

Abstract
The loss of Bacillus subtilis penicillin-binding protein (PBP) 2a, encoded by pbpA, was previously shown to slow spore outgrowth and result in an increased diameter of the outgrowing spore. Further analyses to define the defect in pbpA spore outgrowth have shown that (i) outgrowing pbpA spores exhibited only a slight defect in the rate of peptidoglycan (PG) synthesis compared to wild-type spores, but PG turnover was significantly slowed during outgrowth of pbpA spores; (ii) there was no difference in the location of PG synthesis in outgrowing wild-type and pbpA spores once cell elongation had been initiated; (iii) outgrowth and elongation of pbpA spores were dramatically affected by the levels of monovalent or divalent cations in the medium; (iv) there was a partial redundancy of function between PBP2a and PBP1 or -4 during spore outgrowth; and (v) there was no difference in the structure of PG from outgrowing wild-type spores or spores lacking PBP2a or PBP2a and -4; but also (vi) PG from outgrowing spores lacking PBP1 and -2a had transiently decreased cross-linking compared to PG from outgrowing wild-type spores, possibly due to the loss of transpeptidase activity.
Bacillus subtilis is a gram-positive bacterium that undergoes sporulation when deprived of an essential nutrient or nutrients. The resulting elliptical spore is metabolically dormant and resistant to a variety of harsh environmental conditions. However, in the presence of specific germinant molecules, such as l-alanine, the spore can resume metabolic activity and eventually return to vegetative growth through the processes of spore germination, which does not require macromolecular synthesis, and spore outgrowth, which begins with the resumption of macromolecular synthesis (35). An essential event during spore outgrowth is the synthesis of new cell wall peptidoglycan (PG). Much of the spore’s PG is degraded during the first minutes of germination (8). However, the spore PG which is not degraded comprises the germ cell wall, which is thought to provide the template for PG synthesis during spore outgrowth (3). The PG generated during the latter period then generates the rod-shaped vegetative cell.
One group of enzymes essential for PG synthesis is the penicillin-binding proteins (PBPs), which catalyze PG strand elongation (transglycosylase activity) and regulate PG side chain cross-linking through transpeptidase and d,d-peptidase activities (reviewed in reference 9). A number of PBPs are present in growing B. subtilis cells, and some are in the dormant spore; the initiation of transcription of a number of pbp genes also occurs very early in spore outgrowth (17, 18, 20, 30). However, the specific function of most individual PBPs in vegetative growth and spore outgrowth has been difficult to discern, as mutations in one or more pbp genes often result in few phenotypic effects, probably because many of these proteins perform redundant functions (31).
There are, however, several PBPs whose loss results in a clear phenotype (5, 18, 30, 37). One such PBP is the class B high-molecular-weight (HMW) PBP2a encoded by pbpA, as the loss of this putative monofunctional transpeptidase results in both delayed spore outgrowth and an increased cell diameter early in spore outgrowth (18). In contrast, the loss of PBP2a does not cause phenotypic changes during vegetative growth (18). In this report we further characterize the outgrowth of spores lacking PBP2a and examine PG metabolism during this period. The importance of other HMW PBPs during spore outgrowth was also studied by constructing strains lacking PBP2a and additional HMW PBPs and examining the properties of these strains during spore outgrowth, as well as during vegetative growth and sporulation.
MATERIALS AND METHODS
Bacterial strains, growth, sporulation, and spore germination and outgrowth.
The B. subtilis strains used are shown in Table 1; all strains are derived from PS832, a derivative of strain 168. B. subtilis was transformed with either chromosomal DNA or plasmid DNA as previously described (1), and transformants were selected on 2× SG plates (14) by their resistance to appropriate antibiotics: chloramphenicol (Cmr) (3.0 μg/ml); the macrolides (MLSr) (lincomycin, 12.5 μg/ml; erythromycin, 0.5 μg/ml); or spectinomycin (Spr) (100 μg/ml). B. subtilis was routinely grown and sporulated at 37°C in antibiotic-free 2× SG medium (14). The spores were then purified by repeated washing in water as described previously (21). Purified spores were germinated at 37°C in a number of different media, including 2× YT (31), Penassay broth (PAB) (Difco), and Luria broth (LB) (containing [per liter] 10 g of tryptone, 5 g of yeast extract, and 10 g of NaCl), after a 30-min heat shock in water at 70°C as described previously (21); all germination media were supplemented with l-alanine (4 mM) to stimulate the initiation of spore germination. To remove molecules released during the initial stages of germination, all spores outgrowing in PAB medium were harvested by centrifugation at 3,000 × g for 5 min at 24°C 30 min after the initiation of spore germination and the pellet was resuspended in an equal volume of prewarmed (37°C) fresh medium. The optical density at 600 nm (OD600) of all vegetative cells and germinating and outgrowing spores was monitored with a Genesys 5 spectrophotometer.
TABLE 1.
B. subtilis strains used
| Strain | Genotypea | Transformation donor | Transformation recipient | Source or reference |
|---|---|---|---|---|
| PS832 | Prototrophic revertant of 168 | Laboratory stock | ||
| PS1869 | ΔpbpF::Erm | 28 | ||
| PS2022 | ΔpbpD::Erm | 29 | ||
| PS2062 | ΔponA::Spr | 30 | ||
| PS2328 | pbpC::pTMM4 Spr | 17 | ||
| PS2465 | pbpA::pTMA4 Cmr | 18 | ||
| PS2466 | pbpA::pTMA4 CmrΔponA::Spr | PS2062 | PS2465 | This work |
| PS2467 | pbpA::pTMA4 CmrpbpC::pTMM4 Spr | PS2328 | PS2465 | This work |
| PS2468 | pbpA::pTMA4 CmrΔpbpD::Erm | PS2022 | PS2465 | This work |
| PS2478 | pbpA::pTMA4 CmrΔpbpF::Erm | PS2465 | PS1869 | This work |
Abbreviations for antibiotic resistance: Erm, lincomycin (12.5 μg/ml) plus erythromycin (0.5 μg/ml); Cm, chloramphenicol (3 μg/ml); Sp, spectinomycin (100 μg/ml).
Membrane isolation, penicillin-binding assays, spore heat resistance, and analysis of PG structure.
Membranes were isolated from B. subtilis cells in log-phase growth in 2× SG medium at an OD600 of approximately 0.5 or from spores outgrowing in 2× YT medium 2 h after the initiation of spore germination, as previously described (31). The purified membranes were incubated with fluorescein-hexanoic acid 6-aminopenicillanic acid (FLU-C6-APA), the proteins were separated by sodium dodecyl sulfate–10% polyacrylamide gel electrophoresis, and labeled PBPs were visualized with a FluorimagerSI (Vistra), as described previously (31).
