

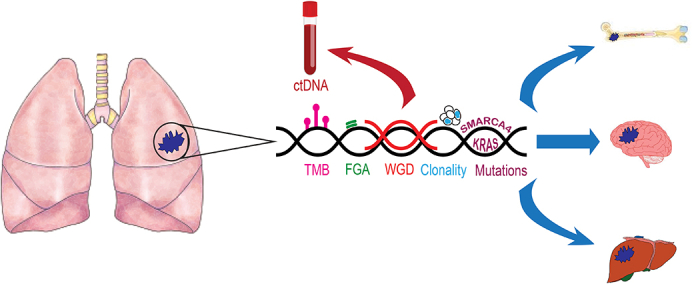
Next-generation sequencing can be used to assess risk of metastasis in NSCLC.
Central Message.
Genomic profiling in patients with operable, early-stage NSCLC can enhance our understanding of tumor biology and metastatic risk.
Next-generation sequencing (NGS) and its applications have provided tremendous insight into the biologic landscape of a variety of solid tumors, including non–small cell lung cancer (NSCLC). The genomic characterization of NSCLC has not only altered treatment paradigms but has also aided investigations into tumor biology and the risk of developing metastatic disease. Complete surgical resection with or without chemotherapy and radiation has been the cornerstone of treatment for stage I-III NSCLC; however, up to 50% of patients with operable, early-stage NSCLC are at risk of metastasis after surgical resection.1,2 Thus, recent investigations have applied the knowledge gained in the stage IV NSCLC setting to earlier-stage disease. Studies are now focused on leveraging tumor genomic data (DNA and RNA) to elucidate the biologic mechanisms associated with disease recurrence and to potentially identify patients with a higher risk of recurrence for additional adjuvant therapies.
Diagnostic molecular pathologic analysis and related tumor genomic and transcriptomic analyses are presently being applied to several solid tumors. An obvious example is the use of Oncotype Dx, a 21-gene expression assay for patients with ER-positive/HER2-negative stage I-IIIA breast cancer. Oncotype Dx tests for genes related to cell proliferation, metastasis, HER2 expression, and sex hormone production.3,4 Current National Comprehensive Cancer Network (NCCN) guidelines recommend Oncotype Dx as the preferred panel for prognosis and prediction of benefit from adjuvant chemotherapy.5 The broad applicability of gene assays such as Oncotype Dx in NSCLC has been limited by the substantial histologic and molecular heterogeneity of NSCLC. In this report, we outline the current role of genomics in the management of NSCLC, including risk-stratification of patients, and provide foundational knowledge for thoracic surgeons caring for patients with NSCLC.
NSCLC Heterogeneity Contributes to Metastatic Risk
Recent publications by the TRACERx Lung consortium have highlighted the presence of significant intratumoral subclonal somatic copy number alterations (SCNA) and mutations using whole-exome sequencing data on surgically resected NSCLC.6,7 In a cohort of treatment-naive patients with stage I-III NSCLC who underwent complete surgical resection, tumors with a greater proportion of subclonal mutations were associated with an increased risk of recurrence or death.6 Subclonal whole-genome doubling, recent subclonal expansion, and a high level of SCNA intratumor heterogeneity (ITH) were also predictive of relapse; however, only high SCNA ITH was independently prognostic of early (<12 months after surgery) and extrathoracic recurrences.7 Wu and colleagues8 demonstrated similar findings in a spatial analysis of the ITH of lung adenocarcinoma in which they identified 2 distinct patterns—clustered and random geographic diversification—using both proteomic and genomic data. When clinicopathologic features were controlled for, the random proteomic geographic diversification pattern was associated with a greater risk of recurrence or death. These studies, which used NGS, offer important therapeutic insights into the mechanisms of resistance and vulnerability in NSCLC tumors, and future studies will continue to improve our appreciation of the diverse intratumoral environment.
