

Abstract
Glioblastoma (GBM) tumors consist of multiple cell populations, including self-renewing glioblastoma stem cells (GSCs) and immunosuppressive microglia. Here we identified Kunitz-type protease inhibitor TFPI2 as a critical factor connecting these cell populations and their associated GBM hallmarks of stemness and immunosuppression. TFPI2 promotes GSC self-renewal and tumor growth via activation of the c-Jun N-terminal kinase-signal transducer and activator of transcription (STAT)3 pathway. Secreted TFPI2 interacts with its functional receptor CD51 on microglia to trigger the infiltration and immunosuppressive polarization of microglia through activation of STAT6 signaling. Inhibition of the TFPI2-CD51-STAT6 signaling axis activates T cells and synergizes with anti-PD1 therapy in GBM mouse models. In human GBM, TFPI2 correlates positively with stemness, microglia abundance, immunosuppression and poor prognosis. Our study identifies a function for TFPI2 and supports therapeutic targeting of TFPI2 as an effective strategy for GBM.
Glioblastoma (GBM) is a devastating primary brain tumor. The current standard of care for GBM offers only modest survival benefit, and almost all eventually recur, in part due to the presence of glioblastoma stem cells (GSCs)1. Mice implanted with GSCs produce tumors that are less responsive to chemotherapy than tumors established from more differentiated GBM cells, a finding that supports a crucial role of GSCs in treatment resistance2. GSCs account for ~10% of total cancer cells in tumor tissues. However, the GSC subpopulation is known for self-renewal that is important for tumor growth and recurrence2. Given the inverse correlation between GSC stemness and survival3, targeting GSCs holds a great potential for GBM therapy.
Despite advances in our understanding of GSC biologic properties, substantial knowledge gaps remain, including those involving relationships between GSCs and additional cell populations of the tumor microenvironment (TME)1,4. Within the GBM TME, tumor-associated macrophages and microglia (TAMs) are abundant and account for up to 50% of total cells in the tumor mass5,6. TAMs are usually polarized towards an immunosuppressive phenotype that can inhibit the infiltration and antitumor activation of cytotoxic T cells and induce immunotherapy resistance5. Increasing evidence supports a symbiotic interaction between GSCs and TAMs1,4,7,8. For instance, our recent studies have demonstrated that GSC expression of CLOCK transcriptionally upregulates olfactomedin-like 3 (OLFML3) and legumain (LGMN), which, in turn, promote microglia infiltration and immunosuppressive polarization9,10. These findings provide a framework from which to identify therapeutic targets intercepting the GSC-immune cell crosstalk. In the current study, we have conducted a comprehensive analysis aimed at identifying the factors that contribute to GSC-immune cell symbiosis. In this investigation, tissue factor pathway inhibitor 2 (TFPI2) has been identified as a key symbiosis effector.
TFPI2 is a member of the Kunitz-type serine proteinase inhibitor family. Despite its name and structural similarity to TFPI1, a potent inhibitor for factors Xa and VIIa/tissue factor (TF) complex in the extrinsic pathway of blood coagulation, TFPI2 has little inhibitory effect on TFs11,12. Instead, TFPI2 is recognized as a critical factor that can inhibit extracellular matrix degradation via suppressing plasmin-dependent activation of pro-matrix metalloproteinase 1 (MMP1) and pro-MMP13 (ref. 12). Since extracellular matrix degradation is important for tumor progression13, TFPI2 has been considered as a tumor suppressor14–16. However, contrasting results indicate a protumor activity of TFPI2 in multiple cancers, including hepatocellular carcinoma17, melanoma18,19 and ovarian clear cell carcinoma20. In total, these findings suggest that the role of TFPI2 in tumor biology may be context and cancer type dependent. In GBM, TFPI2 has been shown to inhibit the growth, survival, migration and invasion of differentiated GBM cells21–23. However, the potential roles of TFPI2 in GSC stemness and GSC–immune symbiosis have not been investigated.
In this Article, we identified an unexpected function for TFPI2 in supporting stemness maintenance and tumor growth via activation of the c-Jun N-terminal kinase (JNK)–signal transducer and activator of transcription (STAT)3 pathway in GSCs. Moreover, secreted TFPI2 contributes to an immunosuppressive TME by promoting microglia infiltration and polarization via activation of CD51 (also known as integrin alpha V, ITGAV)–STAT6 signaling pathway in microglia. We show that inhibition of the TFPI2–CD51–STAT6 pathway in GBM mouse models activates antitumor immunity that extends the survival of tumor-bearing mice. This antitumor immune activation can be coupled with immune checkpoint inhibitor (ICI) to further extend survival. In combination with results from our analysis of patient tumor and plasma samples, our investigation points to TFPI2 as a potential therapeutic target for treating GBM.
Results
TFPI2 overexpression promotes GSC self-renewal
Pan-cancer analyses have shown that high stemness of cancer cells is correlated positively with increased immunosuppressive pathways24. To identify potential associations between GSCs and immunity, we performed correlation analyses between tumor stromal and immune cell scores25 and GSC signature26 using The Cancer Genome Atlas (TCGA) GBM dataset. We found that GSC signature correlated positively with stromal and immune scores (Fig. 1a). To identify the key gene/factor(s) that could mediate the GSC–immune cell crosstalk, we used the following approaches: (1) start with the 1,708 human genes that encode secreted proteins27; (2) determine the subset of these genes whose expression inversely correlates with TCGA GBM patient survival; (3) determine the subset of these genes whose expression correlates positively with stemness signature24; and (4) rank the expression of these genes in CD45− GBM cells/GSCs using the tumor immune microenvironment (TIME) dataset28. Following these analyses, TFPI2 and LOXL2 were identified (Fig. 1b and Extended Data Fig. 1a–e). Analysis of TCGA GBM and low-grade glioma (LGG) data for these two genes revealed that TFPI2, but not LOXL2, is amplified in approximately 4% of GBM cases, 1.4% of GBM/LGG merged cases and 0.4% of LGG cases (Fig. 1c), and the enhanced TFPI2 gene copy number correlated positively with increased TFPI2 messenger RNA level in GBM (Fig. 1d and Extended Data Fig. 1f). In addition, the expression of TFPI2 was found to be higher in GBM than LGG (Extended Data Fig. 1g,h). With respect to a commonly used transcriptional subclassification, mesenchymal GBM showed the highest average level of TFPI2 expression (Extended Data Fig. 1i).
Fig. 1: TFPI2 overexpression promotes GSC self-renewal.
