 2.
TABLE 3.
Plasma PK parameters of SAR443820 (PK population).
| Parameter | SAD (part 1a) | PK‐CSF (part 1b) | ||||
|---|---|---|---|---|---|---|
| SAR443820 | SAR443820 | |||||
| 10 mg (n = 6) | 20 mg (n = 6) | 30 mg (n = 6) | 40 mg (n = 6) | 10 mg (n = 6) | 40 mg (n = 6) | |
| C max (ng/mL), geometric mean (%CV) | 116 (27.2) | 241 (20.5) | 417 (34.2) | 555 (28.7) | 111 (31.0) | 374 (22.0) |
| T max (h), median (range) | 1.02 (1.00, 3.08) | 1.50 (1.00, 3.00) | 1.00 (0.50, 2.00) | 1.50 (1.00, 2.00) | 4.24 (3.00, 6.00) | 4.50 (2.00, 6.00) |
| AUClast (h*ng/mL), geometric mean (%CV) | 1120 (40.1) | 2360 (19.1) | 2420 (30.2) | 5120 (34.5) | 1170 (36.6) | 3960 (30.7) |
| AUC (h*ng/mL), geometric mean (%CV) | 1170 (40.9) | 2410 (19.0) | 2440 (30.2) | 5180 (34.6) | 1370 (42.6) | 4740 (42.5) |
| T 1/2z (h), geometric mean (%CV) | 7.97 (29.0) | 7.48 (21.4) | 5.70 (31.8) | 6.29 (20.6) | 7.97 (19.4) | 8.03 (29.5) |
| Vz/F (L), geometric mean (%CV) | 98.4 (20.2) | 89.4 (14.2) | 101 (12.1) | 70.1 (23.7) | 83.6 (25.6) | 97.7 (14.3) |
| CL/F (L/h), geometric mean (%CV) | 8.56 (37.8) | 8.28 (21.4) | 12.3 (44.7) | 7.72 (36.1) | 7.27 (29.7) | 8.43 (34.2) |
| MAD (Part 2) | ||||
|---|---|---|---|---|
| SAR443820 a | ||||
| 10 mg q.d. | 20 mg q.d. | 15 mg b.i.d. | 20 mg b.i.d. | |
| Day 1 | n = 8 | n = 8 | n = 8 | n = 8 |
| C max (ng/mL), geometric mean (%CV) | 157 (20.7) | 240 (20.2) | 211 (44.2) | 205 (20.3) |
| T max (h), median (range) | 1.00 (0.50, 2.00) | 1.5 (1.00, 2.00) | 1.00 (0.50, 2.00) | 2.00 (1.00, 4.00) |
| AUCtau (h*ng/mL), geometric mean (%CV) | 1080 (24.2) | 1860 (22.5) | 1100 (33.1) | 1350 (18.4) |
| Day 14 | n = 7 | n = 8 | n = 8 | n = 7 |
| C max (ng/mL), geometric mean (%CV) | 158 (31.9) | 250 (22.7) | 268 (36.3) | 353 (19.9) |
| T max (h), median (range) | 1.00 (1.00, 2.00) | 2.00 (0.60, 2.00) | 1.00 (1.00, 2.00) | 1.00 (0.50, 2.00) |
| AUCtau (h*ng/mL), geometric mean (%CV) | 1410 (27.4) | 2130 (21.4) | 1640 (32.5) | 2160 (25.9) |
| T 1/2z (h), geometric mean (%CV) | 8.87 (23.2) | 8.53 (19.0) | 7.21 (15.6) | 7.37 (23.0) |
| CLss/F (L/h), geometric mean (%CV) | 7.08 (25.9) | 9.40 (22.2) | 9.16 (30.0) | 9.27 (24.9) |
Abbreviations: AUClast, area under the plasma concentration versus time curve from time zero to the real time t last; b.i.d., twice daily; CL/F, apparent total body clearance of a drug from the plasma after single dose; CLss/F, apparent total body clearance of a drug from the plasma at steady‐state; C max, maximum plasma concentration; CSF, cerebrospinal fluid; CV, coefficient of variation; MAD, multiple ascending dose; PK, pharmacokinetic; q.d., once daily; SAD, single ascending dose; T last, time corresponding to the last concentration above the limit of quantification, C last; T max, time to reach C max; T 1/2z, terminal half‐life; Vz/F, apparent volume of distribution during the terminal phase.
