

ABSTRACT
The human gut microbiome plays a significant role in health and disease. The viral component (virome) is predominantly composed of bacteriophages (phages) and has received significantly less attention in comparison to the bacteriome. This knowledge gap is largely due to challenges associated with the isolation and characterization of novel gut phages, and bioinformatic hurdles such as the lack of a universal phage marker gene and the absence of sufficient numbers of homologs in viral databases. Here, we describe the isolation from human feces of a novel lytic phage with siphovirus morphology, φPDS1, infecting Parabacteroides distasonis APCS2/PD, and classified within a newly proposed Sagittacolavirus genus. In silico and biological characterization of this phage is presented in this study. Key to the isolation of φPDS1 was the antibiotic-driven selective enrichment of the bacterial host in a fecal fermenter. Despite producing plaques and lacking genes associated with lysogeny, φPDS1 demonstrates the ability to coexist in liquid culture for multiple days without affecting the abundance of its host. Multiple studies have shown that changes in Parabacteroides distasonis abundance can be linked to various disease states, rendering this novel phage-host pair and their interactions of particular interest.
KEYWORDS: Bacteriophage, Parabacteroides, phage-bacteria interaction, phage isolation, fecal fermentation, gut microbiome, phage characterization
1. Introduction
Bacteriophages (phages) constitute the majority of the human gut virome and are believed to play an important role in shaping the composition and functionality of the microbiome, either through direct interactions with members of the gut bacterial community, or indirectly by interacting with the host immune system.1–3 Several biological factors (i.e., age, diet, geographic location, ethnicity, etc.) and multiple disease states are associated with alterations in the human gut virome composition.4–7 For example, in patients with inflammatory bowel disease (IBD) there is a shift in virome composition from predominantly virulent phages toward induction of temperate phages.8
Despite this revived interest in phages, the diversity of the gut virome remains understudied. While recent efforts to sequence, assemble, and catalog the genomes of uncultured human gut phages resulted in databases of over 50,000 viral operational taxonomic units (OTUs) representing up to 50% of previously un-annotated gut phage DNA (“the viral dark matter”),9,10 most of these genomes remain unclassified and not assigned to any bacterial hosts. The key obstacles include the absence of a universal phylogenetic marker gene in phage genomes, often a lack of sequence homology with known phages included in the present International Committee on Taxonomy of Viruses (ICTV) taxonomy, and the absence of a widely accepted universal framework for classification of novel and uncultured virus taxa.11,12 Isolation of novel gut phages in culture can be hampered by: (i) difficulties in culturing host bacteria; (ii) the inadequacy of traditional screening methods involving plaque or spot assays; (iii) an inability to mimic the specific gut conditions required for phage replication; and (iv) the rapid emergence of resistance in bacterial hosts, e.g. through phase variation.13–15
The isolation of novel phage-host pairs from the human gut provides new insights into phage-host dynamics and the role of phages in this ecosystem. Order Bacteroidales is one of the most abundant bacterial taxonomic groups in a healthy human gut, with Bacteroides and Parabacteroides being two of the most abundant genera.16,17 Analyses of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) spacer sequences, which reveal information about previous encounters between bacteria and genetic elements such as viruses or plasmids, suggest that a significant proportion of persistent virulent phages in the human virome infect hosts of the Bacteroidales order.18 The Bacteroides-infecting Crassvirales (crAss-like phages), the most abundant phage order in the human gut, provide one of the most notable examples of how the isolation and characterization of a phage originally detected in silico was key in gaining insights into its biological properties.19–21
Here, we conducted an ex vivo selective antibiotic enrichment of a fecal bacterial community to promote the growth of Bacteroidales and to permit the parallel expansion of associated phages. We report the isolation of a novel Parabacteroides distasonis phage; φPDS1, which is classified in the newly candidate genus Sagittacolavirus. This phage is the first lytic P. distasonis-infecting siphovirus isolated from human feces to be characterized and to have its genome sequenced. Given that P. distasonis has been linked to certain human diseases,17,22 characterization of its phages takes on an added significance.
2. Results
2.1. Bacterial composition in fecal fermentation following antibiotic enrichment
A graphical overview of the steps taken to screen novel Bacteroidales phages is presented in Figure 1. A fecal sample from a healthy donor (subject ID: 924) was used to initiate batch fermentations, using two chemostat vessels in parallel, one of which contained vancomycin (7.5 μg/ml) and kanamycin (100 μg/ml) with the aim of inhibiting facultative anaerobes and gram-positive strict anaerobes, thus favoring the growth of strictly anaerobic gram-negative Bacteroidales. These fermentations were performed three times.
Figure 1.
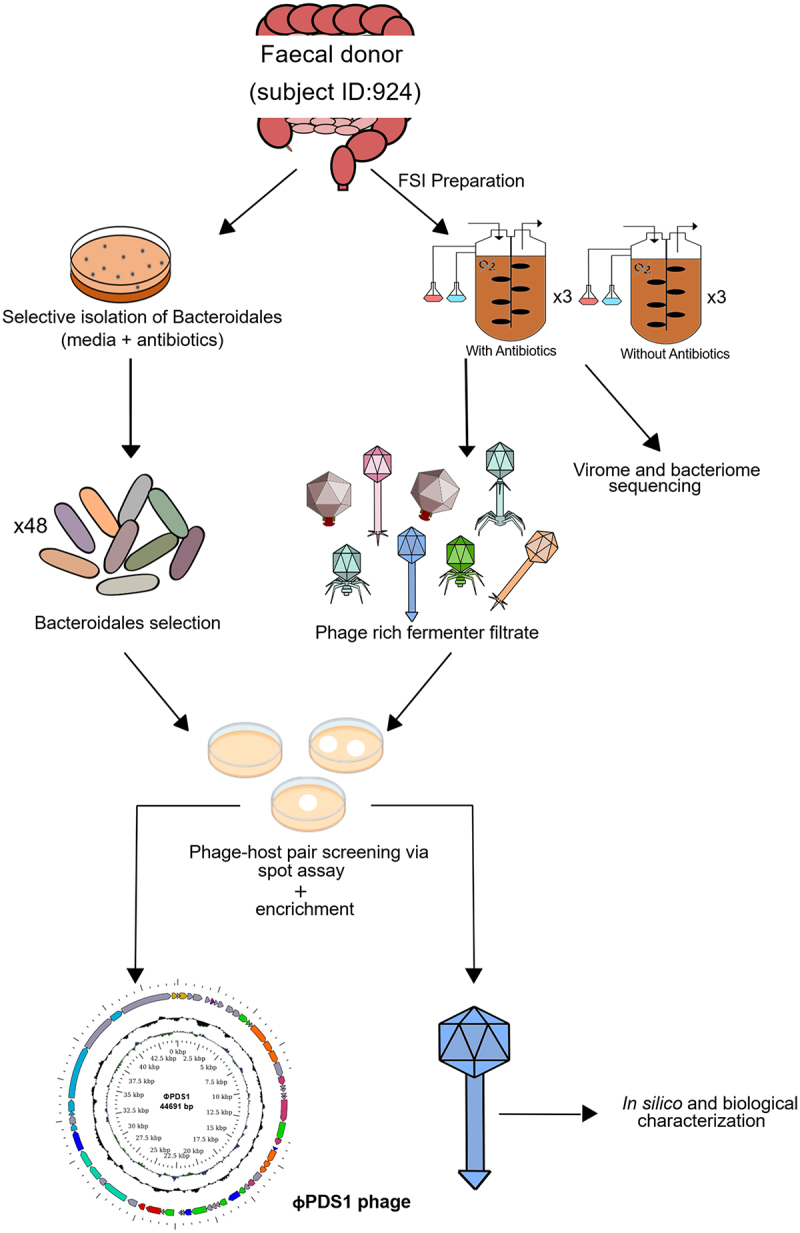
Schematic overview of the key experimental steps carried out to screen for novel phage-host pairs targeting Bacteroidales order bacteria.
16S rRNA sequencing of total DNA extracted from the two vessels at different time points was already published in our previous work23 revealing a significant difference in relative abundance of bacterial genera (Figure S1). At T0 hours both vessels were dominated by the orders Clostridiales (~50%), Bacteroidales (~30%) including the genera Parabacteroides and Bacteroides, and Selenomonadales (~12.5%). By T4.5 hours, a significant change in the composition of bacterial communities was observed. In the vessel treated with antibiotics, the balance shifted toward the order Bacteroidales (~65%), while the vessel without antibiotics became dominated by gram-positive Catenibacterium (~75%). This observation supported the efficacy of vancomycin in inhibiting the growth of gram-positive anaerobic bacteria in our chemostat model. By T17.5 hours, the vessel treated with antibiotics showed a remarkable predominance of Bacteroides and Parabacteroides, representing approximately 90% of the relative abundance. In contrast, these genera contributed less than 20% of the relative abundance in the control vessel (without antibiotics). In the vessel without antibiotics an increase in Faecalibacterium was observed, accompanied by a reduction in the relative abundance of Catenibacterium. Subsequently, from T17.5 hours to T24 hours, no substantial shifts in the relative abundance of the genera were observed in either of the two vessels.
2.2. Identification of potential phage-host candidates targeting Bacteroidales order
In conjunction with the fecal fermentation, individual Bacteroidales isolates were obtained from the frozen standard inoculum (FSI) on various media (FAA, CBA, and YCFA-agar) containing the same antibiotics. To screen for novel phage-host pairs, the phage-enriched fermentation filtrate was screened against the isolated Bacteroidales species using spot-on lawn assays. A total of 48 colonies were selected based on differences in colony morphology. 16S rRNA gene sequencing was performed to obtain an initial estimate of the potential bacterial genus isolated and revealed that most of them belonged to the genus Bacteroides: B. uniformis (25 colonies), B. dorei (7), B. fragilis (1), B. xylanisolvens (1) and B. ovatus (1), and there was also a notable enrichment of Parabacteroides distasonis (13 colonies) (Table S1). These findings were consistent with the high prevalence of Bacteroides and Parabacteroides in the fecal fermentation with antibiotics (~90%). The T21-hour phage-rich fecal fermentate was spotted on lawns of the 48 isolated Bacteroidales species, resulting in consistent clearing specifically on the lawns of P. distasonis (Figure S2). Furthermore, the phage-rich fecal fermentate collected from the antibiotic vessel formed more readily visible spots during screening for phage-host pairs as compared to the non-antibiotic vessel, confirming that phages were also enriched using this approach (Figures S2B and S2C). These results show that the antibiotic selection facilitated the phage-host pair screening process.
2.3. Genome analysis of the novel isolated siphovirus φPDS1 and its host Parabacteroides distasonis APCS2/PD
The phage that produced clearing areas on P. distasonis, designated as φPDS1, was subsequently propagated on its host, and subjected to shotgun genome sequencing. Reads were assembled into a circularized genome of 44,691 bp (GenBank MN929097; Figure 2). The GC content of the φPDS1 genome was determined to be 45.28 mol%, which closely matched the GC content of the host at 45.30 mol%. ORF prediction in the φPDS1 genome identified 61 putative protein-coding genes (CDS), and approximately half could be assigned functions using HHpred, VIGA and BLASTp (Figure 2, Table S2). These annotations included two structural head proteins, five tail-associated proteins, five DNA replication genes, three recombinase genes, two large terminase genes, and two lysis genes. Notably, the two large terminase genes represent the two domains of the large terminase split into two CDS (Figure S3). This observation raises the possibility of either a distinct codon table specific to the φPDS1 genome or a potential issue at the assembly level. The right-hand side of the genome (0−22.5 kbp) contained genes encoding functions associated with DNA replication and maintenance, while the left-hand side (22.5−44.7 kbp) was dominated by phage structural genes, along with genes associated with packaging and assembly. Lysogenic gene modules, which are typically composed of a serine or tyrosine integrase, a repressor of the lytic cycle and excisionase,24 were not identified in the φPDS1 genome.
Figure 2.