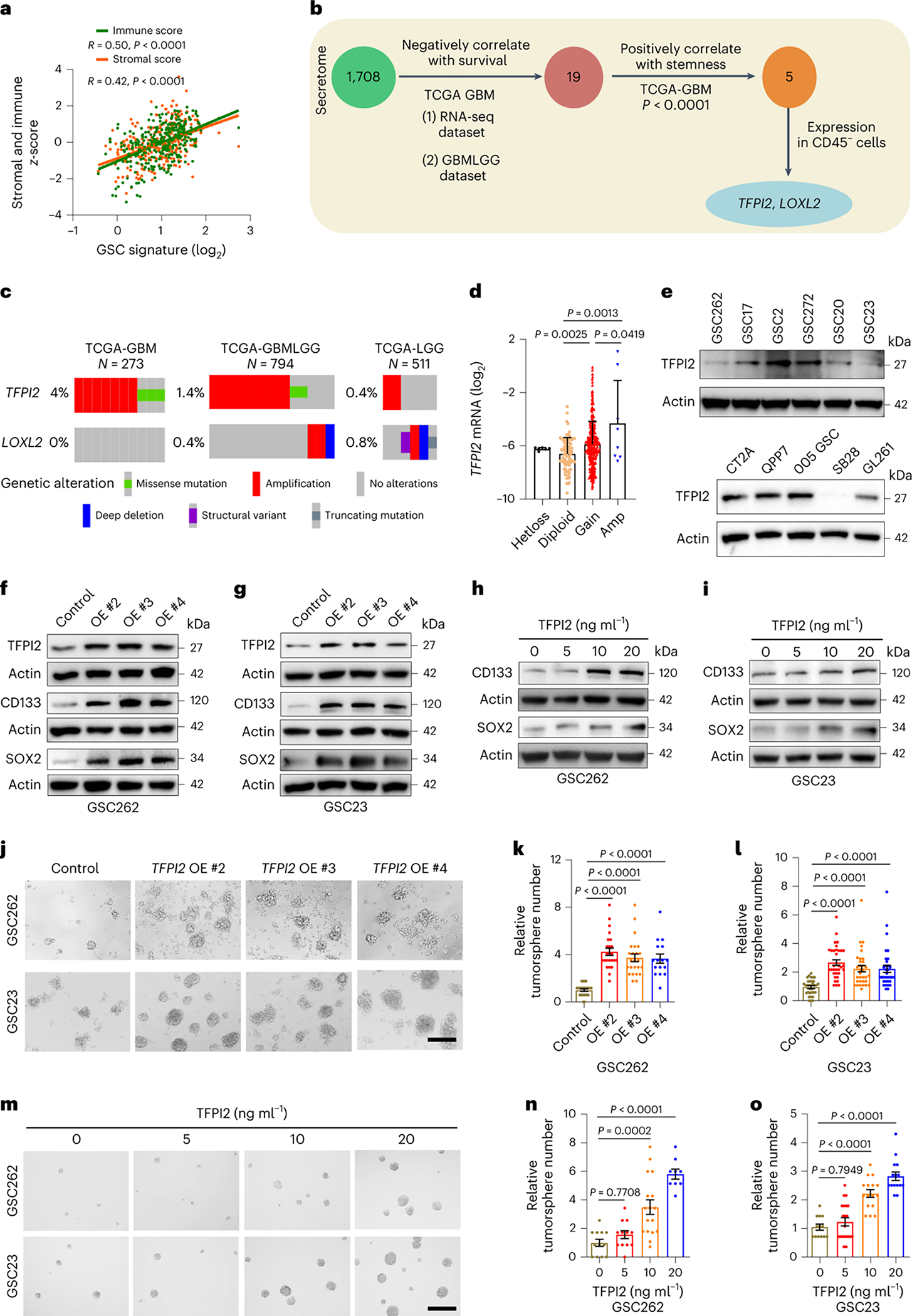
a, The correlation between GSC signature and immune/stromal scores in TCGA GBM dataset (n = 300). R and P values were determined by Pearson correlation. b, Strategy for identification of TFPI2 and LOXL2 that encode secreted proteins, correlate negatively with patient survival and positively with the stemness signature in TCGA GBM patient tumors, and are highly expressed in CD45− cells of GBM tumors. c, Amplification of TFPI2 and LOXL2 in GBM and LGG patient tumors. d, TFPI2 gene copy number correlates with TFPI2 mRNA level in TCGA GBM patient tumors under the Agilent-4502A platform. n = 6, 83, 372 and 8 biological replicates for Hetloss, Diploid, Gain and Amp, respectively. e, Immunoblot for TFPI2 in cell lysates of GBM patient-derived GSCs (top) and mouse-derived GSCs and GBM cells (bottom). Based on the expression levels, GSC272, GSC2, QPP7 GSC, CT2A and 005 GSC are considered as TFPI2-high cells. f,g, Immunoblots for TFPI2, CD133 and SOX2 in cell lysates of GSC262 (f) and GSC23 (g) cells with or without TFPI2 overexpression (OE). h,i, Immunoblots for CD133 and SOX2 in cell lysates of GSC262 (h) and GSC23 (i) cells treated with or without TFPI2 recombinant protein at indicated concentrations for 24 h. j, Representative of tumorspheres of GSC262 and GSC23 cells expressing control and TFPI2 OE plasmids. Scale bar, 400 μm. k,l, Quantification of tumorspheres of GSC262 (k) and GSC23 (l) cells expressing control and TFPI2 OE plasmids. In k, n = 24 and 16 biological replicates for control, OE #2 and OE #3 groups and OE #4 group, respectively. In l, n = 33 biological replicates. m, Representative of tumorspheres of GSC262 and GSC23 cells treated with TFPI2 recombinant protein at indicated concentrations. Scale bar, 400 μm. n,o, Quantification of tumorspheres of GSC262 (n) and GSC23 (o) cells treated with TFPI2 recombinant protein. In n, n = 12, 17 and 9 biological replicates for TFPI2 at 0 and 5, 10 and 20 ng ml−1, respectively. In o, n = 13, 21, 16 and 15 biological replicates for TFPI2 at 0, 5, 10 and 20 ng ml−1, respectively. Error bars indicate mean ± s.d. or s.e.m. One-way ANOVA test.
To investigate a possible relationship between TFPI2 expression and GSC self-renewal, we analyzed TCGA GBM data by comparing TFPI2-amplified patient tumors versus non-amplified tumors for stemness signature. The result showed that the stemness signature was enriched in TFPI2-amplified tumors (Supplementary Fig. 1a). We next analyzed single-cell RNA sequencing (scRNA-seq) data from 28 patient GBMs29 and found that the stemness signature was enriched in TFPI2-high GBM cells (Extended Data Fig. 1j–n). To address the role of TFPI2 in regulating GSC stemness through experimentation, we overexpressed TFPI2 or added TFPI2 recombinant protein to patient-derived GSCs with relatively low TFPI2 expression, such as GSC262, GSC23 and GSC20 (Fig. 1e). Western blotting showed that ectopic TFPI2 expression (Fig. 1f,g) and TFPI2 recombinant protein treatment (Fig. 1h,i and Extended Data Fig. 1o) increased the expression of stemness-associated factors CD133 and SOX2. The ectopic expression of TFPI2 increased GSC self-renewal (Fig. 1j–l) while decreasing GSC apoptosis (Extended Data Fig. 1p–r). Enhanced self-renewal was also observed in GSCs upon TFPI2 recombinant protein treatment (Fig. 1m–o). Together, these findings demonstrate that TFPI2 is amplified in GBM, and its overexpression promotes GSC self-renewal.
TFPI2 depletion impairs GSC stemness and tumor growth
To address the effects of reducing cellular TFPI2 in GSC stemness maintenance, TFPI2 was targeted by short hairpin RNA (shRNA)-mediated knockdown (Fig. 2a and Supplementary Fig. 2a) in human and mouse GSCs with relatively high endogenous TFPI2 expression (Fig. 1e), such as GSC272 and GSC2 (patient-derived GSCs), QPP7 GSCs (a GSC line derived from an engineered GBM mouse model with null alleles for Qki, Pten and Trp53 (ref. 30)) and CT2A cells (a mouse line isolated from a carcinogen-induced glioma possessing a GSC-like phenotype9,31). TFPI2 depletion resulted in reduced CD133 and SOX2 expression (Fig. 2b and Supplementary Fig. 2a), self-renewal (Fig. 2c–f, Extended Data Fig. 2a,b and Supplementary Fig. 2b,c) and proliferation (Fig. 2g–j and Extended Data Fig. 2c,d) in both human and mouse GSCs. Conversely, cellular apoptosis was increased upon TFPI2 knockdown in GSCs (Extended Data Fig. 2e–k). Reexpression of shRNA-resistant TFPI2 complementary DNA rescued TFPI2 depletion-induced impairment of CD133 and SOX2 expression as well as self-renewal (Fig. 2k and Extended Data Fig. 2l,m). Lastly, the role of TFPI2 in promoting GSC stemness was reinforced by an additional genetic approach showing that clustered regularly interspaced short palindromic repeats (CRISPR)-mediated TFPI2 knockout (KO) in GSC272 reduced CD133 and SOX2 expression as well as self-renewal (Fig. 2l and Extended Data Fig. 2n–p).
Fig. 2: TFPI2 depletion impairs stemness and extends survival of GSC-bearing mice.