The dosing interval tau is 12 h for the b.i.d. cohorts and 24 h for the q.d. cohorts.
FIGURE 2.
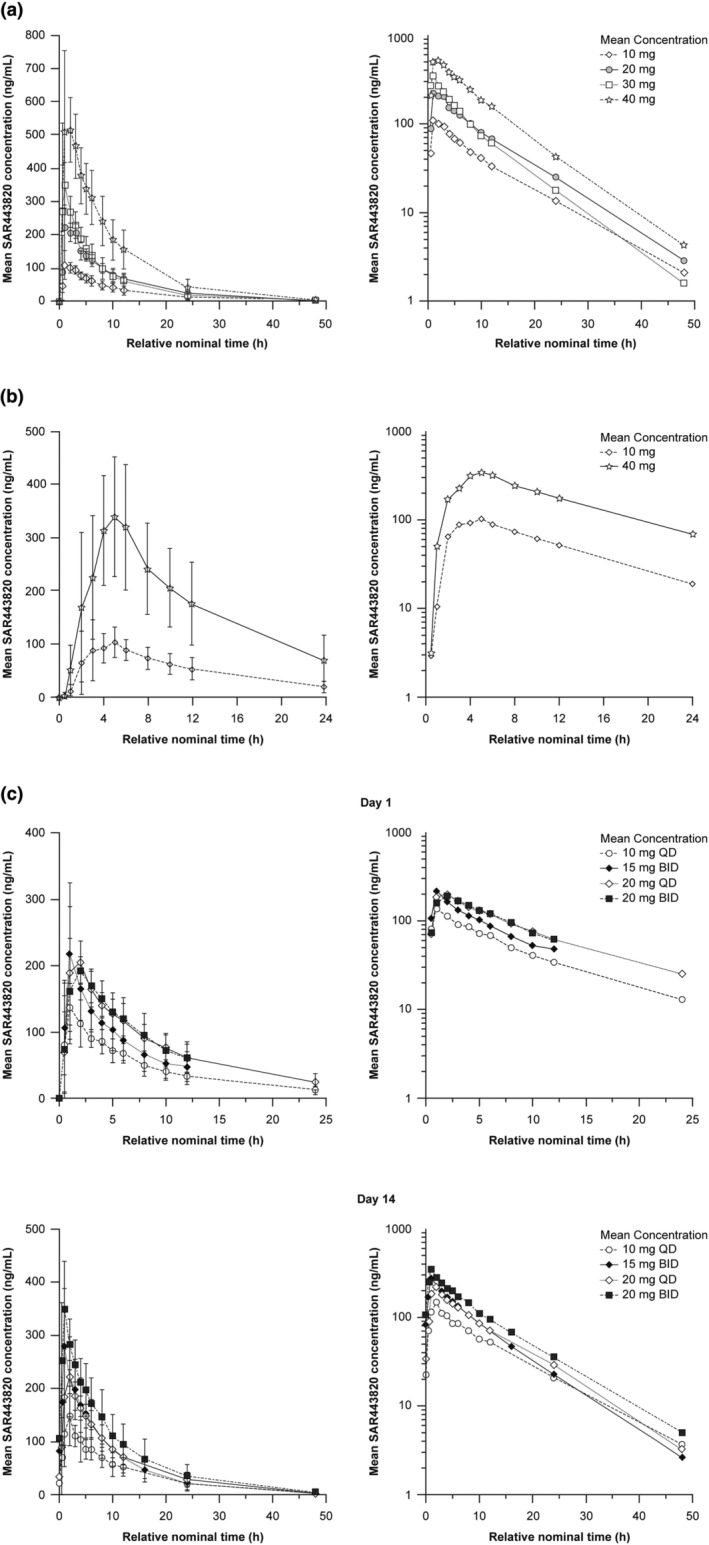
Mean plasma concentration‐time profiles of SAR443820 in healthy participants – Pharmacokinetic population (a) SAD – part 1a, (b) PK‐CSF – part 1b, and (c) MAD – part 2 (day 1 and day 14). The error bars depict the standard deviations. Left panel represents linear scale and right panel represents semilogarithmic scale. BID, twice daily; CSF, cerebrospinal fluid; MAD, multiple ascending dose; QD, once daily; PK, pharmacokinetic; SAD, single ascending dose.
