

Abstract
Recently, attention mechanism and derived models have gained significant traction in drug development due to their outstanding performance and interpretability in handling complex data structures. This review offers an in-depth exploration of the principles underlying attention-based models and their advantages in drug discovery. We further elaborate on their applications in various aspects of drug development, from molecular screening and target binding to property prediction and molecule generation. Finally, we discuss the current challenges faced in the application of attention mechanisms and Artificial Intelligence technologies, including data quality, model interpretability and computational resource constraints, along with future directions for research. Given the accelerating pace of technological advancement, we believe that attention-based models will have an increasingly prominent role in future drug discovery. We anticipate that these models will usher in revolutionary breakthroughs in the pharmaceutical domain, significantly accelerating the pace of drug development.
Keywords: drug discovery, attention mechanism, Artificial Intelligence, molecular representation, molecule generation, transformer
INTRODUCTION
Drug development has traditionally been a time-consuming, intricate and capital-intensive process [1]. Taking a drug from discovery to market often spans over a decade and requires billions of dollar in investments [2]. This complex journey is significantly hampered by inefficiencies in primary screening, the intricacies of biological tests and the unpredictability of clinical trials [3]. However, with the growing sophistication of Artificial Intelligence (AI), its role in drug development is being increasingly recognized, gradually reshaping conventional paradigms [4–6]. Particularly under the influence of deep learning, AI demonstrates its outstanding performance in handling vast amounts of data, enhancing prediction accuracy and automating intricate workflows [7]. Throughout the drug discovery and clinical research, AI reveals its extensive application potential (Figure 1): from swiftly screening numerous compounds, assisting in drug synthesis design, to clinical drug trials, risk assessment of medicines and personalized medication [8–10]. Furthermore, by analyzing extensive chemical data, AI unveils the intrinsic relationship between efficacy and molecular structure, thereby generating novel drug molecule candidates [11–13]. Data-driven approaches also empower researchers to delve deeper into the molecular mechanisms of diseases, paving the way for more targeted treatments [14].
Figure 1.
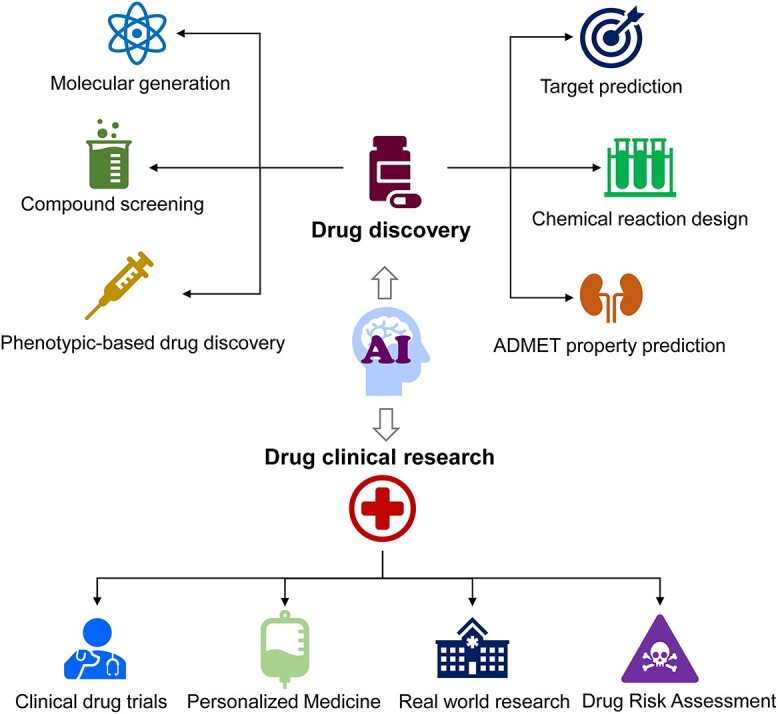
The application of AI in drug discovery and clinical drug research.
Among the vast AI model spectrum, the attention mechanism and its derived models such as Transformer, Graph Attention Networks (GATs), Bidirectional Encoder Representations from Transformers (BERT) and Generative Pre-trained Transformer (GPT) have gained substantial traction in drug development in recent years [15–20]. The attention mechanism dynamically focuses on crucial data segments during processing, significantly boosting model expressiveness and prediction accuracy [21]. Notably, attention-based models, compared with traditional deep learning techniques, are more interpretable, allowing researchers to discern which molecular sections the model prioritizes—an invaluable insight for examining structure–activity relationships [22, 23]. Even more critical is that, thanks to their self-attention properties, these models can process data in parallel, greatly enhancing computational efficiency [24].
This review centers on the applications and emerging trends of the attention mechanism and its related models in small molecule drug development, excluding nucleic acid and protein biopharmaceuticals. We first introduce the foundational principles of the attention mechanism and its extended models (e.g. GAT, Transformer, BERT, GPT), followed by their specific applications in drug development. Subsequently, we provide a comprehensive review of these techniques’ applications in six key tasks in drug development: Drug–Drug Interaction (DDI), Synergistic Drug Combinations, Molecular Generation, Molecular Property Prediction, Drug Response and Drug–Target Interaction (DTI). Finally, we discuss the challenges faced by these technologies in the realm of drug development and look forward to future trends.
ATTENTION-BASED MODELS AND THEIR ADVANTAGES IN DRUG DISCOVERY
The emergence of the attention mechanism can be traced back to its initial use in machine translation tasks, effectively addressing long-distance dependencies in sequences [25]. This brought about a more flexible and efficient mode of operation for deep learning models. Then, the introduction of the Transformer architecture amplified the capabilities of attention, heralding significant changes in the research domain [26]. Transformers not only elegantly handle complex sequence information but also introduce unparalleled parallel processing capabilities. This evolution spawned giants in the pre-trained model world, such as BERT [27] and GPT [28], which have exhibited outstanding performance across numerous Natural Language Processing (NLP) tasks, ushering in the era of pre-trained models. Notably, the rise of large language models (LLMs) such as ChatGPT (https://chat.openai.com/) further propelled the popularity of such models, spurring extensive research and discussions [29].
However, the story does not end here. Attention-based models have displayed promising potential in the realms of biomedicine and drug discovery [21, 30]. These models can precisely capture intricate interactions between drug molecules, achieving commendable results in drug binding site prediction, toxicity forecasting and drug interaction forecasting [31–35]. Their applications in protein structure and gene sequence studies have also carved a new chapter for precision medicine and personalized treatments [36–42]. In this section, we delve into the evolution of the attention mechanism, its various derived models and architectures, and underscore their superiorities and application scenarios in drug discovery.
The attention mechanism
The inception of the attention mechanism dates back to its application in machine translation by Bahdanau et al. in 2014 [25]. During sequence data processing, the model dynamically allocates attention to various sequence positions, effectively capturing the significance of different parts. This mechanism mirrors human attention focus, allowing it to hone in on different sections of the input sequence. In short, the essence of attention lies in assigning distinct weights to different input data, allowing the model to ‘focus’ on pivotal information.
In the attention mechanism, sequence or word vectors map to Query (Q), Key (K) and Value (V) vectors (Figure 2A). Attention scores are then computed to measure the relevance between Q and K, typically using methods such as dot product, scaled dot product, additive, general, concat, etc. This mechanism allows the model to adjust its attention based on different parts of the input sequence, improving its data representation and retrieval capacities. Using the self-attention mechanism as an example, Q, K and V can be derived from word vectors multiplied by three distinct weight matrices wq, wk and wv. For a specific word vector, the computation of similarity between Q and K in Scaled Dot-Product attention is
Figure 2.
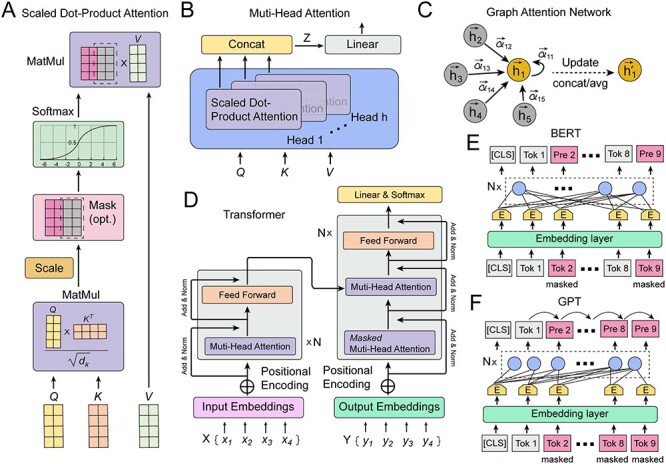
Illustrations of attention mechanism and derived models. (A) Illustration of attention mechanism. (B) Illustration of multi-head attention. (C) Illustration of GAT. (D) Illustration of transformer. (E) Illustration of Bert. (F) Illustration of GPT.
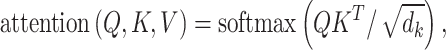 |
(1) |
where QKT denotes the correlation between different input word vectors, and dk represents the dimension of the input vector.
As the effectiveness of the attention mechanism in processing sequence data became evident, researchers sought to further refine and optimize it. This led to the introduction of multi-head attention, centered on considering multiple subspace representations of data (Figure 2B). Multi-head attention does not just apply the attention operation once. Instead, it concurrently performs this operation across multiple different representation spaces or ‘heads’. Each ‘head’ has its unique weights, implying they might focus on different parts of the input data. Outputs from all heads are computed in parallel and then consolidated into a unified output
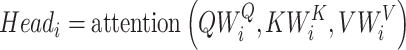 |
(2) |
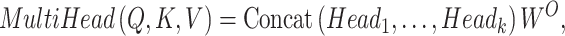 |
(3) |
WO is an additional weight matrix.
Attention operates by dynamically weighing input information in the model. This capability allows the model to emphasize certain relevant or crucial sections during data processing. In drug discovery, drug molecules often exhibit intricate structure–activity relationships, where certain sections are more pivotal than others. The attention mechanism enables the model to autonomously weigh the importance of these parts, improving prediction accuracy. For instance, predicting DTIs is inherently complex. The attention mechanism lets the model focus on the most crucial parts of the interactions, such as key binding sites or active pockets, offering invaluable insights for activity prediction and drug design [43, 44].
In fields of drug development, researchers typically integrate various attention variants, such as cross attention, substructure attention, feature-wise attention (FA) and hierarchical attention, to precisely represent data. For instance, Qian et al. [45] and Kurata et al. [46] adopted the cross-attention mechanism to encode and integrate features of drug molecules and their target proteins for DTI prediction. Due to its bidirectional interactions between drug and target features, cross attention achieved enhanced feature fusion, notably improving DTI prediction accuracy. Additionally, Ma et al. [47] and Yang et al. [48] employed the substructure attention mechanism to automatically extract drug substructures for DDI prediction. This is because it can capture and allocate unique weights for substructures of varying sizes and shapes, precisely representing key substructures related to DDI. Zhu et al. [49], aiming to bolster molecular property prediction accuracy, introduced a module termed FA. This module, by modeling relationships among feature dimensions, automatically adjusts atomic representations, enhancing the precision of drug molecule representation.
Graph attention networks
As advancements in bioinformatics and chemoinformatics unfold, increasing attention is being paid to complex structured data, such as molecular chemical structures and biological molecular networks such as protein–protein interactions (PPIs). These structures are often represented as graphs where nodes symbolize entities such as atoms or proteins, and edges signify chemical bonds or interactions between them. Graph Neural Networks (GNNs) have shown significant efficacy for these non-Euclidean data by learning the representation of nodes, edges or the entire graph through information propagation and aggregation [50–52]. Traditional GNNs, such as Graph Convolutional Networks (GCNs), often assume that the contribution from all neighboring nodes is uniform, or based on predefined weights, potentially overlooking intricate inter-node interactions and dependencies. To address this, GATs were introduced [53]. GAT employs attention mechanisms to provide different attention weights to different neighbors. The importance of each neighboring node may vary depending on the task or the specific context of the central node. This is particularly relevant in drug discovery, where data often naturally appear as graphs [54]. For example, a molecule can be seen as a graph where atoms are nodes, and chemical bonds are edges. GAT can capture intra-molecular interactions more precisely, such as covalent and hydrogen bonds, resulting in a more comprehensive and accurate molecular representation [55].
GATs are implemented by stacking simple graph attention layers (Figure 2C). In this setup, the attention score ei,j reflects the similarity between two nodes and is calculated as
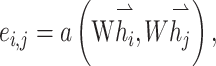 |
(4) |
where hi and hj are the vector representations of nodes i and j, W is the corresponding weight parameter and a is a function that computes the similarity between two nodes, which could theoretically be any differentiable function. These attention scores are then normalized to obtain attention coefficients 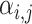 that represent each neighbor’s weighted contribution
that represent each neighbor’s weighted contribution
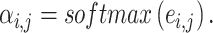 |
(5) |
The final output feature for each node is calculated as a linear combination of the features of its neighbors, weighted by the attention coefficients
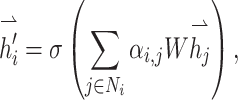 |
(6) |
where Ni denotes the neighbors of the node, and σ is a sigmoid function. For multi-head attention mechanisms extracting different node features, features from different ‘heads’ can be concatenated
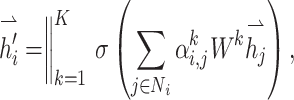 |
(7) |
where || represents vector concatenation.
In the drug discovery domain, GATs are one of the most commonly used deep models. GATs can allocate weights based on each node’s context, considering both local substructures and global information for a more accurate molecular representation. Furthermore, GATs offer an intuitive way to understand why a particular prediction was made, enabling scientists to easily identify the most critical atoms or molecular fragments for a given prediction—an invaluable feature for compound design and optimization [56].
Current research extensively employs GAT-based models and their variants to represent molecular structures and biological molecular networks in various drug discovery studies, such as DTI, drug response (DR), molecular property prediction and synergistic drug combinations. For instance, Xiong et al. [54] built a compound molecular representation method, Attentive FP, based on GAT, displaying excellent performance in multiple molecular property prediction tasks. Liu et al. [57] proposed an attention-wise graph masking strategy, utilizing GAT as a molecular graph encoder and the learned attention weights as masking guides to generate enhanced molecular graphs for property prediction. Wang et al. [58] modeled relationships between drug combinations and cell lines as heterogeneous graphs, then used a heterogeneous GAT to encode these relationships for predicting synergistic drug combinations. Jiang et al. [59] decomposed drug molecules into multiple fragments containing pharmacophores, retaining inter-fragment reaction information to construct heterogeneous molecular graphs, then used a heterogeneous GAT for molecular feature representation. Moreover, numerous methods in the field integrate various other GNN models with attention mechanisms, such as combining GCN with supervised Attention for drug–target affinity (DTA) prediction [60], or integrating self-attention with GNNs to extract 1D substructure sequence information and 2D chemical structure graphs for adverse drug reaction (ADR) prediction [61].