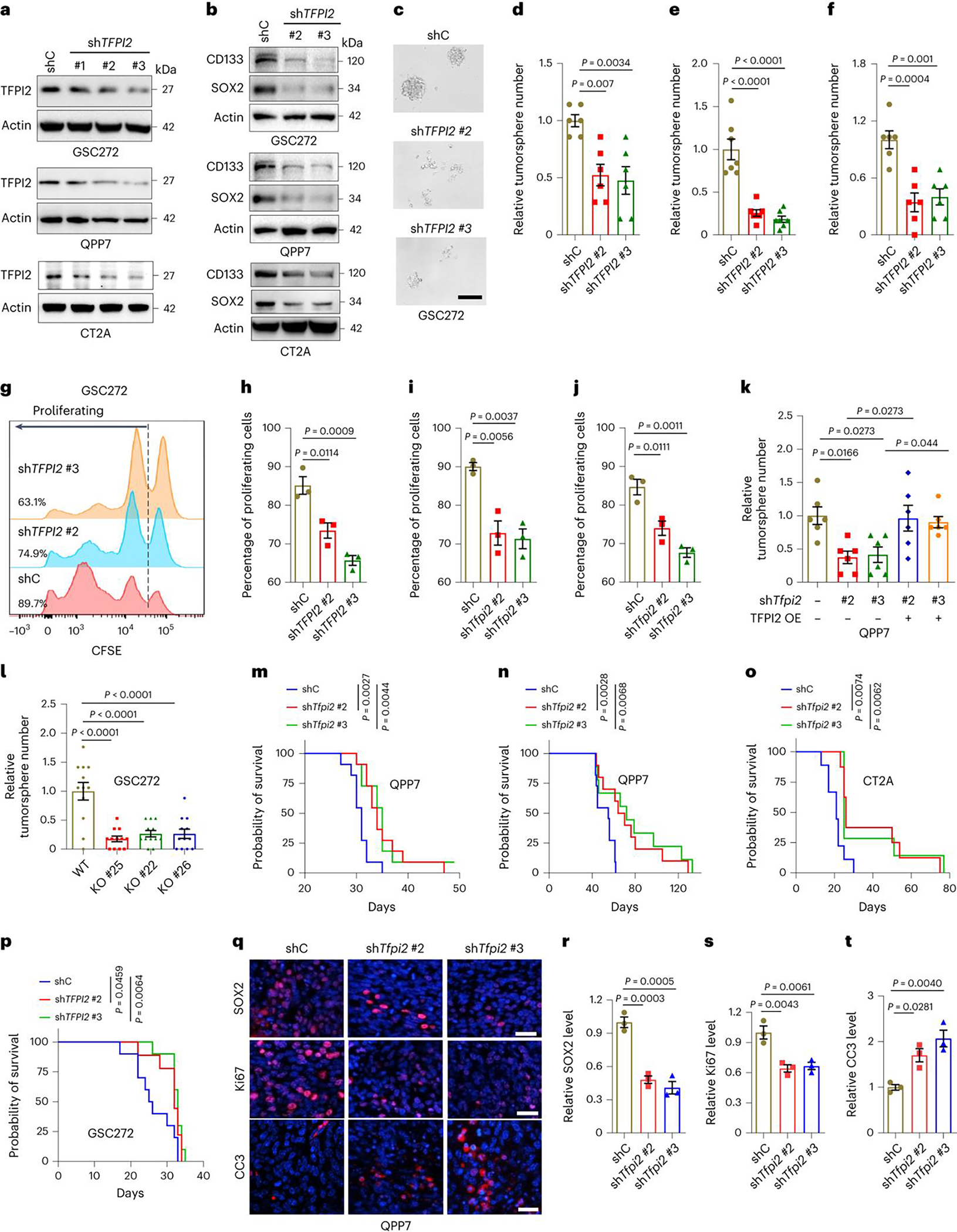
a,b, Immunoblots for TFPI2 (a), CD133 and SOX2 (b) in cell lysates from human GSC272, and mouse QPP7 GSCs and CT2A cells expressing shRNA control (shC) and TFPI2 shRNAs (shTFPI2). c,d, Representative (c) and quantification (d) of tumorspheres in GSC272 cells expressing shC and shTFPI2. n = 6 biological replicates. Scale bar, 200 μm. e,f, Quantification of tumorspheres in QPP7 GSCs (e, n = 6) and CT2A cells (f, n = 7) expressing shC and shTfpi2. g,h, Representative (g) and quantification (h) of proliferation in GSC272 cells expressing shC and shTFPI2. The percentage of proliferating cells for each group is indicated. n = 3 biological replicates. i,j, Quantification of proliferation in QPP7 GSCs (i) and CT2A cells (j) expressing shC and shTfpi2. n = 3 biological replicates. k, Quantification of tumorspheres in QPP7 GSCs expressing shTfpi2 with or without reexpression of shRNA-resistant TFPI2 cDNA. n = 6 biological replicates. l, Quantification of tumorspheres in TFPI2 WT and CRISPR KO GSC272 cells. n = 12 biological replicates. m,n, Survival curves of C57BL/6 mice implanted with 2 × 105 (m) or 2 × 104 (n) QPP7 GSCs expressing shC and shTfpi2. n = 11 biological replicates. o, Survival curves of C57BL/6 mice implanted with 2 × 104 shC and shTfpi2 CT2A cells. n = 7–9 biological replicates. p, Survival curves of nude mice implanted with 2 × 105 shC and shTFPI2 GSC272 cells. n = 9–10 biological replicates. q, Representative of immunofluorescence staining of SOX2, Ki67 and cleaved caspase 3 (CC3) in tumors from C57BL/6 mice intracranially implanted with shC and shTfpi2 QPP7 GSCs. Scale bar, 50 μm. r–t, Quantification of the relative expression levels of SOX2 (r), Ki67 (s) and CC3 (t) in tumors from C57BL/6 mice intracranially implanted with QPP7 GSCs expressing shC and shTfpi2. n = 3 biological replicates. Error bars indicate mean ± s.e.m. One-way ANOVA test. In m–p, log-rank test was carried out.
To further investigate the role of TFPI2 in GBM tumor in vivo, we utilized the shRNA knockdown system to deplete TFPI2 in QPP7 and CT2A tumors implanted into C57BL/6 mice and in GSC272 tumors implanted into nude mice. TFPI2 depletion significantly inhibited tumor growth (Supplementary Fig. 2d–i) and extended the survival of tumor-bearing mice (Fig. 2m–p). Immunohistochemical analysis revealed that GSC markers SOX2 and CD133 (Fig. 2q,r and Extended Data Fig. 2q–t) and proliferation marker Ki67 (Fig. 2q,s and Extended Data Fig. 2u,v) were significantly reduced, whereas apoptosis marker cleaved caspase 3 (Fig. 2q,t and Extended Data Fig. 2w,x) was increased, in tumors established from TFPI2-depleted GSCs. Collectively, these results confirm the essential role of TFPI2 in promoting GSC stemness and GBM progression.
TFPI2 activates JNK–STAT3 signaling to promote stemness and tumor growth
To determine the molecular basis of TFPI2’s effect on GSC self-renewal, we performed RNA sequencing (RNA-seq) profiling on GSC272 cells with or without TFPI2 knockdown. Gene set enrichment analysis (GSEA) on oncogenic signatures resulted in identification of eight signatures that were influenced by TFPI2 (Fig. 3a). Among them, JNK (one of the top two pathways), that has been associated with tumor cell stemness32,33, was examined by immunoblotting with results showing that increasing TFPI2 expression promoted corresponding increases in JNK and P-JNK; and the opposite effect was apparent in TFPI2 knockdown GSCs (Fig. 3b,c). Pharmacologic inhibition of JNK with its inhibitor JNK-IN-8 negated self-renewal (Fig. 3d–f), proliferation (Extended Data Fig. 3a–d) and apoptotic (Extended Data Fig. 3e–h) effects of increasing cellular TFPI2 in GSCs.
Fig. 3: TFPI2 promotes stemness and tumor growth by activating JNK–STAT3 signaling.