In addition to molecular heterogeneity, NSCLC displays considerable histologic heterogeneity. The most common histologic type of NSCLC is adenocarcinoma, and further division into predominant subtypes (ie, lepidic, acinar, papillary, micropapillary, and solid) has provided unique genomic insights into the risk of metastasis for these patients.9 Caso and colleagues10 described changes at the gene and pathway levels that are associated with histologic subtype. A greater proportion of lepidic-predominant tumors had alterations in EGFR, RBM10, and TERT, compared with other subtypes, whereas the more-aggressive histologic subtypes (micropapillary and solid) had enrichment of BRAF, TP53, SETD2, MGA, and SMARCA4 mutations. The presence of an aggressive histologic subtype was also associated with greater chromosomal instability, and in the TRACERx cohort, greater chromosomal instability was associated with shorter disease-free survival (DFS) and an increased risk of metastasis.6 Oncogenic pathway alterations also vary according to histologic subtype, with less-invasive lepidic-predominant tumors associated with RTK/RAS pathway changes and more-invasive micropapillary- and solid-predominant tumors associated with Myc, p53, and Wnt pathway changes.11
Genomic Changes Associated With Lung Adenocarcinoma Metastasis
Several genes and oncogenic pathways have been associated with metastasis in early-stage lung adenocarcinoma. Lengel and colleagues12 performed NGS on 766 primary lung adenocarcinoma samples and compared surgically resected primary tumors that metastasized with those that did not (Figure 1). Alterations in several genes, including TP53, KEAP1, CDKN2A, MDM2, PIK3CA, NKX2-1, RB1, MYC, SMARCA4, and FOXA1, were more common in primary lung adenocarcinomas that metastasized. Other studies have confirmed some of these findings in similar cohorts of patients.10,13,14 Conversely, RBM10 is more common in lung adenocarcinoma that does not metastasize.12 In surgically resected tumors that metastasize, specific genomic alterations also influence survival; MDM2, MYC, SMARCA4, and TP53 are associated with shorter metastasis-free survival, whereas EGFR and NF1 are associated with longer survival after relapse.
Figure 1.

Oncoprint comparing nonmetastatic (NM) and ever-metastatic (EM) primary lung adenocarcinoma tumors. The oncoprint displays clinical features and genes altered at significantly different frequencies between primary lung adenocarcinomas that metastasized (EM) and those that did not (NM). Hx, History; pStage, pathologic stage. (From Lengel and colleagues.12 Reprinted with permission).
Certain pathway alterations also increase a patient's risk of NSCLC metastasis, including alterations in the p53, PI3K, cell cycle, and transforming growth factor-beta pathways.12 Changes in the Wnt signaling pathway have also been associated with poor outcomes in lung adenocarcinoma without a clear oncogenic driver.15 Cui and colleagues15 showed that differential expression of 4 Wnt pathway genes (CTNNB1, SOB9, DVL3, and WNT2B) can be used to partition patients into low- and high-risk groups with respect to overall survival. This finding is supported by the work of Kim and colleagues,13 who identified CTNNB1 as an independent predictor of recurrence in early-stage lung adenocarcinoma. Finally, the total number of oncogenic pathways altered is also important. An increased number of pathways altered is not only associated with high-risk clinicopathologic features—such as maximum standardized uptake value on positron emission tomography, aggressive histologic subtype, and lymph node involvement—but is also an independent predictor of worse DFS.16
There has been a focus on driver alterations in NSCLC, and targeted therapies for an increasing number of these mutations have been developed.13 However, activating mutations in EGFR and their association with prognosis in early-stage lung adenocarcinoma is unclear. Kim and colleagues13 found that the presence of an EGFR mutation portended a better prognosis, whereas others have found no association between EGFR mutation and presence or absence of metastasis.12,13 Deng and colleagues17 reported that EGFR mutations were associated with metastatic organotropism to the brain and bone in a cohort of 1531 patients who underwent NGS; however, Lengel and colleagues12 did not find a link between EGFR alterations and metastatic organotropism. The diverging results on the association between EGFR and metastasis may be attributable to the differing frequencies of EGFR mutations in Asian and North American populations.18 KRAS mutations have also been implicated in recurrence of early-stage NSCLC. KRAS G12C alterations are associated with shorter recurrence-free survival (RFS), compared with other KRAS mutations, in surgically resected lung adenocarcinoma.19 Furthermore, co-mutation of any KRAS mutation with STK11, ATM, or LRRIQ3 was associated with shorter RFS, compared with KRAS mutation alone.14 KRAS alterations have also been associated with early metastasis, which is defined as a recurrence within 2 years of surgical resection.17