Transformer
The Transformer model has emerged as a significant innovation in the deep learning domain in recent years. Unique in its design, it exclusively relies on attention mechanisms, sidestepping traditional Recurrent Neural Networks (RNNs) or Convolutional Neural Networks (CNNs). The core principle of the Transformer is its capacity to allow data at any position to interact directly, effectively capturing long-distance dependencies. Since its inception, the architecture of Transformer has been rapidly adopted across various NLP tasks and has gradually penetrated fields such as life sciences and drug discovery [62, 63].
At its core, the Transformer model boasts a classic encoder–decoder structure (Figure 2D). Within the encoder, the Feed Forward Network (FFN) essentially consists of two fully connected layers, represented by the following formula:
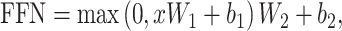 |
(8) |
where x is the input vector, and W1, W2, b1 and b2 are the respective weight matrices and biases for the two layers. Furthermore, the encoder connects add and layer normalization after both the attention layer and the FFN. The addition, or residual network, adds the pre-input and post-input vectors together, efficiently counteracting the vanishing gradient issue. Layer normalization, on the other hand, accelerates model convergence, as shown in the following formula:
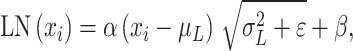 |
(9) |
where α and β are learnable parameters, while σ and μ represent the standard deviation and mean of the input word vectors, respectively. Compared with the encoder, the decoder adds an encoder–decoder attention layer before the self-attention layer, offering context for generative tasks such as sequence or molecule generation.
When representing drug molecules, the Transformer’s self-attention mechanism captures potentially long-distance interactions between atoms, especially in larger molecules or proteins. This precision offers invaluable insights for drug design [64, 65]. Compared with RNNs, Transformers boast parallelism, processing vast datasets rapidly—a critical attribute for large-scale chemical library screening. They can also concurrently learn various drug-related tasks, such as hydrophilicity, lipophilicity and bioactivity prediction, enhancing model generalization and accuracy. Moreover, the Transformer can be trained as a generative model for de novo drug design, learning chemical space distributions and generating promising new molecules [64, 66]. Its superior interpretability, through attention weights, allows researchers to more transparently understand the decision-making process, identifying key molecular structures or groups [67].
Currently, the Transformer finds myriad applications in various domains of small molecule drugs. For example, Lin et al. [68] employed the Transformer encoder to merge different drug combination features, predicting DDI. Schwarz et al. [69] utilized a Siamese transformer model to represent and integrate various drug molecules for DDI prediction. Jiang et al. [70] harnessed the Transformer to represent drug molecular features combined with transcriptomic data to predict cancer drug reactions. Wang et al. [71] used the Transformer encoder to represent protein–ligand interaction features at character and fragment levels, predicting the affinity between proteins and ligands. As a generative model, the Transformer not only excels in data representation but also in molecular generation. Liu et al. [72] constructed a molecular structure generator, DrugEx v3, based on the graph transformer model. Harnessing the Transformer’s prowess with sequence tasks, Kim et al. [73] learned the molecular structure and properties information in the latent space of compound SMILES Language. Using a decoder, they sampled molecular information from this latent space and given conditions, generating novel molecules with desired attributes. Mao et al. [74] designed TransAntivirus based on the Transformer model, facilitating effective and enhanced sampling of the molecular chemical space for antiviral drug virtual screening and molecular design.
Bidirectional encoder representations from transformers
BERT is a pre-trained model based on the Transformer architecture that has achieved remarkable success in the realm of NLP (Figure 2E). BERT’s distinguishing feature is its bidirectional contextual modeling, complemented by a pre-training stage. During its training, BERT uses a Masked Language Model approach, where a portion of the input data is randomly masked, and the model aims to predict these occluded sections. This deep and bidirectional representation opens up substantial opportunities for applications in drug discovery.
Firstly, BERT’s bidirectional nature enables it to capture nuanced information within molecular structures, including interactions among atoms and the molecule’s overall configuration. This capability offers a highly detailed representation for complex drug molecules [75–77]. Furthermore, BERT employs a pre-train/fine-tune paradigm, allowing it to be initially trained on large datasets and later fine-tuned for specific tasks. In drug discovery, this means leveraging existing large-scale biomolecular data for pre-training and subsequently fine-tuning the model for specialized tasks. Although deep learning models are often perceived as ‘black boxes’, BERT’s attention mechanism offers a window into understanding the model’s decisions. For instance, one can identify which atoms and bonds the model focuses on when processing drug molecules. Owing to BERT’s ability to capture the sequential relationships within molecules, the resulting molecular representations are stable and consistent, thereby enhancing the accuracy of downstream tasks such as activity prediction and drug optimization [78, 79].
Currently, in drug discovery, BERT models are primarily employed for molecular representation, forming the backbone of pre-trained models that are applied to various downstream prediction tasks. For example, Wu et al. [80] developed a knowledge-based BERT model involving three pre-training tasks: atomic feature prediction, molecular feature prediction and contrastive learning. This significantly enhanced BERT’s capability in molecular feature extraction, showing promising potential across multiple tasks, including molecular generation and property prediction. Zhang et al. [81] employed BERT to represent 1D, 2D and 3D information of compound molecules for molecular property prediction. Chithrananda et al. [82], in 2020, trained ChemBERT, a large-scale self-supervised model based on BERT, on a dataset comprising over 77 million SMILES, focusing on molecular property prediction. The team subsequently released an updated version of ChemBERT in 2022, further augmenting the model’s performance across a variety of downstream tasks [83].
Generative pre-trained transformer
GPT, developed by OpenAI, is a model grounded in the Transformer architecture, designed for pre-training on vast amounts of unlabeled data and subsequent fine-tuning across various tasks. The GPT model fully adopts the decoder structure of the Transformer and is pre-trained upon it. GPT’s pre-training task revolves around a left-to-right language modeling task, where the model predicts the subsequent word or token, and this structure involves multiple stacked Transformer decoders (Figure 2F).
GPT possesses distinct advantages for de novo drug design [30]. Firstly, as a generative model, GPT is especially adept at molecular generation tasks. It can learn and discern latent patterns within molecular representations and generate novel molecules potentially possessing desired characteristics [84]. Furthermore, similar to BERT, GPT’s pre-train/fine-tune paradigm allows it to initially be trained on expansive molecular libraries and then tailored for specific generation tasks, aiding the model in better understanding and producing molecules with the desired properties. Moreover, researchers can adapt the structure and size of GPT according to specific drug discovery tasks, such as designing custom versions of GPT for particular activity predictions or drug optimization.
In early 2023, the debut of ChatGPT garnered widespread global attention, particularly in the field of drug development, where it quickly became a focal point among researchers. Industry experts generally agree that ChatGPT’s value in drug discovery primarily lies in assisting researchers with in-depth literature searches, efficient data analysis and the development of innovative hypotheses. This tool can swiftly process and analyze large volumes of scientific literature, effectively aiding researchers in uncovering new drug targets and mechanisms of action. Additionally, ChatGPT excels in assisting with the design of experimental plans and the optimization of molecular structures of drugs, significantly accelerating the drug development process and greatly enhancing the efficiency and innovation of drug discovery [85, 86].
Currently, in drug discovery, GPT models are primarily applied for de novo design and other molecular generation tasks. For instance, Bagal et al. [87] employed the GPT model to learn SMILES sequences of molecules for drug molecule generation. Wang et al. [88], working from a conditional GPT architecture, studied the SMILES characters of drugs, aiming to generate SMILES strings of drug-like compounds with or without specified targets. Taking a different approach, Wang et al. [89] combined the SMILES information of drug molecules with physicochemical properties and amino acid information of target proteins, leveraging the GPT model to guide molecular generation. Hu et al. [90] utilized GPT-2 to learn from ~1.9 million bioactive molecules and then designed novel molecular inhibitors for the SARS-CoV-2 3C-like protease. Liang et al. [91] developed DrugChat, a prototype system combining GNNs and LLMs, similar to ChatGPT, to enable interactive Q&A and textual descriptions of drug molecular graphs. DrugChat aims to revolutionize interactions with drug molecular graphs, accelerating drug discovery, predicting drug properties and offering suggestions for drug design and optimization.
APPLICATIONS OF ATTENTION-BASED MODELS IN DRUG DISCOVERY
The attention mechanisms, inspired by the human brain’s selective focus, allow AI algorithms to allocate varying degrees of attention to different elements within a dataset, amplifying their significance. This heightened focus has far-reaching implications for drug research, providing solutions to numerous challenges faced by the pharmaceutical industry. As shown in Figure 3, this section will review the applications of attention mechanism in drug research, mainly covering six types of tasks: DDI, DTI, DTA, Molecular Property Prediction, Molecular Generation, DR and Synergistic Drug Combinations.
Figure 3.
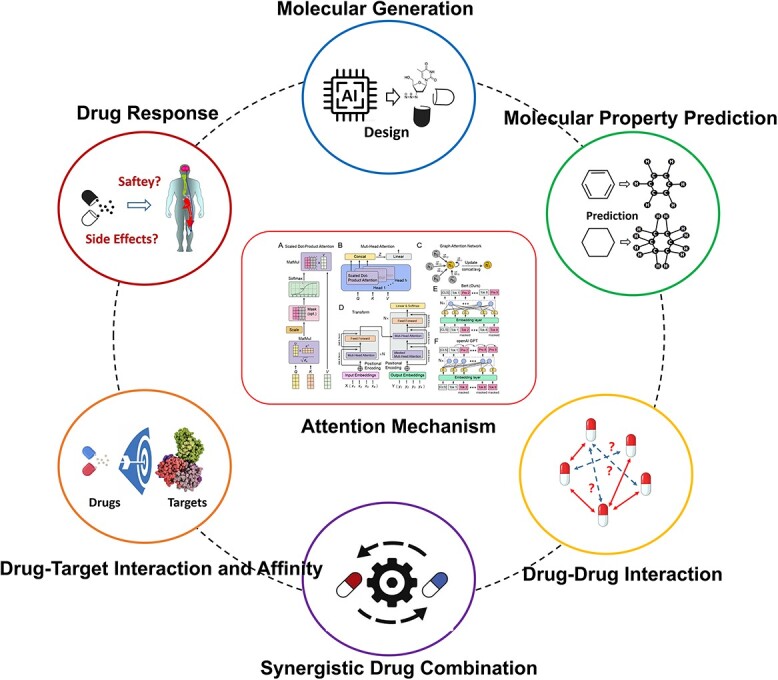
Applications of attention-based models in drug discovery.
DDI prediction
In the complex world of pharmaceutical research and healthcare, understanding and predicting DDIs are crucial for ensuring patient safety and optimizing treatment outcomes. This section explores the role of attention mechanisms in DDIs research.
The role of attention mechanisms in DDIs research
Attention mechanisms have emerged as indispensable tools in DDIs research. They empower models to discern subtle yet significant patterns and connections among drugs, genes and biological pathways, shedding light on potential DDIs. For example, the Att-BLSTM model, enriched with attention-based components, significantly improves DDI detection and classification [92]. By highlighting salient words in biomedical texts related to specific drug pairs, it enhances overall accuracy. Another study by Yu et al. [93] utilized biomedical knowledge graphs (KGs) to detect DDIs, introducing the SumGNN (knowledge summarization GNN) method. SumGNN efficiently identified relevant subgraphs within KGs, improving multi-typed DDI predictions through self-attention-based subgraph summarization. Additional research explored novel approaches such as DDKG framework, using attention mechanisms to enhance DDIs prediction accuracy [94, 95]. These models achieved commendable results across various metrics and datasets. In multi-classification scenarios, the MSEDDI model employed self-attention to fuse features from different channels, demonstrating superior performance in predicting DDIs for new drugs [96]. Other model such as MuFRF integrated drug molecular structure and biomedical KGs for DDIs prediction, excelling in both binary and multi-class tasks [48, 97]. All these models excelled in both binary and multi-class DDIs prediction tasks, underlining the significance of combining molecular structure and semantic information in DDIs research.
Substructure attention mechanisms for enhanced DDIs prediction
Substructure attention mechanisms offer a granular view of chemical substructures, accommodating variations in size and shape commonly observed in molecules. The SA-DDI model, for example, introduces a substructure-aware GNN equipped with substructure attention [48]. This adaptable approach captures evolving substructures and enhances the understanding of complex relationships driving DDIs. Another innovative approach, DGNN-DDI, leverages dual GNNs and substructure attention [47]. These networks collaboratively extract molecular substructure features and determine the significance of various substructure features in predicting drug pair interactions. Its predictive capabilities are validated against real datasets.
Graphing insights with attention
Graph attention mechanisms have ushered in a transformative era in DDIs research, allowing researchers to unveil intricate patterns and relationships within vast biomedical KGs, molecular structures and drug attributes. DGAT-DDI, a directed GAT, addresses the often-neglected asymmetrical roles of drugs in interactions [98]. It adeptly learns drug embeddings for source, target and self-roles, offering insights into how drugs influence and are influenced within DDIs. Concurrently, GNN-DDI employs a multi-layer GAT to derive concise drug feature representations from chemical molecular graphs [98]. This multi-layer approach effectively captures diverse substructure functional groups, enhancing feature representation. Lastly, LaGAT, the link-aware graph attention method for DDIs prediction, generates different attention pathways for drug entities based on different drug pair links. Experimental results on two datasets demonstrate the effectiveness of this model compared with several state-of-the-art works [48].
Transformer models for DDIs prediction
The ability of Transformer to capture intricate long-range dependencies and relationships between drugs, genes and molecular components positions it as a formidable tool for processing large-scale datasets and unraveling complex DDI mechanisms. AttentionDDI, a Siamese self-attention multi-modal neural network, exemplifies this transformation [69]. By integrating various drug similarity measures derived from drug characteristic comparisons, it strikes a balance between predictive accuracy and model explainability. Addressing the need for accurate DDI prediction, the MDF-SA-DDI approach harnesses multi-source drug fusion, multi-source feature fusion and the Transformer self-attention mechanism [68]. AMDE (Attention-Based Multidimensional Feature Encoder), a novel attention-mechanism-based multidimensional feature encoder for DDIs prediction. Specifically, in AMDE, drug features are encoded from multiple dimensions, including information from both Simplified Molecular-Input Line-Entry System sequence and atomic graph of the drug. Experimental results show that AMDE performs better than other classic machine learning and deep learning strategies [94].
In conclusion
In DDIs research, attention mechanisms have brought significant advancements. This section began with a discussion of their fundamental role and then explored their application in substructure and graph analysis. Finally, it highlighted the effectiveness of Transformer models. For a quick reference, please consult Table 1, summarizing the discussed models. These case studies collectively emphasize the pivotal role of attention mechanisms in elevating the accuracy, interpretability and scientific rigor of DDI research outcomes.