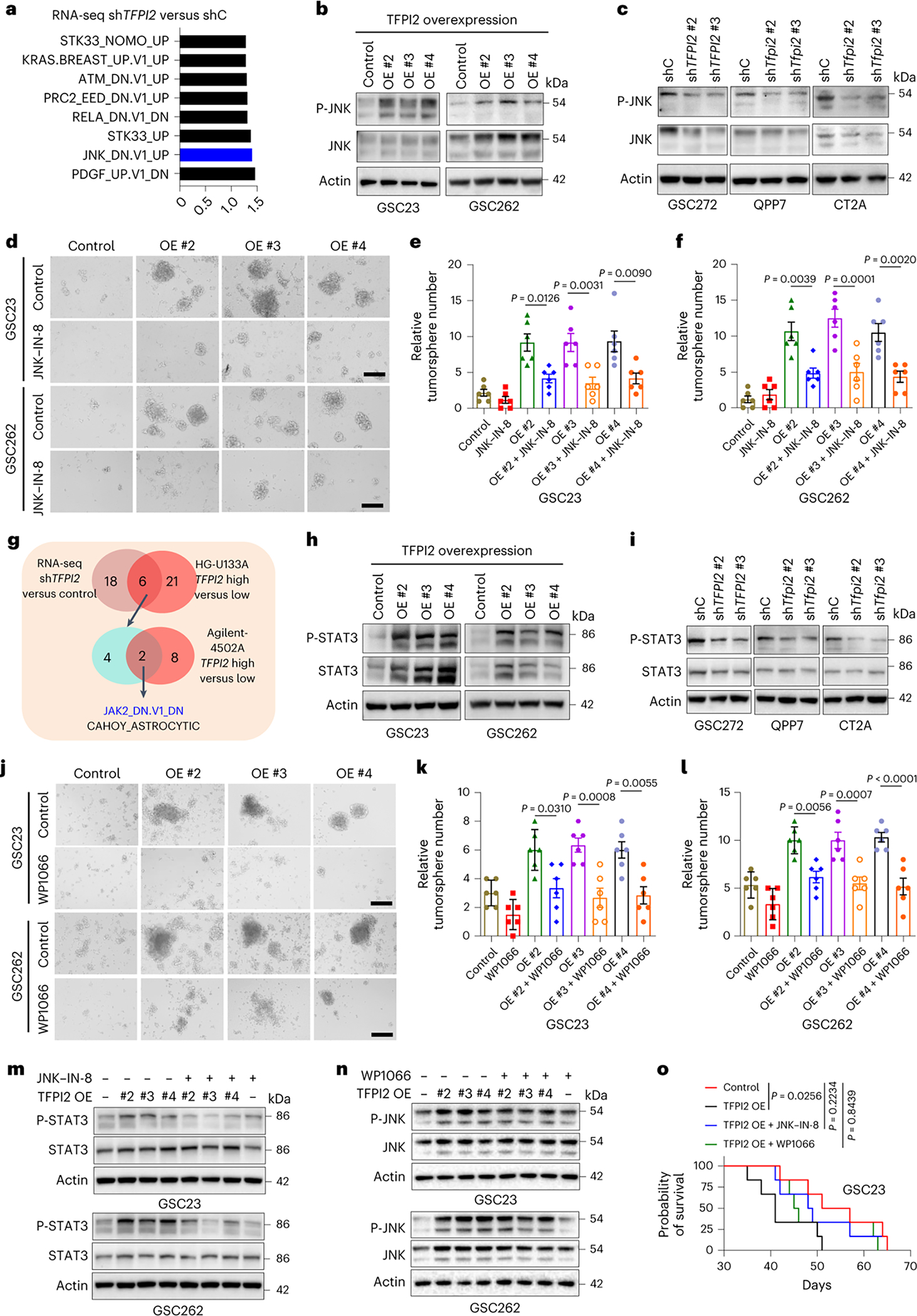
a, Transcriptomic profiling in GSC272 cells following TFPI2 depletion and GSEA analysis shows the eight oncogenic pathways affected by TFPI2 depletion. b, Immunoblots for P-JNK and JNK in GSC23 and GSC262 cells expressing control and TFPI2 overexpression (OE) plasmid. c, Immunoblots for P-JNK and JNK in GSC272, QPP7 GSCs and CT2A cells expressing shRNA control (shC) and TFPI2 shRNAs (shTFPI2). d, Representative images of tumorspheres in GSC23 and GSC262 expressing control and TFPI2 OE plasmid treated with or without JNK inhibitor JNK-IN-8 (10 nM). Scale bar, 400 μm. e,f, Quantification of tumorspheres in GSC23 (e) and GSC262 (f) expressing control and TFPI2 OE plasmid treated with or without JNK-IN-8 (10 nM). n = 6 biological replicates. g, Identification of two oncogenic pathways (as indicated) in distinct datasets with three comparisons (GSC272 RNA-seq data: shC versus shTFPI2 and two TCGA GBM datasets: TFPI2-high versus TFPI2-low). h, Immunoblots for P-STAT3 and STAT3 in GSC23 and GSC262 expressing control and TFPI2 OE plasmid. i, Immunoblots for P-STAT3 and STAT3 in GSC272, QPP7 GSCs and CT2A cells expressing shC and shTFPI2. j, Representative images of tumorspheres in GSC23 and GSC262 expressing control and TFPI2 OE plasmid treated with or without STAT3 inhibitor WP1066 (20 nM). Scale bar, 400 μm. k,l, Quantification of tumorspheres in GSC23 (k) and GSC262 (l) expressing control and TFPI2 OE plasmid treated with or without WP1066 (20 nM). n = 6 biological replicates. m, Immunoblots for P-STAT3 and STAT3 in GSC23 and GSC262 expressing control and TFPI2 OE plasmid treated with or without JNK inhibitor JNK-IN-8 (10 nM). n, Immunoblots for P-JNK and JNK in GSC23 and GSC262 expressing control and TFPI2 OE plasmid treated with or without STAT3 inhibitor WP1066 (20 nM). o, Survival curves of nude mice implanted with 2 × 104 GSC23 expressing control and TFPI2 overexpression (OE) plasmid. Mice were treated with JNK inhibitor JNK-IN-8 (30 mg kg−1 body weight, i.p., every day) or STAT3 inhibitor WP1066 (30 mg kg−1 body weight, i.p., every day) on day 7. n = 6 biological replicates. Error bars indicate mean ± s.e.m. One-way ANOVA test. In o, log-rank test was carried out.
Next, we analyzed TCGA GBM datasets with TFPI2-high versus TFPI2-low patient tumors and GSC272 RNA-seq data with shTFPI2 versus shRNA control (shC). GSEA on oncogenic signatures resulted in identification of two key signatures that were influenced by TFPI2 expression (Fig. 3g). One of these, JAK2/STAT3, has been described as being essential for GSC self-renewal34,35. Consistent with our results from the JNK pathway analysis, we found elevated STAT3 and P-STAT3 in association with increased TFPI2 expression in GSCs, whereas TFPI2 depletion had an opposite effect (Fig. 3h,i). Treatment of GSCs with STAT3 inhibitor WP1066 negated the self-renewal (Fig. 3j–l), proliferation (Extended Data Fig. 3i–l) and apoptotic (Extended Data Fig. 3m–p) effects of elevating cellular TFPI2. To reveal the connection between JNK and STAT3, TFPI2-overexpressing or TFPI2 recombinant protein-conditioned GSCs were treated with or without JNK-IN-8 or WP1066. Immunoblot analysis revealed that TFPI2-induced STAT3 activation was blocked by JNK inhibition (Fig. 3m and Supplementary Fig. 3a). However, STAT3 inhibition had no effect on TFPI2-induced JNK activation (Fig. 3n and Supplementary Fig. 3b). In vivo, TFPI2 overexpression shortened the survival of GSC23 tumor-bearing mice, an effect that was abolished by the treatment with JNK-IN-8 or WP1066 (Fig. 3o). Together, these findings suggest that the JNK–STAT3 signaling is essential for TFPI2-induced GSC self-renewal and tumor growth.
TFPI2 promotes microglia infiltration by activating STAT6
Emerging evidence demonstrates that cancer cell stemness directly correlates with immunosuppression across cancer types, including GBM4,24. Our recent studies have shown that CLOCK is amplified in about 5% of GBM cases and can act as a positive regulator for GSC stemness and microglia inflitration9,10. Surprisingly, analysis of TCGA GBM and LGG datasets demonstrated that TFPI2 amplification is mutually exclusive with CLOCK amplification (Extended Data Fig. 4a), which indicates that TFPI2 and CLOCK may share similar functions and/or are associated with a common signaling pathway36. GSEA on hallmark, Gene Ontology Biological Process, Gene Ontology Molecular Function and Kyoto Encyclopedia of Genes and Genomes pathways revealed a prominent representation of immune suppressive signatures, cytokine and chemokine signatures, and immune response and leukocyte migration signatures in TFPI2-high GBM patient tumors (Extended Data Fig. 4b and Supplementary Table 1). Moreover, microglia signature was enriched in TFPI2-amplified patient tumors compared with control tumors (Supplementary Fig. 1b).
Since TFPI2 is a secreted protein, we hypothesized that GSC-derived TFPI2 might promote microglia infiltration. Using transwell migration assays, we found that recombinant TFPI2-supplemented medium dramatically increased the migration of distinct types of microglia (Fig. 4a,b), including mouse (SIM-A9) and human (HMC3) microglia cell lines, and primary human microglia (PrhMG, isolated from postmortem human brains37). To further investigate the role of TFPI2 in promoting microglia infiltration in the context of GBM, we examined the effects of conditioned medium (CM) from TFPI2-overexpressing GSC262 and GSC23, with results showing enhanced migration of PrhMG (Fig. 4c and Extended Data Fig. 4c). CM from TFPI2-depleted GSC272 and QPP7 GSCs reduced the migration of human and mouse microglia (Fig. 4d and Extended Data Fig. 4d). The impaired microglia migration was partially rescued when CM from TFPI2-depleted GSCs transfected with shRNA-resistant TFPI2 cDNA was used (Supplementary Fig. 4a,b). To investigate TFPI2’s effect on microglia infiltration in vivo, we determined microglia abundance in GBM tumors established from GSCs with and without shTFPI2. Flow cytometry results showed that TFPI2 depletion significantly reduced the population of CD45lowCD11b+ cells, but had little effect on CD45highCD11b+ cells in CT2A tumors (Extended Data Fig. 4e,f). The reduction effect was specific to CD45lowCD11b+CX3CR1+ microglia (Fig. 4e,f), but not CD45highCD11b+CD68+ macrophages (Extended Data Fig. 4g,h). Immunofluorescence for CX3CR1 confirmed that TFPI2 depletion reduced microglia abundance in QPP7 tumors (Fig. 4g,h) and GSC272 tumors (Fig. 4i,j). These findings demonstrate that TFPI2 is a chemokine triggering microglia infiltration into the GBM TME.