Finally, individual genomic alterations are not the sole determinant of prognosis in lung adenocarcinoma, as other genomic parameters play a major role. Multiple studies, including a recent report of TRACERx data, found that high tumor mutation burden, fraction of genome altered, and whole-genome doubling were all more common in primary lung adenocarcinoma tumors that metastasized than in those that did not.6,12 Tumor mutation burden is an important biomarker, as studies have demonstrated its ability to predict response to immunotherapy in NSCLC.20 These observations strongly suggest that NGS panels should investigate changes at the chromosomal level in addition to individual gene alterations.21
Validated Models Predicting Recurrence in Lung Adenocarcinoma
The identification of significant clinical, pathologic, and genomic features that influence metastasis and survival is an important aspect of properly risk-stratifying patients and provides useful information for decisions regarding treatment. Yet these factors in isolation are often inadequate, and models incorporating multiple variables to predict patient outcomes are more clinically applicable. In a cohort of patients with surgically resected early-stage lung adenocarcinoma (75% pathologic stage I), a multivariable analysis including clinicopathologic and genomic variables showed that fraction of genome altered and alterations in SMARCA4 and TP53 were associated with worse RFS.22 Based on association analyses, Jones and colleagues created a recurrence prediction model, PRecur, using a machine-learning framework (Table 1).22, 23,E1 When PRecur was used to stratify patients on the basis of predicted RFS, it outperformed the standard prognostic tumor, node and metastasis model (concordance probability estimate, 0.73 vs 0.61; P < .001) (Figure 2).22 Interestingly, the PRecur model correctly classified 83% of patients with stage I who developed a recurrence.
Table 1.
Genes included in select risk prediction models
| Prediction model | Genes included |
|---|---|
| Kratz et al23 | BAG1, BRCA1, CDC6, CD2AP1, ERBB3, FUT3, IL11, LCK, RND3, SH3BGR, WNT3A, ESD, TBP, YAP1 |
| ORACLEE1 | ANLN, ASPM, CDCA4, ERRFI1, FURIN, GOLGA8A, ITGA6, JAG1, LRP12, MAFF, MRPS17, PLK1, PNP, PPP1R13L, PRKCA, PTTG1, PYGB, RPP25, SPEP1, SCL46A3, SNX7, TPBG, XBP1 |
| PRecur22 | ALK, APC, ARDI1A, ARID2, ATM, B2M, BAP1, BCOR, BRAF, BRCA2, CDK4, CDKN2A, CDKN2B, CTNNB1, EGFR, ERBB2, FAT1, FOXA1, GLI1, GNAS, KEAP1, KIT, KRAS, MDM2, MED12, MET, MGA, MYC, NF1, NF2, NKX2.1, NTRK1, PIK3CA, PIK3R1, PTPRD, PTPRT, RB1, RBM10, RET, ROS1, SETD2, SMAD4, SMARCA4, STK11, TERT, TP53, U2AF1 |
Figure 2.

Computational machine-learning prediction model (PRecur) for relapse-free survival applied to 2 patient scenarios. A, Patient with a small, 1.8-cm tumor (pT1bN0M0, stage IA). Three-year RFS curves were predicted by PRecur versus the TNM model for all patients with pT1bN0M0 in the study cohort (n = 136). B, Patient with a large, 5.1-cm tumor (pT3N0M0, stage IIB). Three-year RFS curves were predicted by PRecur versus the TNM model for all patients with pT3N0M0 in the study cohort (n = 53). SUVmax, Maximum standardized uptake value; TMB, tumor mutation burden; FGA, fraction of genome altered; LVI, lymphovascular invasion; RFS, recurrence-free survival; TNM, tumor, node and metastasis. (From Jones and colleagues.22 Reprinted with permission).
In a similar cohort of patients with nonsquamous NSCLC who underwent surgical resection, a smaller, 14-gene assay (Encore Clinical) identified patients at high risk of recurrence (Table 1).23 This assay has been validated in both North American and Asian populations, and it outperformed tumor, node and metastasis staging and the NCCN high-risk guidelines for prediction of recurrence.24,25 Furthermore, in a prospective cohort of 100 patients with stage I-IIA disease, 5-year DFS was 92% among molecularly high-risk patients who received adjuvant chemotherapy, compared with 49% among molecularly high-risk patients who did not receive adjuvant chemotherapy.24 Based on these results, panels like this may be able to identify early-stage patients who would benefit from adjuvant therapy.