Spore wet-heat resistance was measured by incubating purified spores in water at 85°C for 15 min and plating serial dilutions of unheated and heated spores on LB plates (27). Spore cortex PG was isolated, hydrolyzed, reduced, and analyzed by reverse-phase high-performance liquid chromatography (HPLC) as previously described (2, 25). PG from outgrowing spores of various strains was isolated 30, 60, and 90 min after the initiation of spore germination and analyzed in a similar manner.
Light microscopy and viability assays.
Outgrowing spores (0.5 ml) were fixed in 2.5% glutaraldehyde for 20 min at 22°C, washed in 0.5 ml of phosphate-buffered saline (PBS; 137 mM NaCl, 3 mM KCl, 5.4 mM Na2HPO4, 1.7 mM KH2PO4 [pH 7.3]), placed on 0.01% polylysine-coated coverslips, and visualized by differential interference contrast microscopy with a Noran confocal laser scanning microscope employing a 100× Plan-APO chromatic oil immersion lens (Zeiss) as described previously (31). To visualize the cell walls and DNA of outgrowing spores, the fixed cells on coverslips were treated with lysozyme (1 mg/ml) at 24°C for 30 s, rinsed several times in PBS, and incubated with 2% bovine serum albumin in PBS for 1 h, and wheat germ agglutinin conjugated to Oregon green (Molecular Probes) was added along with 4′,6-diamidino-2-phenylindole (DAPI) to final concentrations of 5 and 1.25 μg/ml, respectively (22). After a 2-h incubation, the cells were rinsed eight times with PBS, placed on slides by using the SlowFade light antifade kit (Molecular Probes), and viewed at the appropriate wavelength (22).
Spore viability was assessed by plating purified spores on either LB plates or LB plates supplemented with 10 mM MgCl2. The plates were incubated overnight at 37°C, and the colonies were counted. The LIVE/DEAD BacLight bacterial viability kit (Molecular Probes) was also used as directed by the manufacturer to assess viability during spore outgrowth in 2× YT medium. Samples (0.5 ml) of outgrowing spores were harvested at various times after the initiation of spore germination and incubated with 1.5 μl of LIVE/DEAD stain for 2 to 3 min. An aliquot of the suspension was placed on a 1% agarose-coated slide, a coverslip was gently applied, and the cell fields were examined with the fluorescein filter on an Olympus B-max microscope employing a 40× Dplan-APO lens. The live cells were counted, and then the rhodamine filter was used to assay the number of dead cells in the same field. At each time point, at least 300 cells were examined.
Measurement of macromolecular synthesis during spore outgrowth.
To assess PG synthesis during spore outgrowth in a medium where cells remained viable and yet morphological changes were evident, 12.5 ml of prewarmed (37°C) 2× YT medium containing 4 mM l-alanine, 100 μM N-acetyl-d-glucosamine, and 5 μCi of [3H]N-acetyl-d-glucosamine (6.6 Ci/mmol) were inoculated with heat-shocked dormant spores to an initial OD600 of approximately 0.5. Duplicate 0.5-ml samples were harvested throughout subsequent spore germination and outgrowth, added to 0.5 ml of 10% trichloroacetic acid (TCA) at 4°C, and incubated on ice for at least 1 h, and the TCA precipitate was harvested by filtration through GF/C membranes (Whatman) (10, 24). The membranes were washed five times with 5 ml of 5% TCA and four times with 5 ml of 95% ethanol and dried under a heat lamp; 3.5 ml of Biosafe II counting fluid was added, and the samples were counted for 5 min with a Delta 300 scintillation counter (Searle). To determine the extent to which PG synthesis required new protein synthesis, spore germination and outgrowth was carried out as described above but with a final total N-acetyl-d-glucosamine concentration of 10 μM to improve the sensitivity of the assay and with the addition of chloramphenicol (100 μg/ml) (20). In all of these experiments, the percentage of [3H]N-acetyl-d-glucosamine incorporated into the TCA-insoluble fraction was determined and divided by the initial OD of the spore culture to correct for differences in the sizes of the initial inocula.
To confirm that the incorporation of [3H]N-acetyl-d-glucosamine into a TCA-insoluble form did represent PG synthesis, we used the method previously described (24). Samples were harvested as described above but were boiled for 10 min in 5% TCA to denature proteases which might interfere with subsequent reactions. Then the TCA was removed by centrifugation (10,000 × g; 24°C) and the pellet was washed with 1.0 M Tris HCl (pH 8.0) and dissolved in 1 ml of 20 mM Tris-HCl (pH 8.0) for digestion with either lysozyme (0.5 mg/ml) or trypsin (0.1 mg/ml, with 10 mM CaCl2). After incubation overnight at 37°C, the digests were centrifuged for several minutes at 10,000 × g and aliquots of both the pellet and supernatant fraction were counted as described above. More than 90% of the counts were solubilized by lysozyme, while less than 10% of the counts were solubilized by trypsin treatment (data not shown). Consequently, the great majority of [3H]N-acetyl-d-glucosamine incorporated into TCA-insoluble form was in cell wall material. Previous work has demonstrated that more than 90% of radiolabeled N-acetyl-d-glucosamine incorporated into TCA-insoluble material in growing B. subtilis cells is incorporated into the cell wall, with 70 to 75% incorporated into the PG fraction and the remainder into glucosamine-containing teichoic acids (7, 24).
In order to measure PG turnover during outgrowth of wild-type and pbpA spores, 12.5 ml of prewarmed (37°C) 2× YT medium (containing 4 mM l-alanine) and 5 μCi of [3H]N-acetyl-d-glucosamine (6.6 Ci/mmol) was inoculated with heat-shocked spores to an OD600 of 0.5. Thirty minutes after the initiation of spore germination, the outgrowing spores were harvested by centrifugation and resuspended in 12.5 ml of prewarmed (37°C) 2× YT medium containing 1 mM unlabeled N-acetyl-d-glucosamine, and TCA-insoluble radioactivity was measured during subsequent incubation as described above (10). PG turnover during vegetative growth was assayed by adding 5 μCi of [3H]N-acetyl-d-glucosamine (6.6 Ci/mmol) to log-phase wild-type B. subtilis (OD600, 0.5) in 12.5 ml of 2× YT medium, incubating the cells for 10 min, harvesting the cells by centrifugation, resuspending the cell pellet in fresh 2× YT medium containing 1 mM unlabeled N-acetyl-d-glucosamine, and analyzing aliquots for TCA-precipitable radioactivity as described above.
DNA synthesis during spore germination and outgrowth was measured in 12.5-ml cultures of 2× YT medium containing 6.25 μCi of [3H-methyl]thymidine (6.7 Ci/mmol), 10 μM unlabeled thymidine, and 4 mM 2-deoxyadenosine (32). Protein synthesis was measured similarly but by the addition of 12.5 μCi of l-[3,4,5-3H(N)]leucine (179 Ci/mmol) to 12.5 ml of 2× YT medium without additional unlabeled leucine (32). In both cases TCA-precipitable radioactivity was measured as described above.
Electron microscopy and autoradiography.