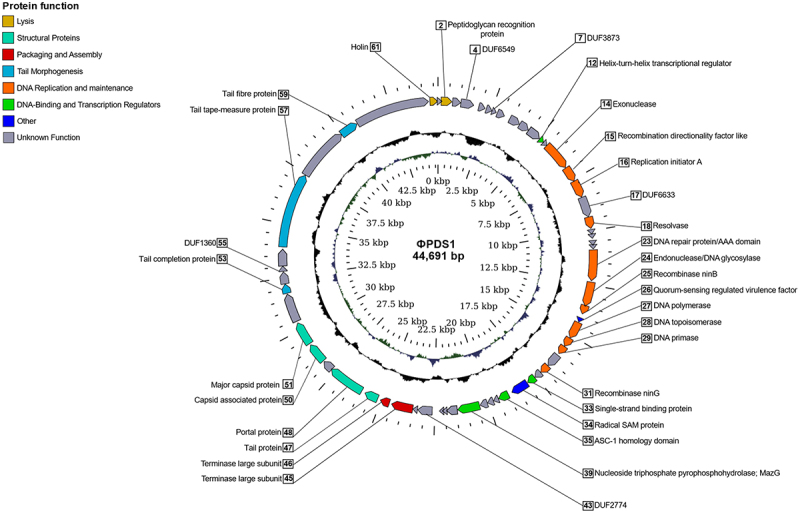
Circular genome map of the novel phage φPDS1 with a genome size of 44,691 bp. The innermost ring represents the GC skew (blue for the positive strand and green for the negative strand) while the central ring (black) displays the GC content. The outermost circle illustrates the coding genes (CDS) with HHpred predicted function as labels. The coloration of the CDS corresponds to their general functions as indicated in the legend. Genes whose function could not be determined are colored gray and remain unlabeled.
The Parabacteroides genus was predicted as a potential host for φPDS1 with a score cutoff of 90% through the iPHoP analysis, which integrates different approaches to infer host genus of phages. P. distasonis and P. sp900552465 were detected at the species level with higher confidence scores (92.9% and 91.1%, respectively). Notably, P. sp900552465 represents an uncultured human gut Parabacteroides species (GenBank: GCA_900552465.1, Bioproject: PRJEB26432), derived from a metagenome-assembled genome isolated from human feces.25 Regarding the different methods applied, BLAST was concordant with these two species, while CRISPR-spacers targeting φPDS1 were found in P. distasonis and in Odoribacter splanchnicus (Table S3).
Whole genome sequencing of the φPDS1 host, designated as Parabacteroides distasonis APCS2/PD, revealed a single circular chromosome (GenBank CP042285) with a genome size of 5,344,828 bp (Figure S4A). Additionally, one associated circular plasmid, pPDS2–1, measuring 4,148 bp (GenBank CP042284), was identified (Figure S4B) and its annotation revealed that it carries genes encoding for a toxin-antitoxin system and vesicle mobilization. The host taxonomy was determined using the Genome Taxonomy Database (GTDB) and the associated taxonomic classification toolkit (GTDB-Tk), which classified the host as belonging to the Parabacteroides species with the closest phylogenetically related strain being P. distasonis DSM 20701, sharing an average nucleotide identity (ANI) of 97.32%.
2.4. φPDS1 is classified in a newly candidate genus, Sagittacolavirus, within the candidate family Paboviridae
A new candidate family Paboviridae, within the class Caudoviricetes, has been recently proposed.26 This family infects bacteria belonging to the Parabacteroides, Alistipes, and other genera within the Bacteroidales order in the human gut. To determine whether φPDS1 could be classified within this candidate family Paboviridae, we followed a similar approach to Shen and colleagues.26 From the Gut Phage Database (GPD), we selected phage genomes with a completeness of at least 95% (as assigned by CheckV) for which we were able to detect the large terminase subunit (n = 313), along with the two experimental phages of their study, Parabacteroides phage PD491P1 and Alistipes phage AS73P1, to investigate phylogenetic relationships. Analyses conducted using VIRIDIC and VICTOR tools confirmed that φPDS1 belongs to the candidate family Paboviridae (Figure 3 and Table S4). We proceeded to perform a VIRIDIC analysis, based on intergenomic nucleotide similarities and which implements the traditional algorithm used by ICTV, to explore whether we could classify φPDS1 into a genus within this family. Out of the 316 total sequences analyzed, VIRIDIC detected a total of 38 genus clusters (Table S5). φPDS1 was grouped at the genus level (>70% similarity) with the PD491P1 phage and 58 other phages from the candidate family Paboviridae. Furthermore, it revealed a potential species cluster with uvig_202125 (97.01% similarity) (Table S5). We also inferred the phylogenetic tree using the VICTOR classifier. It grouped the representative phage genomes and φPDS1, within the same family (Figure 3). The VICTOR tree confirmed that φPDS1 belongs phylogenetically to the candidate Paboviridae family. However, the taxonomic annotation obtained for the genera of the outgroup phage genomes contradicted the ICTV genus annotation of the selected outgroup. We confirmed that the outgroup phage genomes were correctly clustered at the genus level using VIRIDIC. Therefore, we made the decision to disregard the genus annotation provided by VICTOR and exclusively relied on the annotation performed by VIRIDIC.
Figure 3.
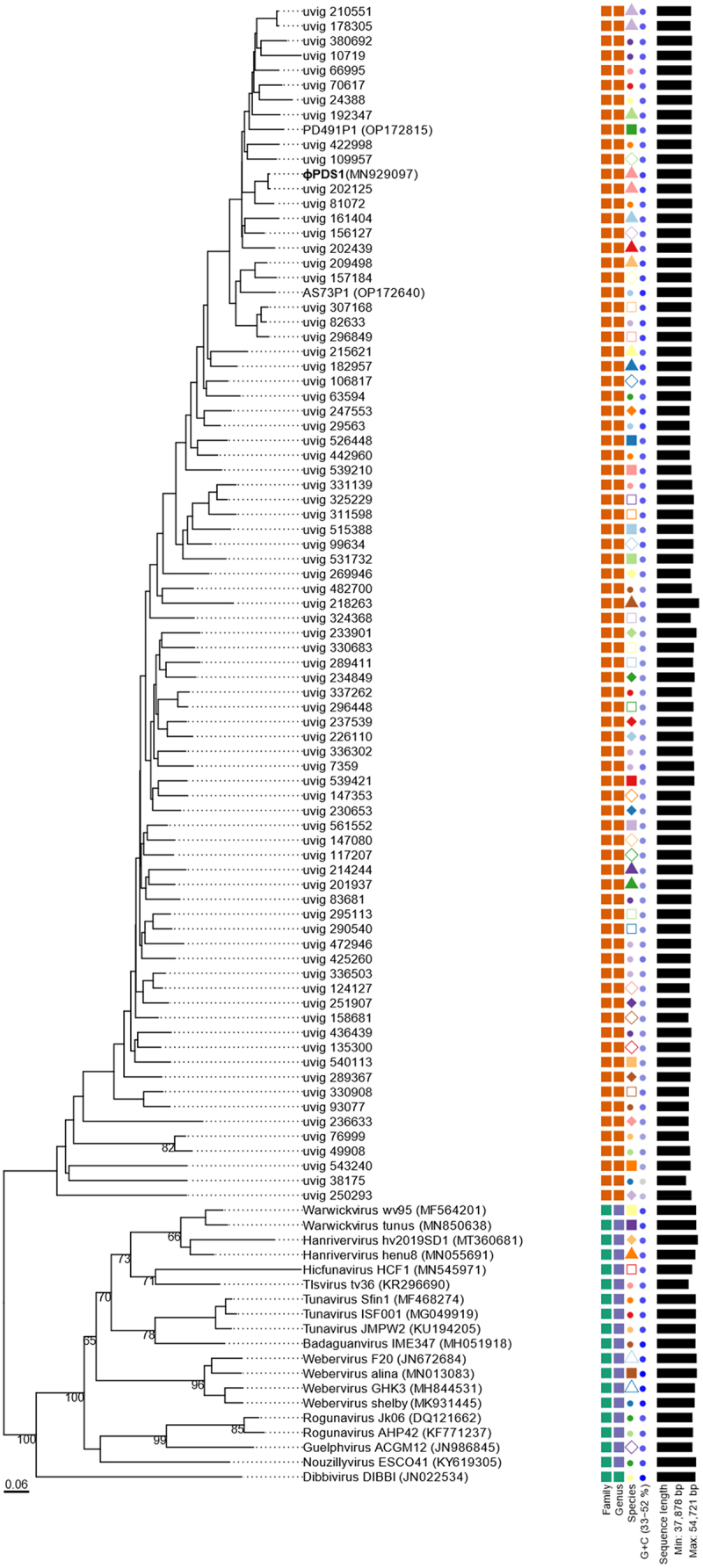
Phylogenomic genome BLAST distance phylogeny (GBDP) tree representing φPDS1, 78 members of the new candidate Paboviridae family, and using 19 phages from the Drexlerviridae family as outgroup. The tree was generated using the VICTOR tool with the D0 formula. The numbers above the branches indicate GBDP pseudo-bootstrap support values from 100 replicates. The branch lengths are scaled in terms of the respective distance formula used. Family, genus, species are tentatively grouped by their phylogenetic relationships when the same shape and color coincide. The GC content is represented in blue; the darker the color, the higher the content. Accession numbers are provided in brackets.
This leads us to propose the new candidate genus Sagittacolavirus, which includes the two isolated phages, φPDS1 and PD491P1, and 58 other phages from the candidate Paboviridae family (Table S6). Based on the CRISPR-spacer targeting results from Shen et al. (2023),26 these 58 phages, clustered within the new potential genus Sagittacolavirus, were found to target hosts in Bacteroides (47), Parabacteroides (9), and 2 unassigned genera, in addition to PD491P1 whose host is Parabacteroides. These phages exhibited a similar range of GC content, varying from 43.09 to 45.28 mol%, and had genome sizes ranging from 42,828 to 46,430 bp. Additionally, the percentage of similarity compared to φPDS1 ranged from 75.47% to 97.01%. The closest relative of φPDS1 was identified as uvig_202125 and the most distant uvig_70617.
A member of each genus cluster (38) from the candidate Paboviridae family, together with φPDS1 and a member from an outgroup (KY619305; Nouzillyvirus ESCO41) were selected, and their genomes were aligned to observe the genetic synteny among them (Figures 4 and S5). Additionally, the intergenomic nucleotide similarities of these clusters were represented through VIRIDIC (Figure S6). The overall genome organization and genetic synteny were found to be largely conserved across all the clusters, except for the groups at the bottom of Figure 4, which showed lower percentages of identity. While most of the sequences showed a high percentage of identity between all the clusters, there was a region corresponding to a protein with an unknown function in which most of the clusters differed. The functions of the proteins encoded by the surrounding genes are related to tail morphogenesis, which suggests that it may have a related function. The member of the outgroup confirmed that there was no genomic similarity with these phages.
Figure 4.
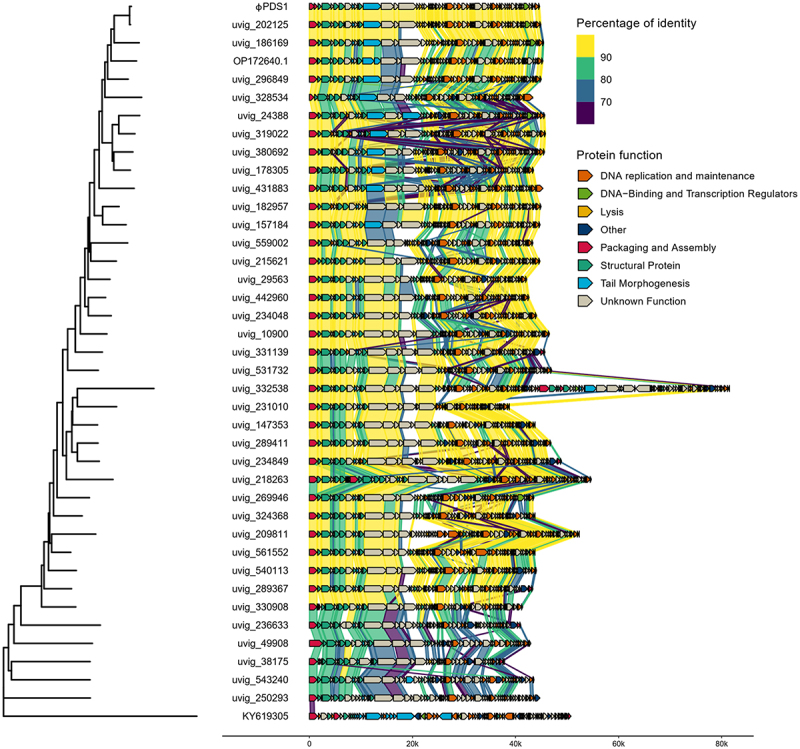
Whole genome comparisons of φPDS1 against members of the different genus clusters within the candidate Paboviridae family identified by VIRIDIC, along with one outgroup member from the Drexlerviridae family (KY619305; Nouzillyvirus ESCO41). The figure shows the percentage of identity of different proteins compared to the closest phylogenetic phage genome. The comparison was performed using Diamond. The color between two genes represents the percentage of identity between both at the protein level. The color of the gene is based on the annotation given by Pharokka. The left-side tree was inferred using the VICTOR tool and the figure was created using the R packages gggenomes and ggtree.
