

Abstract
The compound material titanium oxycarbide (TiOC) is found to be an effective electrocatalyst for the electrochemical oxidation of ethanol to CO2. The complete course of this reaction is one of the main challenges in direct ethanol fuel cells (DEFCs). While TiOC has previously been investigated as catalyst support material only, in this study we show that TiOC alone is able to oxidize ethanol to acetaldehyde without the need of expensive noble metal catalysts like Pt. It is suggested that this behavior is attributed to the presence of both undercoordinated sites, which allow ethanol to adsorb, and oxygenated sites, which facilitate the activation of water. This is a milestone in DEFC research and development and opens up innovative possibilities for the design of catalyst materials for intermediate temperature fuel cells.
Keywords: titanium oxycarbide, compound material, DEMS, EOR, energy conversion, platinum-free
Alternative energy conversion systems are considered crucial for mitigating challenges related to limited fossil fuel supplies and a rapid transition toward the implementation of renewable energies.1 While electrification is considered to be the method of choice for most applications, aircraft, ships, and even trucks will most likely continue to run mainly on carbon-neutral, synthetic solar fuels derived by hydrogenation of CO2.2 Accordingly, alternative energy scenarios are outlined, which may become critical for addressing climate change, while at the same time they will likely result in trillions of dollars in net energy cost savings.3,4
Ethanol is one of the most promising alternatives to gasoline and can be obtained via solar-energy-driven conversion of biomass to bioethanol that offers economy, environment, and energy benefits.5 Accordingly, direct ethanol fuel cells (DEFCs) are regarded as important energy conversion devices to power mobile and portable applications and have been subject to numerous studies in recent years.6,7
The anodic reaction in DEFCs, i.e., the ethanol oxidation reaction (EOR), is a multielectron transfer process that suffers from slow kinetics of the ethanol electrooxidation reaction and its poor selectivity toward complete oxidation to CO2.8,9 Up to now, mainly Pt- and Pd-based electrocatalysts have been employed for the EOR in DEFCs, which has hampered their economic success because of the high costs of the noble metals.10,11 The highest faradaic efficiency was, to the best of our knowledge, achieved by a SnO2-Rh nanosheet catalyst, which demonstrated a 72.8% conversion rate of ethanol to CO2 at 0.78 VRHE in an alkaline electrolyte.12 Under acidic conditions, a PtAuSn/W2C catalyst achieved the highest Faraday efficiency with 6.5% at 0.65 VRHE.13 Various studies have suggested and proved that efficient EOR with low ethanol concentrations requires the use of elevated temperatures > 60 °C.9,14,15 This causes issues concerning the stability and durability of the catalyst materials that are usually mainly based on carbon-supported Pt (alloys). Carbon is prone to corrosion under conventional DEFC operation conditions, which can result in detachment and agglomeration of catalyst nanoparticles.16 Higher efficiencies in DEFCs, specifically higher selectivity toward complete oxidation to CO2, could potentially initiate a paradigm change in the use of this technology because it might enable the operation at lower temperatures, which leads to milder fuel cell operation conditions.
The central reason for the slow kinetics and low efficiency toward full oxidation to CO2 is the complexity of the electrochemical EOR that has been thoroughly mechanistically described in the past decades.8,17,18 The EOR consists of multiple steps with a number of adsorbed intermediates and side products.19,20 The total oxidation to CO2 proceeds via two types of adsorbed intermediates, C1ad and C2ad, which are fragments with one and two carbon atoms. To elucidate the mechanism of the EOR in acidic environment, differential electrochemical mass spectrometry (DEMS),15,21−25 Fourier transform infrared spectroscopy (FTIR),20,25−29 gas chromatography (GC), high-performance liquid chromatography (HPLC),30−32 and liquid-state nuclear magnetic resonance spectroscopy (NMR)33 have been employed.
The higher structural and electronic complexity of compound materials offers equivalent possibilities to engineer adsorption sites and thereby break the known metal scaling relations,34,35 as through promotion or alloying of metal electrodes. PtRu and PtSn are the best investigated solid solutions for the EOR. In the case of PtRu, intermediates are found to be more weakly adsorbed, and the dissociative adsorption of ethanol seems to be inhibited by Pt, while Ru seems to enhance the oxidation of strongly bound adsorbed intermediates through the activation and dissociation of water to give a higher relative yield of CO2 than on pure Pt.