In SAD, the median T max was reached between 1.00 and 1.50 h postdose with no apparent dose effect. The geometric mean T 1/2z was similar across single doses and ranged from 5.7 to 8.0 h. The between‐subject variability (percent coefficient of variation [%CV]) of SAR443820 C max, and AUCs ranged between 19.0% and 40.9%. SAR443820 exposures increased with dose with no major deviation from dose proportionality between 10 mg and 40 mg. Four‐fold increase in dose from 10 mg to 40 mg led to 4.79‐fold increase (90% CI: 3.61–6.36) for C max, 4.43‐fold increase (90% CI: 3.16–6.23) for AUC, and 4.56‐fold increase (90% CI: 3.24–6.42) for AUClast (Table S5).
In MAD, the median time to steady‐state was reached within 1.5–2 days after the first administration irrespective of the dose or the dosing regimen. After single or repeated SAR443820 administration in fasting conditions, median T max ranged between 1.00 and 2.00 h postdose. At steady‐state, C max and AUCtau increased with the dose for q.d. and b.i.d. regimen, T 1/2z remained similar across doses and dosing regimen and geometric mean values ranged between 7.2 and 8.9 h. With two‐fold increase in dose from 10 mg q.d. to 20 mg q.d., C max and AUCtau increased by 1.58‐fold (90% CI: 1.26–1.99) and 1.51‐fold (90% CI: 1.21–1.88) at day 14, respectively. With 1.33‐fold increase in dose from 15 mg b.i.d. to 20 mg b.i.d., C max and AUCtau increased by 1.31‐fold (90% CI: 1.00–1.73) and 1.32‐fold (90% CI: 1.01–1.72) at day 14, respectively (Table S5). Almost no or limited accumulation was observed after repeated oral dosing of SAR443820 in q.d. cohorts; accumulation ratios between day 14 and day 1 were around 1.03 for C max and 1.22 for AUCtau. In b.i.d. cohorts, an overall accumulation around 1.5‐fold was observed for both C max and AUCtau (Table S6).
In the PK‐CSF part of the study, a delay was observed in plasma T max with median values of 4.24 and 4.50 h postdose at 10 mg and 40 mg dose levels, respectively, as compared to the plasma T max of 1.02 and 1.50 h postdose at 10 mg and 40 mg dose levels, respectively, in a fasting state, as observed in SAD. The geometric mean T 1/2z was similar across doses and was about 8 h. A similar between‐subject variability (%CV) ranging between 22.0% and 42.6% for SAR443820 C max and AUCs was observed in fed conditions, irrespective of the dose.
Overall, by pooling the data from two dose level groups of SAR443820 (10 mg and 40 mg) in the PK‐CSF part, the CSF‐to‐unbound plasma concentration ratio was about 0.8 for samples collected during the time intervals 1–3 h and 4–6 h postdose, and the ratio was around 1.3 during the time interval 8–10 h postdose.
Point estimates (90% CI) of the day 14/day 1 4β‐hydroxycholesterol ratio were 0.88 (0.82–0.95) and 1.02 (0.91–1.14) for pooled q.d. and b.i.d. regimen, respectively. The 4β‐hydroxycholesterol plasma level measured after 14 days of multiple q.d. or b.i.d. doses did not increase as compared to baseline value, suggesting that there is no potential for CYP3A4 induction by SAR443820 after repeated doses (up to 20 mg b.i.d. dose; Table S7).