The prevalence of φPDS1 in the human gut was also studied based on the mapping of the φPDS1 genome to metagenomic reads from 965 human gut metagenomes with a coverage higher than 30% (Table S7). φPDS1 showed a prevalence of 31.4% in these samples, suggesting a high prevalence of this phage in the gut. The highest prevalence (59.6%) was observed in samples from Danish individuals (n = 109), while the lowest (22.6%) was noted in samples from Italian individuals (n = 62). No detected φPDS1 was determined in a Tanzanian cohort (n = 40).
2.5. Relative and absolute abundance of φPDS1 in the fecal fermenter
The relative and absolute abundance of φPDS1 in the fermenters was determined under both conditions (with antibiotics vs no antibiotics). Relative abundance was obtained using the shotgun metaviromics dataset already published in our previous work,23 while the absolute abundance was measured by qPCR with the standard curve method. It was evident that the selective enrichment of the host greatly aided the propagation and expansion of φPDS1, which facilitated its isolation (Figure 5). The relative abundance of φPDS1, in relation to the total viral reads sequenced, showed that φPDS1 contributed to ~ 14% of the sequenced reads in the later time point samples collected from the antibiotic-treated vessel (Figure 5(a)). In contrast, less than 1% of reads belonged to φPDS1 in the untreated antibiotic vessel. Absolute quantification using qPCR also confirmed an increase in φPDS1 abundance over time. At the beginning of the fermentation (T0), the abundance of the phage was similar in both vessels (~1 × 104 copies/ml). However, at the later time points the phage achieved a titer of ~ 6 × 108 copies/ml in the antibiotic-treated vessel, while the titer in the non-treated antibiotic vessel was two logs lower at ~ 5 × 106 copies/ml (Figure 5(b)). The results obtained for the whole virome, which were already published,23 support the φPDS1 selection via antibiotic treatment, as a reduction in virome diversity indices was documented in the presence of antibiotics.
Figure 5.
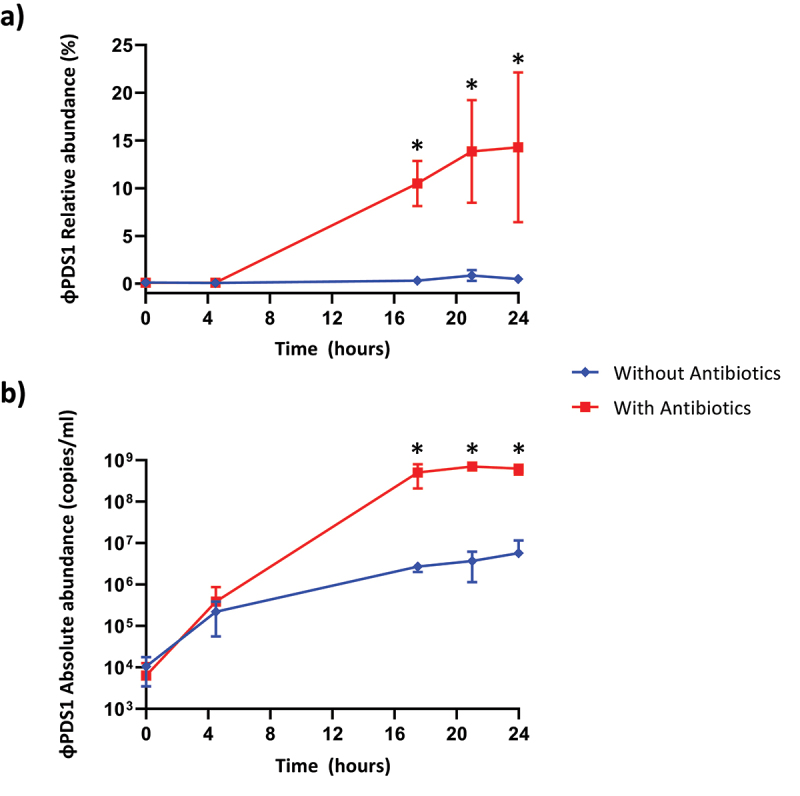
Abundance of φPDS1 in the fermenter vessel with and without selective conditions showing that the selective conditions greatly aided φPDS1 propagation. (a) Relative abundance of φPDS1 with and without antibiotics obtained from the total viral reads sequenced. (b) Absolute quantification of φPDS1 via qPCR targeting a segment of the phage DNA polymerase. Titre determined in copies/ml using the standard curve method. Error bars represent standard deviation (n = 3). * indicates statistically significant differences between vessels, with a p-value <0.05 after performing an unpaired t-test or the Mann – Whitney U test, depending on the data distribution (normal or non-normal, respectively).
2.6. Biological characterization of φPDS1
Transmission electron micrographs of the φPDS1-rich lysate showed that this phage has a characteristic siphovirus-like morphology with a long, non-contractile tail (Figure 6(a)). The arrow-shaped tail tip of the phage was unusual and larger than typically observed for other siphoviruses. The capsid diameter was approximately 53 ± 2.0 nm and tail length 150 ± 10.0 nm.
Figure 6.
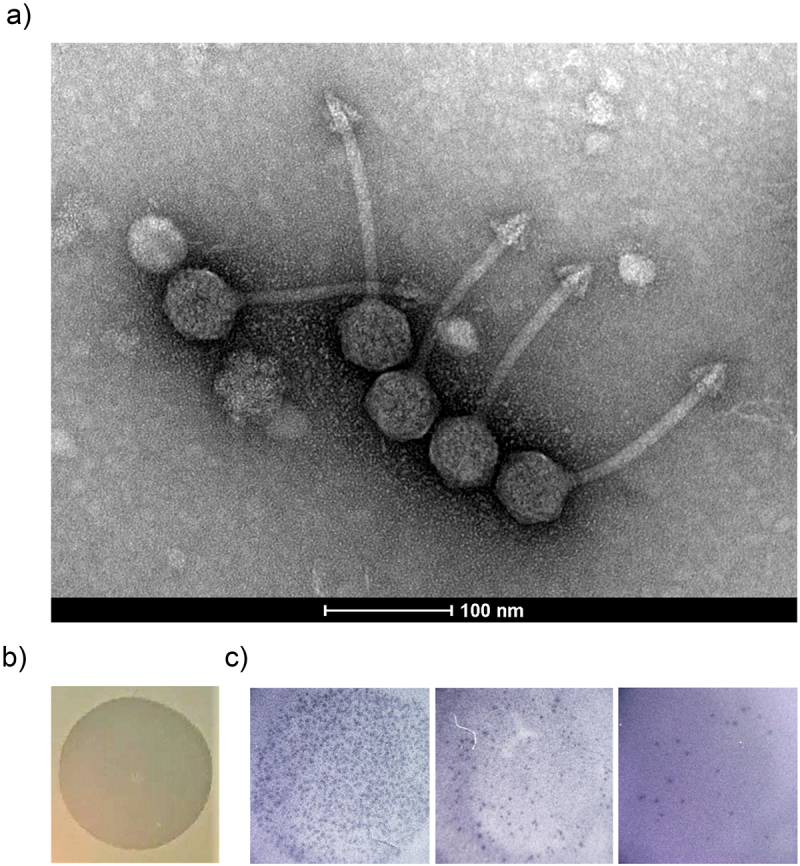
Characterisation and visualization of φPDS1 and formed spots and plaques. (a) Transmission electron micrograph of φPDS1 generated from the enriched lysate, stained with uranyl acetate showed that φPDS1 has a siphovirus morphology. The capsid diameter is approximately 53 ± 2.0 nm, and the tail length is 150 ± 10.0 nm. (b) Spot morphology with incomplete clearing. (c) Visualization of φPDS1 plaques from a spot assay at different titers using a stereoscopic microscope.
Spots on lawns produced by high titers of the phage were readily visible, but incomplete clearing was also observed (Figure 6(b)). Colonies within zones of incomplete clearing were picked and streaked up to five times. PCR analysis was performed to examine the presence of φPDS1, but all colonies tested were negative for the phage. φPDS1 also produces plaques on its sensitive host, but they are pinprick in size making visualization and enumeration challenging to the naked eye. Individual plaques after a spot assay were visualized through a stereoscopic microscope allowing for rough pfu enumeration (Figure 6(c)). However, qPCR allows for a more precise quantification.
A φPDS1 one-step growth curve was performed in triplicate with a MOI of ~ 1 (Figure 7(a)). The phage has a latent period of approximately 90 minutes with a relatively small burst size of ~ 23 copies per infected cell. Continuous co-culture of φPDS1 with its bacterial host showed that after 24 hours the phage titer recovered to ~ 1 × 1010 copies/ml following initial reductions consistent with dilution associated with sub-culturing (Figure 7(b)). By day two, the titer reduced to ~ 4 × 108 copies/ml and remained at this approximate titer for subsequent rounds of sub-culturing (Figure 7(b)). This suggests that the presence of the phage selects for an increase in non-phage permissive host variants but does not render the culture fully resistant. The bacterial pellet collected at the end of the experiment was serially streaked three times. PCR confirmed that all colonies were φPDS1 negative.
Figure 7.
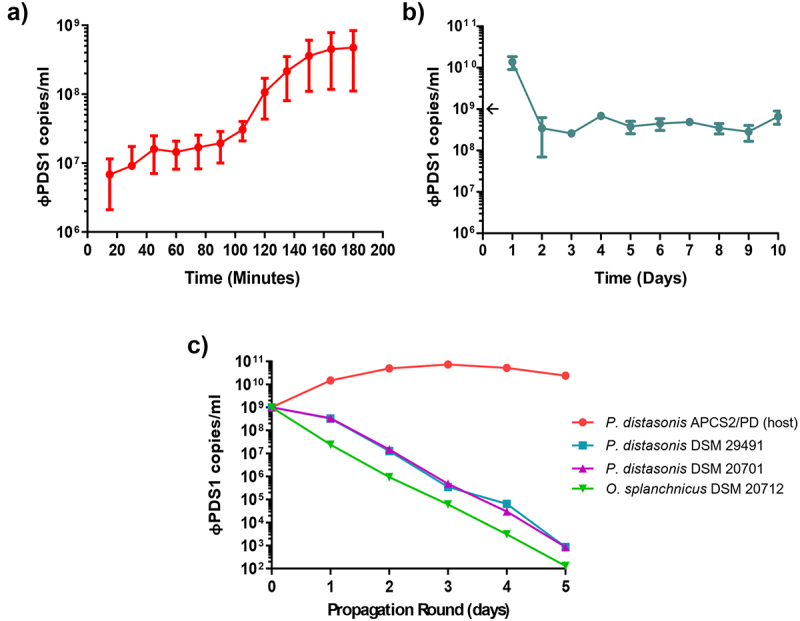
Biological characterization of φPDS1. (a) φPDS1 one-step growth curve. Sampling was performed every 15 minutes over 3 hours. The latent period was 90 minutes, and the burst size was ~ 23 copies per infected cell. (b) The titer of φPDS1 following continuous co-culture on the host over 10 days showing the persistence of the phage with its host. (c) Liquid propagation of φPDS1 on two commercial P. distasonis strains (DSM 29491 and DSM 20701) and O. splanchnicus (DSM 20712) was examined over five days with host P. distasonis APCS2/PD used as a control. Titres were determined in copies/ml via qPCR for all the experiments. Arrows on the y-axis indicate phage titer, after dilution, on initiation of the experiment. Error bars indicate standard deviation (n = 3).