It has been theoretically shown that the presence of novel interface sites between two materials with different scaling properties offers unique possibilities for bifunctional activity gains.36,37 Furthermore, oxygenated titanium species improve the CO tolerance of platinum because of their ability to supply oxygenated species (OH) at low potentials.38 This bifunctional mechanism was proposed in the case of TiC-supported Pt catalysts to explain their enhanced activity toward the methanol oxidation reaction (MOR).38−40 In case of the EOR studied in the present paper, the oxidation of adsorbed intermediates, such as CO, CHx, and CHxO (all C1 species), and of acetaldehyde through the activation of water forming adsorbed OH at the surface is key to forming CO2 via the so-called C1 pathway.33 The activation of H2O is significantly enhanced on the titanium oxycarbide (TiOC) support because it promotes H2O dissociation and surface oxidation.41,42
Titanium oxycarbide (TiOC) has been shown to be suited as a stable and synergistic support for Pt nanoparticles during the EOR in acidic electrolytes.41 A significant enhancement of the CO2 efficiency was observed in combination with a surface passivation of the TiOC support and with respect to the reference catalyst material Pt/C Vulcan. The difference in CO2 efficiency during the EOR and the enhanced oxidation activity toward adsorbed CO was found to be related to the presence of TiO2 and OH groups on the oxycarbide support,41 which improve the CO tolerance of the Pt catalyst through this bifunctional mechanism.43,44 However, the potential catalytic activity of the TiOC support material, itself, has not been anticipated or reported to date.
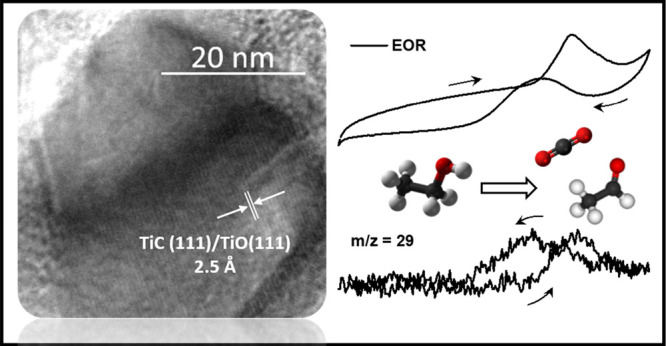
In this work, we report on the performance of the compound material TiOC as an electrocatalyst for the electrochemical oxidation of ethanol without the addition of any noble metal catalyst. DEMS and subtractively normalized interfacial Fourier transform infrared spectroscopy (SNIFTIRS) reveal the formation of acetaldehyde via online and in situ detection during electrocatalytic EOR operation. Prior to the electrocatalytic and electroanalytic evaluation of the compound material, the morphology and structure of the TiOC are thoroughly studied with transmission electron microscopy (TEM). Figure 1 reveals individual differently oriented grains displaying strong diffraction contrast (black-and-gray areas) in the overview bright field image (Figure 1a). The selected area electron diffraction pattern (inset in Figure 1a) can be explained by the presence of TiC and/or TiO [khamrabaevite TiC (face-centered cubic structure, a = 4.325 Å) or cubic TiO (face-centered cubic structure, a = 4.117 Å)]. The presence of TiOC is inferred through the combination of the structural with the compositional data, shown in Figure 1b, that reveals the homogeneous distribution of carbon, oxygen, and titanium. Most of the grains reveal distinct particle outlines, thereby indicating defined morphologies. The observed particle shapes are characteristic for projected truncated pyramidal particles in [100] or [110] direction,45 which—because of the well-defined particle outlines and the strong diffraction contrast in the respective dark-field images (Figure 1c)—directly reveal their three-dimensional truncated pyramidal shape by exposing the characteristic contour lines.46 Finally, Figure 1d confirms the presence of the TiO/TiC material by revealing (111) lattice planes of TiOC [experimentally, 2.5 Å; fcc khamrabaevite TiC (theoretical spacing, 2.49 Å) or fcc TiO (2.41 Å)].
Figure 1.

Transmission electron microscopy (TEM) images of the TiOC powder material. (a) Overview bright-field image with the corresponding selected area electron diffraction (SAED) pattern (inset). (b) High-angle annular dark-field (HAADF) image (top left) and individual EDX maps of the C (top right), O (bottom left), and Ti K-edges (bottom right). (c) Dark-field image of the TiOC powder directly showing diffraction contrast caused by differently oriented grains. For two selected individual grains (boxes), contour lines of equal height are clearly visible. (d) High-resolution (HR) TEM image of a TiOC grain revealing TiC/TiO (111) lattice fringes.