Pharmacodynamics – target engagement
The reduction of pS166‐RIPK1 measured in PBMC lysates is considered as an indication of target engagement by SAR443820. During SAD, the percent change from baseline was evaluated at multiple timepoints up to 24 h postdose (Figure 3). No inhibition was detected in the placebo group. Median pS166‐RIPK1 inhibition of greater than 80% was observed in all SAR443820 dose groups at as early as 1 h postdose and was maintained over at least 5 h postdose. At 5 h postdose, maximum median peripheral pS166‐RIPK1 inhibition ranged between 82% and 97% among the SAR443820 single dose groups.
FIGURE 3.
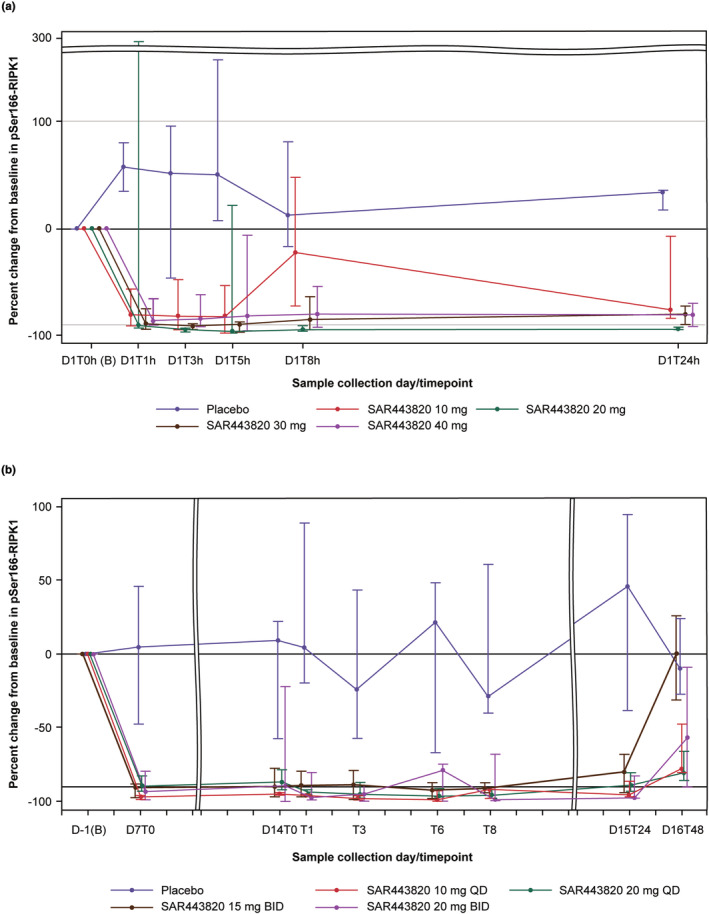
Line plot of median (Q1–Q3) percent change from baseline in pSer166‐RIPK1 in PBMC lysates over the course of the study by dose level group – Pharmacodynamic population (a) SAD – part 1a and (b) MAD – part 2. B, baseline; BID, twice daily; D, day; MAD, multiple ascending dose; QD, once daily; pSer166‐RIPK1, phosphorylated receptor‐interacting serine/threonine protein kinase 1 at Serine 166; SAD, single ascending dose; T, time.
Similarly, in the MAD part of the study, the percent change was evaluated at multiple timepoints from baseline up to 48 h after administration of the final dose on day 14 (Figure 3). Administration of SAR443820 doses from 10 mg q.d. to 20 mg b.i.d. in healthy participants led to a marked target engagement with median pS166‐RIPK1 inhibition from baseline close to 90% at steady‐state before morning dosing (C trough) on day 7 (first post‐baseline timepoint) and similarly at day 14 (predose). On day 14, the inhibition of greater than 80% was maintained up to 24 h post last morning dosing in all dose level groups. At 48 h post last dosing, less inhibition of RIPK1 phosphorylation was observed in all dose level groups.