φPDS1 showed stability within a temperature range of −80°C to 45°C, and a pH range of 3 to 11. Its activity decreased at 55°C and 65°C, resulting in the production of less clear spots on host lawns. The activity was completely lost at 80°C, and under extremely acidic (pH 1–2) and alkaline (pH 12–14) pH conditions.
We examined the ability of the φPDS1 to propagate on two P. distasonis strains obtained from a culture collection, DSM 29491 and DSM 20701, and an O. splanchnicus strain, DSM 20712, chosen following host-genus prediction analyses. The phage failed to propagate on the tested strains despite efficient propagation on P. distasonis APCS2/PD, as determined by qPCR (Figure 7(c)). The host range of φPDS1 was also examined using these strains, along with other strains from the Bacteroidales order (P. goldsteinii JCM13446, B. intestinalis APC919/174, B. xylanisolvens APCS1/XY and B. thetaiotaomicron VPI-5482), and no activity was observed against any of them.
The resistant mutation rate test found that ~ 22% of P. distasonis APCS2/PD cells are resistant on initial exposure to φPDS1. This is consistent with lack of clearing of broth cultures and hazy spots on lawns. Additionally, we tested whether the percentage of resistant colonies was maintained when the bacteria had been in contact with the phage previously. The same experiment was carried out by plating P. distasonis APCS2/PD that had been co-cultured with φPDS1 for 48 hours. The results showed that the percentage of resistant colonies after being in contact with the phage for 48-hours increased by approximately ~ 22% to ~ 95%. This finding suggests that a phase variable mechanism may be involved in the persistence of the phage without impacting the number of bacteria.
In the absence of the phage, the host appeared to form two colony morphologies: larger round creamy colonies and smaller granular colonies. A greater count of the former morphology type was present on the phage-seeded plate, suggesting that possible phenotypic alterations have a role to play in morphology changes and the observed resistance. When φPDS1 was screened against lawns of these creamy colonies the zones of clearing were cloudier compared to spots that formed on lawns of P. distasonis APCS2/PD which was not recently exposed to the phage. The resistant colonies were re-streaked and colony PCR confirmed that all were φPDS1 negative.
3. Discussion
In recent years, many studies have revealed interesting insights into the human gut virome. However, most phages continue to exist only as sequence “dark matter”. Phages that have solely been characterized in silico can only provide limited information about their biological properties. Isolating and characterizing these phages is crucial for advancing our understanding of phage-host interactions in the human gut. The human gut virome is largely composed of dsDNA phages belonging to the recently established order Crassvirales,12 as well as ssDNA phages of the Microviridae family.18,27,28 However, the bacterial hosts for the majority of these phages remain unidentified. CRISPR-spacer analyses of virulent gut phages have indicated a notable association with hosts from the Clostridiales and Bacteroidales orders.18 To address this knowledge gap and to potentially shed light on phage-host interactions, the current study aimed to isolate novel human gut phages, with a specific focus on Bacteroidales due to the significant role and prevalence of this bacterial order within the human gut ecosystem. The majority of Bacteroidales species are classified within the genera Bacteroides and Parabacteroides, which are of significant importance and highly abundant in the human gut, where they have been associated with both health and disease.16,17 Although there have been multiple reports on phages infecting Bacteroides,14,20,23,29–33 only a limited number of studies have examined phages infecting Parabacteroides. Two Parabacteroides-associated prophages, ϕParabacteroides YZ-2015a and ϕParabacteroides YZ-2015b, were first detected in Sphagnum-dominated peat viromes. These prophages infect P. distasonis and P. merdae, respectively, and both belong to the ssDNA Microviridae family.34 Additionally, seven more Parabacteroides prophages of the Microviridae family were discovered in the gut of Ciona robusta, a marine vertebrate.35 Recently, Shen et al. (2023) have isolated 22 Parabacteroides phages from a collection of gut phage isolates.26 These phages were found to infect P. faecis, P. distasonis, and P. merdae and were isolated mainly from municipal sewage. Notably, one of these phages, PD491P1, showed a high prevalence in the human gut after screening 1,333 publicly available human gut metagenomic samples.26 This confirms the relevance of studying Parabacteroides phages in the human gut and highlights the limited knowledge currently available.
The selective enrichment method implemented in our study proved effective in facilitating the growth of Bacteroidales (Figure S1), resulting in the successful isolation of a novel lytic P. distasonis phage, φPDS1. This phage represents the first P. distasonis-targeting siphovirus to be isolated from human feces, biologically characterized, and deposited in NCBI taxonomy databases (NCBI:txid2709316). P. distasonis is a gram-negative, obligate anaerobe with rod-shaped morphology that is predominantly associated with the human gut.36 These bacteria have been recognized as important commensals with potential anti-inflammatory roles in the gut, contributing to the alleviation of metabolic disorders and obesity.17,37–39 Reduced abundance of the Parabacteroides genus has also been observed in the feces of patients suffering from IBD, as well as in patients with multiple sclerosis and rheumatoid arthritis.40–43 These findings could suggest potential for P. distasonis as a next-generation probiotic.44 However, several other studies have also linked P. distasonis to disease states, suggesting its potential role as an opportunistic pathogen.17,22 It has been reported that P. distasonis is the most abundant bacterial species in fecal samples collected from individuals suffering from Crohn’s disease (CD) and has been associated with enhanced colitis in DSS-induced mice.45,46 Furthermore, P. distasonis has been isolated from a lesion removed from the ileum of a CD patient.47 Considering the health and disease implications associated with this bacterial species, P. distasonis and φPDS1 should prove to be an intriguing phage-host pair for further study.
Annotation of the φPDS1 genome revealed that nearly half of the genes remain unidentified (Figure 2). However, among the successfully annotated genes, there were several with noteworthy functions. One of particular interest is gp_26, which encodes a quorum sensing (QS)-regulated transcription factor associated with virulence. QS is a bacterial phenomenon that allows cells to control the expression of specific genes in a population-density-dependent manner through cell-to-cell communication.48 This mechanism plays a crucial role in various processes such as virulence and biofilm formation. Furthermore, φPDS1 gene gp_34 encodes radical SAM enzymes, which are associated with the synthesis and metabolism of many cell compounds, and also they are involved in pathways that lead to the synthesis of the key players of QS molecules, known as autoinducers.48 Various QS response regulator homologs have been identified in sequenced phage genomes deposited in NCBI databases,49 and several examples of phages hijacking this process have been described. For instance, the Vibrio cholerae infecting phage, VP882, encodes a QS receptor homologous to that of its host. As a result, the phage can “listen in” on its hosts and monitor population densities thus allowing an informed lytic/lysogenic lifestyle switch. When the host cell density is high, the phage initiates lysis resulting in optimal propagation.50 Additionally, QS has been demonstrated to interfere with host phage defenses, aiding the infection process in a lytic Pseudomonas aeruginosa phage.51 Furthermore, φPDS1 encodes a single-stranded binding protein (SSB) which suggests that it does not rely on a host SSB for replication.52 Moreover, genes gp_25 and gp_31 encode Nin family recombinases, also known as orf and rap, which are associated with lytic growth.53,54 Another interesting feature is the presence of an auxiliary metabolic gene, MazG, encoded by gene gp_39. Such genes are of host origin and allow phages to alter host metabolic processes to their advantage. MazG genes have been mostly detected in marine phages. They are associated with both lytic and temperate lifestyles, and have been linked to maintenance of phage propagation in starved host cells by aiding metabolism.55,56 Based on the information presented, it is possible that φPDS1 possesses mechanisms that could potentially impact its host through the alteration of host metabolic processes and interference with host phage defenses. However, further studies are needed to accurately determine which mechanisms are truly involved in the interaction with its host.
Host-genus prediction analyses suggested that φPDS1 has evolved to infect P. distasonis. CRISPR spacers detected another potential member of the Bacteroidales order, Odoribacter splanchnicus. Some specific strains of P. distasonis (DSM 20701 and DSM 29491) and O. splanchnicus (DSM 20712) were obtained from DSMZ to test φPDS1 strain specificity. However, none of these strains were able to support the propagation of φPDS1 (Figure 7(c)), or produce clear spots on the lawns of these Bacteroidales strains. This suggests that φPDS1 is highly strain-specific in its infection strategy and has specialized to propagate on the P. distasonis APCS2/PD strain from which it was isolated.
A candidate phage family, Paboviridae, within the Caudoviricetes class, has been recently proposed.26 This family infects bacteria belonging to the Parabacteroides, Alistipes, and other genera within the Bacteroidales order in the human gut. Shen and colleagues identified this family based on the large terminase subunit of two isolated phages, PD491P1 and AS73P1, which infect Parabacteroides and Alistipes, respectively. Shen et al. included 258 complete phage genomes from the Gut Phage Database (GDP) in this candidate family. However, despite comparing the sequence identity of these two phages with φPDS1, they did not incorporate φPDS1 into their phylogenetic analysis. Our phylogenetic analysis has shown that φPDS1 could also be included in the candidate family Paboviridae, and we propose the formation of a new candidate genus, Sagittacolavirus, within this family after performing a VIRIDIC analysis. This genus includes φPDS1, PD491P1, and other 58 phage complete genomes from the GDP. While they found a 97.37% sequence identity by BLASTn between φPDS1 and PD491P1, our VIRIDIC analysis based on intergenomic similarities, revealed 78.5%, suggesting they could be clustered within the same genus but not at the species level. Regarding phage morphology, φPDS1 and PD491P1 are very similar, both exhibiting the characteristic arrowhead-like tail tip. This suggests that this feature may be a distinctive characteristic of this phage genus. Shen and colleagues described a similarity percentage of 82.82% between Alistipes phage AS73P1 and φPDS1 using BLASTn.26 However, our VIRIDIC results showed 66.83% of similarity, which prevented its inclusion within the proposed genus Sagittacolavirus. This difference could be related to the distinct host genera target (Alistipes and Parabacteroides), and differences in phage morphology, since although both are long-tailed, it appears that AS73P1 does not possess the characteristic arrowhead-like tail tip. The prevalence of φPDS1 in the human gut (31.4%) was shown to be higher than that of PD491P1 (29.5%) and AS73P1 (19.5%)26 after performing the same analysis. The only difference was that the number of human gut metagenomes used to map our phage, which was slightly lower (965 vs 1,333). This result supports the notion that this family of phages appears to be very prevalent in the human gut.26
φPDS1 failed to clear liquid cultures of their host despite efficient propagation, and zones of clearing remained slightly hazy. The phage was also unable to form stable lysogens, and no integrase was detected in its genome, strongly indicating that the phage is lytic. φPDS1 also demonstrated the ability to stably coexist with its host during serial co-culturing. Interestingly, previous studies have observed similar behavior in apparently lytic phages, where they fail to clear the liquid culture of their hosts. For instance, the lytic phage φcrAss002, persists and attains high titers but lacks the ability to form plaques or clear liquid cultures of its host B. xylanisolvens.23 More similar to φPDS1, the B. intestinalis phage, φcrAss001,15,20 and B. thetaiotaomicron phages14,57 can form large plaques on their hosts and propagate at high titers, but also do not produce clear liquid cultures. This phenomenon could be explained by the phase-variable expression of host capsule polysaccharide loci and surface features, which create transient phenotypic heterogeneity within an isogenic population.15 This heterogeneity results in a mixture of phage-permissive and non-permissive host variants, thus allowing both the phage and host to coexist.14,15 This could explain the observed difference in the percentage of resistant cells before (~22%) and after a 48-h co-culture with φPDS1 (~95%). A potential phase variable mechanism may be promoting the growth of bacterial subpopulations that are resistant, leading to a reduction in the sensitive subpopulations. However, this level of resistance may be sufficient for φPDS1 and its host to coexist. Phase-variable regions have been previously reported among gut P. distasonis strains.58–60 Phase variation of surface structures involves the reversible inversion of DNA regions containing promoters, resulting in the expression or suppression of downstream genes depending on the orientation.61 Invertible promotor regions are characteristic of Bacteroidales species residing in the human gut and are generally not conserved among Bacteroidales occupying other niches.59,61 This highlights the importance of host factors in influencing phage-host interactions. Further investigation, such as transcriptomics analysis, may provide insights into the role of these features and help understand the mechanisms mediating the interaction between φPDS1 and P. distasonis APCS2/PD.