The stability range of TiOC under anodic polarization is limited by the formation of TiO2 and CO2 at potentials > 0.9 VSHE.41,47 The CO2 evolution without ethanol in the electrolyte can be tracked online with DEMS to verify this and to set the background for the EOR studies (see Figure S1 in the Supporting Information). It has, however, previously been found that TiOC corrosion is delayed in the presence of ethanol in the electrolyte. Ethanol molecules adsorb on the TiOC surface and inhibit its oxidation, which is also expected to favor the EOR in the presence of suitable catalyst nanoparticles supported on TiOC.47 Therefore, the stability range is likely larger toward more anodic potentials under the EOR operation. DEMS is further employed to qualitatively identify the reaction products forming during the EOR in 0.5 M H2SO4 and 0.1 M EtOH at 20 °C (see Figure 2). Potentiodynamic EOR studies enable qualitative and online identification of the reaction products through the occurrence of specific ionic signals. The formation onsets are determined at the potential where the respective ionic currents deviate from the background signal (see Figure S2 in the Supporting Information for details). Acetaldehyde formation is visible through the occurrence of a signal at m/z = 29 (CHO+) at around 0.6 VSHE, which is well in line with the ionic current observed upon potential cycling, where a slight increase in the m/z = 29 signal can be observed at 0.6 VSHE (Figure 2b). The very subtle deviation from the background at m/z = 22 (CO2+2) even indicates the formation of very small amounts of CO2 at the same onset potential. Prior to these EOR investigations, the TiOC was preconditioned by measuring a blank CV (see Figure S3a in the Supporting Information) between 0.03 VSHE and 1.1 VSHE followed by three CO stripping sweeps at 2 mV s–1 (see Figure S3b in the Supporting Information).
Figure 2.
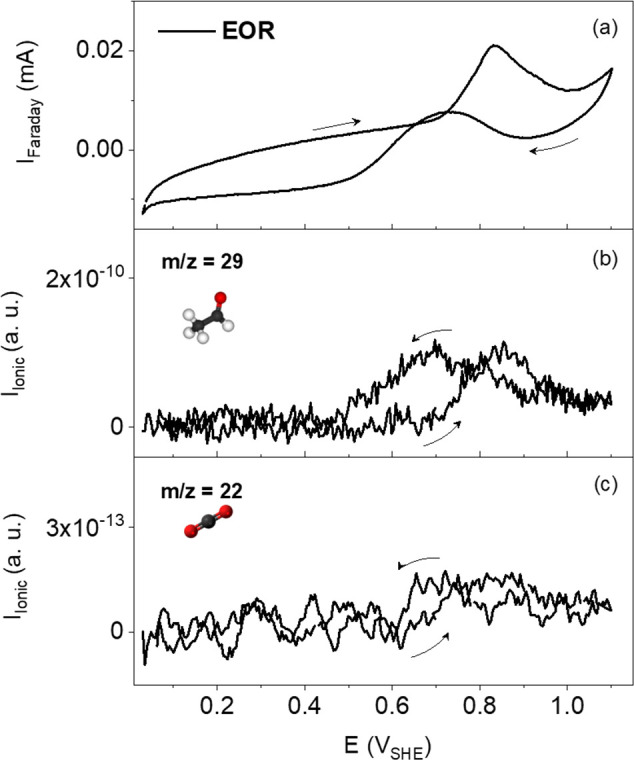
EOR of TiOC powder ink. (a) CV recorded in 0.5 M H2SO4 and 0.1 M EtOH at 20 °C and mass spectrometric cyclic voltammograms (MSCVs) for (b) m/z = 29 and (c) m/z = 22. Scan rate: 2 mV s–1. Specific gravimetric peak current at 0.83 VSHE: 0.01 mA mg–1.
SNIFTIR spectra were recorded at increasing anodic potentials from 0.3 VSHE to 1.0 VSHE (Figure 3). The formation of acetaldehyde (dashed gray boxes) starts at 0.7 VSHE. The band at 3080–2650 cm–1 (C–H stretching mode) and at 1750–1530 cm–1 (C=O vibrational mode) correspond to the vibrational modes of the aldehyde group, which overlaps with the stretching and bending modes of the methyl group in the acetaldehyde.48 The bands at 2360 cm–1 and 2331 cm–1 are assigned to gaseous CO2 (R and P branch),49 which arise because of residual ambient atmosphere in the beam path. Significantly, at 1.0 VSHE, the spectral profile undergoes a notable transformation characterized by the emergence of upward features. These features manifest as a band at 1640 cm–1, associated with the bending mode of water, and a broad band in the range 3100–3700 cm–1. This wave exhibits prominent peaks at 3100 cm–1 and 3635 cm–1, which can be assigned to the stretching vibration of water and the Ti-OH, respectively.50 The reason for this can be attributed to the substantial reduction in surface OH groups (Ti-OH) stemming from the oxidation of TiOC and the simultaneous decrease in the water content resulting from the oxidation reaction.
Figure 3.