The location of newly synthesized cell wall components was examined during spore outgrowth by inoculating 5 ml of prewarmed (37°C) 2× YT medium with heat-shocked wild-type or pbpA spores to an OD600 of 1.5. After approximately 60 min, 500 μCi of [3H]N-acetyl-d-glucosamine (6.9 Ci/mmol) was added to each culture, and 2 min later 250 μg of cold N-acetyl-d-glucosamine was added. After a 10-min chase (72 min after the initiation of spore germination), the samples were harvested by centrifugation for 5 min (7,500 × g), fixed for 20 min in 1 ml of 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4), washed in 0.1 M sodium cacodylate buffer, and embedded in low-melting-temperature agarose. The agarose pellets were postfixed in 1% osmium tetroxide–0.8% potassium ferricyanide in 0.1 M cacodylate buffer, stained with 0.5% aqueous uranyl acetate, dehydrated in graded ethanol solutions, and embedded in epoxy resin (34).
Thin sections were cut with a diamond knife, placed on celloidin-coated glass slides, and stained with uranyl acetate and lead citrate, and then the slides were carbon coated in a vacuum evaporator, dipped in Ilford L4 emulsion diluted 1:3.5 with water, dried, and stored in lighttight boxes at 4°C for 20 weeks (11). The autoradiographs were then developed in Agfa-Gevaert solution physical developer (12) and fixed in 24% sodium thiosulfate, and sections collected on copper specimen grids were viewed with a Philips CM10 transmission electron microscope.
To determine if the cell wall was the source of most of the developed silver grains in the electron microscopic autoradiographs described above, the shortest distance from the center of each grain to the center of the nearest cell wall was measured with a 7× magnifying eyepiece. Where several grains were clustered in a circle equivalent to the average diameter of an Ilford L4 silver bromide crystal (140 nm [12]), the center of the cluster was considered the center of the grain. To confirm that developed silver grains were derived from the cell wall, the distance on either side of the center of the cell wall that included 50% of the counted grains (the half-distance) was determined (6). This analysis gave half-distances for wild-type and pbpA spores of 80 and 89 nm, respectively, compared to a value of 83 nm obtained in previous studies of cell wall synthesis in Bacillus megaterium (6). These values are also consistent with the published value for the half-distance for tritium when samples are prepared by similar methods (13). Consequently, silver grains which were within the half-distance were considered to be derived from the wall and only those grains were further analyzed with regard to distribution along the outgrowing spore (see below). At least 200 grains within the half-distance were analyzed in both the outgrowing wild-type and pbpA spores, with 15 elongating wild-type spores and 23 elongating pbpA spores analyzed.
To determine the location of the newly synthesized PG for each strain, the position of a silver grain (within the half-distance from the wall) was used to determine the position of labeled PG in the wall by taking the shortest distance between the grain and the wall; the distance between this position and the center of the cell or of the forming septum was then measured and expressed as a percentage of total cell length (see Fig. 3). Due to the increased diameter of the pbpA spores, a higher percentage of wall material is present at the poles of pbpA spores than in wild-type spores. To correct for this difference, an average cell length and width were determined for each strain (see Fig. 3); for this analysis an outgrowing spore was considered a rectangle with semicircles at each pole. The average outgrowing spore for each strain was then divided into 10 bins with identical amounts of wall material, and the grains were placed into each bin based upon their distance to the center of the cell as a percentage of cell length. As the cell is symmetrical, bins at equal distances from the cell center were combined, so the grains were grouped into five compartments. Grains associated with forming septa were excluded from the analysis; outgrowing spores, which are round, do not provide a reference point to analyze grain distribution and were also excluded. All statistical analyses employed Excel software (Microsoft).
FIG. 3.


Electron microscopic autoradiography of wild-type and pbpA spores. The spores were heat shocked and inoculated into 2× YT medium with 4 mM l-alanine, as described in Materials and Methods. Sixty minutes after the initiation of spore germination, 500 μCi of [3H]N-acetyl-d-glucosamine was added and the culture was incubated for 2 min, followed by the addition of unlabeled N-acetyl-d-glucosamine to 1 mM. After 10 min of further incubation, the cells were harvested and processed for electron microscopic autoradiography, as described in Materials and Methods. Examples of wild-type (A) and pbpA (C) outgrowing spores are shown, with arrows indicating the labeled wall. The statistical analysis of silver grain distribution was done as described in Materials and Methods and is shown for the average wild-type (B) and pbpA (D) outgrowing spore after half of each cell was divided into 5 bins, with equal amounts of wall material in each bin. Bars, 1 μM.
RESULTS
Macromolecular synthesis, PG turnover, and location of PG synthesis during spore outgrowth.
The alteration in cell morphology and the slowed outgrowth of pbpA spores seen previously (18) suggested that PG synthesis might be altered during outgrowth of spores lacking PBP2a. Indeed, consistent with the slower outgrowth of pbpA spores compared to that of wild-type spores (Fig. 1A), PG synthesis during spore outgrowth in 2× YT medium was also slightly slowed by the loss of PBP2a (Fig. 1B). In this medium [3H]thymidine incorporation into DNA began to increase significantly between 60 and 90 min after the initiation of germination of wild-type spores, pbpA spores, and spores of all other pbp mutants examined and then paralleled the changes in the OD600 during subsequent outgrowth (data not shown). The relative rates of protein synthesis were also similar to those found for PG synthesis (data not shown). Chloramphenicol addition blocked (>98%) PG synthesis during the first 120 min of germination and outgrowth of both wild-type and pbpA spores (data not shown), indicating that PG synthesis during outgrowth requires protein synthesis.
FIG. 1.

Spore outgrowth (A) and PG synthesis (B) in strains lacking HMW PBPs. The spores were heat shocked and inoculated into 2× YT medium with 4 mM l-alanine containing [3H]N-acetyl-d-glucosamine, as described in Materials and Methods. The cells were harvested, and TCA-insoluble radioactivity was measured as described in Materials and Methods. The percentage of total radioactivity in the sample that was TCA insoluble was divided by the initial OD600 to give the units in Fig. 1B. The kinetics of spore outgrowth shown are from the same experiment in which PG synthesis was monitored. □, PS832 (wild type); ○, PS2465 (pbpA); ◊, PS2466 (pbpA ponA); ▵, PS2468 (pbpA pbpD).
PG turnover was also measured during outgrowth of wild-type and pbpA spores; for wild-type spores, the newly synthesized PG was stable for approximately 30 min, which is consistent with our measurements of PG turnover in vegetative cells as well as with previous work (Fig. 2) and data not shown [16, 23]). However, newly synthesized PG in pbpA spores was stable for longer than 30 min, and subsequent PG turnover was faster in the outgrowing wild-type spores, consistent with their higher rate of outgrowth (Fig. 2). Interestingly, measurements of cell wall width from electron micrographs of spores harvested 75 min after the initiation of spore germination revealed that during outgrowth pbpA spores possess a thicker wall than wild-type spores (59 ± 10 nm for pbpA spores versus 45 ± 7.4 nm for wild-type spores; n > 100 spores for each strain; P < 0.05 by the two-tailed Student’s t test). It is possible that the increased thickness of the outgrowing pbpA spore wall is due at least in part to the slower PG turnover in pbpA spores (see Discussion).