In conclusion, we report the isolation, biological and in silico characterization of φPDS1. This phage is the first characterized lytic siphovirus to be isolated from the human gut that infects P. distasonis, and it has been deposited in the NCBI Taxonomy database, making it the first of its type to be documented. Additionally, we propose that φPDS1 may be classified in the new candidate genus Sagittacolavirus within the candidate family Paboviridae and the class Caudoviricetes. Considering the health and disease associations of P. distasonis, this phage-host pair merits further investigation which may also provide interesting insights into phage-host interactions in the human gut.
4. Material and methods
4.1. Donor recruitment and sample collection
A healthy female donor in her forties, designated as subject ID: 924, was recruited for fecal sample donation in accordance with the study protocol APC055 and ethics approved by Cork Research Ethics Committee. This individual had been previously identified as a persistent carrier of crAss-like phages, which infect commensal bacteria of the order Bacteroidales.18,23,62 Given these findings, the subject was deemed a potential donor for the in vitro isolation of potential novel phages that infect hosts of the Bacteroidales order.
4.2. Fecal fermentation with selective antibiotic enrichment
The fecal sample was processed immediately upon receipt into frozen standard inoculum (FSI) as described elsewhere.23 Fermentation medium was prepared as detailed in Guerin et al. (2018).62 The FSI was aliquoted into three volumes to perform the fermentations in triplicate, and they were run in batch format over 24 hours at 37°C following the same conditions outlined in Guerin et al. (2021).23 Two fermenter vessels were established in parallel, one with the addition of antibiotics (7.5 μg/ml vancomycin (Sigma-Aldrich, MO, USA) and 100 μg/ml kanamycin (Sigma-Aldrich, MO, USA)) to the YCFA-GSCM broth post-autoclaving, while the other served as a control without the incorporation of antibiotics. The antibiotics were chosen to selectively promote the growth of Bacteroidales via the elimination of gram-positive bacteria (vancomycin) as well as to limit faster growing facultative anaerobes (kanamycin).23 Samples were collected at specific time points: 0, 4.5, 17.5, 21, and 24 hours and were directly processed through centrifugation at 4,700 rpm at 4°C for 10 minutes. The resulting supernatants were filtered through a 0.45 μm pore polyethersulfone (PES) membrane filter (Sarstedt, Nümbrecht, Germany) and were stored at 4°C. The remaining bacterial-rich pellets were stored at −80°C.
4.3. Extraction of total DNA, 16S rRNA gene sequencing library preparation, and analysis of 16S rRNA gene sequencing data
Fecal pellets generated following centrifugation of fermentation samples were used to extract total DNA as described elsewhere.23 PCR amplification of hypervariable regions V3-V4 of bacterial 16S rRNA gene and sequencing library preparation were carried out following the procedure outlined in Shkoporov et al. (2018).63 FastQC (v0.11.5) was utilized to visualize the quality of the raw reads. Trimmomatic (v0.36) was employed to perform quality filtering on the reads.64 The filtered reads were imported into R (v3.4.3) and subjected to error analysis with the DADA2 package (v1.6.0).65 Detected errors were corrected through additional quality filtering and trimming, resulting in the generation of unique Ribosomal Variant Sequences (RSVs). Chimera filtering was performed on the RSVs using both the de novo and reference-based chimera filtering implemented in USEARCH (v8.1.1861) with the ChimeraSlayer gold database (v20110519).66 The remaining RSVs were classified with mothur (v1.34.4)67 against the RDP database (v11.4), as well as classified with SPINGO to species level.68 The resulting RSVs were further analyzed to assess the relative abundance of bacterial genera within the antibiotic- and non-antibiotic-containing vessels. R package ggplot2 (v2.2.1) was used to visualize the abundance data.
4.4. Novel phage-host pair screening
To selectively isolate anaerobic Bacteroidales species, ten-fold serial dilutions of the FSI were prepared using fresh Fastidious Anaerobe Broth (FAB) (Neogen, MI, USA). 100 μl of each was spread plated onto plates with different media containing vancomycin (7.5 µg/ml) and kanamycin (100 µg/ml). The media used were: Fastidious Anaerobe Agar (FAA) (Neogen, MI, USA), YCFA-Agar, and Columbia Blood Agar (CBA) (Oxoid, Hampshire, UK) with 5% sheep blood (TCS Biosciences, Birmingham, UK) supplemented with 25 μg/ml hemin (Sigma-Aldrich, MO, USA) and 100 μg/ml vitamin K (Sigma-Aldrich, MO, USA). The dilution plates were incubated anaerobically at 37°C for 48 hours. Colonies were restreaked, and the isolated colonies were incubated both aerobically and anaerobically to ensure the absence of aerobes. An estimation of species identification was carried out via Sanger sequencing of the 16S rRNA region using the universal bacterial primers 27F primer 5′-AGAGTTTGATCCTGGCTCAG-3′ and 1492 R primer 5′-GGTTACCTTGTTACGACTT-3′. Samples were prepared following the guidance for the LightRun Tube service (GATC Biotech AG, Ebersberg, Germany). BLASTn analysis was conducted on the obtained sequences against the NCBI 16S ribosomal RNA sequences (Bacteria and Archaea) database using standard parameters.
48 pure cultures of Bacteroidales were used in the phage-host pair screening. Overnight cultures were prepared in 10 ml of FAB and an agar overlay method was used for screening. From each of these cultures, 300 µl was added to 0.3% FAA agar overlay (0.3% agar w/v), with MgSO4 and CaCl2 (1 mM final concentration). This mixture was poured onto pre-prepared FAA base agar (1.5% agar w/v), and 5 µl of the filtered fermentation supernatants collected from the antibiotic and non-antibiotic containing vessels at 21 h were spotted onto the lawns of each culture and dried. The original fecal filtrate, prepared from subject ID: 924 feces, was also spotted on the lawns. The plates were incubated anaerobically at 37°C for 48 hours. Formed spots were picked using an inoculation loop, placed into 100 µl of SM buffer (1 M Tris HCl pH 7.5, 5 M NaCl, 1 M MgSO4), vortexed and incubated at room temperature for 5 hours. The resuspended spots were spun in a desktop centrifuge at maximum speed for 10 minutes, and the supernatant was filtered through a 0.45 μm pore PES membrane filter. Ten-fold serial dilutions of the lysates were prepared in SM buffer. Spot assays were then repeated using each lysate on lawns of the culture on which it was originally spotted on. Plaque assays were also performed using 3 ml of 0.3% FAA agar (0.3% agar w/v), with the addition of MgSO4 and CaCl2 (1 mM final concentration), 300 µl of each overnight culture on which spots formed and 50 µl of phage dilution. Following this, spot picking and spot assays were repeated a third time. Spot formation was consistently observed in all cultures identified as Parabacteroides distasonis through Sanger sequencing of the 16S rRNA region. Four P. distasonis cultures, on which spot formation was least cloudy, were chosen for phage enrichment. These cultures were designated as FAA-S8, FAA-B5, FAA-B8, and CBA-S2 (Table S1).
4.5. Novel phage enrichment and shotgun sequencing of the phage and fermenter virome
Six rounds of serial enrichment of the phage were performed over six days on the P. distasonis cultures to increase the titer. Each round of propagation was performed using P. distasonis at OD600 = ~0.2 which was reached approximately 5 to 6 hours post sub-culturing of 100 μl of overnight culture in 10 ml of fresh FAB plus MgSO4 and CaCl2 (1 mM final concentration) with anaerobic incubation at 37°C. Once the culture was at the desired optical density, 1 ml of phage lysate was added at an unknown titer to the culture and incubated anaerobically overnight at 37°C. The propagations were centrifuged at 4,700 rpm at 4°C for 10 minutes and passed once through a 0.45 μm PES membrane syringe filter. Filtrates were then stored at 4°C. Each round of propagation was performed following the same procedure with 1 ml of the prior phage lysate added to P. distasonis culture at OD600 = ~0.2. To ensure maintenance of phage throughout the serial propagations, spot assays were performed as described above. On completion of the enrichment, spot and plaque assays were repeated to check for improved clearing.
Nucleic acids were extracted from 10 ml of filtered phage lysate generated from the final round of enrichment on each of the P. distasonis cultures. The virus-like particle (VLP) purification and DNA extraction steps were performed as described elsewhere.63 The extracted DNA was then purified using the DNeasy Blood & Tissue Kit according to the manufacturer’s instructions with a final elution volume of 50 μl. The DNA was quantified using Qubit dsDNA HS Assay kit (ThermoFisher Scientific, Vilnius, Lithuania) and was directly subjected to random shotgun library preparation using Nextera XT DNA Library Preparation Kit (Illumina) without preliminary multiple displacement amplification (MDA). Normalization was performed according to the manufacturer’s protocol using the bead-based method. The prepared libraries were sequenced using 2 × 300bp paired-end chemistry on an Illumina MiSeq platform (Illumina, CA, USA) at GATC Biotech AG (Ebersberg, Germany). The quality of the raw reads was analyzed using FastQC (v0.11.5). Removal of Nextera adaptors was performed with Trimmomatic (v0.36)64 with the following parameters: minimum length of 60, a sliding window size of 4 and a minimum Phread score of 33. The trimmed and filtered reads were then assembled into contigs using SPAdes (v1.13.1).69
Additionally, extraction of VLPs after enrichment from 10 ml of collected filtered fermentation supernatants was carried out, following the established protocol described by Shkoporov et al. (2018).63 Sequencing of the fermenter virome and its analysis are described in Guerin et al. (2021).23 The relative abundance of the reads generated following sequencing of the fermenter samples were aligned to the novel phage. Results were expressed as novel phage reads relative to the total number of reads.