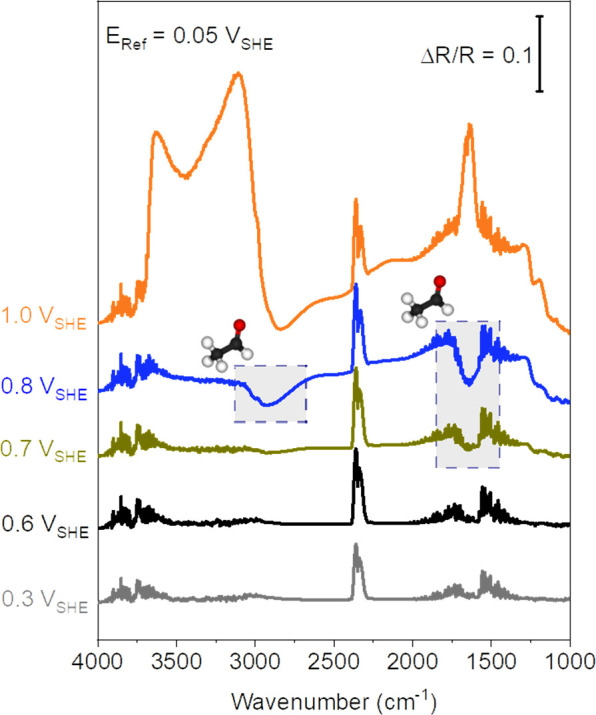
SNIFTIR spectra at increasing anodic potentials from 0.3 VSHE to 1.0 VSHE recorded in 0.5 M H2SO4 and 1 M EtOH. The bands, indicating acetaldehyde formation, begin to grow at a potential of ∼0.7 VSHE (dashed gray boxes). The reference spectrum was taken at 0.05 VSHE .
For further evaluation of the CO2 conversion efficiency at TiOC, current transients of the EOR are recorded together with the corresponding DEMS signals for the m/z ratios of 29 and 22 (Figure S4).
High-resolution XPS spectra (Ti 2p and C 1s regions) of the TiOC electrodes have been recorded after the SNIFTIRS studies to confirm the continuous presence of carbide and, consequently, the long-term stability of the TiOC material (Figure S5). Moreover, analysis of the Ti 2p region implies that, in addition to the species associated with TiO2, TiC, and TiOC, reduced TiO2–x species are present, which signifies the existence of oxygen vacancies. This conclusion gains further support from Raman experiments conducted on the powder catalyst (Figure S6). TiOC is known to decompose both thermodynamically and at contact with ambient air to TiO2 and carbon at its outermost surface.51 At potentials larger than 0.9 VSHE, where more TiO2 and carbon are formed at the solid/liquid interface, TiOC starts to passivate. In the presence of ethanol, however, this passivation is partly inhibited, which has been suggested to be caused by ethanol adsorption at the TiO2-terminated oxycarbide surface.47 Therefore, we propose the following pathway for the electrooxidation of ethanol on this material, which is, at the current stage of investigation, highly speculative: initially, ethanol adsorbs on the catalyst surface, preferably binding to undercoordinated sites, such as oxygen vacancies.52 It is well known that TiO2 sites, which are inherently present on the surface because of partial oxidation of the TiO2–x sites, can supply oxygenated species (−OH groups) through their ability to dissociate the water from the electrolyte. These OH groups can further react with the adsorbed ethanol to form acetaldehyde. The combination of the present −OH groups and the abundance of oxygen vacancies resulting from the composite nature of the material (see Figure 1) might account for the observed exceptional catalytic activity.
In this work, the electrocatalytic activity of TiOC toward the EOR is investigated under acidic electrolyte conditions at room temperature via DEMS and SNIFTIRS investigations. TEM analysis reveals three-dimensional truncated pyramidal-shaped TiOC particles, and EDX and XPS confirm the stability of the TiOC material under both electrochemical conditions and exposure to ambient atmosphere. We show, for the first time, that TiOC is able to oxidize ethanol to acetaldehyde without the need for expensive noble metals. The catalytic activity is attributed to the presence of both undercoordinated sites for ethanol adsorption and oxygenated sites facilitating the oxidation of adsorbed ethanol. While it is unlikely that TiOC becomes a valuable catalyst by itself, TiOC may serve as an important cocatalyst and catalyst support simultaneously, thereby adding improved activity to the system by enabling bifunctional activity gains.
Acknowledgments
We thank Alexander Thöny and Thomas Lörting for providing the Raman experiments (Figure S6). We gratefully acknowledge the support of the Austrian Science Fund (FWF) within the FWF projects P34233, P35510, and I-4114, as well as the support of the Austrian Research Promotion Agency (FFG) within the FFG projects 877095 and 870523.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c04097.