Given the complexity of ITH in NSCLC, Biswas and colleaugesE1 tested previously published RNA sequencing (RNA-seq)-based and microarray-based prognostic assays on multiple tumor regions in 48 patients. They recognized that whether a patient was classified as low risk or high risk on RNA-seq prognostic models was dependent on the tumor region analyzed for a given patient, with discordance rates of 29% to 43%. When microarray-based prognostic models were used, median discordance was 50% among 9 models, which means that half of patients could be misclassified by these models, depending on the area of the tumor selected for analysis. Therefore, using RNA-seq data to identify genes associated with intratumoral homogeneity but high variability between tumors, the authors developed a 23-gene prognostic signature, ORACLE (Table 1). ORACLE had a dramatically lower rate of discordance of 11%. Furthermore, this model has been predictive of overall survival in several external validation cohorts.E2,E3
Last, recent work by Sorin and colleaguesE4 has highlighted the predictive role that the tumor immune microenvironment plays with respect to patient outcomes. They used highly multiplexed imaging mass cytometry to perform spatial analysis of immune cells and their activation states and, using deep learning, identified patients who will have recurrence after surgical resection. While this method is not widely available at this time, it is a tremendous advancement that could serve as a valuable tool for future investigations.
Genomic Profiling in Lung Squamous Cell Carcinoma
NGS has identified far fewer distinct genomic profiles in lung squamous cell carcinoma than in lung adenocarcinoma. Genomic alterations in lung squamous cell carcinoma appear to resemble alterations in squamous cell carcinomas of other solid organs, rather than alterations in lung adenocarcinoma.E5 Sanchez-Vega and colleagues11 analyzed the oncogenic signaling pathways altered in 502 lung adenocarcinoma and 464 lung squamous cell carcinoma samples from The Cancer Genome Atlas (TCGA). Squamous cell carcinoma samples more frequently had alterations in the p53 (86% vs 61%) and PI3K (68% vs 38%) pathways and less frequently had alterations in the RTK/RAS pathway (54% vs 77%), compared with adenocarcinoma samples. Compared with genomic alterations in lung adenocarcinoma, alterations in squamous cell carcinoma have been investigated less frequently; however, several of these commonly altered genes have been associated with recurrence in patients with squamous cell carcinoma. Alterations in TP53 have been associated with an increased risk of recurrence, whereas PI3KCA mutations exhibit increased time to recurrence and better overall survival.E6,E7 BCL6 and ARID1A alterations have also been shown to be associated with shorter DFS and overall survival, respectively, in patients with lung squamous cell carcinoma.E8
The tumor immune microenvironment appears to play a significant role in recurrence of early-stage squamous cell carcinoma, as Fan and colleagues uncovered differentially expressed immune-related genes.E9 They developed a signature comprising 17 immune-related genes to predict overall survival in patients with early-stage lung squamous cell carcinoma; this gene signature has been externally validated. A summary of genes associated with both lung adenocarcinoma and squamous cell carcinoma is shown in Table 2.
Table 2.
Genes associated with recurrence in non–small cell lung cancer
Using Circulating Tumor DNA (ctDNA) to Assess Metastatic Risk
When considering metastasis in NSCLC, providers face the challenge of risk-stratifying patients with early-stage NSCLC and how to accurately surveil them postoperatively. At present, the NCCN guidelines recommend that all patients undergo a history, physical examination, and computed tomography of the chest with or without contrast every 6 months for 2 to 3 years, and a low-dose noncontrast computed tomography of the chest annually thereafter.E10 One recent development that provides an assessment of a patient's status in real time is ctDNA. LUNGCA-1, a prospective multicenter study of perioperative ctDNA in patients with NSCLC undergoing surgery, demonstrated that a high concentration of ctDNA before resection was a negative predictor of RFS.E11 Conversely, in the CheckMate 816 cohort, ctDNA clearance before the last cycle of nivolumab and chemotherapy was associated with pathologic complete response, compared with those who remained ctDNA positive (46% vs 0%); pathologic complete response was associated with better event-free survival.E12 In a similar cohort of patients, Abbosh and colleaguesE13 found that the presence of preoperative ctDNA was more common in patients with nonadenocarcinoma lung cancer (compared with lung adenocarcinoma [92% vs 42%]), more smoking pack-years, and clinically occult mediastinal disease in lung adenocarcinoma. A landmark analysis of these patients 120 days postoperatively revealed that 93% of patients with minimal residual disease (MRD) detected by the presence of ctDNA developed recurrence. They also used ctDNA to determine that clonal expansion predicted metastasis.