4.6. In silico analyses and characterisation of φPDS1
Annotation of the novel phage genome, denoted as φPDS1, was performed using de novo viral genome annotator VIGA.70 Manual functional analyses were performed on the predicted protein-coding sequences using HHPred and BLASTp to generate a more detailed annotation. HHPred annotations were performed with the following databases: PDB_mm_CIF70_18_Jun, Pfam-A_v35, NCBI_CD_v3.19, and TIGRFAMs_v15.0.71 A genomic map of the φPDS1 genome was generated using the Proksee tool with annotations incorporated.72
The genome sequence of φPDS1 was compared with 506 phage genomes from the Gut Phage Database (GPD) of the candidate Paboviridae family. This family is characterized as infecting Parabacteroides, Alistipes, and other bacterial genera within Bacteroidales order in the human gut.26 CheckV (v1.0.1) was used to ensure the completeness of all the genomes.73 The genomes with a completion of at least 95% were selected to minimize bias in the subsequent genomic analysis, resulting in a total of 313 complete phage genomes. VIRIDIC74 was used to calculate virus intergenomic similarities of all complete phage genomes (n = 313) belonging to the candidate family Paboviridae obtained from GPD. The number of complete genomes exceeded the maximum limit allowed in the VICTOR classifier, which is 100 genomes. To address this, we decided to reduce the number of phages for comparison while preserving the maximum diversity in the sample. To achieve this, large terminase genes were identified using Pharokka (v1.3.2).75 Subsequently, the 313 terminases were aligned using MAFFT (v7.520) (linsi algorithm, using the options – dash and – originalseqonly).76 The protein alignment was trimmed using clipkit (v1.3.0) (options, -m kpic).77 Maximum likelihood trees were then inferred from the curated alignment using IQ-TREE (v2.2.0.3) (options -nm 4,000).78 Node support values were assessed using the options -bb 1,000 for ultrafast bootstraps and -alrt 1,000 for SH-aLRT. The best evolutionary model was selected using ModelFinder (BIC criterion).79 The resulting tree was used to select the 80 most diverse genomes (representative genome) with Treemmer (v0.3).80 These representative genomes, along with φPDS1 and 19 Drexlerviridae phages (used as an outgroup), were compared using the VICTOR classifier81 to investigate whether φPDS1 belongs to the candidate family Paboviridae, resulting in the construction of a phylogenetic tree inferred by the Genome-BLAST Distance Phylogeny method with default parameters and D0 formula. A sequence from each of the genus-level clusters obtained in the VIRIDIC analysis was selected as a representative to perform a pairwise comparison, in order to examine synteny and relatedness among these phages and φPDS1. iPHoP tool was used for computational prediction of the host genus of φPDS1 based on its genome sequence with a score cutoff of 90%.82 This tool infers candidate phage-host pairs integrating results from multiple host prediction approaches (blast, CRISPR, WIsH, VHM, PHP and RaFAH).82
To compare the genomic organization of the different phages within the genera defined by VIRIDIC, we randomly selected one genome per genus as a representative of the cluster, and we added one phage as the representative of the outgroup. All phages were annotated using Pharokka v1.3.2.75 Specifically, coding sequences (CDS) were predicted using PHANOTATE. Functional annotation was generated by matching each CDS to the PHROGs, VFDB, and CARD databases using MMseqs2 and PyHMMER. The different genomes were compared with each other using BLASTn (v2.14.0) (options -task blastn, -evalue 1, -max_target_seqs 25,000) for nucleotide full genome comparison and Diamond v2.1.8 (options blastp, -k0, –ultra-sensitive – algo 1) for protein comparison. A phylogenetic tree was inferred using the reference genomes with the VICTOR classifier,81 to arrange the genomes by order of phylogenetic proximity. The comparison figure was generated using the R packages gggenomes and ggtree.83
The prevalence of φPDS1 in the human gut was analyzed by mapping the φPDS1 genome to metagenomic reads from 965 human gut samples from five different publicly available studies (PRJEB5224, PRJEB6997, PRJNA422434, PRJNA392180, PRJNA553191)84–88 using BBmap89 with default parameters. A φPDS1 genome with a coverage of more than 30% was considered to be present in the metagenomic samples. Accession numbers for the sequence read archives and information related to the human gut metagenomic samples are detailed in Table S7.
The host’s complete genome was used to determine its taxonomy using the Genome Taxonomy Database (GTDB) (v214.1) and the associated taxonomic classification toolkit (GTDB-Tk) (v2.3.0) with an average nucleotide identity (ANI) radius > 95%.90
4.7. Transmission electron microscopy
A 50 ml pool of φPDS1 lysate prepared from the serial enrichment was concentrated via ultra-centrifugation using a F65L-6×13.5 rotor (Thermo Scientific, MA, USA) at 120,000 g for a total of 3 hours at 4°C. Finally, the pellets were resuspended in a final volume of 5 ml of SM buffer. The suspensions were then applied to a step gradient of 5 M and 3 M cesium chloride (CsCl) solutions followed by centrifugation at 105,000 g for 2.5 hours at 4°C. The band containing viral particles was collected and subjected to CsCl clean-up steps.62 A spot assay was performed to confirm the presence and titer of φPDS1 in the concentrated purified fraction. Five microliter aliquots of the concentrated viral fraction were applied to Formvar/Carbon 200 Mesh, Cu grids (Electron Microscopy Sciences, PA, USA) with subsequent removal of excess sample by blotting. Grids were then negatively contrasted with 0.5% (w/v) uranyl acetate and examined at UCD Conway Imaging Core Facility (University College Dublin, Dublin, Ireland) by Tecnai G2 12 BioTWIN transmission electron microscope.
4.8. Quantitative real-time PCR (qPCR), primer and standard development and quantification of φPDS1
The φPDS1 major capsid protein was chosen for qPCR primer and standard development. Primers (Fwd 5‘GGAACAACGGGACGATTG-3’ and Rev 5’-CAATCACGGACGCAATAGG-3’) were designed using PERL Primer software and CLC Sequence Viewer 8.0. These primers detect φPDS1 producing a 193 bp PCR product with the following conditions: initial denaturation at 95°C for 5 minutes, then 35 cycles of 95°C for 20 seconds, 60°C for 20 seconds, 72°C for 20 seconds and a final cycle at 72°C for 1 minute. To develop standards for qPCR calibration curves, products from these primers were cloned into pCR2.1-TOPO TA vector (ThermoFisher Scientific, Vilnius, Lithuania). Extracted plasmids were quantified using Qubit dsDNA BR Assay kit (ThermoFisher Scientific, Vilnius, Lithuania) and diluted to 109 copies/μl based on molar mass of DNA. Ten-fold serial dilutions of the plasmids were used to build a standard calibration curve. To quantify φPDS1 in the fecal fermenters, qPCR of viral nucleic acids from filtered fermentation supernatants was performed at all time points in triplicate. This was carried out in a 15 μl reaction volume using SensiFAST SYBR No-ROX mastermix (Bioline, London, UK) in a LightCycler 480 thermocycler (Roche) with the following conditions: initial denaturation at 95°C for 5 minutes, then 45 cycles of 95°C for 20 seconds, 60°C for 20 seconds and 72°C for 20 seconds. Resulting Ct-values were converted to copies/ml based on the generated calibration curves.
4.9. Shotgun sequencing of P. distasonis APCS2/PD using Illumina and Oxford Nanopore platforms
Bacterial genomic DNA was extracted from 10 ml overnight cultures of P. distasonis APCS2/PD using phenol/chloroform with precipitation in 3 M sodium acetate and cold absolute ethanol for the generation of long- and short-read sequences for hybrid assembly. DNA was quantified using Qubit BR DNA Assay Kit. Short-read shotgun sequencing of the extracted DNA was carried out using the Accel-NGS 1S Plus DNA Library Kit (Swift Biosciences, MI, USA) and Illumina HiSeq 4000 technology following manufacturer’s instructions. Long-read Oxford Nanopore library preparation was performed according to the manufacturer protocol for Rapid Barcoding Sequencing Kit (SQK-RPK004; Oxford Nanopore Technologies, UK) with the adaptions described by Guerin et al., (2021).23 Pooled samples were loaded into SpotON Flow Cell (Oxford Nanopore Technologies, UK) and MinION sequenced for 48 hours (Oxford Nanopore Technologies,UK). Hybrid assembly of quality-filtered and trimmed Illumina and raw Nanopore reads was conducted by hybridSPAdes (v1.13.1).91 Nine scaffolds greater than 1 kb were generated and were manually curated, joined, and circularized using CLC Sequence Viewer. The assembled and circularized genome and associated plasmid were then submitted to NCBI Prokaryotic Genome Annotation Pipeline. The GenBank file for the genome and associated plasmid was visualized using Proksee.72
4.10. Biological characterization of φPDS1
A one-step growth curve was performed in triplicate to determine the latent period and burst size of φPDS1. An early logarithmic phase culture of P. distasonis APCS2/PD was infected with φPDS1 with a multiplicity of infection (MOI) of ~ 1. Following incubation at room temperature for 5 minutes, the infected culture was centrifuged at 5,000 rpm for 15 minutes at 20°C, and the pellet was resuspended with FAB. The phage-host pair were maintained at 37°C under anaerobic conditions for 3 hours with 1 ml sample collection every 15 minutes. Samples were centrifuged and the supernatants were filtered through 0.45 µM pore syringe filters. Analysis was performed using absolute qPCR with the standard curve method and primers as described above.
The calculation of the resistant mutation rate was carried out by pouring 3 ml of 0.3% FAA molten overlay containing different 10-fold serial dilutions of an overnight culture of P. distasonis APCS2/PD (100 µl), along with 100 µl of high-titer φPDS1 lysate (>1010 copies/ml) on FAA agar plates. Negative controls were prepared in the same manner, excluding phage. The plates were incubated anaerobically for 48 hours at 37°C. The resistant mutation rate was calculated as the percentage of colonies on the phage-containing plate versus counts for the equivalent negative control. To assess the maintenance after the cohabitation of P. distasonis APCS2/PD with φPDS1, the same experiment was conducted plating P. distasonis APCS2/PD that had been co-cultured with φPDS1 for 48 hours. Colony morphology was observed, and sixteen colonies were picked and restreaked four times. Standard PCR was performed using primers specific to φPDS1 as described above to check for the potential integration of the phage into the host genome. Spot assays were always performed to check for host resistance after phage exposure.
The thermolability and pH tolerance of φPDS1 were examined after treating it at different temperatures (−80°C, −20°C, 4°C, 25°C, 37°C, 45°C, 55°C, 65°C, and 80°C) and pH (1–14) ranges for 24 hours. The determination of the phage’s stability was based on its ability to produce spots using the overlay method after the treatments.
Co-culturing of φPDS1 and P. distasonis APCS2/PD was performed by serial sub-culturing over ten days in triplicate. This was initiated using 1 ml of φPDS1 lysate at 1010 copies/ml that was introduced into 10 ml of P. distasonis APCS2/PD culture in FAB at OD600 = 0.2. Subsequent rounds of sub-culturing were performed by introducing the prior co-culture into 10 ml fresh FAB at a ratio of 1:50. φPDS1 titer was quantified using qPCR. The bacterial pellet collected, following generation of the final phage lysate on day ten, was serially streaked three times, and the resultant colonies were examined for the presence of the phage by standard PCR.
The ability of the φPDS1 to infect other commercially available P. distasonis strains, DSM 29491 and DSM 20701 (ATCC 8503), and Odoribacter splanchnicus DSM 20712 was investigated via liquid propagation using the same method as described above for phage enrichment. Propagation of φPDS1 on P. distasonis APCS2/PD was performed in parallel as a control. Propagation of the phage on the other bacterial strains was examined via absolute qPCR as described above. The host range of φPDS1 was also tested by spotting serial dilutions of the phage suspension onto FAA overlays seeded with these bacterial strains. The host range test also included other strains from the Bacteroidales order: P. goldsteinii JCM13446 (EU136697), B. intestinalis APC919/174 (CP041379), B. xylanisolvens APCS1/XY (CP042282) and B. thetaiotaomicron VPI-5482 (CP092641).
Supplementary Material
Funding Statement
This research was performed with the financial support of the Science Foundation Ireland (SFI) under Grant Numbers [SFI/12/RC/2273]; Science Foundation Ireland Spokes Programme which is co-funded under the European Regional Development Fund under Grant Number [SFI/14/SP APC/B3032] and a research grant from Janssen Biotech, Inc, and the European Union’s Horizon 2020 Research and Innovation Programme under the INSPIRE COFUND Marie Skłodowska Curie grant agreement No. [101034270].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Author contributions
A.C.M. conducted wet lab work, data analysis and wrote the manuscript; E.G. conducted wet lab work; R.D. and S.R.S. performed bioinformatics and data analysis; L.A.D. managed the project; R.P.R., A.N.S. and C.H. secured funding and supervised the project. All authors reviewed and edited the manuscript.