Experimental methods, TiOC powder ink blank measurement (Figure S1), exemplary onset determination (Figure S2), blank CV measurements and CO stripping (Figure S3), current transients and corresponding mass spectrometric ionic current signals (Figure S4), high-resolution XP spectra (Figure S5) and Raman spectra of the TiOC powder catalyst (Figure S6) (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Turner J. A. A Realizable Renewable Energy Future. Science (80-.) 1999, 285 (5428), 687–689. 10.1126/science.285.5428.687. [DOI] [PubMed] [Google Scholar]
- Bergero C.; Gosnell G.; Gielen D.; Kang S.; Bazilian M.; Davis S. J. Pathways to Net-Zero Emissions from Aviation. Nat. Sustain. 2023, 6 (4), 404–414. 10.1038/s41893-022-01046-9. [DOI] [Google Scholar]
- Way R.; Ives M. C.; Mealy P.; Farmer J. D. Empirically Grounded Technology Forecasts and the Energy Transition. Joule 2022, 6, 2057–2082. 10.1016/j.joule.2022.08.009. [DOI] [Google Scholar]
- Hoegh-Guldberg O.; Jacob D.; Taylor M.; Guillén Bolaños T.; Bindi M.; Brown S.; Camilloni I. A.; Diedhiou A.; Djalante R.; Ebi K.; Engelbrecht F.; Guiot J.; Hijioka Y.; Mehrotra S.; Hope C. W.; Payne A. J.; Pörtner H.-O.; Seneviratne S. I.; Thomas A.; Warren R.; Zhou G. The Human Imperative of Stabilizing Global Climate Change at 1.5°C. Science (80-.) 2019, 365 (6459), eaaw6974 10.1126/science.aaw6974. [DOI] [PubMed] [Google Scholar]
- Tabah B.; Pulidindi I. N.; Chitturi V. R.; Reddy Arava L. M.; Varvak A.; Foran E.; Gedanken A. Solar-Energy-Driven Conversion of Biomass to Bioethanol: A Sustainable Approach. J. Mater. Chem. A 2017, 5 (30), 15486–15506. 10.1039/C7TA03083E. [DOI] [Google Scholar]
- Staffell I.; Scamman D.; Velazquez Abad A.; Balcombe P.; Dodds P. E.; Ekins P.; Shah N.; Ward K. R. The Role of Hydrogen and Fuel Cells in the Global Energy System. Energy Environ. Sci. 2019, 12 (2), 463–491. 10.1039/C8EE01157E. [DOI] [Google Scholar]
- Altarawneh R. M. Overview on the Vital Step toward Addressing Platinum Catalyst Poisoning Mechanisms in Acid Media of Direct Ethanol Fuel Cells (DEFCs). Energy & Fuels 2021, 35 (15), 11594–11612. 10.1021/acs.energyfuels.1c00453. [DOI] [Google Scholar]
- Friedl J.; Stimming U. Model Catalyst Studies on Hydrogen and Ethanol Oxidation for Fuel Cells. Electrochim. Acta 2013, 101, 41–58. 10.1016/j.electacta.2012.12.130. [DOI] [Google Scholar]
- Sun S.; Halseid M. C.; Heinen M.; Jusys Z.; Behm R. J. Ethanol Electrooxidation on a Carbon-Supported Pt Catalyst at Elevated Temperature and Pressure: A High-Temperature/High-Pressure DEMS Study. J. Power Sources 2009, 190 (1), 2–13. 10.1016/j.jpowsour.2009.01.073. [DOI] [Google Scholar]
- Lyu F.; Cao M.; Mahsud A.; Zhang Q. Interfacial Engineering of Noble Metals for Electrocatalytic Methanol and Ethanol Oxidation. J. Mater. Chem. A 2020, 8 (31), 15445–15457. 10.1039/D0TA03199B. [DOI] [Google Scholar]
- Ioroi T.; Siroma Z.; Yamazaki S.; Yasuda K. Electrocatalysts for PEM Fuel Cells. Adv. Energy Mater. 2019, 9 (23), 1801284 10.1002/aenm.201801284. [DOI] [Google Scholar]
- Bai S.; Xu Y.; Cao K.; Huang X. Selective Ethanol Oxidation Reaction at the Rh–SnO2 Interface. Adv. Mater. 2021, 33 (5), 2005767 10.1002/adma.202005767. [DOI] [PubMed] [Google Scholar]