Fig. 4: TFPI2 activates STAT6 to promote microglia infiltration.
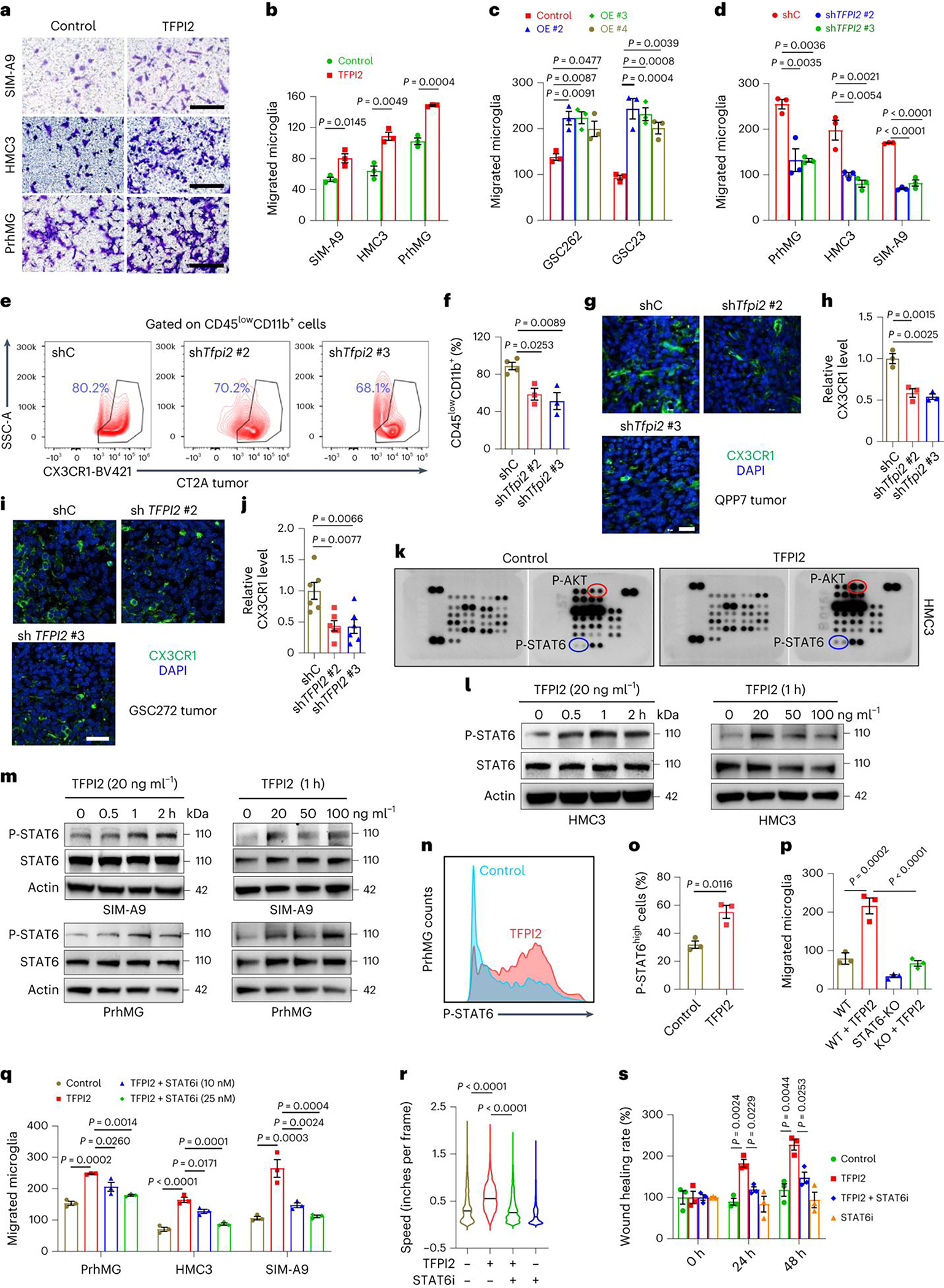
a,b, Representative (a) and quantification (b) of relative migration of SIM-A9, HMC3, and primary human microglia (PrhMG) following stimulation with TFPI2 recombinant protein. Scale bars, 200 μm for HMC3 and SIM-A9, and 400 μm for PrhMG. c, Quantification of transwell migration of PrhMG following stimulation with CM from GSC262 or GSC23 cells expressing control and TFPI2 overexpression (OE) plasmid. d, Quantification of transwell migration of PrhMG, HMC3 and SIM-A9 following stimulation with CM from GSC272 cells (for PrhMG and HMC3) or QPP7 cells (for SIM-A9) expressing control shRNA (shC) and TFPI2 shRNAs (shTFPI2). e,f, Representative images (e) and quantification (f) of flow cytometry for the percentage of CD45lowCD11b+CX3CR1+ microglia in size-matched shC and shTfpi2 CT2A tumors. n = 4 and 3 for shC and shTfpi2, respectively. g,h, Immunofluorescence (g) and quantification (h) of CX3CR1 in size-matched shC and shTfpi2 QPP7 tumors. Scale bar, 25 μm. i,j, Immunofluorescence (i) and quantification (j) of CX3CR1 in size-matched shC and shTFPI2 GSC272 tumors. Scale bar, 25 μm. n = 6 biological replicates. k, Representative of human phospho-kinases in HMC3 cells treated with or without TFPI2 protein. Affected kinases are indicated. l,m, Immunoblots for P-STAT6 and STAT6 in cell lysates of HMC3 (l), SIM-A9 and PrhMG (m) incubated with TFPI2 protein at indicated concentrations and timepoints. n,o, Representative (n) and quantification (o) of flow cytometry for P-STAT6 expression in PrhMG treated with or without TFPI2 protein. p, Quantification of transwell migration of mouse primary microglia isolated from WT and STAT6 KO mice following stimulation with TFPI2 protein. q, Quantification of transwell migration of PrhMG, HMC3, and SIM-A9 microglia following stimulation with TFPI2 protein in the presence or absence of STAT6 inhibitor AS1517499 (25 nM). r,s, Quantification of incucyte live-cell (r) and scratch wound healing (s) migration of HMC3 microglia following TFPI2 protein stimulation with or without AS1517499 (25 nM). In r, n = 6,289, 2,934, 7,966 and 34,440 cells for control, TFPI2, TFPI2 + STAT6i, and STAT6i, respectively. In b–d, h, o–q and s, n = 3 biological replicates. TFPI2 protein was used at 20 ng ml−1 unless indicated. Error bars indicate mean ± s.e.m. A two-tailed Student’s t-test was performed for comparisons between two groups. A one-way ANOVA followed by a Tukey test was performed to compare more than two groups.
To investigate the molecular basis of TFPI2-induced microglia migration, we performed human phospho-kinase antibody array with results showing that STAT6 and AKT were activated in HMC3 microglia in association with TFPI2 protein treatment (Fig. 4k). Bioinformatics analyses in TCGA GBM tumors demonstrated that STAT6, but not AKT1 and AKT2, positively correlated with microglia signature (Extended Data Fig. 4i). Moreover, STAT6 showed a stronger positive correlation with microglia signature than AKT3 (Extended Data Fig. 4i). Immunoblotting and flow cytometry results confirmed increased phosphorylation of STAT6 in human and mouse microglia upon the treatment with TFPI2 protein (Fig. 4l–o).
To further investigate the role of STAT6 in mediating TFPI2-induced microglia migration, we isolated microglia from wild-type (WT) and STAT6-KO mice and used them in a transwell migration assay, the results of which showed that microglia isolated from STAT6-KO mice had impaired migration ability compared with microglia isolated from WT mice in response to TFPI2 treatment (Fig. 4p and Extended Data Fig. 4j). Pharmacologic inhibition of STAT6 with AS1517499 (ref. 38) suppressed TFPI2-induced transwell migration of human and mouse microglia (Fig. 4q and Extended Data Fig. 4k). The impairment of TFPI2-induced HMC3 microglia migration from AS1517499 treatment was also confirmed by the investigations using incucyte live-cell and scratch assays (Fig. 4r,s, Supplementary Fig. 5 and Supplementary Video 1). Together, these findings support a critical role for STAT6 signaling in mediating TFPI2-directed microglia infiltration into the GBM TME.