Table 1.
The attention-based models in DDIs prediction
| Model name | Attention-based model | Jointly used model or strategy | Reference |
|---|---|---|---|
| Att-BLSTM | Attention | LSTM | [92] |
| DDKG | Attention | Bi-LSTM | [95] |
| MuFRF | Multi-head Attention | CNN/Auto-encoder | [97] |
| SumGNN | Self-attention | GNN | [93] |
| MSEDDI | Self-attention | GNN | [96] |
| SSIM | Substructure Attention | MPNN | [48] |
| DGNN-DDI | Substructure Attention | DMPNN | [47] |
| DGAT-DDI | Directed GAT | [98] | |
| LaGAT | Link-aware GAT | [99] | |
| GNN-DDI | GAT | [100] | |
| AttentionDDI | Transformer | Siamese network | [69] |
| MDF-SA-DDI | Transformer | Siamese network, CNN/AE | [68] |
| AMDE | Transformer/MPAN | [94] |
DTI and DTA prediction: accelerating discovery
Understanding DTI and DTA is fundamental in drug discovery and development. Specifically, DTI primarily investigates whether a drug interacts with its target. DTA focuses on the strength of the interaction between the drug and the target, that is, the affinity. In simple terms, DTI asks ‘Is there an interaction?’ while DTA asks ‘How strong is the interaction?’. Recent advancements in deep learning and attention mechanisms have revolutionized predictive accuracy and interpretability in this domain. This section delves into pivotal roles played by attention mechanisms in deciphering the intricate web of DTIs, DTA and Compound–Protein Interaction (CPI), shedding light on their applications, advantages and implications in drug discovery and precision medicine.
Attention mechanism in DTIs prediction
Several models employ attention mechanisms to enhance feature representation in DTI prediction. For instance, CSConv2d extends DEEPScreen, utilizing 2D structural representations of compounds with Convolutional Block attention Modules for effective interaction prediction [101]. MHSADTI leverages GATs and multi-head self-attention to extract distinctive features from drugs and proteins, significantly improving accuracy and interpretability [22]. Furthermore, the HyperAttentionDTI innovatively uses 1D-CNN layers to learn feature matrices from SMILES strings of drugs and amino acid sequences of proteins, enhancing feature representation and DTI prediction performance [102].
On the other hand, graph-based models offer unique advantages for DTI prediction, and several related methods have also been proposed in recent years. Among these graph-based strategies, GVDTI integrates various pairwise representations, utilizing a graph convolutional autoencoder (AE) to predict drug–protein interactions effectively [103]. Meanwhile HGDTI introduces a heterogeneous GNN, aggregating data from diverse sources to significantly enhance DTI prediction accuracy [104]. GCDTI, incorporating attention mechanisms in a GNN, captures multi-type neighbor topologies for drug and protein nodes, demonstrating superior performance over existing methods [105]. Additionally, AttentionSiteDTI, a graph-based model, incorporates structural features of small molecules and protein binding sites, effectively identifying regions likely to bind drugs for superior prediction performance [106]. Another notable method, DTiGNN, introduces a multifaceted approach, one that considers multiple perspectives in feature learning. This model’s ability to identify previously unknown drug–target pairs is a testament to the power of comprehensive feature exploration [107].
Incorporating multimodal information can also enrich DTI prediction. MCL-DTI integrates multimodal information related to drugs, capturing their characteristics from various perspectives through semantic learning and bidirectional cross-attention [45]. ICAN focuses on attention mechanisms, specifically cross-attention, considering sub-sequence interactions between drugs and proteins, demonstrating superior prediction accuracy and paving the way for advancements in DTI prediction [46].
Another noteworthy approach involves a classification workflow. A recent novel machine learning-based multiclass classification workflow categorizes DTIs into active, inactive and intermediate pairs. This involves transforming drug molecules, protein sequences and molecular descriptors into machine-interpretable embeddings, resulting in models that significantly outperform baseline methods [108].
The method of hierarchical attention mechanism is also applied in the prediction of DDIs. Multiview Heterogeneous Information Network Embedding with hierarchical attention mechanisms (MHADTI) constructs distinct similarity networks for drugs and targets, amalgamating multisource information and incorporating the known DTI network [109]. Hierarchical attention mechanisms are employed, enhancing the quality of drug and target embeddings.
Leveraging GAT in DTIs prediction
In the quest to enhance DTIs prediction, graph attention mechanisms have been instrumental in achieving breakthroughs. Here, we delve into various models, each offering a unique perspective on how attention can elevate prediction accuracy. The model DTI-MGNN distinguishes itself by harnessing the power of multi-channel GCNs and graph attention mechanisms. This combination effectively melds structural and semantic features, offering a holistic approach to DTI prediction [110]. Shifting gears, DTI-HETA frames DTI prediction as a link prediction challenge. This model constructs a heterogeneous graph and deploys a graph CNN, enriched with attention mechanisms, to navigate this complex landscape [111]. Taking an end-to-end deep collaborative learning approach, EDC-DTI skillfully integrates various drug-target-related information. At its core is an enhanced GAT, seamlessly incorporating heterogeneous data to enhance DTI prediction [112]. Lastly, IMCHGAN introduces a two-level neural attention mechanism, leveraging latent features from a heterogeneous network. This innovative approach demonstrates its prowess in predicting DTIs, highlighting the potential of dual-level attention [113].
Advancing DTIs prediction with transformer models
Transformer-based models have emerged as game-changers in the prediction of DTIs. These models offer innovative approaches to overcome traditional limitations. MolTrans leads the way by addressing existing DTI prediction challenges [114]. It employs a knowledge-inspired sub-structural pattern mining algorithm and an interaction modeling module to significantly enhance prediction accuracy and interpretability. Another model, FastDTI takes on the complexities of computational load and multimodal representation in DTI prediction with its unique approach [115]. By leveraging advancements from NLP and GNNs, FastDTI unifies diverse drug and protein modalities into an efficient single model. It streamlines sequence and graph representations, reducing computational complexity, and elevates prediction accuracy through the incorporation of drug and protein properties. On the other hand, TransDTI introduces a transformer-based language model framework for multiclass classification and regression of DTIs. Molecular docking and simulation analyses validate TransDTI’s predictions, highlighting its potential in advancing personalized therapy and clinical decision-making. These transformer-based models showcase the potential of end-to-end models in predicting DTIs. By employing self-attention layers, these models effectively capture biological and chemical contexts, providing more accurate predictions compared with traditional methods.
DTA prediction by attention mechanisms
Attention mechanisms significantly refine binding affinity predictions. The model based on attention demonstrates the effectiveness of attention in capturing compound substructure and protein sub-sequence relationships, leading to enhanced prediction accuracy. Several models leverage graph attention mechanisms to enhance DTA predictions. GEFA, for instance, represents drugs as nested atom graphs within larger complex graphs and employs pre-trained protein embeddings for accurate modeling [116]. SAG-DTA addresses the intricacies of drug and protein representations by harnessing self-attention on drug molecular graphs [117]. Another notable model, AttentionDTA, employs attention mechanisms to identify key subsequences within drug and protein sequences during affinity prediction [118]. MultiscaleDTA employs self-attention to boost feature relevance, making accurate predictions based solely on primary sequence data [119]. Additionally, GSATDTA utilizes self-attention to capture compound substructure and protein sub-sequence relationships, exhibiting adaptability across diverse protein 3D structures [120]. GraphATT-DTA takes a unique approach by emphasizing local-to-global interactions through an attention-based framework, efficiently processing raw drug graph data and protein amino acid sequences with the aid of 1D CNNs [121].
In a distinct category, several models employ unique attention strategies for protein-ligand affinity prediction. CAPLA introduces a cross-attention mechanism that enhances protein–ligand binding affinity prediction by capturing mutual interaction features between protein pockets and ligands [122]. Meanwhile, SEGSA_DTA adopts a novel approach by learning feature representations from protein and ligand graph structures using multiple supervised attention blocks, providing valuable insights for structure-based lead optimization [60]. BindingSite-AugmentedDTA improves DTA predictions by identifying probable protein binding sites, enhancing prediction efficiency while maintaining interpretability. Another noteworthy model, NHGNN-DTA offers a dynamic acquisition of feature representations for drugs and proteins, facilitating information interaction at the graph level. Conversely, ArkDTA introduces an explainable deep model for predicting DTIs, featuring NCI-aware attention regularization [123]. By adjusting attention weights to distinguish between active and inactive residues, ArkDTA enhances interpretability while maintaining prowess.
Transformer models in DTA research
In recent DTA research, innovative Transformer-based models have emerged, showing promise in enhancing DTA predictions. DTITR utilizes self-attention and cross-attention layers to represent biological and chemical contexts of proteins and compounds, achieving competitive performance [17]. ELECTRA-DTA employs unsupervised learning to train contextual embedding models for amino acids and compound SMILES strings, showcasing superiority on challenging datasets and potential for drug repurposing [124]. The Multigranularity Protein-Ligand Interaction (MGPLI) model leverages Transformer encoders to capture character and fragment-level features, leading to substantial improvements in prediction performance [71]. These models highlight the potential of Transformer architectures in advancing DTA research.
Attention mechanism in CPI research
In predicting CPIs, early work utilized GNN and CNN, showcasing the potential of low-dimensional neural networks and attention mechanisms for superior performance and clearer visualizations without traditional feature engineering [125]. BACPI, another model, employed GAT and CNN, integrating compound and protein representations to focus on local effective sites and enhance interpretability [126]. SSGraphCPI framework based on attention mechanism improved accuracy by combining GCNN and BiGRU to extract molecular information and local chemical background, further enhanced in SSGraphCPI2 by incorporating protein amino acid sequence data [127]. Lastly, the Perceiver CPI network employed cross-attention mechanisms and extended-connectivity fingerprints, exhibiting commendable performance and advancing CPI prediction through attention mechanisms [128]. These studies collectively highlight the potential of attention-based approaches in CPI prediction and their impact on bioinformatics.
Several studies have explored the application of Transformer and BERT models in CPIs prediction. One research endeavor introduces DISAE, a deep learning framework that leverages evolutionary insights and self-supervised learning to predict chemical binding onto poorly annotated proteins [129]. The CAT-CPI model combines CNN and transformer encoders to enhance molecular image learning and protein sequence representation [130]. CAT-CPI utilizes Feature Relearning to capture interaction features, achieving optimal outcomes and extending its application to DDI tasks. Additionally, the MDL-CPI approach employs a hybrid architecture integrating BERT, CNN and GNNs for protein and compound feature extraction [131]. MDL-CPI’s unified feature space demonstrates superior predictive performance, highlighting the value of learned interactive information between compounds and proteins in enhancing accuracy. These studies collectively showcase the potential of Transformer and BERT models in advancing CPI prediction and exploring chemical landscapes across sequenced genomes.
In conclusion
The section on attention mechanisms in DTI, DTA and CPI research provides a comprehensive exploration of cutting edge techniques. It begins by introducing the transformative potential of attention mechanisms within deep learning frameworks, especially emphasizing the role of the Transformer model. Several case studies illustrate how attention mechanisms revolutionize predictive accuracy and model interpretability in these critical domains (Table 2). For instance, in DTI prediction, the section discusses how attention-based approaches enhance feature representation, facilitating more accurate predictions. Similarly, in DTA prediction, the utilization of attention mechanisms greatly refines binding affinity predictions, especially when combined with transformer-based models. Furthermore, attention mechanisms in CPI research extract intricate relationships and improve information extraction between compounds and proteins. Overall, this section underscores the transformative impact of attention mechanisms and the Transformer model in advancing predictive capabilities and understanding complex interactions in DTI, DTA and CPI research.
Table 2.
The attention-based models in DDI, DTA and CPI research
| Tool | Attention-based model | Jointly used model or strategy | Task | Reference |
|---|---|---|---|---|
| HyperAttentionDTI | Attention | CNN | DTI | [102] |
| GVDTI | Attention | VAE, GCN | DTI | [103] |
| HGDTI | Attention | Bi-LSTM | DTI | [104] |
| / | Attention | GCNN/Bi-LSTM | DTI | [108] |
| GCDTI | Attention | GNN/CNN | DTI | [105] |
| AttentionSiteDTI | Attention/GAT | GNN | DTI | [106] |
| GCHN-DTI | Attention | GCN | DTI | [132] |
| CSConv2d | Channel Attention/Spatial Attention | CNN | DTI | [101] |
| MHSADTI | Multi-Head Self-Attention/GAT | DTI | [22] | |
| MHADTI | Hierarchical Attention | Multi-view Learning | DTI | [109] |
| MCL-DTI | Cross-attention/Multi-head Self-attention | DTI | [45] | |
| ICAN | Cross-attention | CNN | DTI | [46] |
| DTI-MGNN | GAT | GCN | DTI | [110] |
| DTI-HETA | GAT | GCN | DTI | [111] |
| EDC-DTI | GAT | RWR | DTI | [112] |
| DTiGNN | attention | GNN/CNN | DTI | [107] |
| IMCHGAN | Heterogeneous GAT | DTI | [113] | |
| MolTrans | Augmented Transformer | DTI | [114] | |
| TransDTI | Transformer | DTI | [115] | |
| FastDTI | Transformer | Multimodality | DTI | [133] |
| GEFA | Self-attention | GCN | DTA | [116] |
| MultiscaleDTA | Self-attention | CNN | DTA | [119] |
| GSATDTA | Graph–sequence Attention/Transformer | BiGRU/GNN | DTA | [120] |
| GraphATT-DTA | Attention | CNN/GNN | DTA | [121] |
| AttentionDTA | Attention | CNN | DTA | [118] |
| BindingSite-AugmentedDTA | Self-attention/GAT | DTA | [134] | |
| NHGNN-DTA | Multi-head Self-attention | BiLSTM | DTA | [135] |
| ArkDTA | Cross-attention | DTA | [123] | |
| CAPLA | Cross-attention | DTA | [122] | |
| SEGSA_DTA | Supervised Attention | GCN | DTA | [60] |
| SAG-DTA | Self-attention | GCN | DTA | [117] |
| DTITR | Transformer/Cross-attention | DTA | [17] | |
| ELECTRA-DTA | Transformer | DTA | [124] | |
| MGPLI | Transformer | CNN/Highway Network | DTA | [71] |
| CPI prediction | Attention | GNN/CNN | CPI | [125] |
| SSGraphCPI | Attention | GNN/CNN | CPI | [127] |
| Perceiver CPI | Cross-attention/Self-attention | CNN | CPI | [128] |
| BACPI | GAT | CNN | CPI | [126] |
| DISAE | Transformer/ALBERT | CPI | [129] | |
| CAT-CPI | Transformer | CNN | CPI | [130] |
| MDL-CPI | BERT | CNN/GNN/AE | CPI | [131] |
Molecular property prediction
In the rapidly evolving landscape of Molecular Property Prediction, attention mechanisms have ushered in a profound paradigm shift, significantly influencing the analysis and prediction of molecular characteristics. Inspired by human cognitive processes, attention mechanisms have surpassed their initial domains of NLP and computer vision, offering a transformative approach to model and comprehend intricate molecular structures. The application of attention mechanisms in Molecular Property Prediction encompasses several key areas, each contributing to the enhancement of predictive accuracy and interpretability, thus promising a future of enhanced precision in pharmacokinetics and related fields.