FIG. 2.

PG turnover during outgrowth of wild-type and pbpA spores. The spores were heat shocked and germinated in 2× YT medium with 4 mM l-alanine for 30 min in the presence of [3H]N-acetyl-d-glucosamine. Then the cells were harvested and resuspended in fresh 2× YT medium with unlabeled N-acetyl-d-glucosamine, and TCA-insoluble radioactivity was measured, as described in Materials and Methods. The amount of TCA-insoluble radioactivity immediately after resuspension was set at 100%. □, OD600 of PS832 (wild type); ○, OD600 of PS2465 (pbpA); ■, percent incorporated [3H]N-acetyl-d-glucosamine remaining in PS832 (wild type); •, percent incorporated [3H]N-acetyl-d-glucosamine remaining in PS2465 (pbpA).
An alternative explanation is that the wall may be thicker in only some areas of pbpA spores during outgrowth due to a change in the location of PG incorporation compared to that in wild-type spores. To examine this possibility, wild-type and pbpA spores were pulse labeled with [3H]N-acetyl-d-glucosamine 60 min after the initiation of spore germination and subjected to electron microscopic autoradiography to determine the sites of PG synthesis during spore elongation (Fig. 3A and C). Initial analyses confirmed that in these autoradiographs the silver grains were derived from label in the cell wall (see Methods). Further statistical analysis of grain distribution along the wall of the outgrowing spores revealed that when cell shape was corrected for by dividing each cell into compartments containing equal amounts of wall, the grain distribution was similar for both wild-type and pbpA spores and appeared uniform along the cell (Fig. 3B and D). This suggests that after the initiation of spore elongation, sites of PG synthesis are similar for the wild-type and pbpA spores, although this analysis does not address sites of earlier PG synthesis (i.e., when outgrowing spores are still round).
The loss of PBP1 or PBP4 further delays outgrowth of spores lacking PBP2a.
Previous work has suggested that HMW PBPs perform partially redundant functions in B. subtilis (31). Given the unique phenotype associated with the loss of PBP2a, we were interested in determining the effect of the loss of other HMW PBPs in addition to PBP2a. Consequently, mutations inactivating the genes encoding the predominant class A HMW putative bifunctional transglycosylases and transpeptidases, PBP1, -2c, and -4, and the class B putative monofunctional transpeptidase PBP3 were combined with a mutation in pbpA (Table 1). That these mutant strains lacked the appropriate PBPs was confirmed by analysis of PBP profiles in membranes isolated from the appropriate strains (Fig. 4 and data not shown). In 2× SG or 2× YT medium, the vegetative growth rates of strains lacking PBP2a and either PBP2c, PBP3, or PBP4 were essentially identical to those of the wild-type strain while the strain lacking PBP2a and PBP1 (encoded by ponA) grew at a rate identical to that of a strain lacking only PBP1 (data not shown) (30). All of these multiple pbp mutant strains produced dormant spores with heat resistance properties and PG cortex structures essentially identical to those of wild-type spores (data not shown).
FIG. 4.
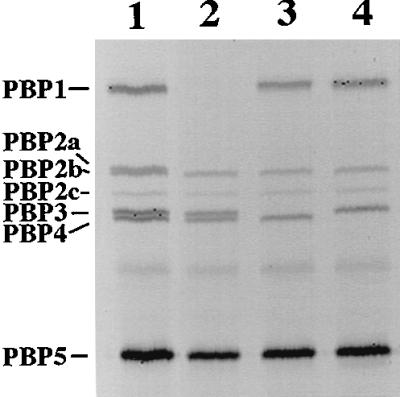
PBP profiles from membranes of various strains. Membranes were isolated from vegetative cells and labeled with FLU-C6-APA, as described in Materials and Methods. Approximately 10 μg of total membrane protein was run on sodium dodecyl sulfate–10% polyacrylamide gel electrophoresis for 4 h at 100 V, and labeled PBPs were visualized with a FluorimagerSI. The PBP pattern of wild-type cell membranes is like that previously found (lane 1) (18). Lane 1, PS832 (wild type); lane 2, PS2466 (pbpA ponA); lane 3, PS2467 (pbpA pbpC); lane 4, PS2468 (pbpA pbpD). The PBPs were designated as previously described (31).
Spores of all multiple pbp mutants initiated germination similarly to wild-type spores (Fig. 1A and data not shown). The loss of either PBP2c or PBP3 in addition to PBP2a did not further alter either the rate of spore outgrowth, the previously observed morphological changes or the rate of PG synthesis during spore outgrowth, or spore viability compared to the effects of the loss of PBP2a alone (data not shown). However, the loss of either PBP4 or PBP1 in a pbpA background further slowed spore outgrowth in 2× YT medium (Fig. 1A) and decreased PG synthesis comparably to the decrease in the rate of spore outgrowth (Fig. 1B); these effects were much greater than the effects of loss of either PBP1 or -4 alone (data not shown) (29, 30). Examination of spores 120 min after the initiation of spore germination in 2× YT medium revealed that 45% of outgrowing spores lacking PBP4 and PBP2a had the increased diameter observed in spores lacking only PBP2a while the remaining spores showed no elongation and no increase in diameter (n > 200 spores; data not shown). In contrast, 85% of spores lacking only PBP2a exhibited the increased diameter 120 min after the initiation of spore outgrowth. No spores lacking PBP1 and PBP2a outgrowing in 2× YT medium showed the increased cell diameter exhibited by outgrowing pbpA spores, and outgrowth of pbpA ponA spores was accompanied by severe bending (Fig. 5C and Table 2).
FIG. 5.

Morphologies of cells and outgrowing spores of the wild-type strain and strains lacking PBP2a or PBP2a and PBP1. The spores were heat shocked and inoculated into either PAB medium, PAB medium with 500 mM NaCl, or 2× YT medium, all of which contained 4 mM l-alanine. Outgrowing spores were harvested 120 min after the initiation of spore germination unless otherwise noted. The following strains (genotypes) and growth or outgrowth conditions are shown: (A) PS2062 (ponA), vegetative cells in PAB medium with 50 μM MgCl2; (B) PS2466 (pbpA ponA), vegetative cells in PAB medium with 50 μM MgCl2; (C) PS2466 (pbpA ponA), spores outgrowing in 2× YT medium with no additions, harvested 150 min after the initiation of spore germination; (D) PS832 (wild type), spores outgrowing in PAB medium with no additions; (E) PS2465 (pbpA), spores outgrowing in PAB medium with no additions; (F) PS2465 (pbpA ponA), spores outgrowing in PAB medium with no additions; and (G) PS2465 (pbpA), spores outgrowing in PAB medium with 500 mM NaCl. Bars, 10 μM.