TFPI2–STAT6 axis promotes microglia immunosuppressive polarization
Microglia are a heterogeneous population of immune cells in the GBM TME and can be classified as immunostimulatory or immunosuppressive phenotype5. Flow cytometry results showed that CM from GSC262 and GSC23 polarized PrhMG towards an immunosuppressive phenotype by increasing the percentage of CD206+ cells, and this effect was increased when using CM from GSC262 and GSC23 modified for increasing TFPI2 expression (Fig. 5a–c and Extended Data Fig. 5a). In contrast, microglia immunosuppressive polarization was inhibited when treating with CM from TFPI2-depleted GSCs (Fig. 5d,e and Extended Data Fig. 5b,c). The impaired microglia immunosuppressive polarization was rescued by reexpression of shRNA-resistant TFPI2 cDNA in shTFPI2 GSCs (Fig. 5e and Extended Data Fig. 5c). In vivo, TFPI2 depletion in tumors established from CT2A cells presented reduced numbers of CD45lowCD11b+CX3CR1+CD206+ immunosuppressive microglia (Fig. 5f,g), while not affecting CD45highCD11b+CD68+CD206+ immunosuppressive macrophages (Extended Data Fig. 5d,e). Co-immunofluorescence for CX3CR1 and immunosuppressive markers (for example, CD206, CD163 and ARG1) confirmed that immunosuppressive microglia were reduced upon TFPI2 depletion in tumors established from QPP7 GSCs (Fig. 5h–j and Supplementary Fig. 6a,b), CT2A cells (Extended Data Fig. 5f–i and Supplementary Fig. 6c,d) and GSC272 (Extended Data Fig. 5j–m and Supplementary Fig. 6e,f). These findings demonstrate that GSC-derived TFPI2 is essential for microglia immunosuppressive polarization in GBM.
Fig. 5: TFPI2–STAT6 signaling axis triggers microglia immunosuppressive polarization.

a,b, Representative (a) and quantification (b) of flow cytometry for the percentage of CD206+ PrhMG treated with CM from GSC262 expressing control and TFPI2 overexpression (OE) plasmid. c, Quantification of flow cytometry for the percentage of CD206+ PrhMG treated with CM from control and TFPI2 OE GSC23. d, Quantification of flow cytometry for the percentage of CD206+ cells in HMC3 microglia treated with CM from GSC272 expressing shRNA control (shC) and TFPI2 shRNAs (shTFPI2). e, Quantification of flow cytometry for the percentage of CD206+ cells in SIM-A9 microglia treated with CM from shC and shTfpi2 QPP7 GSCs with or without reexpression of shRNA-resistant TFPI2 cDNA (OE). f,g, Representative images (f) and quantification (g) of flow cytometry for the percentage of CD45lowCD11b+CX3CR1+CD206+ microglia in size-matched shC and shTfpi2 CT2A tumors. n = 4 and 3 for shC and shTfpi2, respectively. h–j, Immunofluorescence (h) and quantification of CX3CR1+ CD206+ (i) or CX3CR1+CD163+ (j) cells in size-matched shC and shTfpi2 QPP7 tumors. Scale bar, 25 μm. n = 6 biological replicates. k, Quantification of flow cytometry for the percentage of CD206+ cells in primary microglia isolated from WT and STAT6 KO mice following stimulation with TFPI2 protein. l,m, Representative (l) and quantification (m) of flow cytometry for arginase 1 (ARG1) in primary microglia isolated from WT and STAT6 KO mice following stimulation with TFPI2 protein. n,o, Quantification of flow cytometry for the percentage of CD206+ cells (n) and ARG1 expression (o) in SIM-A9 microglia treated with TFPI2 protein in the presence or absence of STAT6i AS1517499. p,q, Quantification of flow cytometry for the percentage of CD206+ cells (p) and ARG1 expression (q) in HMC3 microglia treated with TFPI2 protein combining with or without AS1517499. r, Relative mRNA expression of indicated genes in SIM-A9 cells treated with TFPI2 protein in the presence or absence of AS1517499. s, Relative mRNA expression of indicated genes in HMC3 cells treated with TFPI2 protein in the presence or absence of AS1517499. n = 3 or 4 biological replicates. In b–e, k and m–r, n = 3 biological replicates. Error bars indicate mean ± s.e.m. One-way ANOVA test. TFPI2 protein at 20 ng ml−1 and AS1517499 at 25 nM were used.
We further investigated whether TFPI2-induced microglia immunosuppressive polarization is regulated by STAT6 signaling using flow cytometry analysis for CD206 and ARG1 expression, with results showing that immunosuppressive microglia were increased by TFPI2 protein treatment. This increase was negated by genetic depletion of STAT6 in primary mouse microglia (Fig. 5k–m and Extended Data Fig. 5n) and by pharmacologic inhibition of STAT6 using AS1517499 in both mouse and human microglia (Fig. 5n–q and Extended Data Fig. 5o–r). This result is consistent with data obtained from quantitative real-time polymerase chain reaction (RT–qPCR) for immunosuppressive marker expression in mouse and human microglia (Fig. 5r,s). We also examined the potential role of the TFPI2–STAT6 signaling pathway in regulating microglia immunostimulatory polarization using RT–qPCR, with results showing that TFPI2 protein treatment suppressed the expression of immunostimulatory microglia markers (for example, MIF, IL23, CXCL11, IL6, TNFA, LFNG and HLA-DR) in microglia, which was countered by treatment with STAT6 inhibitor AS1517499 (Fig. 5r,s and Extended Data Fig. 5s). Together, these findings suggest that STAT6 signaling is involved in TFPI2-induced microglia immunosuppressive polarization in GBM.
CD51 is a receptor on microglia for TFPI2
To investigate whether TFPI2 exerts migratory and polarization effects through interaction with membrane proteins, PrhMG were incubated with TFPI2 recombinant protein (Fig. 6a). Membranous PrhMG proteins were subsequently extracted and TFPI2 was immunoprecipitated (Fig. 6a). The major protein band revealed by denaturing gel electrophoresis of immunoprecipitates was analyzed by LC–MS, with results showing 395 co-precipitated proteins. Sixty-one of them exhibited a ≥2-fold change in TFPI2 immunoprecipitation (IP) group versus IgG group. Searching for these proteins in a membrane protein database39 revealed 15 candidates as potentially interacting with TFPI2 (Fig. 6a). With further filtering using the TIME dataset of isocitrate dehydrogenase (IDH)-WT GBM patient tumors28 as well as data from CT2A tumor model, we identified CD51 as the only potential receptor exhibiting higher expression in microglia compared with CD45− GBM cells, macrophages, lymphocytes and other immune cells (Extended Data Fig. 6a,b). The interaction between CD51 and TFPI2 was confirmed by immunoblotting of mouse and human microglia immunoprecipitates with an anti-CD51 antibody (Fig. 6b,c) or anti-TFPI2 antibody (Extended Data Fig. 6c,d). To determine whether CD51 affects TFPI2 signaling in microglia, we utilized a calcium mobilization assay, which has been widely used to measure the activation of receptors, including chemokine receptors40, with results showing that genetic and pharmacologic inhibition of CD51 decreased TFPI2-triggered calcium mobilization in mouse and human microglia (Fig. 6d,e and Extended Data Fig. 6e,f). TFPI2-induced phosphorylation of PLCγ1 and PKCζ, known mediators in the calcium signaling pathway, were also inhibited by genetic depletion and pharmacologic inhibition of CD51 in microglia (Fig. 6f and Extended Data Fig. 6g,h).
Fig. 6: CD51 is a membrane receptor for TFPI2 on microglia.