Data availability statement
All data generated or analyzed during this study are included in this report and in the supplementary material. The genome of φPDS1 is deposited into GenBank under accession MN929097 (assembled and annotated genome). The genome of the bacterial host of φPDS1, Parabacteroides distasonis APCS2/PD, is deposited under the following accession codes: BioProject PRJNA556872, GenBank CP042285 (assembled and annotated genome) and associated plasmid pPDS2–1, GenBank CP042284.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2023.2298254
References
- 1.Van Belleghem JD, Dąbrowska K, Vaneechoutte M, Barr JJ, Bollyky PL.. Interactions between bacteriophage, bacteria, and the mammalian immune system. Viruses. 2018;11(1):10. doi: 10.3390/v11010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gogokhia L, Buhrke K, Bell R, Hoffman B, Brown DG, Hanke-Gogokhia C, Ajami NJ, Wong MC, Ghazaryan A, Valentine JF, et al. Expansion of bacteriophages is linked to aggravated intestinal inflammation and colitis. Cell Host Microbe. 2019;25:285–299.e8. doi: 10.1016/j.chom.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shkoporov AN, Turkington CJ, Hill C. Mutualistic interplay between bacteriophages and bacteria in the human gut. Nat Rev Microbiol. 2022;20(12):737–24. doi: 10.1038/s41579-022-00755-4. [DOI] [PubMed] [Google Scholar]
- 4.Zuo T, Sun Y, Wan Y, Yeoh YK, Zhang F, Cheung CP, Chen N, Luo J, Wang W, Sung JJY, et al. Human-gut-DNA virome variations across geography, ethnicity, and urbanization. Cell Host Microbe. 2020;28(5):741–751.e4. doi: 10.1016/j.chom.2020.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Gregory AC, Zablocki O, Zayed AA, Howell A, Bolduc B, Sullivan MB. The gut virome database reveals age-dependent patterns of virome diversity in the human gut. Cell Host Microbe. 2020;28(5):724–740.e8. doi: 10.1016/j.chom.2020.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shareefdeen H, Hill C. The gut virome in health and disease: new insights and associations. Curr Opin Gastroenterol. 2022;38(6):549–554. doi: 10.1097/MOG.0000000000000885. [DOI] [PubMed] [Google Scholar]
- 7.Tobin CA, Hill C, Shkoporov AN. Factors affecting variation of the human gut phageome. Annu Rev Microbiol. 2023;77:363–379. doi: 10.1146/annurev-micro-032421-105754. [DOI] [PubMed] [Google Scholar]
- 8.Clooney AG, Sutton TDS, Shkoporov AN, Holohan RK, Daly KM, O’Regan O, Ryan FJ, Draper LA, Plevy SE, Ross RP, et al. Whole-virome analysis sheds light on viral dark matter in inflammatory bowel disease. Cell Host Microbe. 2019;26(6):764–778.e5. doi: 10.1016/j.chom.2019.10.009. [DOI] [PubMed] [Google Scholar]
- 9.Camarillo-Guerrero LF, Almeida A, Rangel-Pineros G, Finn RD, Lawley TD. Massive expansion of human gut bacteriophage diversity. Cell. 2021;184(4):1098–1109.e9. doi: 10.1016/j.cell.2021.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nayfach S, Páez-Espino D, Call L, Low SJ, Sberro H, Ivanova NN, Proal AD, Fischbach MA, Bhatt AS, Hugenholtz P, et al. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nat Microbiol. 2021;6(7):960–970. doi: 10.1038/s41564-021-00928-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bin Jang H, Bolduc B, Zablocki O, Kuhn JH, Roux S, Adriaenssens EM, Brister JR, Kropinski AM, Krupovic M, Lavigne R, et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat Biotechnol. 2019;37(6):632–639. doi: 10.1038/s41587-019-0100-8. [DOI] [PubMed] [Google Scholar]
- 12.Turner D, Shkoporov AN, Lood C, Millard AD, Dutilh BE, Alfenas-Zerbini P, van Zyl LJ, Aziz RK, Oksanen HM, Poranen MM, et al. Abolishment of morphology-based taxa and change to binomial species names: 2022 taxonomy update of the ICTV bacterial viruses subcommittee. Arch Virol. 2023;168(2):74. doi: 10.1007/s00705-022-05694-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hyman P. Phages for phage therapy: isolation, characterization, and host range breadth. Pharm (Basel). 2019;12(1):35. doi: 10.3390/ph12010035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porter NT, Hryckowian AJ, Merrill BD, Fuentes JJ, Gardner JO, Glowacki RWP, Singh S, Crawford RD, Snitkin ES, Sonnenburg JL, et al. Phase-variable capsular polysaccharides and lipoproteins modify bacteriophage susceptibility in Bacteroides thetaiotaomicron. Nat Microbiol. 2020;5(9):1170–1181. doi: 10.1038/s41564-020-0746-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shkoporov AN, Khokhlova EV, Stephens N, Hueston C, Seymour S, Hryckowian AJ, Scholz D, Ross RP, Hill C. Long-term persistence of crAss-like phage crAss001 is associated with phase variation in Bacteroides intestinalis. BMC Biol. 2021;19(1):163. doi: 10.1186/s12915-021-01084-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wexler AG, Goodman AL. An insider’s perspective: Bacteroides as a window into the microbiome. Nat Microbiol. 2017;2:17026. doi: 10.1038/nmicrobiol.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui Y, Zhang L, Wang X, Yi Y, Shan Y, Liu B, Zhou Y, Lü X. Roles of intestinal Parabacteroides in human health and diseases. FEMS Microbiol Lett. 2022;369(1):fnac072. doi: 10.1093/femsle/fnac072. [DOI] [PubMed] [Google Scholar]
- 18.Shkoporov AN, Clooney AG, Sutton TDS, Ryan FJ, Daly KM, Nolan JA, McDonnell SA, Khokhlova EV, Draper LA, Forde A, et al. The human gut virome is highly diverse, stable, and individual specific. Cell Host & Microbe. 2019;26(4):527–541.e5. doi: 10.1016/j.chom.2019.09.009. [DOI] [PubMed] [Google Scholar]
- 19.Dutilh BE, Cassman N, McNair K, Sanchez SE, Silva GG, Boling L, Barr JJ, Speth DR, Seguritan V, Aziz RK, et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat Commun. 2014;5:4498. doi: 10.1038/ncomms5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shkoporov AN, Khokhlova EV, Fitzgerald CB, Stockdale SR, Draper LA, Ross RP, Hill C. ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat Commun. 2018;9(1):4781. doi: 10.1038/s41467-018-07225-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith L, Goldobina E, Govi B, Shkoporov AN. Bacteriophages of the order Crassvirales: what do we currently know about this keystone component of the human gut virome? Biomolecules. 2023;13(4):584. doi: 10.3390/biom13040584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ezeji JC, Sarikonda DK, Hopperton A, Erkkila HL, Cohen DE, Martinez SP, Cominelli F, Kuwahara T, Dichosa AEK, Good CE, et al. Parabacteroides distasonis: intriguing aerotolerant gut anaerobe with emerging antimicrobial resistance and pathogenic and probiotic roles in human health. Gut Microbes. 2021;13(1):1922241. doi: 10.1080/19490976.2021.1922241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guerin E, Shkoporov AN, Stockdale SR, Comas JC, Khokhlova EV, Clooney AG, Daly KM, Draper LA, Stephens N, Scholz D, et al. Isolation and characterisation of ΦcrAss002, a crAss-like phage from the human gut that infects Bacteroides xylanisolvens. Microbiome. 2021;9(1):89. doi: 10.1186/s40168-021-01036-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo E, Aylward FO, Mende DR, DeLong EF. Bacteriophage distributions and temporal variability in the ocean’s interior. mBio. 2017;8(6):e01903–e01917. doi: 10.1128/mBio.01903-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Almeida A, Mitchell AL, Boland M, Forster SC, Gloor GB, Tarkowska A, Lawley TD, Finn RD. A new genomic blueprint of the human gut microbiota. Nature. 2019;568(7753):499–504. doi: 10.1038/s41586-019-0965-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen J, Zhang J, Mo L, Li Y, Li Y, Li C, Kuang X, Tao Z, Qu Z, Wu L, et al. Large-scale phage cultivation for commensal human gut bacteria. Cell Host Microbe. 2023;31(4):665–677.e7. doi: 10.1016/j.chom.2023.03.013. [DOI] [PubMed] [Google Scholar]
- 27.Minot S, Bryson A, Chehoud C, Wu GD, Lewis JD, Bushman FD. Rapid evolution of the human gut virome. Proc Natl Acad Sci U S A. 2013;110(30):12450–51245. doi: 10.1073/pnas.1300833110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim ES, Zhou Y, Zhao G, Bauer IK, Droit L, Ndao IM, Warner BB, Tarr PI, Wang D, Holtz LR. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat Med. 2015;21(10):1228–1234. doi: 10.1038/nm.3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benler S, Cobián-Güemes AG, McNair K, Hung SH, Levi K, Edwards R, Rohwer F. A diversity-generating retroelement encoded by a globally ubiquitous Bacteroides phage. Microbiome. 2018;6(1):191. doi: 10.1186/s40168-018-0573-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hedžet S, Rupnik M, Accetto T. Novel Siphoviridae bacteriophages infecting Bacteroides uniformis contain diversity generating retroelement. Microorganisms. 2021;9(5):892. doi: 10.3390/microorganisms9050892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benler S, Yutin N, Antipov D, Rayko M, Shmakov S, Gussow AB, Pevzner P, Koonin EV. Thousands of previously unknown phages discovered in whole-community human gut metagenomes. Microbiome. 2021;9(1):78. doi: 10.1186/s40168-021-01017-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bean EM, Morella NM, Dey N, Roux S. Complete genome sequences of two Bacteroides uniformis bacteriophages. Microbiol Resour Announc. 2022;11(10):e0061022. doi: 10.1128/mra.00610-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bakuradze N, Merabishvili M, Kusradze I, Ceyssens PJ, Onsea J, Metsemakers WJ, Grdzelishvili N, Natroshvili G, Tatrishvili T, Lazvliashvili D, et al. Characterization of a bacteriophage GEC_vB_Bfr_UZM3 active against Bacteroides fragilis. Viruses. 2023;15(5):1042. doi: 10.3390/v15051042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quaiser A, Dufresne A, Ballaud F, Roux S, Zivanovic Y, Colombet J, Sime-Ngando T, Francez AJ. Diversity and comparative genomics of Microviridae in Sphagnum- dominated peatlands. Front Microbiol. 2015;6:375. doi: 10.3389/fmicb.2015.00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Creasy A, Rosario K, Leigh BA, Dishaw LJ, Breitbart M. Unprecedented diversity of ssDNA phages from the family Microviridae detected within the gut of a protochordate model organism (Ciona robusta). Viruses. 2018;10(8):404. doi: 10.3390/v10080404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakamoto M, Benno Y. Reclassification of Bacteroides distasonis, Bacteroides goldsteinii and Bacteroides merdae as Parabacteroides distasonis gen. nov., comb. nov., Parabacteroides goldsteinii comb. nov. and Parabacteroides merdae comb. nov. Int J Syst Evol Microbiol. 2006;56(Pt 7):1599–1605. doi: 10.1099/ijs.0.64192-0. [DOI] [PubMed] [Google Scholar]
- 37.Koh GY, Kane A, Lee K, Xu Q, Wu X, Roper J, Mason JB, Crott JW. Parabacteroides distasonis attenuates toll-like receptor 4 signaling and Akt activation and blocks colon tumor formation in high-fat diet-fed azoxymethane-treated mice. Int J Cancer. 2018;143(7):1797–1805. doi: 10.1002/ijc.31559. [DOI] [PubMed] [Google Scholar]
- 38.Wang K, Liao M, Zhou N, Bao L, Ma K, Zheng Z, Wang Y, Liu C, Wang W, Wang J, et al. Parabacteroides distasonis alleviates obesity and metabolic dysfunctions via production of succinate and secondary bile acids. Cell Rep. 2019;26(1):222–235.e5. doi: 10.1016/j.celrep.2018.12.028. [DOI] [PubMed] [Google Scholar]
- 39.Cuffaro B, Boutillier D, Desramaut J, Jablaoui A, Werkmeister E, Trottein F, Waligora-Dupriet AJ, Rhimi M, Maguin E, Grangette C. Characterization of two Parabacteroides distasonis candidate strains as new live biotherapeutics against obesity. Cells. 2023;12(9):1260. doi: 10.3390/cells12091260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, Lee D, Bittinger K, Bailey A, Friedman ES, Hoffmann C, et al. Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in pediatric Crohn’s disease. Cell Host & Microbe. 2015;18(4):489–500. Erratum in: Cell Host Microbe. 2017;22(2):247. doi: 10.1016/j.chom.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, Kanner R, Bencosme Y, Lee YK, Hauser SL, et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci U S A. 2017;114(40):10713–10718. Erratum in: Proc Natl Acad Sci U S A. 2017 Oct 17;114(42):E8943. doi: 10.1073/pnas.1711235114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olbjørn C, Cvancarova Småstuen M, Thiis-Evensen E, Nakstad B, Vatn MH, Jahnsen J, Ricanek P, Vatn S, Moen AEF, Tannæs TM, et al. Fecal microbiota profiles in treatment-naïve pediatric inflammatory bowel disease - associations with disease phenotype, treatment, and outcome. Clin Exp Gastroenterol. 2019;12:37–49. doi: 10.2147/CEG.S186235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun H, Guo Y, Wang H, Yin A, Hu J, Yuan T, Zhou S, Xu W, Wei P, Yin S, et al. Gut commensal Parabacteroides distasonis alleviates inflammatory arthritis. Gut. 2023;72(9):1664–1677. doi: 10.1136/gutjnl-2022-327756. [DOI] [PubMed] [Google Scholar]