DISCUSSION
RIPK1 has emerged as a promising therapeutic target for a spectrum of neuroinflammatory diseases in humans. However, there is still a limited understanding of the specific modalities of RIPK1 involvement in different diseases. 1 Given that RIPK1 activation and necroptosis have now been genetically and mechanistically linked with several human neurodegenerative diseases, a highly selective, safe, brain penetrant inhibitor of RIPK1 may provide a unique opportunity by targeting neuroinflammation and cell death, which drive various neurologic conditions, such as multiple sclerosis, Alzheimer's disease, and ALS and altering the disease progression. 10 , 14 , 16 , 25
Overall, SAR443820 was well‐tolerated in this study, without raising any safety concerns in the dose escalation and safety profile was consistent with that of other brain penetrant RIPK1 inhibitors. 9 , 26 Mild abnormalities in laboratory values were observed related to SAR443820, however, the safety data do not suggest a dose‐dependent trend. Two cases of asymptomatic ALT increase (≥2‐fold ULN but limited <3‐fold ULN) were reported in this study, but none were considered as clinically relevant. Similar to SAR443820, other RIPK1 inhibitors, such as SAR443060, 9 GSK2982772, 20 and GFH312, 26 were found to be well‐tolerated with no reports of ALT abnormalities in healthy participants. Taken together, it may be assumed that the liver function abnormalities may not be a mechanism‐based consideration.
Overall, few PCSAs for the laboratory values, vital signs, and ECG parameters were observed, however, none of them were considered clinically relevant. Furthermore, a thorough time‐matched concentration‐ECG analysis revealed no clinically relevant trends in ECG parameters.
SAR443820 showed a favorable PK profile in healthy volunteers. The rapid absorption in the fasted condition suggests that there are no barriers to SAR443820 absorption as median T max reached between 1.00 h and 1.50 h postdose following single doses up to 40 mg and between 1.00 h and 2.00 h after repeated doses across the dose level groups. There were no major deviations from dose proportionality over the range of doses tested. A slight delay in absorption rate was observed when SAR443820 was administered as a capsule in fed conditions; however, there was no evidence that the extent of absorption was impacted by food. The elimination half‐lives (geometric mean) ranged between 5.7–8.0 h and 7.2–8.9 h following single and repeated oral doses of SAR443820, respectively, with no apparent dose effect. Consistently, the accumulation ratio for AUC0‐24 observed after q.d. or b.i.d. dosing was limited with a value of 1.2 and 1.5, respectively, and a median time to steady‐state reached within 1.5–2 days.
The CSF‐to‐unbound plasma concentration ratio (0.8–1.3) indicates that SAR443820 has high brain penetrance following its oral administration because CSF can be considered as a surrogate of brain penetration based on nonclinical data obtained in rats and monkeys showing similar values of CSF/unbound plasma ratio and brain/unbound plasma ratio.
Based on preclinical in vivo mouse models, 16 brain concentration above the 90% inhibitory concentration (IC90; in vitro pS166‐RIPK1 inhibition in mouse microglia) correlates with better disease efficacy in rodents. In the present study, CSF concentrations reached values above the IC90 after 10 or 40 mg single administration, thus suggesting an appropriate exposure at central level in humans for achieving high level of target engagement that is required for treatment effectiveness. Furthermore, previous evidence suggests that high levels (>90%) of RIPK1 inhibition are needed to exhibit clinical efficacy. 9 , 17 , 18 , 19 , 20 Thus, one of the objectives of the SAD and MAD parts of this study was to determine the magnitude of pS166‐RIPK1 inhibition that could be achieved by each dose as measured in the collected PBMC lysates. In this study, the maximum median peripheral pS166‐RIPK1 inhibition of greater than 80% was observed at as early as 1 h postdose and was maintained up to 5 h postdose following single SAR443820 doses, whereas with repeated doses, it was close to 90% before morning dosing at C trough day 7 and at predose day 14, reflecting a marked target engagement. These effects were consistent with PD results from previous clinical studies of brain penetrant RIPK1 inhibitors. 8 , 9 Overall, these data met the targeted pS166‐RIPK1 inhibition.