TABLE 2.
Spore outgrowth phenotypes of HMW PBP mutantsa
| Strain | Morphology (outgrowth rateb) in:
|
||||
|---|---|---|---|---|---|
| PAB | PAB + 500 mM NaCl | PAB + 10 mM Mg2+ | 2× YTc | 2× YT + 10 mM Mg2+ | |
| PS832 (wtd) | wte (115) | Elongation, then lysis (150) | wt (107) | wt (105) | wt (103) |
| PS2465 (pbpA) | ↑f Cell widthe (>180) | Spherical, then lysise (150) | ↑ Cell width (108) | ↑ Cell width (108) | ↑ Cell width (110) |
| PS2466 (pbpA ponA) | ↓ Cell width; poor elongatione (>180) | No ↑ in OD | ↓ Cell width (125) | ↓ Cell width; bendinge (>150) | ↓ Cell width (123) |
| PS2468 (pbpA pbpD) | ↑ Cell width (>180) | ↑ Cell width; elongation, then lysis (150) | ↑ Cell width (111) | ↑ Cell width (140) | ↑ Cell width (113) |
Germination was done at 37°C with the inclusion of 4 mM l-Ala in the medium, as described in Materials and Methods. Spores in PAB medium were centrifuged 30 min after the initiation of spore germination, as described in Materials and Methods.
Values in parentheses are the minimum times in minutes needed for the initial OD600 of a culture to double after the addition of spores to the germination medium.
See Fig. 1A.
wt, wild type.
See Fig. 5.
↑, increased; ↓, decreased.
In both this and previous work we observed that when we plated equal numbers of wild-type and pbpA spores, the pbpA spores gave 50% fewer colonies than the wild type (18) (Table 3). However, spores lacking PBP2a and PBP1 had greatly decreased viability, while those lacking either PBP1 or PBP1 and PBP4 had viabilities essentially identical to that of wild-type spores (Table 3 and data not shown). The addition of 10 mM Mg2+ to LB plates significantly increased the viability of spores lacking PBP2a and PBP1 (Table 3), suggesting that these spores require increased levels of divalent cations for outgrowth. This is consistent with recent work, which demonstrated that decoated spores lacking PBP1 require increased concentrations of Mg2+ for outgrowth (19). However, colony formation from spores also requires cell growth, and ponA cells require a high concentration of Mg2+ for growth (19).
TABLE 3.
Viability of spores from strains lacking HMW PBPsa
| Strain | Dormant spore viability (%)b
|
% Dead cells among outgrowing sporesc
|
|||
|---|---|---|---|---|---|
| No Mg2+ | Mg2+ | 60 min | 90 min | 120 min | |
| PS832 (wild type) | 100 | 100 | 1.1 | <1 | <1 |
| PS2465 (pbpA) | 44 | 53 | 1.9 | 1.6 | 2.8 |
| PS2466 (pbpA ponA) | 1d | 20 | 2.4 | 8.3 | 15.4 |
| PS2468 (pbpA pbpD) | 23 | 67 | 2.3 | 5.7 | 4.3 |
Dormant and outgrowing spore viabilities were determined as described in Materials and Methods.
The number of colonies produced by wild-type spores per OD600 was set at 100%. Particle counts indicated that the number of spores per OD unit was similar for all strains. MgCl2 was added to LB plates at a final concentration of 10 mM.
Times given are those after the initiation of spore germination. The percentages of dead cells at 120 min are actually underestimates, as cell division has occurred.
Values in boldface are the most dramatic changes observed.
To determine when during outgrowth spores were no longer viable, LIVE/DEAD assays were performed during spore outgrowth in 2× YT medium. After 90 min of spore outgrowth, and even more so after 120 min, a significant number of dead cells were observed with the strain lacking PBP2a and PBP1 (Table 3), this cell death could explain the extremely slow outgrowth of spores of this strain. The percentage of dead cells given at 120 min is actually a significant underestimate of the true number, since chains of cells were present at this time, suggesting that significant division of the cells that remained viable had already occurred (data not shown). In contrast to the results with spores lacking PBP1 and -2a, outgrowth of spores lacking PBP2a or PBP2a and -4 was not accompanied by significant cell death (Table 3).
PG structure during outgrowth of spores lacking HMW PBPs.
While the rate of PG synthesis was decreased similarly in outgrowing spores lacking PBP1 and -2a or PBP4 and -2a, the viabilities of the two strains during spore outgrowth were drastically different. One explanation for this difference is that the structures of the PGs being synthesized by the two strains might differ significantly. To explore this possibility, PG was purified from spores of a variety of HMW PBP mutants after 30, 60, and 90 min of outgrowth in 2× YT medium and the PG was digested and analyzed by HPLC. The amounts of PG synthesized by 30 and 60 min are not dramatically different in various mutant strains (Fig. 1B), so PG from comparable amounts of outgrowing spores was loaded onto the HPLC column for each mutant strain. However, for samples analyzed at 90 min of outgrowth, PG from twice as many spores lacking PBP2a and either PBP1 or -4 was loaded than from spores of other strains. The pattern of muropeptide peaks varied significantly among the 30-, 60-, and 90-min samples (data not shown), but at each time point similar patterns were found in the wild-type strain and most PBP mutants examined, even though distinct morphological differences were observed between some strains at 90 min. However, 60 min after the initiation of spore germination, spores lacking PBP1 and PBP2a exhibited significant changes in PG structure, as determined by reverse-phase HPLC (Fig. 6). Cochromatography with known standards, amino acid and amino sugar analysis, and mass spectrometry of peaks 4, 5, and 6 revealed that in the strain lacking PBP1 and PBP2a (Fig. 6A), there was an increase in levels of disaccharide tetrapeptide (peak 4) and a corresponding decrease in a disaccharide tetrapeptide cross-linked to either a disaccharide tetrapeptide with a glycine present in the cross-link or a disaccharide pentapeptide with glycine in either the fourth or fifth position of the side chain (peak 5/6) compared to a strain lacking only PBP1 (Fig. 6B) or to the wild-type strain (data not shown). Peaks 5 and 6 had identical amino acid and amino sugar compositions, so we suspect the split peak is due to a partial modification of the muropeptide, possibly partial amidation of the peptide side chain. The ratio of cross-linked species to monomer (Fig. 6A, ratio of peak 5/6 to peak 4) was 1.10 in ponA spores compared to 0.72 in pbpA ponA spores. Similar (within 7%) results were seen in duplicate analyses of two independent spore preparations of both strains. This suggests that the percentage of total PG cross-linking is decreased during outgrowth of spores lacking PBP1 and PBP2a compared to that in wild-type spores. Since these changes in PG structure were not detected in the strain lacking PBP1 and PBP2a either 30 or 90 min after initiation of spore germination, this suggests that the change in PG cross-linking is transient.
FIG. 6.