Currently, there is no standard ctDNA assay, and the basis for detection in these assays can be either tumor-specific or tumor-independent (ie, epigenetic features), with variable limits of detection.E14 While tumor-independent methods have the benefit of lower costs and do not require tissue acquisition, tumor-specific approaches have better limits of detection and sensitivity. In the MRD setting, sensitivity remains an issue, as a landmark analysis of a variety of ctDNA assays across multiple solid tumors revealed a median sensitivity of only 56%.E15 Chaudhuri and colleaguesE16 improved on the previous methodology with the use of a tumor-informed NGS platform (CAPP-seq) that detects commonly mutated lung cancer genes at a lower limit of detection of 0.002%. In a cohort that included patients with stage I-III NSCLC treated with curative intent and healthy controls, receiver operating characteristic analysis revealed an area under the curve of 0.97 and maximal sensitivity and specificity of 93% and 96%, respectively. Similarly, Kurtz and colleaguesE17 greatly improved the limit of detection of ctDNA and thus assay sensitivity using an alternative method called phased variant enrichment and detection sequencing (PhasED-seq), which detects multiple somatic mutations in individual DNA fragments. Although these assays are not widely available in clinical practice, these 2 studies offer the most promising ctDNA assays that should be incorporated in future trials.
Concerns have also been raised regarding the predictive value of MRD ctDNA assays in the context of recurrence. The IMpower010 trial, which investigated atezolizumab versus best supportive care (BSC) after chemotherapy in patients with resected stage IB-IIIA NSCLC, used a different commercially available ctDNA assay (Signatera; Natera) to detect MRD postoperatively.E18 Patients who were ctDNA-negative had better DFS than ctDNA-positive patients, regardless of which treatment they received (atezolizumab vs BSC). These results demonstrate the strong positive predictive value of these assays. However, in the BSC arm, nearly two-thirds of recurrences occurred in patients who were ctDNA negative, highlighting the low negative predictive value of the current ctDNA platforms. The IMpower010 authors also found that ctDNA positivity increased with stage (IB = 9%, II = 14%, IIIA = 29%). The ctDNA assays presented here are only three of several platforms available. Thus, ctDNA is an exciting development in the fight against metastasis in NSCLC, and subsequent studies will need to build on the current assays to improve the applicability of this biomarker in the clinical setting.
Conclusions
Tumor genomic profiling is now a standard-of-care tool for early-stage NSCLC and has several clinical implications. Models incorporating tumor genomic alterations serve as important predictors of metastasis and assist in our ability to risk-stratify patients. However, genomic analyses have also shown that patients with NSCLC are a diverse group who cannot be separated in neatly organized silos. Currently, clinicians caring for these patients face the challenge of synthesizing all the data that genomics has provided to make treatment decisions. Strategies incorporating the complexity of ITH in NSCLC into these prediction models will strengthen their utility and clinical applicability. As researchers better understand genetic alterations in NSCLC, additional treatments tailored to the individual patient will become available. Further, ctDNA assays that detect ctDNA preoperatively or the presence of MRD postoperatively represent a promising development for the identification of patients with early-stage disease who may benefit from neoadjuvant or adjuvant therapy. Overall, genomic profiling has the potential to move us away from a one-size-fits-all treatment approach to a personalized approach that offers patients a greater benefit to survival.
Conflict of Interest Statement
David R. Jones serves as a consultant or speaker for or has received grant support from Merck, AstraZeneca, Genentech, More Health, and DAVA Oncology. All other authors reported no conflicts of interest.