Enhancing interpretability through attention mechanisms
The realm of molecular property prediction has seen the emergence of several innovative models that focus on enhancing interpretability and precision. The Self-attention-based Message-Passing Neural Network (SAMPN) stands as a significant milestone, dynamically assigning importance levels to substructures during the learning phase, effectively revolutionizing property predictions [136]. Moreover, models such as attention MPNN and Edge Memory NN, introduced by Withnall et al. [137], have proven to be formidable competitors against traditional techniques, elegantly leveraging the molecular graph structure for improved predictions. On the other hand, Substructure-Mask Explanation (SME) offers a distinctive approach, identifying pivotal substructures within molecules, thereby providing deeper insights into the mechanisms influencing property predictions [56]. Addressing the challenges of leveraging unlabeled data, the Cascaded Attention Network and Graph Contrastive Learning (CasANGCL) presents a pre-training and fine-tuning model, substantially enhancing prediction performance in downstream tasks [138]. Lastly, the Hierarchical Informative Graph Neural Network (HiGNN) utilizes co-representation learning from molecular graphs and the chemical synthesis of retrosynthetically interesting chemical substructure (BRICS) fragments, offering powerful deep learning assistance to chemists and pharmacists.
Graph attention mechanisms in molecular property prediction
Graph attention mechanisms have played a pivotal role in advancing precision in the field of molecular property prediction. FraGAT, a fragment-oriented multi-scale graph attention model, excels in capturing diverse views of molecule features, especially emphasizing functional groups that play a crucial role in a molecule’s properties [139]. ATMOL, on the other hand, introduces attention-wise graph masking, significantly enhancing molecular representation and consequently, downstream molecular property prediction tasks [57]. FP-GNN represents a notable stride by effectively combining information from molecular graphs and fingerprints, resulting in precise molecular property prediction [140]. Furthermore, the advent of Multi-Order Graph Attention Network (MoGAT) has significantly enhanced predictions related to water solubility, allowing for a deeper understanding of atom importance [141]. In a distinctive approach, PredPS, an attention-based GNN, demonstrates exceptional utility in binary class prediction of compound plasma stability [142].
BERT model in molecular property prediction
The integration of BERT models into Molecular Property Prediction research has brought a dynamic and efficient approach to encoding molecular structures, signifying a pivotal stride in the field. Among these strategies, Molecular Graph BERT (MG-BERT) masterfully amalgamates local message passing mechanisms from GNNs with the BERT model [81]. This fusion enables self-supervised pretraining on extensive unlabeled molecular data, yielding crucial contextual insights for precise property predictions. K-BERT, another model, distinguishes itself through three distinct pre-training tasks [80]. This differentiation empowers K-BERT to exhibit exceptional performance across a diverse array of 15 pharmaceutical datasets, extending the horizons of molecular property prediction research. MolRoPE-BERT takes a novel approach by utilizing rotary position embeddings instead of absolute position embeddings, broadening the spectrum of molecular representation learning [78]. Fingerprints-BERT (FP-BERT) harnesses self-supervised learning, effectively extracting semantic representations of molecules from SMILES data [143]. The incorporation of 3D parameters in Stereo Molecular Graph BERT (SMG-BERT) enables precise chemical representations for diverse molecules [144]. SMILES-BERT, a semi-supervised model, efficiently generalizes across tasks through pretraining on a large-scale unlabeled dataset using a Masked SMILES Recovery task [145]. These advancements collectively underline the transformative potential of BERT-based models, promising a future of enhanced precision in pharmacokinetics and related fields.
Transformer in molecular property prediction
The infusion of Transformers into molecular property prediction research signals the dawn of a new era, signifying a substantial leap forward. Models such as ABT-MPNN and TranGRU have demonstrated the transformative potential of Transformers in enhancing the understanding of molecular information [146–148]. ABT-MPNN, by seamlessly integrating the self-attention mechanism with MPNNs, refines molecular representation embedding, achieving competitive or superior performance across various datasets in quantitative structure–property relationship tasks. TranGRU, on the other hand, enhances the understanding of both local and global molecular information, positioning itself as a versatile sequence encoder for molecular representation extraction. DHTNN, a novel algorithmic framework, introduces the innovative Beaf activation function and leverages a Transformer with Double-head attention for molecular feature extraction, resulting in a robust approach that ensures model convergence and rational weight assignments [149]. Two strategies, MolHGT and PharmHGT, both of them applied the Heterogeneous Graph Transformer mechanism in molecular property research. MolHGT adeptly accommodates heterogeneous structures, capturing distinct node and edge types, thus providing a comprehensive view of molecular property prediction, while PharmHGT excels in capturing diverse views of heterogeneous molecular graph features, consistently outperforming advanced baselines on benchmark datasets [59]. The introduction of these two HGT-based methods underscores the immense potential of harnessing Transformer technology to advance molecular property prediction. Lastly, GROVER harnessing well-designed self-supervision and highly expressive pre-trained models to achieve significant performance enhancements across challenging benchmarks [150].
In conclusion
In conclusion, these diverse approaches collectively underscore the remarkable potential of attention mechanisms and Transformers in advancing Molecular Property Prediction research. They not only enhance predictive accuracy but also elevate interpretability, promising a future of enhanced precision in pharmacokinetics and related fields (Table 3). With a thorough understanding of molecular structures and properties, these advancements significantly contribute to drug discovery and related scientific domains, setting the stage for a new frontier in pharmacology and molecular research.
Table 3.
The attention-based models in molecular property prediction research
| Tool | Attention-based model | Jointly used model or strategy | Reference |
|---|---|---|---|
| SAMPN | self-attention | MPNN | [136] |
| / | Attention | MPNN | [137] |
| MV-GNN | Attention | GCN/Multi-view Learning | [151] |
| SME | Attention | GCN | [56] |
| EAGCN | Edge Attention | GCN | [152] |
| CasANGCL | Cascaded Attention | Pre-training/Graph Contrastive Learning | [138] |
| HiGNN | Feature-Wise Attention | GNN | [49] |
| MG-BERT | BERT | GNN | [81] |
| K-BERT | BERT | Pre-training/Contrastive Learning/Fine-tuning | [80] |
| MolRoPE-BERT | BERT | Pre-training/Fine-tuning | [78] |
| FP-BERT | BERT | CNN | [143] |
| SMG-BERT | BERT | [144] | |
| ChemBERTa | BERT | Pre-training | [82] |
| ChemBERTa-2 | BERT | Pre-training | [83] |
| SMILES-BERT | BERT | Pre-training | [145] |
| FraGAT | GAT | [139] | |
| ATMOL | GAT | Graph Contrastive Learning | [57] |
| FP-GNN | GAT | [140] | |
| MoGAT | GAT | [141] | |
| PredPS | GAT | [142] | |
| ExGCN | GAT | GCN | [153] |
| ABT-MPNN | Transformer | MPNN | [147] |
| TranGRU | Transformer | BiGRU | [148] |
| DHTNN | Transformer | [149] | |
| MolHGT | Heterogeneous Graph Transformer | [146] | |
| PharmHGT | [59] | ||
| GROVER | GNN Transformer | Pre-training | [150] |
Molecular generation
In the rapidly evolving field of De Novo Drug Design, attention mechanisms have emerged as a transformative innovation, reshaping how we approach molecular generation. Attention mechanisms now provide a sophisticated means to capture intricate relationships between molecular components. These mechanisms empower deep learning models to selectively focus on specific elements within complex molecular structures, enhancing precision, interpretability and the potential to accelerate drug discovery.
Attention mechanism: Illuminating molecular relationships
By selectively focusing on specific elements within complex molecular structures, the attention mechanisms decodes the complexities of molecular structures and properties, paving the way for accelerated drug discovery and the tailored design of molecules with precise attributes. For example, a model named CProMG has been introduced to address the challenge of designing molecules with both high binding affinities and desired physicochemical properties [154]. By combining hierarchical protein perspectives and jointly embedding molecule sequences with their drug-like characteristics, CProMG has the ability to autonomously generate custom-designed molecules with superior binding affinity and drug-like properties. This model relies on the attention mechanism, which integrates fine-grained atomic views with coarse-grained amino acid views to provide a more accurate representation of 3D protein structures (pockets). Additionally, it employs multi-head attention modules to compute the proximity between molecular tokens and protein residues and atoms, thereby capturing crucial interactions between protein pockets and molecules.
GPT model in molecular generation
The GPTmodel has emerged as a powerful player in the field of Molecular Generation. Originally designed for NLP, GPT has been adeptly adapted to tackle complex molecular design challenges. Operating as a language model, GPT predicts the next token or element in a sequence based on learned patterns and relationships, enabling the precise generation of novel molecules. PETrans, is a deep learning approach tailored for generating ligands specific to particular targets [89]. It extracts contextual features of molecules using GPT and employs transfer learning to fine-tune the model for generating molecules with superior binding affinity to target proteins. Similarly, cMolGPT, a conditional Transformer architecture, auto-regressively produces target-specific compounds through fine-tuning on target-specific datasets [112]. Additionally, MolGPT, inspired by the success of GPT models in text generation, performs equivalently in generating valid, unique and novel molecules [87]. Its conditional training capabilities allow control over multiple molecular properties and scaffold generation.
Among other studies using GPT model in De Novo Drug Design, there are two other studies that deserve attention. An efficient pipeline has been proposed to generate novel SARS-CoV-2 3C-like protease inhibitors, leveraging the GPT2 generator and precise multi-task predictors [90]. This approach yields numerous novel compounds, enhancing the chemical space for generation and providing valuable insights for potential therapeutic agents. Another novel model for de novo drug design utilizing the GPT architecture and relative attention has been introduced by Haroon et al. [30]. This model offers enhanced validity, uniqueness and novelty, emphasizing the potential of relative attention and transfer learning within the GPT framework for improved de novo drug design.
Transformer-based approaches for molecular design
Owing to the exceptional capacity to capture intricate relationships between molecular components and exploit the potential of attention mechanisms, Transformers have seamlessly transitioned to the generation of molecular structures. At the beginning, one innovative approach reframes molecular design as a translation task [21]. Using transformer-based models, it translates protein amino acid sequences into molecular structures. These models not only generate valid molecular structures but also predict their affinity for target proteins, aligning with physicochemical characteristics, drug-likeness and synthetic accessibility metrics. They demonstrate the remarkable capability to craft molecules tailored to specific targets. Later, models, such as TransVAE [155], leverage attention mechanisms to explore intricate substructural representations of molecular features. Another noteworthy method named Generative Chemical Transformer (GCT) merges transformer models’ language recognition capabilities with the conditional generative power of variational models [73]. Proficient in understanding chemical semantics and adhering to grammar rules, GCT satisfies multiple preconditions simultaneously, offering profound insights into molecular design within the transformer framework. In another recent study, researches proposed a novel approach named the multi-constraint molecular generation (MCMG). This model can satisfy multiple constraints by combining conditional transformer and reinforcement learning algorithms through knowledge distillation [156].
Some other strategies have also been introduced in the molecular generation with transformer model. One innovative study introduces strategies for warm-starting molecule generation models, aiming to accelerate the process [157]. Conditional Randomized Transformers effectively explore drug-like chemical space, expanding the boundaries of molecular design and offering fresh insights into drug discovery [158]. Incorporating the transformer model, another framework Motif2Mol connections between amino acid sequences and molecular structures [159]. It minimizes irrelevant sequence noise, directly assessing the model’s capability to generate active compounds. Recently, an update to DrugEx model (verson 3) introduces scaffold-based drug design, enabling the design of molecules based on scaffolds comprising multiple fragments as input [72]. This approach employs a novel positional encoding scheme, allowing multiple fragments within a scaffold to grow simultaneously and connect, ensuring the validity of generated molecules. Another noteworthy approach Taiga combines transformers and reinforcement learning for molecule graph generation [160]. This integration enables the generation of molecules with specific properties by fine-tuning a continuous vector space.
The researchers have also developed a series of tools based on the transformer mechanism for molecular design. In recent 3 years, COVID-19 has become a major threat to human health. TransAntivirus, a transformer-based model, explores designing antiviral lead compounds by translating IUPAC names into SMILES strings for molecular optimization, with potential implications for addressing challenges such as SARS-CoV-2 [74]. The BERT model has also been applied, and a notable tool using adaptive training strategy has also been proposed to explore the adaptation of language models to enhance molecule generation for optimization tasks [161]. In summary, these transformer-based approaches collectively redefine molecular design, precision medicine and drug development. Their ability to comprehend, manipulate and generate molecular representations positions them as driving forces behind innovation in these pivotal domains.
In conclusion
Attention mechanisms have emerged as valuable tools, revolutionizing the design and synthesis of molecules. This section illuminated their pivotal role in molecular generation, showcased the versatility of GPT models and delved into the myriad applications of transformers. For a concise overview, please refer to Table 4, summarizing the featured models. Collectively, these case studies underscore the paramount significance of attention mechanisms in reshaping molecular generation research, offering innovative and effective avenues for designing molecules tailored to precise properties and functions.
Table 4.
The attention-based models in molecular generation research
| Tool | Attention-based model | Jointly used model or strategy | Reference |
|---|---|---|---|
| / | GEFA | GGNN/Reinforcement learning | [162] |
| CProMG | Multi-head Attention/Cross-attention | [154] | |
| PETrans | GPT | Transfer Learning | [89] |
| cMolGPT | GPT | Pre-Training | [88] |
| MolGPT | GPT | Pre-Training | [87] |
| / | GPT2 | Pre-Training | [90] |
| / | Relative Attention/GPT | Transfer Learning | [30] |
| / | Transformer | [21] | |
| GCT | Transformer | [73] | |
| / | Transformer | VAE | [155] |
| Motif2Mol | Transformer | [159] | |
| Taiga | Transformer | Reinforcement learning | [160] |
| TransAntivirus | Transformer | [74] | |
| / | ChemBERTa/Protein RoBERTa | Pre-Training | [157] |
| CRTmaccs | Conditional Randomized Transformer | [158] | |
| AlphaDrug | Lmser Transformer | [163] | |
| DrugEx v3 | Graph Transformer | Reinforcement learning | [72] |
| cTransformer | Conditional Transformer | [164] | |
| / | BERT | [161] |
DR and ADRs
Predicting how individuals will respond to specific drugs and anticipating adverse reactions is paramount for personalized medicine. Attention mechanisms enable the precise modeling of patient-specific responses by capturing intricate relationships between molecular features and DRs. This section delves into how attention mechanisms are reshaping our understanding of drug efficacy and safety, ultimately paving the way for tailored treatments.