a, Working flow for identifying TFPI2 receptor on microglia. FC, fold change. b,c, IP with CD51 antibody (CD51 Ab) and immunoblotting for the interaction between TFPI2 and CD51 in SIM-A9 (b) and HMC3 (c) microglia. d, Calcium mobilization triggered by TFPI2 recombinant protein (20 ng ml−1) in SIM-A9 microglia expressing shRNA control (shC) or Itgav shRNAs (shItgav). ΔF, change of fluorescence intensity at the given time; F0, resting fluorescence intensity. e, Calcium mobilization triggered by TFPI2 protein in SIM-A9 microglia treated with or without CD51 inhibitor MK-0429 at indicated concentrations. f, Immunoblots for P-PLCγ1, PLCγ1, P-PKCζ and PKCζ in cell lysates of HMC3 microglia incubated with TFPI2 protein in the presence or absence of MK-0429 (50 nM). g, Immunoblots for P-STAT6 and STAT6 in cell lysates from SIM-A9 microglia expressing shC and shItgav treated with or without TFPI2 protein. h, Immunoblots for P-STAT6 and STAT6 in cell lysates from HMC3 microglia treated with or without TFPI2 protein and MK-0429 (50 nM). i,j, Representative images (i) and quantification (j) of relative migration of mouse SIM-A9 microglia expressing shC and shItgav following stimulation with TFPI2 protein. Scale bars, 400 μm. n = 3 biological replicates. k, Quantification of relative migration of PrhMG, human HMC3 and mouse SIM-A9 microglia treated with or without TFPI2 protein in the presence or absence of MK-0429 at indicated concentrations. n = 3 biological replicates. l,m, Representative (l) and quantification (m) of flow cytometry analysis for the percentage of CD206+ SIM-A9 microglia expressing shC and shItgav following stimulation with or without TFPI2 protein. n = 3 biological replicates. n,o, Representative (n) and quantification (o) of flow cytometry analysis for the percentage of ARG1+ SIM-A9 microglia expressing shC and shItgav following stimulation with or without TFPI2 protein. n = 3 biological replicates. p,q, Quantification of flow cytometry analysis for the percentage of CD206+ (p) and ARG1+ (q) PrhMG treated with TFPI2 protein in the presence or absence of MK-0429 (50 nM). n = 3 biological replicates. Error bars indicate mean ± s.e.m. One-way ANOVA test. TFPI2 protein at 20 ng ml−1 was used.
Next, we examined whether CD51 inhibition influences TFPI2 effects on microglia. We found that TFPI2-induced phosphorylation of STAT6 was impaired by genetic and pharmacologic inhibition of CD51 in mouse and human microglia (Fig. 6g,h and Extended Data Fig. 6i), and this effect was accompanied by reduced microglia migration as indicated by the results from transwell, Incucyte live-cell and scratch assays (Fig. 6i–k, Extended Data Fig. 6j, Supplementary Fig. 7a–c and Supplementary Video 2). CD51 inhibition also reduced TFPI2-induced microglia immunosuppressive polarization as indicated by reduced expression of immunosuppressive markers CD206 and ARG1 in mouse and human microglia (Fig. 6l–q and Extended Data Fig. 6k–t). In summary, these findings support CD51 as a functional receptor that mediates TFPI2 activities in microglia.
TFPI2–CD51–STAT6 axis inhibition synergizes with anti-PD1 therapy
We hypothesized that inhibiting microglia immunosuppressive effects via blockade of the TFPI2–CD51–STAT6 signaling pathway might influence T-cell-mediated antitumor immune responses. To test it, we implanted shC and shTfpi2 QPP7 GSCs and CT2A cells into the brains of immunocompromised nude mice. We found that antitumor effects associated with TFPI2 depletion were reduced in nude mice when compared against results from immunocompetent C57BL/6 mice (Fig. 2n,o and Extended Data Fig. 7a,b). We performed flow cytometry of spleens from CT2A tumor-bearing C57BL/6 mice and found that TFPI2 depletion significantly increased CD3+ (CD45+CD3+), CD8+ (CD45+CD3+CD8+) and CD4+ (CD45+CD3+CD4+) T-cell populations (Fig. 7a–d). Increases of CD8+ and CD4+ T cells were confirmed by immunofluorescence staining of TFPI2-depleted CT2A and QPP7 tumors (Extended Data Fig. 7c–f). Furthermore, the exhausted T cells (for example, CD45+CD3+CD8+PD1high and CD45+CD3+CD4+PD1high cells) were downregulated in the spleens of CT2A tumor-bearing mice upon TFPI2 depletion (Fig. 7e,f and Extended Data Fig. 7g). These findings suggest that TFPI2 depletion increases T-cell infiltration and activation in tumors of immunocompetent GBM mouse models. To confirm the involvement of microglia with these results, we utilized a mouse microglia and CD8+ T-cell coculture approach and found that TFPI2-polarized microglia decreased CD8+ T-cell viability. This effect was negated by treatments with STAT6 inhibitor AS1517499 (Fig. 7g,h). We also found that the percentage of CD8+CD69+ and CD8+IFNγ+ T cells was reduced when they were cocultured with TFPI2-treated microglia. These reductions were prevented by AS1517499 treatments (Fig. 7i–k and Extended Data Fig. 7h). Results from human microglia–T-cell cocultures showed that TFPI2-polarized microglia suppressed JURKAT (human CD4+CD8−) T-cell viability and activation, and these effects were prevented by AS1517499 treatments (Fig. 7l–n and Extended Data Fig. 7i–m). Functionally, TFPI2-polarized microglia suppressed T-cell-mediated tumor cell cytotoxicity compared with control microglia, and this immunosuppressive function was rescued by AS1517499 treatments (Extended Data Fig. 8a–c). Together, these findings suggest that blockade of microglia immunosuppressive polarization via inhibition of the TFPI2-STAT6 signaling can increase T-cell infiltration, activation and cytotoxicity.
Fig. 7: TFPI2–CD51–STAT6 axis inhibition activates antitumor immunity and synergizes with anti-PD1 therapy.

a,b, Representative (a) and quantification (b) of flow cytometry analysis for CD45+CD3+ T cells in the spleens of C57BL/6 mice bearing size-matched CT2A tumors expressing shRNA control (shC) and Tfpi2 shRNAs (shTfpi2). c–f, Quantification of flow cytometry analysis for CD45+CD3+CD8+ T cells (c), CD45+CD3+CD4+ T cells (d), CD45+CD3+CD8+PD1hi T cells (e) and CD45+CD3+CD4+PD1hi T cells (f) in the spleens of C57BL/6 mice bearing size-matched shC and shTfpi2 CT2A tumors. n = 4 and 3 for shC and shTfpi2, respectively. g,h, Representative (g) and quantification (h) of the viability of primary CD8+ T cells cocultured with SIM-A9 microglia treated with or without TFPI2 protein (20 ng ml−1) and STAT6 inhibitor AS1517499 (25 nM). i,j, Representative (i) and quantification (j) of the percentage of CD69+ cells out of total CD8+ T cells cocultured with SIM-A9 microglia treated with TFPI2 protein (20 ng ml−1) in the presence or absence of AS1517499 (25 nM). k, Quantification of the percentage of IFNγ+ cells out of total CD8+ T cells cocultured with SIM-A9 microglia treated with or without TFPI2 protein (20 ng ml−1) and AS1517499 (25 nM). l–n, Quantification the viability (l), CD69+ cells (m) and IFNγ+ cells (n) in JURKAT T cells cocultured with PrhMG treated with or without TFPI2 protein (20 ng ml−1) and AS1517499 (25 nM). In b, h, j, k and l–n, n = 3 biological replicates. o, Survival curves of C57BL/6 mice implanted with shC and shTfpi2 CT2A cells (2 × 104 cells). Mice were treated with anti-PD1 (10 mg kg−1, i.p.) on days 11, 14 and 17. n = 6 for shC and shTfpi2 #2+anti-PD1, 7 for shC+anti-PD1, shTfpi2 #3 and shTfpi2 #3+anti-PD1, and 8 for shTfpi2 #2. p–s, Survival curves of C57BL/6 mice implanted with 005 GSCs (2 × 105 cells) or CT2A cells (2 × 104 cells). Mice were treated with MK-0429 (30 mg kg−1, i.p., every other day; p and q) or AS1517499 (10 mg kg−1, i.p., every other day; r and s) on day 7, and then received anti-PD1 treatment on days 11, 14 and 17. In p, n = 8, 7, 9 and 9; and in q, n = 8, 7, 8 and 7 for control, anti-PD1, MK-0429 and MK-0429+anti-PD1, respectively. In r and s, n = 6 biological replicates. Error bars indicate mean ± s.e.m. One-way ANOVA test. In o–s, log-rank test was carried out.