Differences in structures of PG from outgrowing wild-type spores and spores lacking PBP1 and PBP2a. PG from outgrowing spores was harvested 60 min after the initiation of spore germination and purified, digested, reduced, and separated by HPLC with a 0.1% TFA–20% acetonitrile gradient as described previously (25). The muropeptide profile of a strain lacking both PBP1 and PBP2a (A) is compared to the profile of a strain lacking only PBP1 (B); the latter is identical to that from the wild-type strain (data not shown). The arrows denote peaks whose magnitude is significantly different in the strains lacking PBP1 and PBP1 and -2a. Differences in retention times between the two gradients are the result of slight differences in buffer composition among the different HPLC runs.
Divalent cation sensitivity of outgrowing spores lacking HMW PBPs.
Given that the addition of 10 mM MgCl2 to LB plates restored viability to spores lacking PBP1 and PBP2a, we further explored the spore outgrowth and vegetative growth of multiple HMW PBP mutants in PAB medium, which contains low levels of divalent cations. All strains grew in PAB medium except the strain lacking PBP1 and PBP2a, which, like a strain lacking only PBP1, did not grow (reference 19 and data not shown); however, the strain lacking PBP1 and −2a appeared to require more Mg2+ for growth than the strain lacking only PBP1 (data not shown). The addition of 50 μM Mg2+ to PAB medium allowed some growth of a strain lacking both PBP1 and PBP2a, and microscopic examination of these cells revealed dramatic bending and filamentation, more so than with a strain lacking only PBP1 (Fig. 5A and B), suggesting a role for PBP2a in vegetative growth.
The rate of outgrowth of pbpA spores in PAB medium was significantly lower than that of wild-type spores (Table 2). Ninety minutes after the initiation of spore germination in PAB medium, pbpA cultures had many small round cells while wild-type spores had initiated elongation (data not shown). After 120 min of spore outgrowth an increasing number of pbpA cells did exhibit an increased diameter, but little cell elongation had taken place compared to that with wild-type spores (Fig. 5D and E). It is possible that the release of cations during germination is responsible for pbpA spore outgrowth in PAB medium, as this appears to be the case for spores lacking PBP1 (19). Spores lacking PBP1 and PBP2a outgrowing in PAB medium did not show the increased cell diameter exhibited by outgrowing pbpA spores, and very little elongation occurred compared to that of wild-type spores (Fig. 5D and F).
The addition of 10 mM Mg2+ to PAB medium greatly improved the rate of outgrowth of spores of all pbp mutant strains whose outgrowth was previously found to be severely compromised (Table 2). The bent cells previously observed in the strain lacking PBP2a and PBP1 were no longer present, while straight rods with decreased diameter were (data not shown).
Sensitivity of outgrowing spores lacking HMW PBPs to monovalent cations.
Previously we had found that the inclusion of 500 mM NaCl in PAB medium prevented outgrowth of spores lacking PBP1 and resulted in lysis of wild-type spores 120 to 150 min after the initiation of spore outgrowth (19). While wild-type spores were able to initially elongate in PAB medium with 500 mM NaCl (19), pbpA spores remained round and swollen and no elongation was observed (<1% of spores elongated) (Fig. 5G); however, lysis occurred as with wild-type spores outgrowing in this medium. The inclusion of 1.0 M sorbitol in PAB medium did not give these effects (data not shown). The structure of PG purified from these outgrowing spherical pbpA spores 120 min after the initiation of spore germination was identical to that of PG harvested from wild-type spores which had initiated elongation. The addition of 10 mM MgCl2 improved both the rate of outgrowth and elongation efficiency of pbpA spores in PAB medium with 500 mM NaCl and prevented cell lysis in the presence of high salt but did not affect the formation of cells with increased diameters (Table 2 and data not shown). Spores lacking PBP2a and PBP4 exhibited outgrowth similar to that of pbpA spores in PAB medium with 500 mM NaCl with regard to rate and morphology, while spores lacking PBP1 and -2a did not initiate outgrowth in this medium (Table 2), as was found with spores lacking only PBP1 (19).
DISCUSSION
One difficulty in assessing the function of individual HMW PBPs is that these proteins tend to possess at least partially redundant enzyme activities (31). However, during outgrowth of B. subtilis spores, PBP2a, a class B HMW PBP with putative transpeptidase activity, appears to be uniquely important, at least initially, for cell elongation and the determination of proper cell diameter (18). Studies of the PG metabolism of outgrowing pbpA spores revealed slight decreases in PG synthesis and a significant delay in PG turnover compared to that in wild-type spores, suggesting that the coupling of PG synthesis and degradation may be altered in pbpA spores. However, in both strains no PG turnover was detected for at least 30 min after PG synthesis, consistent with models for PG metabolism during vegetative growth, where it has been established that new PG is inserted along the inner wall and spreads to the outer surface, where it is degraded by autolysins (24).
The slower PG turnover early in pbpA spore outgrowth may also explain, at least in part, the increased thickness of the cell wall in those spores at that time, as the magnitude of the decrease in PG turnover early in outgrowth appears greater than the slight slowing of PG synthesis. However, pbpA spores eventually give rise to vegetative cells which are identical to those of the wild-type strain (18). Consequently, any imbalance between PG synthesis and degradation early in the outgrowth of pbpA spores must be corrected as outgrowth proceeds. An additional explanation for the increased cell wall thickness of pbpA spores early in outgrowth is that, since outgrowing wild-type and pbpA spores have similar volumes (18), newly synthesized PG is inserted into a smaller surface area in the outgrowing spherical pbpA spore than in the more cylindrical outgrowing wild-type spore, as a sphere has a smaller surface area than a cylinder.
pbpA spores which had begun elongation showed no dramatic differences from wild type spores in the distribution of newly synthesized PG, after the data had been normalized for cell shape. This analysis excluded pbpA spores which remained round, where incorporation of PG precursors truly may have been different. Structural analysis of purified PG from outgrowing pbpA spores also revealed no differences from that in outgrowing wild-type spores, even when distinct morphological differences were observed. As we currently have no way of observing and quantitating changes in the three-dimensional structure of PG, we are unable to determine the exact nature of the defect during elongation of pbpA spores.
Both the kinetics and the morphology of outgrowing pbpA spores are sensitive to levels of monovalent and divalent cations in the growth medium. Interestingly, in PAB medium with 500 mM NaCl, pbpA spores remain spheres, with no detectable elongation before lysis, confirming the importance of PBP2a in spore elongation. The inclusion of high levels of divalent cations in this medium promotes spore elongation, even in the absence of PBP2a, but the mechanism of this effect of divalent cations is unclear. Previously we found that cells lacking PBP1 require increased levels of divalent cations for growth (19). The requirement of pbpA spores for elevated Mg2+ levels might be related, as a strain lacking both PBP1 and PBP2a requires higher levels of Mg2+ for spore outgrowth and vegetative growth than either single mutant alone (Table 3 and data not shown). This finding suggests that PBP2a also contributes to PG synthesis during vegetative growth, as has been previously reported (33).