The Journal policy requires editors and reviewers to disclose conflicts of interest and to decline handling or reviewing manuscripts for which they may have a conflict of interest. The editors and reviewers of this article have no conflicts of interest.
Footnotes
This study was supported by the National Institutes of Health/National Cancer Institute (R01CA217169 and R01CA240472 to David R. Jones and Cancer Center Support Grant P30 CA008748 to Memorial Sloan Kettering Cancer Center).
References
- 1.Brandt W.S., Yan W., Zhou J., Tan K.S., Montecalvo J., Park B.J., et al. Outcomes after neoadjuvant or adjuvant chemotherapy for cT2-4N0-1 non–small cell lung cancer: a propensity-matched analysis. J Thorac Cardiovasc Surg. 2019;157:743–753.e743. doi: 10.1016/j.jtcvs.2018.09.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Potter A.L., Costantino C.L., Suliman R.A., Haridas C.S., Senthil P., Kumar A., et al. Recurrence after complete resection for non–small cell lung cancer in the National Lung Screening Trial. Ann Thorac Surg. 2023;116:684–692. doi: 10.1016/j.athoracsur.2023.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Paik S., Shak S., Tang G., Kim C., Baker J., Cronin M., et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 4.Kalinsky K., Barlow W.E., Gralow J.R., Meric-Bernstam F., Albain K.S., Hayes D.F., et al. 21-Gene assay to inform chemotherapy benefit in node-positive breast cancer. N Engl J Med. 2021;385:2336–2347. doi: 10.1056/NEJMoa2108873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.National Comprehensive Cancer Network Breast cancer. Version 4.2023. https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1419
- 6.Jamal-Hanjani M., Wilson G.A., McGranahan N., Birkbak N.J., Watkins T.B.K., Veeriah S., et al. Tracking the evolution of non–small-cell lung cancer. N Engl J Med. 2017;376:2109–2121. doi: 10.1056/NEJMoa1616288. [DOI] [PubMed] [Google Scholar]
- 7.Frankell A.M., Dietzen M., Al Bakir M., Lim E.L., Karasaki T., Ward S., et al. The evolution of lung cancer and impact of subclonal selection in TRACERx. Nature. 2023;616:525–533. doi: 10.1038/s41586-023-05783-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu H.J., Temko D., Maliga Z., Moreira A.L., Sei E., Minussi D.C., et al. Spatial intra-tumor heterogeneity is associated with survival of lung adenocarcinoma patients. Cell Genom. 2022;2:100165. doi: 10.1016/j.xgen.2022.100165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Travis W.D., Brambilla E., Noguchi M., Nicholson A.G., Geisinger K.R., Yatabe Y., et al. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. 2011;6:244–285. doi: 10.1097/JTO.0b013e318206a221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caso R., Sanchez-Vega F., Tan K.S., Mastrogiacomo B., Zhou J., Jones G.D., et al. The underlying tumor genomics of predominant histologic subtypes in lung adenocarcinoma. J Thorac Oncol. 2020;15:1844–1856. doi: 10.1016/j.jtho.2020.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanchez-Vega F., Mina M., Armenia J., Chatila W.K., Luna A., La K.C., et al. Oncogenic signaling pathways in The Cancer Genome Atlas. Cell. 2018;173:321–337.e310. doi: 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lengel H.B., Mastrogiacomo B., Connolly J.G., Tan K.S., Liu Y., Fick C.N., et al. Genomic mapping of metastatic organotropism in lung adenocarcinoma. Cancer Cell. 2023;41:970–985.e973. doi: 10.1016/j.ccell.2023.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim I.A., Hur J.Y., Kim H.J., Park J.H., Hwang J.J., Lee S.A., et al. Targeted next-generation sequencing analysis for recurrence in early-stage lung adenocarcinoma. Ann Surg Oncol. 2021;28:3983–3993. doi: 10.1245/s10434-020-09276-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kadara H., Choi M., Zhang J., Parra E.R., Rodriguez-Canales J., Gaffney S.G., et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Ann Oncol. 2017;28:75–82. doi: 10.1093/annonc/mdw436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cui Y., Fang W., Li C., Tang K., Zhang J., Lei Y., et al. Development and validation of a novel signature to predict overall survival in “driver gene-negative” lung adenocarcinoma (LUAD): results of a multicenter study. Clin Cancer Res. 2019;25:1546–1556. doi: 10.1158/1078-0432.CCR-18-2545. [DOI] [PubMed] [Google Scholar]