Attention in DR
In the context of DR research, attention mechanisms have emerged as valuable tools, significantly enhancing predictive accuracy and interpretability. These mechanisms harness the capabilities of deep learning to decipher intricate interactions among drugs, molecular structures and biological data. By selectively focusing on specific elements within these complex datasets, attention mechanisms facilitate the identification of crucial features for predicting DRs and adverse reactions. Moreover, they shed light on the underlying biological mechanisms driving DRs, thus facilitating the discovery of novel therapeutic interventions and improving patient outcomes. For instance, an attention-based multimodal neural approach has been introduced to enhance the interpretability of drug sensitivity prediction [165]. This approach integrates multiple data modalities, including SMILES string encoding of drug compounds, transcriptomics data from cancer cells and intracellular interactions integrated into a PPI network. A comprehensive comparative study demonstrates the superiority of using raw SMILES strings, especially the newly proposed MCA architecture, for predictive performance. The efficacy of the drug attention mechanism is validated through its strong correlation with established structural similarity measures. A gene attention mechanism focusing on informative genes for IC50 prediction is also introduced, enhancing model explainability. Based on this model, a webserver called PaccMann has also been developed [166]. This gene-based approach offers computational tractability and suggests potential extensions involving pathway scores and associated pathway attention mechanisms to further enhance tumor cell representation.
Transformers in DR prediction
Integration of Transformers into DR research holds the promise of improving drug discovery and development while also providing insights into the intricate mechanisms governing drug efficacy and safety, ultimately advancing the field of personalized medicine. Several innovative models have been introduced to enhance drug sensitivity prediction and improve our understanding of patient-specific responses to clinical treatments. For example, DeepTTA incorporates a transformer architecture with self-attention modules to capture essential drug characteristics from compound substructures [70]. In recent studies, innovative neural network architectures such as GrapTransDRP and TCR have been introduced to enhance DR prediction on cell lines [167, 168]. GrapTransDRP [167] leverages Graph Transformer with a fusion of GAT-GCN to improve drug representation extraction from molecular graphs. Multi-omics data are also incorporated to enhance predictive capabilities. On the other hand, TCR adopts a transformer network with multi-head atom omics attention to model drug atom/substructure interactions alongside multiomics data. It combines GCN and transformer networks, emphasizing the effectiveness of learning to rank with a cross-sampling strategy. Both models demonstrate superior effectiveness in predicting DRs compared with existing methods, underscoring their potential value in precision medicine. Another strategy named DRPreter stands out as an interpretable drug-response prediction model that merges biological and chemical-domain knowledge with deep learning technologies [67]. By integrating cancer-related pathways and cell line networks, it provides detailed representations and insights into drug mechanisms. To against the COVID-19, a novo model, DeepCoVDR was proposed [169]. This framework employs a transfer learning approach using a graph transformer for predicting COVID-19 DR. DeepCoVDR showcases proficiency in regression and classification tasks, and it demonstrates accuracy through the screening of FDA-approved drugs and drug candidates.
Graph attention mechanisms in ADR detection
Innovative models based on Graph attention Mechanisms such as GCRS, iADRGSE and GCAP have been developed to ADDRESs the challenges of ADR detection [61, 170, 171]. GCRS predicts drug-side effect associations by encoding specific topologies, common topologies and pairwise attributes of drugs and side effects. Iadrgse focuses on early-stage ADR identification in drug discovery by combining a self-attentive module and a graph-network module. GCAP predicts the severity of adverse reactions to drugs, encompassing potential drug–ADR interactions and determining classes of serious clinical outcomes. These models demonstrate superior performance, independence from known drug–ADR interactions and broader predictive capabilities, expanding their potential applications in safeguarding patients and ensuring drug safety. On the other hand, the Graph Machine Learning neural network model MultiGML has also been developed to enhance ADR classification [172]. MultiGML significantly outperforms traditional classifiers in terms of performance, particularly in classifying ADRs into multiple categories. Transformers have also made significant contributions to ADR research. DeepPSE, for instance, leverages deep drug pair representations and a self-attention mechanism to predict polypharmacy side effects [173]. By exploring various drug fusion methods, DeepPSE aims to enhance the prediction accuracy of polypharmacy side effect, ultimately contributing to safer drug development and patient care.
In conclusion
In conclusion, the integration of attention mechanisms and Transformers in both DR and ADR research has led to a paradigm shift, vastly improving predictive accuracy and interpretability. These advancements hold immense promise for the field of personalized medicine, enabling tailored treatments and a deeper understanding of drug efficacy and safety. The ability to model intricate relationships between molecular features and DRs offers the potential for highly precise predictions, early detection of adverse events and enhanced patient safety. For a concise overview of the models discussed, please refer to Table 5. As researchers continue to refine and innovate upon these models, the future of pharmacovigilance and drug development appears increasingly bright.
Table 5.
The attention-based models in DR and ADR research
| Tool | Attention-based model | Jointly used model or strategy | Task | Reference |
|---|---|---|---|---|
| / | Attention | RNN, CNN | DR | [165] |
| PaccMann | Attention | DR | [166] | |
| DeepTTA | Transformer | DR | [70] | |
| DRPreter | Transformer | GNN | DR | [67] |
| GraTransDRP | Graph Transformer | GAT-GCN | DR | [167] |
| DeepCoVDR | Graph Transformer/Cross-attention | DR | [169] | |
| TCR | Transformer | GCN | DR | [168] |
| GCAP | Multi-level Graph Attention | CNN | ADR | [171] |
| GCRS | Attention | GCA | ADR | [170] |
| Iadrgse | Self-attention | GNN | ADR | [61] |
| / | Cross-attention | ADR | [174] | |
| MultiGML | GAT | GCN | ADR | [172] |
| DeepPSE | Transformer | CNN/AE/Siamese network | ADR | [173] |
Synergistic drug combinations
The pursuit of potent drug combinations that surpass the efficacy of individual drugs has historically been resource-intensive in pharmaceutical research. However, the integration of AI, particularly attention mechanisms, has transformed this field by allowing for a precise focus on molecular interactions driving synergistic effects. In this section, we explore the applications, advantages and emerging trends of attention mechanisms in synergistic drug research, offering innovative solutions for addressing complex diseases.
Graphs attention in synergistic drug combinations
The fusion of attention mechanisms with GNNs and GCNs has proven to be a potent approach in synergistic drug combination research. This synergy enables the capture of intricate relationships within graph-structured drug data, shedding light on drug, gene and molecular interactions. The introduction of attention mechanisms into these networks empowers researchers to prioritize vital nodes and edges within the graph, improving both predictive accuracy and model interpretability. A notable application of this fusion is seen in DeepDDS, a cutting-edge model for forecasting drug combination effects [94]. DeepDDS utilizes graph-based representations of drug chemical structures and employs GCN and attention mechanisms to compute drug embeddings. The model excels in fusing genomic and pharmaceutical data, allowing precise prediction of synergistic drug combinations tailored to specific cancer cell lines. Another anticancer drug research, GraphSynergy, harnesses GCN and attention mechanisms to identify tailored synergistic drug combinations for specific cancer cell lines, promising significant advancements in precision medicine [175].
However, GNNs and GCNs, while proficient in capturing unique features in specific cell lines, often overlook invariant patterns across cell lines. To address this limitation, SDCNet, a novel GCN-based approach, was introduced [176]. SDCNet predicts cell line-specific synergistic drug combinations without relying on cell line data such as gene expressions. An attention mechanism enhances feature fusion across different network layers, significantly improving predictive performance. Another remarkable approach, AttenSyn, is an attention-based deep GNN that automatically learns high-latent features for predicting drug combination synergies [177]. AttenSyn eliminates the need for manual feature engineering and identifies crucial chemical substructures within drugs through its attention-based pooling module, outperforming classical machine-learning techniques and deep-learning methods. Beyond the prominent models discussed, there are two additional noteworthy applications of attention mechanisms with graph strategy in synergistic drug combination research. MGAE-DC combined attention mechanism and graph AE, which focuses on predicting drug combination synergies across various cell lines, treating additive or antagonistic combinations as distinct channels [178]. This approach incorporates cell-line-specific drug embeddings and an attention mechanism, amplifying its predictive power. CGMS, another innovative approach, utilizes a complete graph framework to identify anti-cancer synergistic drug combinations [58]. By integrating a heterogeneous graph attention network and multi-task learning, CGMS eliminates order dependency, enhances whole-graph embeddings and offers interpretability through its attention mechanism.
BERT in synergistic drug combinations
BERT has emerged as a valuable tool in synergistic drug combination research due to its exceptional ability to capture contextual information in textual data. This contextual understanding extends to molecular properties, DDIs and gene expression data, significantly enhancing predictive capabilities and model interpretability. One example of BERT’s impact is evident in the Dual Feature Fusion Network for Drug–Drug Synergy Prediction (DFFNDDS) [179]. It effectively predicts drug combination synergies using drug SMILES representations, hashed atom pair fingerprints and cell line gene expression data. The incorporation of fine-tuned BERT models for drug feature extraction and a double-view feature fusion mechanism consistently outperforms other methods. Furthermore, intensive research efforts focusing on drug combinations have led to computational predictions of drug synergy. An impressive case is DCE-Dforest [180], which utilizes BERT to encode drug information and employs a deep forest approach to model drug-cell line relationships. This model consistently outperforms other methods, contributing significantly to our understanding of drug synergy in cancer therapy.
Transformer in synergistic drug combinations
The Transformer excels in encoding molecular structures, DTIs and genetic information, making it invaluable for learning intricate patterns and dependencies essential for predicting synergistic drug combinations. The TranSynergy model, developed by Liu et al. [181], enhances synergistic drug combination prediction using a self-attention transformer and deep learning. It models cellular effects of drugs through gene dependencies, interactions, and drug-target interactions. The novel SA-GSEA method further aids in identifying critical genes, improving interpretability. Benchmark studies show TranSynergy's good performance over existing methods, highlighting its potential in mechanism-driven machine learning. Following this groundbreaking approach, a refined dual-transformer-based deep neural network, DTSyn, captures intricate biological associations by leveraging GCN for chemical features and self-attention for extracting key interactions [182]. Moreover, DeepTraSynergy, yet another approach, employed deep learning techniques to predict drug combination synergy scores [183]. With the utilization of multimodal inputs and a multitask framework, it achieved superior performance compared with classical and state-of-the-art models. In addition to the models mentioned above, EGTSyn [184], an Edge-based Graph Transformer stands out for its effectiveness in capturing global structural information and critical chemical bond details using specialized GNNs. Its strong generalization capabilities make it a valuable tool for predicting synergistic drug combinations.
In conclusion
In conclusion, the integration of advanced technologies and deep learning models has revolutionized Synergistic Drug Combination research. Notably, the Transformer and BERT, with their contextual understanding, empower the comprehension of intricate drug interactions. The fusion of attention mechanisms with GNNs and GCNs has emerged as a potent strategy, facilitating the precise prediction of drug combination effects. Models such as DeepDDS and GraphSynergy excel in this domain, advancing precision medicine. SDCNet addresses challenges in capturing patterns across cell lines, emphasizing the significance of attention-enhanced GCNs. Models such as AttenSyn and SANEpool enhance interpretability, contributing to a deeper understanding of complex molecular networks. These advances, summarized in Table 6, highlight the transformative potential of attention mechanisms, contextual encoders and graph-based models. They expedite the identification and optimization of synergistic drug combinations, offering new avenues for enhanced therapeutic strategies in complex diseases.
Table 6.
The attention-based models in synergistic drug combination research
| Tool | Attention-based model | Jointly used model or strategy | Reference |
|---|---|---|---|
| DeepDDS | GAT | GCN | [185] |
| GraphSynergy | Attention | GCN | [175] |
| SDCNet | Attention | GCN | [176] |
| SANEpool | Attention | GNN | [186] |
| AttenSyn | Attention | LSTM/GCN | [177] |
| MGAE-DC | Attention | Graph AE | [178] |
| CGMS | Heterogeneous GAT | [58] | |
| DCE-Dforest | BERT | Deep forest | [180] |
| DFFNDDS | BERT/Multi-Head Attention | Highway network | [179] |
| TranSynergy | Transformer | [181] | |
| DTSyn | Transformer | GCN | [182] |
| DeepTraSynergy | Transformer | Nod2Vec | [183] |
| EGTSyn | Graph Transformer | [184] |
CHALLENGES AND FUTURE DIRECTIONS
Models based on the attention mechanism offer vast opportunities for drug development, but they also present several challenges.
Data quality and availability
The success of drug development hinges on high-quality data [187]. However, appropriate data for training these models are often scarce and face various limitations [188]: Lack of Data for Rare Diseases & Specific Populations: Available data for some rare diseases or specific demographics are minimal, leading to potential under-training and reduced predictive accuracy. Data Diversity: Drug development data come from various experimental methods and conditions, introducing issues such as batch effects and experimental errors, complicating data preprocessing. Annotation Issues: A lot of data may lack accurate labeling, especially concerning vital biological activity or toxicity attributes. Incorrect or incomplete labels impact model training and prediction. Data Imbalance: In some studies, positive samples (e.g. effective drugs) may be significantly outnumbered by negative samples, leading models to be biased toward the majority class and potentially overlooking rare but essential samples [189].
Model interpretability
Model interpretability refers to the explainability and understandability of a model’s predictions. While the attention mechanism has relatively increased the transparency of models, it is especially critical in the field of drug development, and there remain several challenges and needs. Deep learning models, particularly those with multiple layers and a large number of parameters, often have internal mechanisms that can be difficult for non-expert users to comprehend [190, 191]. This complexity might lead researchers and pharmaceutical experts to be skeptical about the model’s predictions. Although the attention mechanism can highlight parts of the data the model focuses on, it is hard to grasp the model’s decision-making logic solely based on weights. For instance, the same weight might have different interpretations in different contexts. Deep learning models often involve a plethora of non-linear operations, making their decision pathways more intricate and hard to trace and explain. Moreover, while we might be able to interpret the model’s decisions for specific inputs, understanding its behavior on a global scale remains a challenge.
Computational constraints
As models such as BERT and GPT grow in scale, the extensive data they require can prolong training times, possibly becoming infeasible for smaller research entities [192]. And massive models demand vast computational and storage resources. Storing model weights, intermediate representations and training data can exhaust significant storage capacities [193]. Meanwhile, despite the inherent parallel computation benefits of attention-based models, optimizing this requires deep familiarity with parallel computing frameworks and algorithms. Moreover, deploying large models on mobile or embedded systems can be impractical due to their size and computational demands, emphasizing the importance of model compression and quantization techniques.