To further define a role for PBP2a in spore outgrowth, we examined strains lacking PBP2a and one of the predominant class A HMW PBPs (PBP1, -2c, or -4) and a strain lacking PBP2a and the class B PBP3. The additional loss of either PBP2c or PBP3 had no effects different from those of the loss of PBP2a alone. However, the loss of PBP4 further slowed spore outgrowth, while the loss of PBP1 had an even more dramatic effect on spore outgrowth and viability. This is consistent with previous work demonstrating that PBP1 and PBP4 perform partially redundant functions, with PBP4 being the subordinate class A HMW PBP and PBP2c having an even more minor role (31).
Previous work has demonstrated that PBP1, a class A HMW PBP with putative transglycosylase and transpeptidase activities, is also involved in establishing the correct diameter of vegetative cells (30). Since PBP1 is expressed early in spore outgrowth (20, 30), it is not surprising that this protein is also important for reestablishing the rod-shaped cell morphology, starting from the elliptical dormant spore. The increased diameter observed in pbpA spores during outgrowth is absent in spores lacking PBP1 and PBP2a, suggesting that the activity of PBP1 is required for the increased diameter. Additionally, analysis of PG from spores lacking PBP1 and PBP2a suggests that the degree of PG cross-linking is transiently decreased 60 min after the initiation of spore germination. One possibility is that wild-type spores also undergo this transient decrease in cross-linking during spore outgrowth, but due to the time points we analyzed, this decrease was not detected. Another possibility is that pbpA ponA spores are transiently subjected to increased PG hydrolytic activity during outgrowth. However, a third and more intriguing possibility is that PBP1 and PBP2a possess a redundant transpeptidase activity which is crucial to proper spore outgrowth, the loss of which results in decreased cross-linking, which is corrected later in outgrowth as levels of other PBPs increase. PBP1 and PBP2a have both been found on the inner forespore membrane of dormant spores, consistent with the idea that these enzymes act in proximity to one another during spore outgrowth (4).
One possible model to explain the different morphology and PG structure of outgrowing spores lacking either PBP1, PBP2a, or both PBPs is that during wild-type spore outgrowth PBP1 synthesizes PG strands of a specific length, whose peptide side chains are then cross-linked by PBP2a and PBP1 to facilitate elongation at the appropriate diameter. Spores lacking PBP1 thus produce shorter glycan strands, which are initial substrates for PBP2a cross-linking, in such a way that a narrower cell diameter is established but elongation still occurs, perhaps facilitated by PBP2a, which may generate cross-links perpendicular to the glycan strands. However, whether the cross-linked glycan strands run parallel to each other or are in a more disorganized nonparallel configuration is not clear. At this time we also cannot determine whether the absence of specific PBPs alters the orientation of cross-linked glycan strands with respect to each other. In the absence of PBP2a, PBP1 polymerizes PG strands of the appropriate length, which are then cross-linked inappropriately, perhaps by other HMW PBPs or PBP1. Thus, instead of elongation, a large sphere forms, whose diameter is slowly reduced as PG remodeling occurs and other HMW PBPs are synthesized (17, 28, 29). In the absence of both PBP1 and -2a, short glycan strands which are cross-linked less efficiently, are made, resulting in poor cell elongation and cell death.
An interesting finding in this work is the presence of glycine in the PG purified from outgrowing spores, as previous studies of PG from vegetative B. subtilis cells did not report the presence of glycine (36, 38). More recently, glycine was detected in the PG of dormant spores lacking CwlD, a putative muramoyl-l-alanine amidase (26), and glycine is an important component in the PG of other gram-positive organisms, such as Staphylococcus aureus (15). It is unclear whether glycine is incorporated into the wall transiently during spore outgrowth or whether there is truly a glycine component of B. subtilis vegetative PG; this may be an important area for further study.
ACKNOWLEDGMENTS
This work was supported by grant GM19698 from the National Institutes of Health.
We are grateful to Kit Pogliano for protocols and to Steven Harris for the use of his microscope.
REFERENCES
- 1.Anagnostopoulos C, Spizizen J. Requirements for transformation in Bacillus subtilis. J Bacteriol. 1961;81:74–76. doi: 10.1128/jb.81.5.741-746.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atrih A P Z, Allmaier G, Foster S. Structural analysis of Bacillus subtilis 168 endospore peptidoglycan and its role during differentiation. J Bacteriol. 1996;178:6173–6183. doi: 10.1128/jb.178.21.6173-6183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buchanan C E, Henriques A O, Piggot P J. Cell wall changes during bacterial endospore formation. In: Ghuysen J-M, Hakenbeck R, editors. Bacterial cell wall. New York, N.Y: Elsevier Science Publishers; 1994. pp. 167–186. [Google Scholar]
- 4.Buchanan C E, Neyman S L. Correlation of penicillin-binding protein composition with different functions of two membranes in Bacillus subtilis forespores. J Bacteriol. 1986;165:498–503. doi: 10.1128/jb.165.2.498-503.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daniel R A, Drake S, Buchanan C E, Scholle R, Errington J. The Bacillus subtilis spoVD gene encodes a mother-cell-specific penicillin-binding protein required for spore morphogenesis. J Mol Biol. 1994;235:209–220. doi: 10.1016/s0022-2836(05)80027-0. [DOI] [PubMed] [Google Scholar]
- 6.de Chastellier C, Hellio R, Ryter A. Study of cell wall growth in Bacillus megaterium by high-resolution autoradiography. J Bacteriol. 1975;123:1184–1196. doi: 10.1128/jb.123.3.1184-1196.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duckworth M, Archibald A R, Baddily J. The location of N-acetyl galactosamine in the wall of Bacillus subtilis 168. Biochem J. 1972;130:691–696. doi: 10.1042/bj1300691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foster S J, Johnstone K. Pulling the trigger: the mechanism of bacterial spore germination. Mol Microbiol. 1990;4:137–141. doi: 10.1111/j.1365-2958.1990.tb02023.x. [DOI] [PubMed] [Google Scholar]
- 9.Ghuysen J-M. Serine β-lactamases and penicillin-binding proteins. Annu Rev Microbiol. 1991;45:37–67. doi: 10.1146/annurev.mi.45.100191.000345. [DOI] [PubMed] [Google Scholar]
- 10.Glaser L, Lindsay B. Relationship between cell wall turnover and cell growth in Bacillus subtilis. J Bacteriol. 1977;130:610–619. doi: 10.1128/jb.130.2.610-619.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kopriwa B M. A reliable, standardized method for ultrastructural electron microscopic radioautography. Histochemistry. 1973;37:1–17. doi: 10.1007/BF00306855. [DOI] [PubMed] [Google Scholar]