- Shakibi Nia N.; Guillén-Villafuerte O.; Griesser C.; Manning G.; Kunze-Liebhäuser J.; Arévalo C.; Pastor E.; García G. W 2 C-Supported PtAuSn—A Catalyst with the Earliest Ethanol Oxidation Onset Potential and the Highest Ethanol Conversion Efficiency to CO2 Known till Date. ACS Catal. 2020, 10 (2), 1113–1122. 10.1021/acscatal.9b04348. [DOI] [Google Scholar]
- Chen B.; Brueckner T. M.; Altarawneh R. M.; Pickup P. G. Composition Dependence of Ethanol Oxidation at Ruthenium-Tin Oxide/Carbon Supported Platinum Catalysts. J. Electrochem. Soc. 2018, 165 (15), J3019–J3025. 10.1149/2.0041815jes. [DOI] [Google Scholar]
- Wang H.; Jusys Z.; Behm R. J. Ethanol Electrooxidation on a Carbon-Supported Pt Catalyst: Reaction Kinetics and Product Yields. J. Phys. Chem. B 2004, 108 (50), 19413–19424. 10.1021/jp046561k. [DOI] [Google Scholar]
- Antolini E.; Gonzalez E. R. Ceramic Materials as Supports for Low-Temperature Fuel Cell Catalysts. Solid State Ionics 2009, 180 (9–10), 746–763. 10.1016/j.ssi.2009.03.007. [DOI] [Google Scholar]
- Antolini E. Catalysts for Direct Ethanol Fuel Cells. J. Power Sources 2007, 170 (1), 1–12. 10.1016/j.jpowsour.2007.04.009. [DOI] [Google Scholar]
- Lai S. C. S.; Kleyn S. E. F.; Rosca V.; Koper M. T. M. Mechanism of the Dissociation and Electrooxidation of Ethanol and Acetaldehyde on Platinum As Studied by SERS. J. Phys. Chem. C 2008, 112 (48), 19080–19087. 10.1021/jp807350h. [DOI] [Google Scholar]
- Snell K. D.; Keenan A. G. Effect of Anions and PH on the Ethanol Electro-Oxidation at a Platinum Electrode. Electrochim. Acta 1982, 27 (12), 1683–1696. 10.1016/0013-4686(82)80164-3. [DOI] [Google Scholar]
- Hitmi H.; Belgsir E. M.; Léger J.-M.; Lamy C.; Lezna R. O. A Kinetic Analysis of the Electro-Oxidation of Ethanol at a Platinum Electrode in Acid Medium. Electrochim. Acta 1994, 39 (3), 407–415. 10.1016/0013-4686(94)80080-4. [DOI] [Google Scholar]
- Rao V.; Cremers C.; Stimming U.; Cao L.; Sun S.; Yan S.; Sun G.; Xin Q. Electro-Oxidation of Ethanol at Gas Diffusion Electrodes A DEMS Study. J. Electrochem. Soc. 2007, 154 (11), B1138. 10.1149/1.2777108. [DOI] [Google Scholar]
- Wang J.; Wasmus S.; Savinell R. F. Evaluation of Ethanol, 1-Propanol, and 2-Propanol in a Direct Oxidation Polymer-Electrolyte Fuel Cell: A Real-Time Mass Spectrometry Study. J. Electrochem. Soc. 1995, 142 (12), 4218–4224. 10.1149/1.2048487. [DOI] [Google Scholar]
- Wang H.; Jusys Z.; Behm R. J. Ethanol Electro-Oxidation on Carbon-Supported Pt, PtRu and Pt3Sn Catalysts: A Quantitative DEMS Study. J. Power Sources 2006, 154 (2), 351–359. 10.1016/j.jpowsour.2005.10.034. [DOI] [Google Scholar]
- Fujiwara N.; Friedrich K. A.; Stimming U. Ethanol Oxidation on PtRu Electrodes Studied by Differential Electrochemical Mass Spectrometry. J. Electroanal. Chem. 1999, 472 (2), 120–125. 10.1016/S0022-0728(99)00282-X. [DOI] [Google Scholar]
- de Souza J. P. I.; Queiroz S. L.; Bergamaski K.; Gonzalez E. R.; Nart F. C. Electro-Oxidation of Ethanol on Pt, Rh, and PtRh Electrodes. A Study Using DEMS and in-Situ FTIR Techniques. J. Phys. Chem. B 2002, 106 (38), 9825–9830. 10.1021/jp014645c. [DOI] [Google Scholar]
- Iwasita T.; Pastor E. A Dems and FTir Spectroscopic Investigation of Adsorbed Ethanol on Polycrystalline Platinum. Electrochim. Acta 1994, 39 (4), 531–537. 10.1016/0013-4686(94)80097-9. [DOI] [Google Scholar]