Future directions
Emerging algorithms and technologies suggest the development of more streamlined and efficient models on the horizon. Techniques such as network pruning or knowledge distillation might yield smaller yet powerful models apt for constrained computational environments. Future models are anticipated to integrate various data types, such as molecular structures, clinical trial data and genomic information, leveraging the power of multi-modal fusion. In drug discovery, this approach allows for the efficient combination of insights from diverse data sources. Not only does it maximize the unique perspectives each dataset offers, but it also paves the way for building more comprehensive and accurate prediction models. For instance, merging gene expression data with protein structure information could unveil previously unknown mechanisms of drug–biological interactions. The application of multi-modal fusion effectively mitigates biases and limitations associated with relying on a single data source. Such holistic analysis methods significantly enhance the predictive accuracy for new drug candidates and potential drug targets. In summary, through multi-modal fusion techniques, the drug discovery process is expected to become more in-depth and holistic, accelerating and optimizing the discovery and development of novel therapeutics. Embedding expert knowledge into models could also bolster model transparency and trustworthiness. Furthermore, future research could emphasize lightweight, efficient models and algorithms suited for varying computational settings while minimizing energy consumption and environmental impact.
CONCLUSIONS
In this review, we delved into the application of the attention mechanism and its associated models in drug development. From drug molecule screening and property prediction to molecular generation, attention has showcased its immense value and potential. While challenges pertaining to data quality, model interpretability, computational resources and complexity persist, continuous research and technological advancements suggest that attention-based models hold a promising future in drug development. As technology advances, we can optimistically anticipate that newer and more potent attention-based models will further accelerate the pace and efficiency of drug research, ushering in breakthroughs for human health and pharmaceutical science.
Although this review provides insightful perspectives on the application of attention mechanisms in small molecule drug discovery, it still has certain limitations. First, due to the scope and focus of the article, it primarily concentrates on small molecule drugs, with less discussion on large molecule drugs or biological agents. Secondly, the article does not delve deeply into critical issues such as data quality, data bias and data privacy. These factors are essential in influencing the performance of models and their application in drug development. Additionally, while the use of AI in drug discovery holds great potential, related ethical and legal issues are not sufficiently addressed and discussed in this review. Addressing these issues is crucial for a comprehensive understanding and evaluation of the impact of this technological field.
Key Points
Attention mechanisms selectively allocate weights to different input data, allowing models to emphasize key information, showcasing significant advantages in drug discovery.
Attention-based models have found broad application in drug development, spanning activities from drug molecule screening, property prediction, to the generation of molecular structures.
However, integrating attention mechanisms in drug development comes with challenges such as ensuring data quality, enhancing model interpretability and managing computational limits.
ACKNOWLEDGEMENTS
We would like to thank the anonymous reviewers for valuable suggestions.
Author Biographies
Yang Zhang is an associate professor of the Innovative Institute of Chinese Medicine and Pharmacy, Academy for Interdiscipline, Chengdu University of Traditional Chinese Medicine, Chengdu, China. His research is in the areas of bioinformatics and system biology.
Caiqi Liu is an attending doctor at the Department of Gastrointestinal Medical Oncology, Harbin Medical University Cancer Hospital. His research is in the areas of the exploration of novel therapeutic regime for gastrointestinal oncology.
Mujiexin Liu is a doctor candidate of the Chongqing Key Laboratory of Sichuan-Chongqing Co-construction for Diagnosis and Treatment of Infectious Diseases Integrated Traditional Chinese and Western Medicine, College of Medical Technology, Chengdu University of Traditional Chinese Medicine, Chengdu, China. His research is in the areas of traditional Chinese medicine and bioinformatics.
Tianyuan Liu is a doctoral student of the Graduate School of Science and Technology, University of Tsukuba, Tsukuba, Japan. His research is in the areas of bioinformatics and system biology.
Hao Lin is a professor of the Center for Informational Biology at the University of Electronic Science and Technology of China. His research is in the areas of bioinformatics and system biology.
Cheng-Bing Huang is a professor at Aba Teachers University. His research is in the areas of machine learning and biological data mining.
Lin Ning is a professor of the School of Health and Medical Technology, Chengdu Neusoft University, Chengdu, China. His research is in the areas of bioinformatics and system biology.
Contributor Information
Yang Zhang, Innovative Institute of Chinese Medicine and Pharmacy, Academy for Interdiscipline, Chengdu University of Traditional Chinese Medicine, Chengdu, China.
Caiqi Liu, Department of Gastrointestinal Medical Oncology, Harbin Medical University Cancer Hospital, No.150 Haping Road, Nangang District, Harbin, Heilongjiang 150081, China; Key Laboratory of Molecular Oncology of Heilongjiang Province, No.150 Haping Road, Nangang District, Harbin, Heilongjiang 150081, China.
Mujiexin Liu, Chongqing Key Laboratory of Sichuan-Chongqing Co-construction for Diagnosis and Treatment of Infectious Diseases Integrated Traditional Chinese and Western Medicine, College of Medical Technology, Chengdu University of Traditional Chinese Medicine, Chengdu, China.
Tianyuan Liu, Graduate School of Science and Technology, University of Tsukuba, Tsukuba, Japan.
Hao Lin, School of Life Science and Technology, University of Electronic Science and Technology of China, Chengdu 610054, China.
Cheng-Bing Huang, School of Computer Science and Technology, Aba Teachers University, Aba, China.
Lin Ning, Yangtze Delta Region Institute (Quzhou), University of Electronic Science and Technology of China, Quzhou, Zhejiang, China; School of Healthcare Technology, Chengdu Neusoft University, Chengdu 611844, China.
FUNDING
National Natural Science Foundation of China (62202069, 62372088); Natural Science Foundation of Sichuan Province (2022NSFSC1610, 2023NSFSC0678, 2023NSFSC0569); China Postdoctoral Science Foundation (2023 M730507); Sichuan Province Postdoctoral Research Project Special Support Foundation (TB2023012).
AUTHOR CONTRIBUTIONS
Conceptualization, L.N., H.L., Y.Z., C.H.; Investigation, Y.Z., C.L., L.N., M.-J.X.L., T.-Y.L.; Writing—Original Draft, L.N., H.L., Y.Z.; Writing—Review & Editing, H.L. H.L., Y.Z.; Funding Acquisition, L.N., H.L., Y.Z.
DECLARATION OF GENERATIVE AI AND AI-ASSISTED TECHNOLOGIES IN THE WRITING PROCESS
During the preparation of this work, the authors used chatGPT 3.5 in order to improve language and readability. After using this service, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
DATA AVAILABILITY
This study does not produce or analyze new data.
References
- 1. Wouters OJ, McKee M, Luyten J. Estimated research and development investment needed to bring a new medicine to market, 2009-2018. JAMA 2020;323:844–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dominguez LW, Willis JS. Research and development costs of new drugs. JAMA 2020;324:516. [DOI] [PubMed] [Google Scholar]
- 3. Sun D, Gao W, Hu H, Zhou S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm Sin B 2022;12:3049–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deng J, Yang Z, Ojima I, et al. Artificial intelligence in drug discovery: applications and techniques. Brief Bioinform 2022;23:bbab430. [DOI] [PubMed] [Google Scholar]
- 5. Bender A, Cortés-Ciriano I. Artificial intelligence in drug discovery: what is realistic, what are illusions? Part 1: ways to make an impact, and why we are not there yet. Drug Discov Today 2021;26:511–24. [DOI] [PubMed] [Google Scholar]
- 6. Wang Y, Zhai Y, Ding Y, et al. SBSM-pro: support bio-sequence machine for proteins. arXiv preprint arXiv:230810275 2023. 10.48550/arXiv.2308.10275. [DOI] [Google Scholar]
- 7. Taye MM. Understanding of machine learning with deep learning: architectures, workflow, applications and future directions. Comput Secur 2023;12:91. [Google Scholar]
- 8. Farghali H, Kutinová Canová N, Arora M. The potential applications of artificial intelligence in drug discovery and development. Physiol Res 2021;70:S715–s722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qureshi R, Irfan M, Gondal TM, et al. AI in drug discovery and its clinical relevance. Heliyon 2023;9:e17575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lv H, Shi L, Berkenpas JW, et al. Application of artificial intelligence and machine learning for COVID-19 drug discovery and vaccine design. Brief Bioinform 2021;22:bbab320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lu M, Yin J, Zhu Q, et al. Artificial intelligence in pharmaceutical sciences. Engineering 2023;27:37–69. [Google Scholar]
- 12. Blanco-González A, Cabezón A, Seco-González A, et al. The role of AI in drug discovery: challenges, opportunities, and strategies. Pharmaceuticals (Basel) 2023;16:891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Han R, Yoon H, Kim G, et al. Revolutionizing medicinal chemistry: the application of artificial intelligence (AI) in early drug discovery. Pharmaceuticals 2023;16:1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seyhan AA, Carini C. Are innovation and new technologies in precision medicine paving a new era in patients centric care? J Transl Med 2019;17:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang S, Fan R, Liu Y, et al. Applications of transformer-based language models in bioinformatics: a survey. Bioinform Adv 2023;3:vbad001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Z, Roberts RA, Lal-Nag M, et al. AI-based language models powering drug discovery and development. Drug Discov Today 2021;26:2593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Monteiro NRC, Oliveira JL, Arrais JP. DTITR: end-to-end drug-target binding affinity prediction with transformers. Comput Biol Med 2022;147:105772. [DOI] [PubMed] [Google Scholar]
- 18. Hu J, Yu W, Pang C, et al. DrugormerDTI: drug Graphormer for drug-target interaction prediction. Comput Biol Med 2023;161:106946. [DOI] [PubMed] [Google Scholar]
- 19. Gao J, Shen Z, Xie Y, et al. TransFoxMol: predicting molecular property with focused attention. Brief Bioinform 2023;24:bbad306. [DOI] [PubMed] [Google Scholar]
- 20. Lecler A, Duron L, Soyer P. Revolutionizing radiology with GPT-based models: current applications, future possibilities and limitations of ChatGPT. Diagn Interv Imaging 2023;104:269–74. [DOI] [PubMed] [Google Scholar]
- 21. Grechishnikova D. Transformer neural network for protein-specific de novo drug generation as a machine translation problem. Sci Rep 2021;11:321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cheng Z, Yan C, Wu FX, et al. Drug-target interaction prediction using multi-head self-attention and graph attention network. IEEE/ACM Trans Comput Biol Bioinform 2022;19:2208–18. [DOI] [PubMed] [Google Scholar]
- 23. Fang K, Zhang Y, Du S, et al. ColdDTA: utilizing data augmentation and attention-based feature fusion for drug-target binding affinity prediction. Comput Biol Med 2023;164:107372. [DOI] [PubMed] [Google Scholar]
- 24. Lin T, Wang Y, Liu X, et al. A survey of transformers. AI Open 2022;3:111–32. [Google Scholar]
- 25. Bahdanau D, Cho K, Bengio Y. Neural machine translation by jointly learning to align and translate. arXiv preprint arXiv:14090473 2014. 10.48550/arXiv.1409.0473. [DOI] [Google Scholar]
- 26. Vaswani A, Shazeer N, Parmar N, et al. Attention is all you need. Adv Neural Inf Process Syst 2017;30:1–11. [Google Scholar]
- 27. Devlin J, Chang M-W, Lee K, et al. Bert: pre-training of deep bidirectional transformers for language understanding. arXiv preprint arXiv:181004805 2018. 10.48550/arXiv.1810.04805. [DOI] [Google Scholar]
- 28. Radford A, Wu J, Child R, et al. Language models are unsupervised multitask learners. OpenAI blog 2019;1:9. [Google Scholar]
- 29. Yenduri G, Srivastava G, Maddikunta PKR, et al. Generative pre-trained transformer: a comprehensive review on enabling technologies, potential applications, emerging challenges, and future directions. arXiv preprint arXiv:230510435 2023. 10.48550/arXiv.1810.04805. [DOI] [Google Scholar]
- 30. Haroon S, Hafsath CA, Jereesh AS. Generative pre-trained transformer (GPT) based model with relative attention for de novo drug design. Comput Biol Chem 2023;106:107911. [DOI] [PubMed] [Google Scholar]
- 31. Zhang L, Wang CC, Chen X. Predicting drug-target binding affinity through molecule representation block based on multi-head attention and skip connection. Brief Bioinform 2022;23:bbac468. [DOI] [PubMed] [Google Scholar]
- 32. Lee I, Nam H. Sequence-based prediction of protein binding regions and drug-target interactions. J Chem 2022;14:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen J, Gu Z, Xu Y, et al. QuoteTarget: A sequence-based transformer protein language model to identify potentially druggable protein targets. Protein Sci 2023;32:e4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tan Z, Zhao Y, Zhou T, et al. Hi-MGT: a hybrid molecule graph transformer for toxicity identification. J Hazard Mater 2023;457:131808. [DOI] [PubMed] [Google Scholar]
- 35. Teng S, Yin C, Wang Y, et al. MolFPG: multi-level fingerprint-based graph transformer for accurate and robust drug toxicity prediction. Comput Biol Med 2023;164:106904. [DOI] [PubMed] [Google Scholar]
- 36. Yuan Q, Chen S, Rao J, et al. AlphaFold2-aware protein-DNA binding site prediction using graph transformer. Brief Bioinform 2022;23:bbab564. [DOI] [PubMed] [Google Scholar]
- 37. Hong Y, Lee J, Ko J. A-Prot: protein structure modeling using MSA transformer. BMC Bioinformatics 2022;23:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang B, Kong L, Wang C, et al. Protein structure prediction: challenges, advances, and the shift of research paradigms. Genomics Proteomics Bioinformatics 2023;S1672-0229(23):00065–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cao Y, Shen Y. TALE: transformer-based protein function annotation with joint sequence-label embedding. Bioinformatics 2021;37:2825–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Clauwaert J, Waegeman W. Novel transformer networks for improved sequence Labeling in genomics. IEEE/ACM Trans Comput Biol Bioinform 2022;19:97–106. [DOI] [PubMed] [Google Scholar]
- 41. Song B, Li Z, Lin X, et al. Pretraining model for biological sequence data. Brief Funct Genomics 2021;20:181–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang Y, Liu T, Hu X, et al. CellCall: integrating paired ligand-receptor and transcription factor activities for cell-cell communication. Nucleic Acids Res 2021;49:8520–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang R, Wang Z, Wang X, et al. MHTAN-DTI: Metapath-based hierarchical transformer and attention network for drug-target interaction prediction. Brief Bioinform 2023;24:bbad079. [DOI] [PubMed] [Google Scholar]