- 16.Zhou J., Sanchez-Vega F., Caso R., Tan K.S., Brandt W.S., Jones G.D., et al. Analysis of tumor genomic pathway alterations using broad-panel next-generation sequencing in surgically resected lung adenocarcinoma. Clin Cancer Res. 2019;25:7475–7484. doi: 10.1158/1078-0432.CCR-19-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng C., Zhang Y., Fu F., Ma X., Wen Z., Ma Z., et al. Genetic-pathological prediction for timing and site-specific recurrence pattern in resected lung adenocarcinoma. Eur J Cardio Thorac Surg. 2021;60:1223–1231. doi: 10.1093/ejcts/ezab288. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y.L., Yuan J.Q., Wang K.F., Fu X.H., Han X.R., Threapleton D., et al. The prevalence of EGFR mutation in patients with non-small cell lung cancer: a systematic review and meta-analysis. Oncotarget. 2016;7:78985–78993. doi: 10.18632/oncotarget.12587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones G.D., Caso R., Tan K.S., Mastrogiacomo B., Sanchez-Vega F., Liu Y., et al. KRAS (G12C) mutation is associated with increased risk of recurrence in surgically resected lung adenocarcinoma. Clin Cancer Res. 2021;27:2604–2612. doi: 10.1158/1078-0432.CCR-20-4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hellmann M.D., Ciuleanu T.-E., Pluzanski A., Lee J.S., Otterson G.A., Audigier-Valette C., et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378:2093–2104. doi: 10.1056/NEJMoa1801946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leader A.M., Grout J.A., Maier B.B., Nabet B.Y., Park M.D., Tabachnikova A., et al. Single-cell analysis of human non–small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell. 2021;39:1594–1609.e1512. doi: 10.1016/j.ccell.2021.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones G.D., Brandt W.S., Shen R., Sanchez-Vega F., Tan K.S., Martin A., et al. A Genomic-pathologic annotated risk model to predict recurrence in early-stage lung adenocarcinoma. JAMA Surg. 2021;156:e205601. doi: 10.1001/jamasurg.2020.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kratz J.R., He J., Van Den Eeden S.K., Zhu Z.H., Gao W., Pham P.T., et al. A practical molecular assay to predict survival in resected non-squamous, non–small-cell lung cancer: development and international validation studies. Lancet. 2012;379:823–832. doi: 10.1016/S0140-6736(11)61941-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woodard G.A., Wang S.X., Kratz J.R., Zoon-Besselink C.T., Chiang C.Y., Gubens M.A., et al. Adjuvant chemotherapy guided by molecular profiling and improved outcomes in early stage, non–small-cell lung cancer. Clin Lung Cancer. 2018;19:58–64. doi: 10.1016/j.cllc.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 25.Haro G.J., Sheu B., Cook N.R., Woodard G.A., Mann M.J., Kratz J.R. Comparison of conventional TNM and novel TNMB staging systems for non–small cell lung cancer. JAMA Netw Open. 2019;2:e1917062. doi: 10.1001/jamanetworkopen.2019.17062. [DOI] [PMC free article] [PubMed] [Google Scholar]
E-References
- Biswas D., Birkbak N.J., Rosenthal R., Hiley C.T., Lim E.L., Papp K., et al. A clonal expression biomarker associates with lung cancer mortality. Nat Med. 2019;25:1540–1548. doi: 10.1038/s41591-019-0595-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djureinovic D., Hallström B.M., Horie M., Mattsson J.S.M., La Fleur L., Fagerberg L., et al. Profiling cancer testis antigens in non-small-cell lung cancer. JCI Insight. 2016;1 doi: 10.1172/jci.insight.86837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okayama H., Kohno T., Ishii Y., Shimada Y., Shiraishi K., Iwakawa R., et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012;72:100–111. doi: 10.1158/0008-5472.CAN-11-1403. [DOI] [PubMed] [Google Scholar]