This first‐in‐human healthy volunteer study comprising of randomized, double‐blind, placebo‐controlled SAD and MAD cohorts, and two separate open‐label cohorts to assess CSF exposure, was designed to determine safety and tolerability of SAR443820, with well‐characterized PK and PD analyses. SAR443820 was generally safe and well‐tolerated in healthy adults receiving single oral doses up to 40 mg and repeated doses up to 20 mg b.i.d. for 14 days. Although a short treatment period of 14 days in healthy participants and PD assessments only at the peripheral level in human PBMCs were among the few limitations of this study, the combination of favorable PK, high brain penetrance, and marked peripheral RIPK1 target engagement, as measured by reduction in pS166‐RIPK1 levels, indicate that SAR443820 could be a promising therapeutic option in several human neurodegenerative diseases. These results support further clinical development of SAR443820 in phase II trials in patients with ALS (NCT05237284) and multiple sclerosis (NCT05630547).
AUTHOR CONTRIBUTIONS
A.H.M., X.N., C.C., R.P., P.L., M.B., D.O., E.K., J.H.N., A.E., and N.A. wrote the manuscript. A.H.M., X.N., R.P., M.B., D.O., E.K., and N.A. designed the research. X.N., R.P., M.B., E.K., J.H.N., A.E., and N.A. performed the research. A.H.M., X.N., C.C., R.P., P.L., M.B., E.K., A.E., and N.A. analyzed the data.
FUNDING INFORMATION
This study was funded by Sanofi.
CONFLICT OF INTEREST STATEMENT
A.H.M., X.N., C.C., R.P., M.B., N.A., D.O., and E.K. are employees of Sanofi, and may hold stock and/or stock options in the company. P.L. is an employee of Ividata Life Sciences (contracted by Sanofi). J.H.N. is an employee of Denali Therapeutics Inc. and may hold stocks in the company. A.E. declared no competing interests for this work.
Supporting information
Data S1
ACKNOWLEDGMENTS
The authors would like to thank all the study participants, investigators, and their study staff for their contribution to this study. Global support in the study management was provided by Nian Tian. The authors thank Brian Fox, Javier de Vicente, and Anthony A. Estrada from Denali for their contribution to the discovery synthesis, profiling, and advancement into nGLP and GLP development, and human dose estimations/safety margin evaluations for SAR443820 (DNL788). The authors acknowledge Alana Wong, PhD (Sanofi) for her contribution to manuscript planning, review, and coordination. Medical writing support for this manuscript was provided by Akshada Deshpande, PhD, of Sanofi.
Hincelin‐Mery A, Nicolas X, Cantalloube C, et al. Safety, pharmacokinetics, and target engagement of a brain penetrant RIPK1 inhibitor, SAR443820 (DNL788), in healthy adult participants. Clin Transl Sci. 2024;17:e13690. doi: 10.1111/cts.13690
DATA AVAILABILITY STATEMENT
Qualified researchers may request access to participant‐level data and related documents, including clinical study report, study protocol with any amendments, blank case report form, and dataset specifications. Participant‐level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi's data sharing criteria, eligible studies, and process for requesting access can be found at https://www.vivli.org.
REFERENCES
- 1. Degterev A, Ofengeim D, Yuan J. Targeting RIPK1 for the treatment of human diseases. Proc Natl Acad Sci U S A. 2019;116:9714‐9722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ofengeim D, Ito Y, Najafov A, et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015;10:1836‐1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1‐mediated neuroinflammation in CNS diseases. Nat Rev Neurosci. 2019;20:19‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14:727‐736. [DOI] [PubMed] [Google Scholar]
- 5. Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138:229‐232. [DOI] [PubMed] [Google Scholar]
- 6. Zhu K, Liang W, Ma Z, et al. Necroptosis promotes cell‐autonomous activation of proinflammatory cytokine gene expression. Cell Death Dis. 2018;9:500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Najjar M, Saleh D, Zelic M, et al. RIPK1 and RIPK3 kinases promote cell‐death‐independent inflammation by toll‐like receptor 4. Immunity. 2016;45:46‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grievink HW, Heuberger J, Huang F, et al. DNL104, a centrally penetrant RIPK1 inhibitor, inhibits RIP1 kinase phosphorylation in a randomized phase I ascending dose study in healthy volunteers. Clin Pharmacol Ther. 2020;107:406‐414. [DOI] [PubMed] [Google Scholar]
- 9. Vissers M, Heuberger J, Groeneveld GJ, et al. Safety, pharmacokinetics and target engagement of novel RIPK1 inhibitor SAR443060 (DNL747) for neurodegenerative disorders: randomized, placebo‐controlled, double‐blind phase I/Ib studies in healthy subjects and patients. Clin Transl Sci. 2022;15:2010‐2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ito Y, Ofengeim D, Najafov A, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353:603‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zelic M, Pontarelli F, LaMorte M, et al. RIPK1 is elevated in ALS patient spinal cords and RIPK1 kinase inhibition delays ALS disease progression in the SOD1G93A mouse model. Poster presented at the 32nd International Symposium on ALS/MND, December 7–10, 2021, Virtual.