- Leung L.-W. H.; Chang S.-C.; Weaver M. J. Real-Time FTIR Spectroscopy as an Electrochemical Mechanistic Probe. J. Electroanal. Chem. Interfacial Electrochem. 1989, 266 (2), 317–336. 10.1016/0022-0728(89)85078-8. [DOI] [Google Scholar]
- Camara G. A.; Iwasita T. Parallel Pathways of Ethanol Oxidation: The Effect of Ethanol Concentration. J. Electroanal. Chem. 2005, 578 (2), 315–321. 10.1016/j.jelechem.2005.01.013. [DOI] [Google Scholar]
- Lamy C.; Rousseau S.; Belgsir E.; Coutanceau C.; Léger J.-M. Recent Progress in the Direct Ethanol Fuel Cell: Development of New Platinum–Tin Electrocatalysts. Electrochim. Acta 2004, 49 (22–23), 3901–3908. 10.1016/j.electacta.2004.01.078. [DOI] [Google Scholar]
- Shimada I.; Oshima Y.; Otomo J. Acceleration of Ethanol Electro-Oxidation on a Carbon-Supported Platinum Catalyst at Intermediate Temperatures. J. Electrochem. Soc. 2011, 158 (4), B369. 10.1149/1.3545280. [DOI] [Google Scholar]
- Aricò A. S. Comparison of Ethanol and Methanol Oxidation in a Liquid-Feed Solid Polymer Electrolyte Fuel Cell at High Temperature. Electrochem. Solid-State Lett. 1999, 1 (2), 66. 10.1149/1.1390638. [DOI] [Google Scholar]
- Rousseau S.; Coutanceau C.; Lamy C.; Léger J.-M. Direct Ethanol Fuel Cell (DEFC): Electrical Performances and Reaction Products Distribution under Operating Conditions with Different Platinum-Based Anodes. J. Power Sources 2006, 158 (1), 18–24. 10.1016/j.jpowsour.2005.08.027. [DOI] [Google Scholar]
- Kim I.; Han O. H.; Chae S. A.; Paik Y.; Kwon S.-H.; Lee K.-S.; Sung Y.-E.; Kim H. Catalytic Reactions in Direct Ethanol Fuel Cells. Angew. Chemie Int. Ed. 2011, 50 (10), 2270–2274. 10.1002/anie.201005745. [DOI] [PubMed] [Google Scholar]
- Nørskov J. K.; Bligaard T.; Rossmeisl J.; Christensen C. H. Towards the Computational Design of Solid Catalysts. Nat. Chem. 2009, 1 (1), 37–46. 10.1038/nchem.121. [DOI] [PubMed] [Google Scholar]
- Greeley J. Theoretical Heterogeneous Catalysis: Scaling Relationships and Computational Catalyst Design. Annu. Rev. Chem. Biomol. Eng. 2016, 7 (1), 605–635. 10.1146/annurev-chembioeng-080615-034413. [DOI] [PubMed] [Google Scholar]
- Andersen M.; Medford A. J.; Nørskov J. K.; Reuter K. Scaling-Relation-Based Analysis of Bifunctional Catalysis: The Case for Homogeneous Bimetallic Alloys. ACS Catal. 2017, 7 (6), 3960–3967. 10.1021/acscatal.7b00482. [DOI] [Google Scholar]
- Andersen M.; Medford A. J.; Nørskov J. K.; Reuter K. Analyzing the Case for Bifunctional Catalysis. Angew. Chemie Int. Ed. 2016, 55 (17), 5210–5214. 10.1002/anie.201601049. [DOI] [PubMed] [Google Scholar]
- Roca-Ayats M.; García G.; Peña M. A.; Martínez-Huerta M. V. Titanium Carbide and Carbonitride Electrocatalyst Supports: Modifying Pt–Ti Interface Properties by Electrochemical Potential Cycling. J. Mater. Chem. A 2014, 2 (44), 18786–18790. 10.1039/C4TA03782K. [DOI] [Google Scholar]
- Ito Y.; Takeuchi T.; Tsujiguchi T.; Abdelkareem M. A.; Nakagawa N. Ultrahigh Methanol Electro-Oxidation Activity of PtRu Nanoparticles Prepared on TiO2-Embedded Carbon Nanofiber Support. J. Power Sources 2013, 242, 280–288. 10.1016/j.jpowsour.2013.05.064. [DOI] [Google Scholar]
- Lv Q.; Yin M.; Zhao X.; Li C.; Liu C.; Xing W. Promotion Effect of TiO2 on Catalytic Activity and Stability of Pt Catalyst for Electrooxidation of Methanol. J. Power Sources 2012, 218, 93–99. 10.1016/j.jpowsour.2012.06.051. [DOI] [Google Scholar]