- 44.Wu WKK. Parabacteroides distasonis: an emerging probiotic? Gut. 2023;72(9):1635–1636. doi: 10.1136/gutjnl-2022-329386. [DOI] [PubMed] [Google Scholar]
- 45.Dziarski R, Park SY, Kashyap DR, Dowd SE, Gupta D, Mizoguchi E. Pglyrp-regulated gut microflora Prevotella falsenii, Parabacteroides distasonis and Bacteroides eggerthii enhance and Alistipes finegoldii attenuates colitis in mice. PLoS One. 2016;11(1):e0146162. doi: 10.1371/journal.pone.0146162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lopetuso LR, Petito V, Graziani C, Schiavoni E, Paroni Sterbini F, Poscia A, Gaetani E, Franceschi F, Cammarota G, Sanguinetti M, et al. Gut microbiota in health, diverticular disease, irritable bowel syndrome, and inflammatory bowel diseases: time for microbial marker of gastrointestinal disorders. Dig Dis. 2018;36(1):56–65. doi: 10.1159/000477205. [DOI] [PubMed] [Google Scholar]
- 47.Yang F, Kumar A, Davenport KW, Kelliher JM, Ezeji JC, Good CE, Jacobs MR, Conger M, West G, Fiocchi C, et al. Complete genome sequence of a Parabacteroides distasonis strain (CavFT hAR46) isolated from a gut wall-cavitating microlesion in a patient with severe Crohn’s disease. Microbiol Resour Announc. 2019;8(36):e00585–19. doi: 10.1128/MRA.00585-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Papenfort K, Bassler BL. Quorum sensing signal-response systems in Gram-negative bacteria. Nat Rev Microbiol. 2016;14(9):576–588. doi: 10.1038/nrmicro.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hargreaves KR, Kropinski AM, Clokie MR, Kaufmann GF. What does the talking?: Quorum sensing signalling genes discovered in a bacteriophage genome. PLoS One. 2014;9(1):e85131. doi: 10.1371/journal.pone.0085131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Silpe JE, Bassler BL. A host-produced quorum-sensing autoinducer controls a phage lysis-lysogeny decision. Cell. 2019;176(1–2):268–280.e13. doi: 10.1016/j.cell.2018.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hendrix H, Zimmermann-Kogadeeva M, Zimmermann M, Sauer U, De Smet J, Muchez L, Lissens M, Staes I, Voet M, Wagemans J, et al. Metabolic reprogramming of Pseudomonas aeruginosa by phage-based quorum sensing modulation. Cell Rep. 2022;38(7):110372. doi: 10.1016/j.celrep.2022.110372. [DOI] [PubMed] [Google Scholar]
- 52.Tang F, Bossers A, Harders F, Lu C, Smith H. Comparative genomic analysis of twelve Streptococcus suis (pro)phages. Genomics. 2013;101(6):336–344. doi: 10.1016/j.ygeno.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 53.Tarkowski TA, Mooney D, Thomason LC, Stahl FW. Gene products encoded in the ninR region of phage lambda participate in Red-mediated recombination. Genes Cells. 2002;7(4):351–363. doi: 10.1046/j.1365-2443.2002.00531.x. [DOI] [PubMed] [Google Scholar]
- 54.De Paepe M, Hutinet G, Son O, Amarir-Bouhram J, Schbath S, Petit MA, Casadesús J. Temperate phages acquire DNA from defective prophages by relaxed homologous recombination: the role of Rad52-like recombinases. PLoS Genet. 2014;10(3):e1004181. doi: 10.1371/journal.pgen.1004181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bryan MJ, Burroughs NJ, Spence EM, Clokie MR, Mann NH, Bryan SJ, Redfield RJ. Evidence for the intense exchange of MazG in marine cyanophages by horizontal gene transfer. PLoS One. 2008;3(4):e2048. doi: 10.1371/journal.pone.0002048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Warwick-Dugdale J, Buchholz HH, Allen MJ, Temperton B. Host-hijacking and planktonic piracy: how phages command the microbial high seas. Virol J. 2019;16(1):15. doi: 10.1186/s12985-019-1120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hryckowian AJ, Merrill BD, Porter NT, Van Treuren W, Nelson EJ, Garlena RA, Russell DA, Martens EC, Sonnenburg JL. Bacteroides thetaiotaomicron-infecting bacteriophage isolates inform sequence-based host range predictions. Cell Host Microbe. 2020;28(3):371–379.e5. doi: 10.1016/j.chom.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fletcher CM, Coyne MJ, Bentley DL, Villa OF, Comstock LE. Phase-variable expression of a family of glycoproteins imparts a dynamic surface to a symbiont in its human intestinal ecosystem. Proc Natl Acad Sci U S A. 2007;104(7):2413–2418. doi: 10.1073/pnas.0608797104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Coyne MJ, Comstock LE. Niche-specific features of the intestinal Bacteroidales. J Bacteriol. 2008;190(2):736–742. doi: 10.1128/JB.01559-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chamarande J, Cunat L, Alauzet C, Cailliez-Grimal C. In silico study of cell surface structures of Parabacteroides distasonis involved in its maintenance within the gut microbiota. Int J Mol Sci. 2022;23(16):9411. doi: 10.3390/ijms23169411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiang X, Hall AB, Arthur TD, Plichta DR, Covington CT, Poyet M, Crothers J, Moses PL, Tolonen AC, Vlamakis H, et al. Invertible promoters mediate bacterial phase variation, antibiotic resistance, and host adaptation in the gut. Sci. 2019;363(6423):181–187. doi: 10.1126/science.aau5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guerin E, Shkoporov A, Stockdale SR, Clooney AG, Ryan FJ, Sutton TDS, Draper LA, González-Tortuero E, Ross RP, Hill C. Biology and taxonomy of crAss-like bacteriophages, the most abundant virus in the human gut. Cell Host Microbe. 2018;24(5):653–664.e6. doi: 10.1016/j.chom.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 63.Shkoporov AN, Ryan FJ, Draper LA, Forde A, Stockdale SR, Daly KM, McDonnell SA, Nolan JA, Sutton TDS, Dalmasso M, et al. Reproducible protocols for metagenomic analysis of human faecal phageomes. Microbiome. 2018;6(1):68. doi: 10.1186/s40168-018-0446-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 67.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Allard G, Ryan FJ, Jeffery IB, Claesson MJ. SPINGO: a rapid species-classifier for microbial amplicon sequences. BMC Bioinform. 2015;16:324. doi: 10.1186/s12859-015-0747-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. Spades: a new genome assembly algorithm and its applications to single-cell sequencing. J Computer Biological. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.González-Tortuero E, Sutton TDS, Velayudhan V, Shkoporov AN, Draper LA, Stockdale SR, Ross RP, Hill C. VIGA: a sensitive, precise and automatic de novo VIral Genome Annotator. bioRxiv. 2018:277509. doi: 10.1101/277509. [DOI] [Google Scholar]
- 71.Zimmermann L, Stephens A, Nam SZ, Rau D, Kübler J, Lozajic M, Gabler F, Söding J, Lupas AN, Alva V. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J Mol Biol. 2018;430(15):2237–2243. doi: 10.1016/j.jmb.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 72.Grant JR, Enns E, Marinier E, Mandal A, Herman EK, Chen CY, Graham M, Van Domselaar G, Stothard P. Proksee: in-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 2023;51(W1):W484–W492. doi: 10.1093/nar/gkad326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nayfach S, Camargo AP, Schulz F, Eloe-Fadrosh E, Roux S, Kyrpides NC. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat Biotechnol. 2021;39(5):578–585. doi: 10.1038/s41587-020-00774-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moraru C, Varsani A, Kropinski AM. VIRIDIC-A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses. 2020;12(11):1268. doi: 10.3390/v12111268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bouras G, Nepal R, Houtak G, Psaltis AJ, Wormald PJ, Vreugde S, Marschall T. Pharokka: a fast scalable bacteriophage annotation tool. Bioinformatics. 2023. Jan 1;39(1):btac776. PMID: 36453861; PMCID: PMC9805569. doi: 10.1093/bioinformatics/btac776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rozewicki J, Li S, Amada KM, Standley DM, Katoh K. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 2019;47(W1):W5–W10. doi: 10.1093/nar/gkz342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Steenwyk JL, Buida TJ 3rd, Li Y, Shen XX, Rokas A. ClipKIT: a multiple sequence alignment trimming software for accurate phylogenomic inference. PLoS Biol. 2020;18(12):e3001007. doi: 10.1371/journal.pbio.3001007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020. May 1;37(5):1530–1534. doi: 10.1093/molbev/msaa015. Erratum in: Mol Biol Evol. 2020;37(8):2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14(6):587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Menardo F, Loiseau C, Brites D, Coscolla M, Gygli SM, Rutaihwa LK, Trauner A, Beisel C, Borrell S, Gagneux S. Treemmer: a tool to reduce large phylogenetic datasets with minimal loss of diversity. BMC Bioinform. 2018;19(1):164. doi: 10.1186/s12859-018-2164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Meier-Kolthoff JP, Göker M. VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics. 2017;33(21):3396–3404. doi: 10.1093/bioinformatics/btx440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roux S, Camargo AP, Coutinho FH, Dabdoub SM, Dutilh BE, Nayfach S, Tritt A. iPHoP: an integrated machine learning framework to maximize host prediction for metagenome-derived viruses of archaea and bacteria. PLoS Biol. 2023;21(4):e3002083. doi: 10.1371/journal.pbio.3002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu G. Using ggtree to visualize data on tree-like structures. Curr Protoc Bioinformatics. 2020;69(1):e96. doi: 10.1002/cpbi.96. [DOI] [PubMed] [Google Scholar]
- 84.Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, Arumugam M, Kultima JR, Prifti E, Nielsen T, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32(8):834–841. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 85.Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, Wu X, Li J, Tang L, Li Y, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med. 2015;21(8):895–905. doi: 10.1038/nm.3914. [DOI] [PubMed] [Google Scholar]
- 86.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 87.Smits SA, Leach J, Sonnenburg ED, Gonzalez CG, Lichtman JS, Reid G, Knight R, Manjurano A, Changalucha J, Elias JE, et al. Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Sci. 2017;357(6353):802–806. doi: 10.1126/science.aan4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rampelli S, Soverini M, D’Amico F, Barone M, Tavella T, Monti D, Capri M, Astolfi A, Brigidi P, Biagi E, et al. Shotgun metagenomics of gut microbiota in humans with up to extreme longevity and the increasing role of xenobiotic degradation. mSystems. 2020;5(2):e00124–20. doi: 10.1128/mSystems.00124-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bushnell B. Bbmap: a fast, accurate, Splice-Aware Aligner. Lawrence Berkeley National Laboratory; 2014. [Google Scholar]
- 90.Chaumeil PA, Mussig AJ, Hugenholtz P, Parks DH, Borgwardt K. GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics. 2022;38(23):5315–5316. doi: 10.1093/bioinformatics/btac672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Antipov D, Korobeynikov A, McLean JS, Pevzner PA. hybridSpades: an algorithm for hybrid assembly of short and long reads. Bioinformatics. 2016;32(7):1009–1015. doi: 10.1093/bioinformatics/btv688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this report and in the supplementary material. The genome of φPDS1 is deposited into GenBank under accession MN929097 (assembled and annotated genome). The genome of the bacterial host of φPDS1, Parabacteroides distasonis APCS2/PD, is deposited under the following accession codes: BioProject PRJNA556872, GenBank CP042285 (assembled and annotated genome) and associated plasmid pPDS2–1, GenBank CP042284.