- 12. Mifflin L, Hu Z, Dufort C, et al. A RIPK1‐regulated inflammatory microglial state in amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2021;118:e2025102118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Caccamo A, Branca C, Piras IS, et al. Necroptosis activation in Alzheimer's disease. Nat Neurosci. 2017;20:1236‐1246. [DOI] [PubMed] [Google Scholar]
- 14. Ofengeim D, Mazzitelli S, Ito Y, et al. RIPK1 mediates a disease‐associated microglial response in Alzheimer's disease. Proc Natl Acad Sci U S A. 2017;114:e8788‐e8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mifflin L, Ofengeim D, Yuan J. Receptor‐interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat Rev Drug Discov. 2020;19:553‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zelic M, Pontarelli F, Woodworth L, et al. RIPK1 activation mediates neuroinflammation and disease progression in multiple sclerosis. Cell Rep. 2021;35:109112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weisel K, Berger S, Papp K, et al. Response to inhibition of receptor‐interacting protein kinase 1 (RIPK1) in active plaque psoriasis: a randomized placebo‐controlled study. Clin Pharmacol Ther. 2020;108:808‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weisel K, Berger S, Thorn K, et al. A randomized, placebo‐controlled experimental medicine study of RIPK1 inhibitor GSK2982772 in patients with moderate to severe rheumatoid arthritis. Arthritis Res Ther. 2021;23:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weisel K, Scott N, Berger S, et al. A randomised, placebo‐controlled study of RIPK1 inhibitor GSK2982772 in patients with active ulcerative colitis. BMJ Open Gastroenterol. 2021;8:e000680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weisel K, Scott NE, Tompson DJ, et al. Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol Res Perspect. 2017;5:e00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. U.S. Food and Drug Administration . Guidance Document. E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non‐Antiarrhythmic Drugs Questions and Answers (R3) Guidance for Industry. 2017. https://www.fda.gov/media/71379/download. Accessed January 17, 2023.
- 22. Darpo B, Garnett C, Benson CT, et al. Cardiac safety research consortium: can the thorough QT/QTc study be replaced by early QT assessment in routine clinical pharmacology studies? Scientific update and a research proposal for a path forward. Am Heart J. 2014;168:262‐272. [DOI] [PubMed] [Google Scholar]
- 23. Garnett C, Bonate PL, Dang Q, et al. Scientific white paper on concentration‐QTc modeling. J Pharmacokinet Pharmacodyn. 2018;45:383‐397. [DOI] [PubMed] [Google Scholar]
- 24. U.S. Food and Drug Administration . Guidance for Industry: Toxicity grading Scale for healthy adult and adolescent volunteers enrolled in preventive vaccine clinical trials. 2007. https://www.fda.gov/media/73679/download. Accessed November 28, 2022.
- 25. Xu D, Jin T, Zhu H, et al. TBK1 suppresses RIPK1‐driven apoptosis and inflammation during development and in aging. Cell. 2018;174:1477‐1491.e1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lickliter J, Wang S, Zhang W, et al. A phase I randomized, double‐blinded, placebo‐controlled study assessing the safety and pharmacokinetics of RIPK1 inhibitor GFH312 in healthy subjects. Clin Transl Sci. 2023;16(9):1691‐1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Data Availability Statement
Qualified researchers may request access to participant‐level data and related documents, including clinical study report, study protocol with any amendments, blank case report form, and dataset specifications. Participant‐level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi's data sharing criteria, eligible studies, and process for requesting access can be found at https://www.vivli.org.