- Shakibi Nia N.; Martucci A.; Granozzi G.; García G.; Pastor E.; Penner S.; Bernardi J.; Alonso-Vante N.; Kunze-Liebhäuser J. DEMS Studies of the Ethanol Electro-Oxidation on TiOC Supported Pt Catalysts–Support Effects for Higher CO2 Efficiency. Electrochim. Acta 2019, 304, 80–86. 10.1016/j.electacta.2019.02.089. [DOI] [Google Scholar]
- Rüdiger C.; Brumbarov J.; Wiesinger F.; Leonardi S.; Paschos O.; Vidal C. V.; Kunze-Liebhäuser J. Ethanol Oxidation on TiO x C y -Supported Pt Nanoparticles. ChemCatChem 2013, 5 (11), 3219–3223. 10.1002/cctc.201300217. [DOI] [Google Scholar]
- Watanabe M.; Motoo S. Electrocatalysis by Ad-Atoms. J. Electroanal. Chem. Interfacial Electrochem. 1975, 60 (3), 275–283. 10.1016/S0022-0728(75)80262-2. [DOI] [Google Scholar]
- Vigier F.; Coutanceau C.; Hahn F.; Belgsir E. M.; Lamy C. On the Mechanism of Ethanol Electro-Oxidation on Pt and PtSn Catalysts: Electrochemical and in Situ IR Reflectance Spectroscopy Studies. J. Electroanal. Chem. 2004, 563 (1), 81–89. 10.1016/j.jelechem.2003.08.019. [DOI] [Google Scholar]
- Hayek K.; Goller H.; Penner S.; Rupprechter G.; Zimmermann C. Regular Alumina-Supported Nanoparticles of Iridium, Rhodium and Platinum Under Hydrogen Reduction: Structure, Morphology and Activity in the Neopentane Conversion. Catal. Lett. 2004, 92, 1–9. 10.1023/B:CATL.0000011081.32980.e0. [DOI] [Google Scholar]
- Yacamän M. J.; OcañA Z T. High-Resolution Dark-Field Electron Microscopy of Small Metal Particles. Phys. Status Solidi 1977, 42 (2), 571–577. 10.1002/pssa.2210420220. [DOI] [Google Scholar]
- Calvillo L.; García G.; Paduano A.; Guillen-Villafuerte O.; Valero-Vidal C.; Vittadini A.; Bellini M.; Lavacchi A.; Agnoli S.; Martucci A.; Kunze-Liebhäuser J.; Pastor E.; Granozzi G. Electrochemical Behavior of TiOxCy as Catalyst Support for Direct Ethanol Fuel Cells at Intermediate Temperature: From Planar Systems to Powders. ACS Appl. Mater. Interfaces 2016, 8 (1), 716–725. 10.1021/acsami.5b09861. [DOI] [PubMed] [Google Scholar]
- National Institute of Standards and Technology (NIST). NIST Standard Reference Database Number 69. In NIST Chemistry WebBook; U.S. Department of Commerce. https://webbook.nist.gov/chemistry/ (accessed 2023-12-06). [Google Scholar]
- Mairegger T.; Li H.; Grießer C.; Winkler D.; Filser J.; Hörmann N. G.; Reuter K.; Kunze-Liebhäuser J. Electroreduction of CO2 in a Non-Aqueous Electrolyte—The Generic Role of Acetonitrile. ACS Catal. 2023, 13, 5780–5786. 10.1021/acscatal.3c00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohandas N.; Narayanan T. N.; Cuesta A. Tailoring the Interfacial Water Structure by Electrolyte Engineering for Selective Electrocatalytic Reduction of Carbon Dioxide. ACS Catal. 2023, 13 (13), 8384–8393. 10.1021/acscatal.3c01223. [DOI] [Google Scholar]
- Calvillo L.; Fittipaldi D.; Rüdiger C.; Agnoli S.; Favaro M.; Valero-Vidal C.; Di Valentin C.; Vittadini A.; Bozzolo N.; Jacomet S.; Gregoratti L.; Kunze-Liebhäuser J.; Pacchioni G.; Granozzi G. Carbothermal Transformation of TiO2 into TiOxCy in UHV: Tracking Intrinsic Chemical Stabilities. J. Phys. Chem. C 2014, 118 (39), 22601–22610. 10.1021/jp506728w. [DOI] [Google Scholar]
- Carchini G.; López N. Adsorption of Small Mono- and Poly-Alcohols on Rutile TiO2: A Density Functional Theory Study. Phys. Chem. Chem. Phys. 2014, 16 (28), 14750. 10.1039/c4cp01546k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.