- 12.Kopriwa B M. A comparison of various procedures for fine grain development in electron microscopic autoradiography. Histochemistry. 1975;44:201–224. doi: 10.1007/BF00491492. [DOI] [PubMed] [Google Scholar]
- 13.Kopriwa B M, Levine G M, Nadler N J. Assessment of resolution by half distance values for tritium and radioiodine in electron microscopic radioautographs using Ilford L4 emulsion developed by “solution physical” or D-19b methods. Histochemistry. 1984;80:519–522. doi: 10.1007/BF02400965. [DOI] [PubMed] [Google Scholar]
- 14.Leighton T J, Doi R H. The stability of messenger ribonucleic acid during sporulation in Bacillus subtilis. J Biol Chem. 1971;246:3189–3195. [PubMed] [Google Scholar]
- 15.Matsuhashi M, Dorman C P, Strominger J L. Incorporation of glycine into the cell wall glycopeptide in Staphylococcus aureus: role of sRNA and lipid intermediates. Proc Natl Acad Sci USA. 1965;54:587–594. doi: 10.1073/pnas.54.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mauck J, Chan L, Glaser L. Turnover of the cell wall of Gram-positive bacteria. J Biol Chem. 1971;246:1820–1827. [PubMed] [Google Scholar]
- 17.Murray T, Popham D L, Setlow P. Identification and characterization of pbpC, the gene encoding Bacillus subtilis penicillin-binding protein 3. J Bacteriol. 1996;178:6001–6005. doi: 10.1128/jb.178.20.6001-6005.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murray T, Popham D L, Setlow P. Identification and characterization of pbpA encoding Bacillus subtilis penicillin-binding protein 2A. J Bacteriol. 1997;179:3021–3029. doi: 10.1128/jb.179.9.3021-3029.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murray T, Popham D L, Setlow P. Bacillus subtilis cells lacking PBP1 require increased levels of divalent cations for growth. J Bacteriol. 1998;180:4555–4563. doi: 10.1128/jb.180.17.4555-4563.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neyman S L, Buchanan C E. Restoration of vegetative penicillin-binding proteins during germination and outgrowth of Bacillus subtilis spores: relationship of individual proteins to specific cell cycle events. J Bacteriol. 1985;161:164–168. doi: 10.1128/jb.161.1.164-168.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicholson W L, Setlow P. Sporulation, germination, and outgrowth. In: Harwood C R, Cutting S M, editors. Molecular biological methods for Bacillus. Chichester, England: John Wiley & Sons Ltd.; 1990. pp. 391–450. [Google Scholar]
- 22.Pogliano K, Hofmeister A E M, Losick R. Disappearance of the ςE transcription factor from the forespore and the SpoIIE phosphatase from the mother cell contributes to the establishment of cell-specific gene expression during sporulation in Bacillus subtilis. J Bacteriol. 1997;179:3331–3341. doi: 10.1128/jb.179.10.3331-3341.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pooley H M. Turnover and spreading of old wall during surface growth of Bacillus subtilis. J Bacteriol. 1976;125:1127–1138. doi: 10.1128/jb.125.3.1127-1138.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pooley H M. Layered distribution, according to age, within the cell wall of Bacillus subtilis. J Bacteriol. 1976;125:1139–1147. doi: 10.1128/jb.125.3.1139-1147.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Popham D L, Helin J, Costello C E, Setlow P. Analysis of the peptidoglycan structure of Bacillus subtilis endospores. J Bacteriol. 1996;178:6451–6458. doi: 10.1128/jb.178.22.6451-6458.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Popham D L, Helin J, Costello C E, Setlow P. Muramic lactam in peptidoglycan of Bacillus subtilis spores is required for spore outgrowth but not for spore dehydration or heat resistance. Proc Natl Acad Sci USA. 1996;93:15405–15410. doi: 10.1073/pnas.93.26.15405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Popham D L, Illades-Aguiar B, Setlow P. The Bacillus subtilis dacB gene, encoding penicillin-binding protein 5*, is part of a three-gene operon required for proper spore cortex synthesis and spore core dehydration. J Bacteriol. 1995;177:4721–4729. doi: 10.1128/jb.177.16.4721-4729.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Popham D L, Setlow P. Cloning, nucleotide sequence, and regulation of the Bacillus subtilis pbpF gene, which codes for a putative class A high-molecular-weight penicillin-binding protein. J Bacteriol. 1993;175:4870–4876. doi: 10.1128/jb.175.15.4870-4876.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Popham D L, Setlow P. Cloning, nucleotide sequence, mutagenesis, and mapping of the Bacillus subtilis pbpD gene, which codes for penicillin-binding protein 4. J Bacteriol. 1994;176:7197–7205. doi: 10.1128/jb.176.23.7197-7205.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Popham D L, Setlow P. Cloning, nucleotide sequence, and mutagenesis of the Bacillus subtilis ponA operon, which codes for penicillin-binding protein (PBP) 1 and a PBP-related factor. J Bacteriol. 1995;177:326–335. doi: 10.1128/jb.177.2.326-335.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Popham D L, Setlow P. Phenotypes of Bacillus subtilis mutants lacking multiple class A high-molecular-weight penicillin-binding proteins. J Bacteriol. 1996;178:2079–2085. doi: 10.1128/jb.178.7.2079-2085.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Setlow B, Hand A R, Setlow P. Synthesis of a Bacillus subtilis small, acid-soluble protein in Escherichia coli causes cell DNA to assume some characteristics of spore DNA. J Bacteriol. 1991;173:1642–1653. doi: 10.1128/jb.173.5.1642-1653.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shohayeb M, Chopra I. Mutations affecting penicillin-binding proteins 2a, 2b and 3 in Bacillus subtilis alter cell shape and peptidoglycan metabolism. J Gen Microbiol. 1987;133:1733–1742. doi: 10.1099/00221287-133-7-1733. [DOI] [PubMed] [Google Scholar]
- 34.Spurr A R. A low viscosity epoxy resin embedding medium for electron microscopy. J Ultrastruct Res. 1969;26:31–43. doi: 10.1016/s0022-5320(69)90033-1. [DOI] [PubMed] [Google Scholar]
- 35.Sussman A S, Halvorson H O. Spores: their dormancy and germination. New York, N.Y: Harper and Row; 1966. Physiological and biochemical changes occurring during germination and outgrowth; pp. 216–269. [Google Scholar]
- 36.Warth A D, Strominger J L. Structure of the peptidoglycan from vegetative cell walls of Bacillus subtilis. Biochemistry. 1971;10:4349–4358. doi: 10.1021/bi00800a001. [DOI] [PubMed] [Google Scholar]
- 37.Yanouri A, Daniel R A, Errington J, Buchanan C E. Cloning and sequencing of the cell division gene pbpB, which encodes penicillin-binding protein 2B in Bacillus subtilis. J Bacteriol. 1993;175:7604–7616. doi: 10.1128/jb.175.23.7604-7616.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Young F E. Variation in chemical composition of cell wall of Bacillus subtilis during growth in different media. Nature. 1965;207:104–105. doi: 10.1038/207104b0. [DOI] [PubMed] [Google Scholar]