- Sorin M., Rezanejad M., Karimi E., Fiset B., Desharnais L., Perus L.J.M., et al. Single-cell spatial landscapes of the lung tumour immune microenvironment. Nature. 2023;614:548–554. doi: 10.1038/s41586-022-05672-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell J.D., Alexandrov A., Kim J., Wala J., Berger A.H., Pedamallu C.S., et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016;48:607–616. doi: 10.1038/ng.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Bakir M., Huebner A., Martínez-Ruiz C., Grigoriadis K., Watkins T.B.K., Pich O., et al. The evolution of non-small cell lung cancer metastases in TRACERx. Nature. 2023;616:534–542. doi: 10.1038/s41586-023-05729-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan M., Hoven A.S., Lund-Iversen M., Solberg S., Helland Å., Hirsch F.R., et al. PIK3CA mutations as prognostic factor in squamous cell lung carcinoma. Lung Cancer. 2017;103:52–57. doi: 10.1016/j.lungcan.2016.11.018. [DOI] [PubMed] [Google Scholar]
- Connolly J.G., Tan K.S., Sanchez-Vega F., Jones G.D., Liu Y., Caso R., et al. A23 A Genomically adjusted clinicopathologic model predicts recurrence in resected early-stage lung squamous cell carcinoma. J Thorac Oncol. 2020;15:S19–S20. [Google Scholar]
- Fan T., Lu Z., Liu Y., Wang L., Tian H., Zheng Y., et al. A novel immune-related seventeen-gene signature for predicting early stage lung squamous cell carcinoma prognosis. Front Immunol. 2021;12 doi: 10.3389/fimmu.2021.665407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Comprehensive Cancer Network Non-Small Cell Lung Cancer. Version 3.2023. https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1450
- Xia L., Mei J., Kang R., Deng S., Chen Y., Yang Y., et al. Perioperative ctDNA-based molecular residual disease detection for non-small cell lung cancer: a prospective multicenter cohort study (LUNGCA-1) Clin Cancer Res. 2022;28:3308–3317. doi: 10.1158/1078-0432.CCR-21-3044. [DOI] [PubMed] [Google Scholar]
- Forde P.M., Spicer J., Lu S., Provencio M., Mitsudomi T., Awad M.M., et al. Neoadjuvant nivolumab plus chemotherapy in resectable lung cancer. N Engl J Med. 2022;386:1973–1985. doi: 10.1056/NEJMoa2202170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbosh C., Frankell A.M., Harrison T., Kisistok J., Garnett A., Johnson L., et al. Tracking early lung cancer metastatic dissemination in TRACERx using ctDNA. Nature. 2023;616:553–562. doi: 10.1038/s41586-023-05776-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestvina C.M., Garassino M.C., Neal J.W., Wakelee H.A., Diehn M., Vokes E.E. Early-stage lung cancer: using circulating tumor DNA to get personal. J Clin Oncol. 2023 doi: 10.1200/JCO.23.00258. [DOI] [PubMed] [Google Scholar]
- Moding E.J., Nabet B.Y., Alizadeh A.A., Diehn M. Detecting liquid remnants of solid tumors: circulating tumor DNA minimal residual disease. Cancer Discov. 2021;11:2968–2986. doi: 10.1158/2159-8290.CD-21-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri A.A., Chabon J.J., Lovejoy A.F., Newman A.M., Stehr H., Azad T.D., et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov. 2017;7:1394–1403. doi: 10.1158/2159-8290.CD-17-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz D.M., Soo J., Co Ting Keh L., Alig S., Chabon J.J., Sworder B.J., et al. Enhanced detection of minimal residual disease by targeted sequencing of phased variants in circulating tumor DNA. Nat Biotechnol. 2021;39:1537–1547. doi: 10.1038/s41587-021-00981-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C., Das Thakur M., Srivastava M.K., Zou W., Xu H., Ballinger M., et al. 2O IMpower010: Biomarkers of disease-free survival (DFS) in a phase III study of atezolizumab (atezo) vs best supportive care (BSC) after adjuvant chemotherapy in stage IB-IIIA NSCLC. Ann Oncol. 2021;32:S1374. [Google Scholar]