- 44. Wen J, Gan H, Yang Z, et al. Mutual-DTI: A mutual interaction feature-based neural network for drug-target protein interaction prediction. Math Biosci Eng 2023;20:10610–25. [DOI] [PubMed] [Google Scholar]
- 45. Qian Y, Li X, Wu J, et al. MCL-DTI: using drug multimodal information and bi-directional cross-attention learning method for predicting drug-target interaction. BMC Bioinformatics 2023;24:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kurata H, Tsukiyama S. ICAN: interpretable cross-attention network for identifying drug and target protein interactions. PLoS One 2022;17:e0276609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ma M, Lei X. A dual graph neural network for drug-drug interactions prediction based on molecular structure and interactions. PLoS Comput Biol 2023;19:e1010812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang Z, Zhong W, Lv Q, et al. Learning size-adaptive molecular substructures for explainable drug-drug interaction prediction by substructure-aware graph neural network. Chem Sci 2022;13:8693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhu W, Zhang Y, Zhao D, et al. HiGNN: A hierarchical informative graph neural network for molecular property prediction equipped with feature-wise attention. J Chem Inf Model 2023;63:43–55. [DOI] [PubMed] [Google Scholar]
- 50. Scarselli F, Gori M, Tsoi AC, et al. The graph neural network model. IEEE Trans Neural Netw 2009;20:61–80. [DOI] [PubMed] [Google Scholar]
- 51. Zhang Z, Chen L, Zhong F, et al. Graph neural network approaches for drug-target interactions. Curr Opin Struct Biol 2022;73:102327. [DOI] [PubMed] [Google Scholar]
- 52. Wan X, Wu X, Wang D, et al. An inductive graph neural network model for compound-protein interaction prediction based on a homogeneous graph. Brief Bioinform 2022;23:bbac073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Velickovic P, Cucurull G, Casanova A, et al. Graph attention networks. Stat 2017;1050:10–48550. [Google Scholar]
- 54. Xiong J, Xiong Z, Chen K, et al. Graph neural networks for automated de novo drug design. Drug Discov Today 2021;26:1382–93. [DOI] [PubMed] [Google Scholar]
- 55. Le T, Noé F, Clevert DA. Equivariant graph attention networks for molecular property prediction. arXiv preprint arXiv:220209891 2022. 10.48550/arXiv.2202.09891. [DOI] [Google Scholar]
- 56. Wu Z, Wang J, Du H, et al. Chemistry-intuitive explanation of graph neural networks for molecular property prediction with substructure masking. Nat Commun 2023;14:2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu H, Huang Y, Liu X, Deng L. Attention-wise masked graph contrastive learning for predicting molecular property. Brief Bioinform 2022;23:bbac303. [DOI] [PubMed] [Google Scholar]
- 58. Wang X, Zhu H, Chen D, et al. A complete graph-based approach with multi-task learning for predicting synergistic drug combinations. Bioinformatics 2023;39:btad351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jiang Y, Jin S, Jin X, et al. Pharmacophoric-constrained heterogeneous graph transformer model for molecular property prediction. Commun Chem 2023;6:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gu Y, Zhang X, Xu A, et al. Protein-ligand binding affinity prediction with edge awareness and supervised attention. iScience 2023;26:105892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cheng X, Cheng M, Yu L, Xiao X. iADRGSE: A graph-embedding and self-attention encoding for identifying adverse drug reaction in the earlier phase of drug development. Int J Mol Sci 2022;23:16216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kalyan KS, Rajasekharan A, Sangeetha S. AMMU: A survey of transformer-based biomedical pretrained language models. J Biomed Inform 2022;126:103982. [DOI] [PubMed] [Google Scholar]
- 63. Liu Z, Lv Q, Yang Z, et al. Recent progress in transformer-based medical image analysis. Comput Biol Med 2023;164:107268. [DOI] [PubMed] [Google Scholar]
- 64. Tong X, Liu X, Tan X, et al. Generative models for De novo drug design. J Med Chem 2021;64:14011–27. [DOI] [PubMed] [Google Scholar]
- 65. Guo J, Ibanez-Lopez AS, Gao H, et al. Automated chemical reaction extraction from scientific literature. J Chem Inf Model 2022;62:2035–45. [DOI] [PubMed] [Google Scholar]
- 66. Yang L, Yang G, Bing Z, et al. Transformer-based generative model accelerating the development of novel BRAF inhibitors. ACS Omega 2021;6:33864–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shin J, Piao Y, Bang D, et al. DRPreter: interpretable anticancer drug response prediction using knowledge-guided graph neural networks and transformer. Int J Mol Sci 2022;23:13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lin S, Wang Y, Zhang L, et al. MDF-SA-DDI: predicting drug-drug interaction events based on multi-source drug fusion, multi-source feature fusion and transformer self-attention mechanism. Brief Bioinform 2022;23:bbab421. [DOI] [PubMed] [Google Scholar]
- 69. Schwarz K, Allam A, Perez Gonzalez NA, et al. AttentionDDI: Siamese attention-based deep learning method for drug-drug interaction predictions. BMC Bioinformatics 2021;22:412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jiang L, Jiang C, Yu X, et al. DeepTTA: a transformer-based model for predicting cancer drug response. Brief Bioinform 2022;23:bbac100. [DOI] [PubMed] [Google Scholar]
- 71. Wang J, Hu J, Sun H, et al. MGPLI: exploring multigranular representations for protein-ligand interaction prediction. Bioinformatics 2022;38:4859–67. [DOI] [PubMed] [Google Scholar]
- 72. Liu X, Ye K, van Vlijmen HWT, et al. DrugEx v3: scaffold-constrained drug design with graph transformer-based reinforcement learning. J Chem 2023;15:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kim H, Na J, Lee WB. Generative chemical transformer: neural machine learning of molecular geometric structures from chemical language via attention. J Chem Inf Model 2021;61:5804–14. [DOI] [PubMed] [Google Scholar]
- 74. Mao J, Wang J, Zeb A, et al. Transformer-based molecular generative model for antiviral drug design. J Chem Inf Model 2023. 10.1021/acs.jcim.3c00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Choi SR, Lee M. Transformer architecture and attention mechanisms in genome data analysis: a comprehensive review. Biology (Basel) 2023;12:1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. He J, Nittinger E, Tyrchan C, et al. Transformer-based molecular optimization beyond matched molecular pairs. J Chem 2022;14:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Agarwal P, Rahman AA, St-Charles P-L, et al. Transformers in reinforcement learning: a survey. arXiv preprint arXiv:230705979 2023. 10.48550/arXiv.2307.05979. [DOI] [Google Scholar]
- 78. Liu Y, Zhang R, Li T, et al. MolRoPE-BERT: an enhanced molecular representation with rotary position embedding for molecular property prediction. J Mol Graph Model 2023;118:108344. [DOI] [PubMed] [Google Scholar]
- 79. Ross J, Belgodere B, Chenthamarakshan V, et al. Large-scale chemical language representations capture molecular structure and properties. Nature Machine Intelligence 2022;4:1256–64. [Google Scholar]
- 80. Wu Z, Jiang D, Wang J, et al. Knowledge-based BERT: a method to extract molecular features like computational chemists. Brief Bioinform 2022;23:bbac131. [DOI] [PubMed] [Google Scholar]
- 81. Zhang XC, Wu CK, Yang ZJ, et al. MG-BERT: leveraging unsupervised atomic representation learning for molecular property prediction. Brief Bioinform 2021;22:bbab152. [DOI] [PubMed] [Google Scholar]
- 82. Chithrananda S, Grand G, Ramsundar B. ChemBERTa: large-scale self-supervised pretraining for molecular property prediction. arXiv preprint arXiv:201009885 2020. 10.48550/arXiv.2010.09885. [DOI] [Google Scholar]
- 83. Ahmad W, Simon E, Chithrananda S, et al. Chemberta-2: towards chemical foundation models. arXiv preprint arXiv:220901712 2022. 10.48550/arXiv.2209.01712. [DOI] [Google Scholar]
- 84. Yuesen L, Chengyi G, Xin S, et al. DrugGPT: A GPT-based strategy for designing potential ligands targeting specific proteins. bioRxiv 2023:2023.2006.2029.543848. 10.1101/2023.06.29.543848. [DOI] [Google Scholar]
- 85. Savage N. Drug discovery companies are customizing ChatGPT: here’s how. Nat Biotechnol 2023;41:585–6. [DOI] [PubMed] [Google Scholar]
- 86. Zhao A, Wu Y. Future implications of ChatGPT in pharmaceutical industry: drug discovery and development. Front Pharmacol 2023;14:1194216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bagal V, Aggarwal R, Vinod PK, et al. MolGPT: molecular generation using a transformer-decoder model. J Chem Inf Model 2022;62:2064–76. [DOI] [PubMed] [Google Scholar]
- 88. Wang Y, Zhao H, Sciabola S, Wang W. cMolGPT: A conditional generative pre-trained transformer for target-specific de novo molecular generation. Molecules 2023;28:4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wang X, Gao C, Han P, et al. PETrans: De novo drug design with protein-specific encoding based on transfer learning. Int J Mol Sci 2023;24:1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hu F, Wang D, Hu Y, et al. Generating novel compounds targeting SARS-CoV-2 main protease based on imbalanced dataset. In: IEEE International Conference on Bioinformatics and Biomedicine, Seoul, Republic of Korea. IEEE: Piscataway, NJ, USA, 2020. pp. 432–6.
- 91. Liang Y, Zhang R, Zhang L, et al. DrugChat: towards enabling ChatGPT-like capabilities on drug molecule graphs. arXiv preprint arXiv:230903907 2023;2023. 10.48550/arXiv.2309.03907. [DOI] [Google Scholar]
- 92. Zheng W, Lin H, Luo L, et al. An attention-based effective neural model for drug-drug interactions extraction. BMC Bioinformatics 2017;18:445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yu Y, Huang K, Zhang C, et al. SumGNN: multi-typed drug interaction prediction via efficient knowledge graph summarization. Bioinformatics 2021;37:2988–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pang S, Zhang Y, Song T, et al. AMDE: a novel attention-mechanism-based multidimensional feature encoder for drug-drug interaction prediction. Brief Bioinform 2022;23:bbab545. [DOI] [PubMed] [Google Scholar]
- 95. Su X, Hu L, You Z, et al. Attention-based knowledge graph representation learning for predicting drug-drug interactions. Brief Bioinform 2022;23:bbac140. [DOI] [PubMed] [Google Scholar]
- 96. Yu L, Xu Z, Cheng M, et al. MSEDDI: multi-scale embedding for predicting drug-drug interaction events. Int J Mol Sci 2023;24:4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wang J, Zhang S, Li R, et al. Multi-view feature representation and fusion for drug-drug interactions prediction. BMC Bioinformatics 2023;24:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Feng YY, Yu H, Feng YH, Shi JY. Directed graph attention networks for predicting asymmetric drug-drug interactions. Brief Bioinform 2022;23:bbac151. [DOI] [PubMed] [Google Scholar]
- 99. Hong Y, Luo P, Jin S, et al. LaGAT: link-aware graph attention network for drug-drug interaction prediction. Bioinformatics 2022;38:5406–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Feng YH, Zhang SW. Prediction of drug-drug interaction using an attention-based graph neural network on drug molecular graphs. Molecules 2022;27:3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wang X, Liu D, Zhu J, et al. CSConv2d: A 2-D structural convolution neural network with a channel and spatial attention mechanism for protein-ligand binding affinity prediction. Biomolecules 2021;11:643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhao Q, Zhao H, Zheng K, et al. HyperAttentionDTI: improving drug-protein interaction prediction by sequence-based deep learning with attention mechanism. Bioinformatics 2022;38:655–62. [DOI] [PubMed] [Google Scholar]
- 103. Xuan P, Fan M, Cui H, et al. GVDTI: graph convolutional and variational autoencoders with attribute-level attention for drug-protein interaction prediction. Brief Bioinform 2022;23:bbab453. [DOI] [PubMed] [Google Scholar]
- 104. Yu L, Qiu W, Lin W, et al. HGDTI: predicting drug-target interaction by using information aggregation based on heterogeneous graph neural network. BMC Bioinformatics 2022;23:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Xuan P, Zhang X, Zhang Y, et al. Multi-type neighbors enhanced global topology and pairwise attribute learning for drug-protein interaction prediction. Brief Bioinform 2022;23:bbac120. [DOI] [PubMed] [Google Scholar]
- 106. Yazdani-Jahromi M, Yousefi N, Tayebi A, et al. AttentionSiteDTI: an interpretable graph-based model for drug-target interaction prediction using NLP sentence-level relation classification. Brief Bioinform 2022;23:bbac272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Muniyappan S, Rayan AXA, Varrieth GT. DTiGNN: learning drug-target embedding from a heterogeneous biological network based on a two-level attention-based graph neural network. Math Biosci Eng 2023;20:9530–71. [DOI] [PubMed] [Google Scholar]
- 108. Kalakoti Y, Yadav S, Sundar D. Deep neural network-assisted drug recommendation Systems for Identifying Potential Drug-Target Interactions. ACS Omega 2022;7:12138–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Tian Z, Peng X, Fang H, et al. MHADTI: predicting drug-target interactions via multiview heterogeneous information network embedding with hierarchical attention mechanisms. Brief Bioinform 2022;23:bbac434. [DOI] [PubMed] [Google Scholar]
- 110. Li Y, Qiao G, Wang K, Wang G. Drug-target interaction predication via multi-channel graph neural networks. Brief Bioinform 2022;23:bbab346. [DOI] [PubMed] [Google Scholar]
- 111. Shao K, Zhang Y, Wen Y, et al. DTI-HETA: prediction of drug-target interactions based on GCN and GAT on heterogeneous graph. Brief Bioinform 2022;23:bbac109. [DOI] [PubMed] [Google Scholar]
- 112. Yuan Y, Zhang Y, Meng X, et al. EDC-DTI: an end-to-end deep collaborative learning model based on multiple information for drug-target interactions prediction. J Mol Graph Model 2023;122:108498. [DOI] [PubMed] [Google Scholar]
- 113. Li J, Wang J, Lv H, et al. IMCHGAN: inductive matrix completion with heterogeneous graph attention networks for drug-target interactions prediction. IEEE/ACM Trans Comput Biol Bioinform 2022;19:655–65. [DOI] [PubMed] [Google Scholar]
- 114. Huang K, Xiao C, Glass LM, et al. MolTrans: molecular interaction transformer for drug-target interaction prediction. Bioinformatics 2021;37:830–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kalakoti Y, Yadav S, Sundar D. TransDTI: transformer-based language models for estimating DTIs and building a drug recommendation workflow. ACS Omega 2022;7:2706–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Nguyen TM, Nguyen T, Le TM, et al. GEFA: early fusion approach in drug-target affinity prediction. IEEE/ACM Trans Comput Biol Bioinform 2022;19:718–28. [DOI] [PubMed] [Google Scholar]
- 117. Zhang S, Jiang M, Wang S, et al. SAG-DTA: prediction of drug-target affinity using self-attention graph network. Int J Mol Sci 2021;22:8993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Zhao Q, Duan G, Yang M, et al. AttentionDTA: drug-target binding affinity prediction by sequence-based deep learning with attention mechanism. IEEE/ACM Trans Comput Biol Bioinform 2023;20:852–63. [DOI] [PubMed] [Google Scholar]
- 119. Chen H, Li D, Liao J, et al. MultiscaleDTA: A multiscale-based method with a self-attention mechanism for drug-target binding affinity prediction. Methods 2022;207:103–9. [DOI] [PubMed] [Google Scholar]
- 120. Yan X, Liu Y. Graph-sequence attention and transformer for predicting drug-target affinity. RSC Adv 2022;12:29525–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Bae H, Nam H. GraphATT-DTA: attention-based novel representation of interaction to predict drug-target binding affinity. Biomedicine 2022;11:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Jin Z, Wu T, Chen T, et al. CAPLA: improved prediction of protein-ligand binding affinity by a deep learning approach based on a cross-attention mechanism. Bioinformatics 2023;39:btad049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Gim M, Choe J, Baek S, et al. ArkDTA: attention regularization guided by non-covalent interactions for explainable drug-target binding affinity prediction. Bioinformatics 2023;39:i448–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Wang J, Wen N, Wang C, et al. ELECTRA-DTA: a new compound-protein binding affinity prediction model based on the contextualized sequence encoding. J Chem 2022;14:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Tsubaki M, Tomii K, Sese J. Compound-protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics 2019;35:309–18. [DOI] [PubMed] [Google Scholar]
- 126. Li M, Lu Z, Wu Y, et al. BACPI: a bi-directional attention neural network for compound-protein interaction and binding affinity prediction. Bioinformatics 2022;38:1995–2002. [DOI] [PubMed] [Google Scholar]
- 127. Wang X, Liu J, Zhang C, Wang S. SSGraphCPI: A novel model for predicting compound-protein interactions based on deep learning. Int J Mol Sci 2022;23:3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Nguyen NQ, Jang G, Kim H, Kang J. Perceiver CPI: a nested cross-attention network for compound-protein interaction prediction. Bioinformatics 2023;39:btac731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Cai T, Lim H, Abbu KA, et al. MSA-regularized protein sequence transformer toward predicting genome-wide chemical-protein interactions: application to GPCRome Deorphanization. J Chem Inf Model 2021;61:1570–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Qian Y, Wu J, Zhang Q. CAT-CPI: combining CNN and transformer to learn compound image features for predicting compound-protein interactions. Front Mol Biosci 2022;9:963912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Wei L, Long W, Wei L. MDL-CPI: multi-view deep learning model for compound-protein interaction prediction. Methods 2022;204:418–27. [DOI] [PubMed] [Google Scholar]
- 132. Wang W, Liang S, Yu M, et al. GCHN-DTI: predicting drug-target interactions by graph convolution on heterogeneous networks. Methods 2022;206:101–7. [DOI] [PubMed] [Google Scholar]
- 133. Boezer M, Tavakol M, Sajadi Z. FastDTI: drug-target interaction prediction using multimodality and transformers. In: Proceedings of the Northern Lights Deep Learning Workshop, Tromsø, Norway. Septentrio Academic Publishing: Tromsø, Norway, 2023;4. 10.7557/18.6788. [DOI] [Google Scholar]
- 134. Yousefi N, Yazdani-Jahromi M, Tayebi A, et al. BindingSite-AugmentedDTA: enabling a next-generation pipeline for interpretable prediction models in drug repurposing. Brief Bioinform 2023;24:bbad136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. He H, Chen G, Chen CY. NHGNN-DTA: a node-adaptive hybrid graph neural network for interpretable drug-target binding affinity prediction. Bioinformatics 2023;39:btad355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Tang B, Kramer ST, Fang M, et al. A self-attention based message passing neural network for predicting molecular lipophilicity and aqueous solubility. J Chem 2020;12:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Withnall M, Lindelöf E, Engkvist O, et al. Building attention and edge message passing neural networks for bioactivity and physical-chemical property prediction. J Chem 2020;12:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Zheng Z, Tan Y, Wang H, et al. CasANGCL: pre-training and fine-tuning model based on cascaded attention network and graph contrastive learning for molecular property prediction. Brief Bioinform 2023;24:bbac566. [DOI] [PubMed] [Google Scholar]
- 139. Zhang Z, Guan J, Zhou S. FraGAT: a fragment-oriented multi-scale graph attention model for molecular property prediction. Bioinformatics 2021;37:2981–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Cai H, Zhang H, Zhao D, et al. FP-GNN: a versatile deep learning architecture for enhanced molecular property prediction. Brief Bioinform 2022;23:bbac408. [DOI] [PubMed] [Google Scholar]
- 141. Lee S, Park H, Choi C, et al. Multi-order graph attention network for water solubility prediction and interpretation. Sci Rep 2023;13:957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Jang WD, Jang J, Song JS, et al. PredPS: attention-based graph neural network for predicting stability of compounds in human plasma. Comput Struct Biotechnol J 2023;21:3532–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Wen N, Liu G, Zhang J, et al. A fingerprints based molecular property prediction method using the BERT model. J Chem 2022;14:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Zhang J, Du W, Yang X, et al. SMG-BERT: integrating stereoscopic information and chemical representation for molecular property prediction. Front Mol Biosci 2023;10:1216765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Wang S, Guo Y, Wang Y, et al. SMILES-BERT: large scale unsupervised pre-training for molecular property prediction. In: Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Niagara Falls, NY, USA. Association for Computing Machinery, NY, United States, 2019. pp. 429–36. [Google Scholar]
- 146. Deng D, Lei Z, Hong X, et al. Describe molecules by a heterogeneous graph neural network with transformer-like attention for supervised property predictions. ACS Omega 2022;7:3713–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Liu C, Sun Y, Davis R, et al. ABT-MPNN: an atom-bond transformer-based message-passing neural network for molecular property prediction. J Chem 2023;15:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Jiang J, Zhang R, Ma J, et al. TranGRU: focusing on both the local and global information of molecules for molecular property prediction. Appl Intell (Dordr) 2023;53:15246–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Song Y, Chen J, Wang W, et al. Double-head transformer neural network for molecular property prediction. J Chem 2023;15:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Rong Y, Bian Y, Xu T, et al. Self-supervised graph transformer on large-scale molecular data. Adv Neural Inf Process Syst 2020;33:12559–71. [Google Scholar]
- 151. Ma H, Bian Y, Rong Y, et al. Multi-view graph neural networks for molecular property prediction. arXiv preprint arXiv:200513607 2020. 10.48550/arXiv.2005.13607. [DOI] [Google Scholar]
- 152. Shang C, Liu Q, Chen K-S, et al. Edge attention-based multi-relational graph convolutional networks. arXiv preprint arXiv: 180204944 2018. 10.48550/arXiv.1802.04944. [DOI] [Google Scholar]
- 153. Meng M, Wei Z, Li Z et al. Property prediction of molecules in graph convolutional neural network expansion. In: 2019 IEEE 10th International Conference on Software Engineering and Service Science (ICSESS), Beijing, China. IEEE: Piscataway, NJ, USA, 2019. pp. 263–6.
- 154. Li JN, Yang G, Zhao PC, et al. CProMG: controllable protein-oriented molecule generation with desired binding affinity and drug-like properties. Bioinformatics 2023;39:i326–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Dollar O, Joshi N, Beck DAC, et al. Attention-based generative models for de novo molecular design. Chem Sci 2021;12:8362–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Wang J, Hsieh C-Y, Wang M, et al. Multi-constraint molecular generation based on conditional transformer, knowledge distillation and reinforcement learning. Nature Machine Intelligence 2021;3:914–22. [Google Scholar]
- 157. Uludogan G, Ozkirimli E, Ulgen KO, et al. Exploiting pretrained biochemical language models for targeted drug design. Bioinformatics 2022;38:ii155–61. [DOI] [PubMed] [Google Scholar]
- 158. Wang J, Mao J, Wang M, et al. Explore drug-like space with deep generative models. Methods 2023;210:52–9. [DOI] [PubMed] [Google Scholar]
- 159. Yoshimori A, Bajorath J. Motif2Mol: prediction of new active compounds based on sequence motifs of ligand binding sites in proteins using a biochemical language model. Biomolecules 2023;13:833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Mazuz E, Shtar G, Shapira B, Rokach L. Molecule generation using transformers and policy gradient reinforcement learning. Sci Rep 2023;13:8799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Blanchard AE, Bhowmik D, Fox Z, et al. Adaptive language model training for molecular design. J Chem 2023;15:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Ranjan A, Kumar H, Kumari D, et al. Molecule generation toward target protein (SARS-CoV-2) using reinforcement learning-based graph neural network via knowledge graph. Netw Model Anal Health Inform Bioinform 2023;12:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Qian H, Lin C, Zhao D, et al. AlphaDrug: protein target specific de novo molecular generation. PNAS Nexus 2022;1:pgac227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Wang W, Wang Y, Zhao H, et al. A pre-trained conditional transformer for target-specific de novo molecular generation. arXiv preprint arXiv:2210.08749, 2022. 10.48550/arXiv.2210.08749. [DOI] [PMC free article] [PubMed]
- 165. Manica M, Oskooei A, Born J, et al. Toward explainable anticancer compound sensitivity prediction via multimodal attention-based convolutional encoders. Mol Pharm 2019;16:4797–806. [DOI] [PubMed] [Google Scholar]
- 166. Cadow J, Born J, Manica M, et al. PaccMann: a web service for interpretable anticancer compound sensitivity prediction. Nucleic Acids Res 2020;48:W502–w508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Chu T, Nguyen TT, Hai BD, et al. Graph transformer for drug response prediction. IEEE/ACM Trans Comput Biol Bioinform 2023;20:1065–72. [DOI] [PubMed] [Google Scholar]
- 168. Gao J, Hu J, Sun W, et al. TCR: A transformer based deep network for predicting cancer drugs response. arXiv preprint arXiv:220704457 2022. 10.48550/arXiv.2207.04457. [DOI] [Google Scholar]
- 169. Huang Z, Zhang P, Deng L. DeepCoVDR: deep transfer learning with graph transformer and cross-attention for predicting COVID-19 drug response. Bioinformatics 2023;39:i475–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Xuan P, Wang M, Liu Y, et al. Integrating specific and common topologies of heterogeneous graphs and pairwise attributes for drug-related side effect prediction. Brief Bioinform 2022;23:bbac126. [DOI] [PubMed] [Google Scholar]
- 171. Zhao H, Ni P, Zhao Q, et al. Identifying the serious clinical outcomes of adverse reactions to drugs by a multi-task deep learning framework. Commun Biol 2023;6:870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Krix S, DeLong LN, Madan S, et al. MultiGML: multimodal graph machine learning for prediction of adverse drug events. Heliyon 2023;9:e19441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Lin S, Zhang G, Wei DQ, et al. DeepPSE: prediction of polypharmacy side effects by fusing deep representation of drug pairs and attention mechanism. Comput Biol Med 2022;149:105984. [DOI] [PubMed] [Google Scholar]
- 174. Deac A, Huang Y-H, Veličković P, et al. Drug-drug adverse effect prediction with graph co-attention. arXiv preprint arXiv:190500534 2019. 10.48550/arXiv.1905.00534. [DOI] [Google Scholar]
- 175. Yang J, Xu Z, Wu WKK, et al. GraphSynergy: a network-inspired deep learning model for anticancer drug combination prediction. J Am Med Inform Assoc 2021;28:2336–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Zhang P, Tu S, Zhang W, Xu L. Predicting cell line-specific synergistic drug combinations through a relational graph convolutional network with attention mechanism. Brief Bioinform 2022;23:bbac403. [DOI] [PubMed] [Google Scholar]
- 177. Wang T, Wang R, Wei L. AttenSyn: an attention-based deep graph neural network for anticancer synergistic drug combination prediction. J Chem Inf Model 2023. 10.1021/acs.jcim.3c00709. [DOI] [PubMed] [Google Scholar]
- 178. Zhang P, Tu S. MGAE-DC: predicting the synergistic effects of drug combinations through multi-channel graph autoencoders. PLoS Comput Biol 2023;19:e1010951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Xu M, Zhao X, Wang J, et al. DFFNDDS: prediction of synergistic drug combinations with dual feature fusion networks. J Chem 2023;15:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Zhang W, Xue Z, Li Z, et al. DCE-DForest: A deep Forest model for the prediction of anticancer drug combination effects. Comput Math Methods Med 2022;2022:8693746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Liu Q, Xie L. TranSynergy: mechanism-driven interpretable deep neural network for the synergistic prediction and pathway deconvolution of drug combinations. PLoS Comput Biol 2021;17:e1008653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Hu J, Gao J, Fang X, et al. DTSyn: a dual-transformer-based neural network to predict synergistic drug combinations. Brief Bioinform 2022;23:bbac302. [DOI] [PubMed] [Google Scholar]
- 183. Rafiei F, Zeraati H, Abbasi K, et al. DeepTraSynergy: drug combinations using multimodal deep learning with transformers. Bioinformatics 2023;39:btad438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Hu J, Zhang X, Shang D, et al. EGTSyn: edge-based graph transformer for anti-cancer drug combination synergy prediction. arXiv preprint arXiv:230310312 2023. 10.48550/arXiv.2303.10312. [DOI] [Google Scholar]
- 185. Wang J, Liu X, Shen S, et al. DeepDDS: deep graph neural network with attention mechanism to predict synergistic drug combinations. Brief Bioinform 2022;23:bbab390. [DOI] [PubMed] [Google Scholar]
- 186. Dong Z, Zhang H, Chen Y, et al. Interpreting the mechanism of synergism for drug combinations using attention-based hierarchical graph pooling. Cancers (Basel) 2023;15:4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Bittner MI, Farajnia S. AI in drug discovery: applications, opportunities, and challenges. Patterns (N Y) 2022;3:100529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Ruan D, Ji S, Yan C, et al. Exploring complex and heterogeneous correlations on hypergraph for the prediction of drug-target interactions. Patterns (N Y) 2021;2:100390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Liang H, Chen L, Zhao X, et al. Prediction of drug side effects with a refined negative sample selection strategy. Comput Math Methods Med 2020;2020:1573543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Jiménez-Luna J, Grisoni F, Schneider G. Drug discovery with explainable artificial intelligence. Nature Machine Intelligence 2020;2:573–84. [Google Scholar]
- 191. Linardatos P, Papastefanopoulos V, Kotsiantis S. Explainable AI: a review of machine learning interpretability methods. Entropy (Basel) 2021;23:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Suzuki J, Zen H, Kazawa H. Extracting representative subset from extensive text data for training pre-trained language models. Inf Process Manag 2023;60:103249. [Google Scholar]
- 193. Sharir O, Peleg B, Shoham Y. The cost of training nlp models: A concise overview. arXiv preprint arXiv:200408900 2020. 10.48550/arXiv.2004.08900. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study does not produce or analyze new data.