Since the CD51–STAT6 signaling plays a prominent role in promoting TFPI2-induced microglia migration, immunosuppressive polarization and antitumor immune response, we explored the impact of this signaling on tumor growth and ICI therapy efficacy in immunocompetent GBM mouse models. Since a recent study reported that QPP7 tumors respond to ICI therapy41, we tested a second immunocompetent GBM mouse model, 005 GSC, that expresses relatively high levels of TFPI2 (Fig. 1e) and has been reported as nonresponsive to immunotherapy31. As expected, genetic and pharmacologic inhibition of the TFPI2–CD51–STAT6 axis extended the survival of C57BL/6 mice implanted with 005 GSCs and CT2A cells (Fig. 7o–s). We and others have shown that PD-L1 can mediate microglia-induced immunosuppression in GBM9,42. TCGA GBM data analysis showed that the expression of PD-L1 and PD-L2 correlated positively with TFPI2, ITGAV and STAT6 expression (Extended Data Fig. 8d,e). These results prompted our investigation of combined TFPI2–CD51–STAT6 axis inhibition plus anti-PD1 therapy in GBM-bearing mice. Our results showed that depletion of TFPI2 in GSCs (Fig. 7o), treatment with CD51 inhibitor MK-0429 (Fig. 7p,q), or treatment with STAT6 inhibitor AS1517499 (Fig. 7r,s) synergized with anti-PD1 therapy to extend the survival of animal subjects bearing 005 GSC and CT2A tumors. Together, these findings indicate that inhibition of the TFPI2–CD51–STAT6 signaling axis activates antitumor immunity that can be exploited through combination with anti-PD1 treatment in GBM.
TFPI2 tracks with GSC stemness and microglia abundance in human GBM
To address the prognostic relevance of our findings, we performed immunohistochemistry staining for TFPI2, SOX2, CD133, CX3CR1, Ki67 and cleaved caspase 3 in GBM patient tumors (Fig. 8a and Extended Data Fig. 9a). Our analysis revealed that the expression of TFPI2 correlated positively with CD133 (Fig. 8b), SOX2 (Fig. 8c), Ki67 (Extended Data Fig. 9b) and CX3CR1 (Fig. 8d), and correlated negatively with cleaved caspase 3 (Extended Data Fig. 9c). Analysis of scRNA-seq data from GBM patient tumors confirmed that elevated TFPI2 expression in GBM cells correlated with increased microglia signature (Fig. 8e). Analysis of TCGA GBM patient data showed that stemness and microglia signatures were enriched in TFPI2-high patient tumors (Fig. 8f,g), whereas activated CD8+ T-cell signature was enriched in TFPI2-low patient tumors (Fig. 8h).
Fig. 8: TFPI2 is a prognostic biomarker correlating with GSC stemness and microglia abundance in GBM.

a, Representative images show low, medium and high expression of TFPI2, CD133, SOX2 and CX3CR1 in human GBM tumor samples based on immunohistochemistry staining. Scale bar, 100 μm. b–d, Quantification of immunohistochemistry staining showing strong positive correlations between TFPI2 and CD133 (b), TFPI2 and SOX2 (c), or TFPI2 and CX3CR1 (d) in human GBM tumor samples (n = 60). R and P values were determined by Pearson correlation. e, Quantification of microglia signature in GBM patient tumors with GBM cell TFPI2 high and low based on scRNA-seq data (EGAS00001004422 and GSE131928). n = 22 and 13 patients for TFPI2 low and high, respectively. f–h, GSEA for stemness signature (f), microglia signature (g) and activated CD8+ T-cell signature (h) in TFPI2-high patient tumors compared with TFPI2-low patient tumors from TCGA GBM dataset. Normalized enrichment scores (NES) and false discovery rate (FDR) are shown. i, ELISA for TFPI2 in the plasma from healthy controls (n = 6) and patients with GBM (n = 55). j, Correlation between Ki67 expression in tumors and TFPI2 plasma concentration in patients with GBM (n = 55). k, Correlation between overall survival and TFPI2 plasma concentration in patients with GBM (n = 35). In j and k, R and P values were determined by Pearson correlation. l, Overall survival of patients with high (n = 23) and low (n = 31) TFPI2 plasma concentration (cut-off at median). Log-rank test and P value is shown. Error bars indicate mean ± s.e.m. A two-tailed Student’s t-test was performed for comparisons between two groups.
Since TFPI2 encodes a secreted protein, we analyzed its protein level in the plasma of 6 healthy controls and 55 patients with GBM, with results showing that plasma levels of TFPI2 were significantly higher in patients with GBM than healthy controls (Fig. 8i). Moreover, correlation analyses revealed that plasma TFPI2 correlated positively with intratumoral Ki67 expression (Fig. 8j) and negatively with patient overall survival (Fig. 8k,l). Our analyses also showed that TFPI2 levels were not correlated with status of recurrence, gender and age of patients with GBM (Extended Data Fig. 9d–f). Together, these human findings support the role of TFPI2 in regulation of GSC stemness, microglia infiltration and T-cell activation, and suggest that TFPI2 is a potential prognostic biomarker for patients with GBM.
Discussion
In this study, we identified TFPI2 as a key GBM-associated factor that promotes GSC stemness and microglia immunosuppression (Extended Data Fig. 10a). TFPI2 promotes GSC self-renewal and tumor growth via activation of the JNK–STAT3 pathway. In addition to its GSC intrinsic effects, secreted TFPI2 binds to CD51 on microglia to activate STAT6 signaling that, in turn, contributes to microglia infiltration and immunosuppressive polarization. Consistent with our previous findings9,10, the activation of TFPI2–STAT6 signaling in microglia suppresses T-cell infiltration and activation, and in so doing promotes tumor growth. In preclinical models, inhibition of the TFPI2–CD51–STAT6 signaling axis generates potent antitumor activity that is heightened when combined with anti-PD1 therapy (Extended Data Fig. 10b). In addition, our analyses of GBM patient tumor and plasma samples support TFPI2 is a potential biomarker whose circulating levels correlate with GBM patient outcome. Together, our work reveals the molecular mechanisms underlying stemness and GSC–microglia symbiosis and informs a responder hypothesis to evaluate the therapeutic strategy by combining GSC–microglia symbiosis inhibition and ICI in GBM.
GSCs are a subpopulation of self-renewing cells that contribute to GBM resistance to treatments1,4. Much of our research has focused on the molecular basis and consequences of GSC activities. However, increasing evidence demonstrates that the phenotypic and functional properties of GSCs are relied on both cell-intrinsic factors and TME niche factors43. Among them, genetic drivers gain added significance as their alterations in GBM cells/GSCs are critical for tumor tumorigenesis and stemness maintenance44,45. We and others have identified CLOCK, which is amplified in about 5% of human GBM10, as an important contributor to GSC maintenance and function, and whose genetic as well as pharmacologic inhibition impairs GSC stemness10,46. In the current study, we extend the concept and identify TFPI2 as a factor that is amplified in about 4% of human GBM and critical for GSC stemness maintenance.
TFPI2 is a 32 kDa protein that inhibits a wide range of proteases11. It has been described as a tumor suppressor in studies of non-small cell lung cancer, breast cancer and pancreatic cancer14–16. However, TFPI2 has also been implicated in having protumor activities in melanoma19 and hepatocellular carcinoma17. Given the prominent heterogeneity of GBM cells and GSCs in brain tumors47,48, they may contradictorily respond to TFPI2 depletion. Several studies of TFPI2 in GBM have yielded results that are complex with respect to labeling TFPI2 activities as protumor versus antitumor. One study has shown that elevated TFPI2 expression in differentiated GBM cells decreases their proliferative and invasive capacity21,49, with the latter consequence potentially due to TFPI2 suppression of MMP activity22. In this study, scRNA-seq analysis of GBM patient tumors and bioinformatics analysis of TCGA GBM tumors revealed that TFPI2 is correlated with GSC signature, suggesting a protumor role of TFPI2 in GBM. Results from our gain- and loss-of-function experiments indicate that increasing TFPI2 expression in TFPI2-low GSCs promotes self-renewal and tumor progression, while reducing TFPI2 in TFPI2-high GSCs has the opposite effects. These findings gain added significance as recent studies have shown that differentiated GBM cell lines may not fully recapitulate the heterogeneity and stem-cell-like properties in GBM50,51. Interestingly, findings from single-cell chromatin accessibility profiling studies indicate that patient-derived GSCs share a similar chromatin identity with putative cancer stem cells from primary brain tumors3. Considering the advantages of GSC models, we profiled TFPI2 expression in different GSC lines and confirmed the protumor effect of TFPI2 expression in GBM tumorigenesis. In revealing the molecular mechanism underlying TFPI2’s role in promoting GSC self-renewal, we observed that TFPI2 activates the JNK–STAT3 signaling pathway that is important for GSC self-renewal and maintenance32–35, highlighting the potential of testing of JNK and STAT3 inhibitors in patients with TFPI2-high GBM.
Another vital factor affecting the role of TFPI2 in GBM, that has not been addressed in previous studies, is the TME. GSCs are known to secrete factors that promote immune cell infiltration and activation in the GBM TME4,9,10,47,52. These immune cells, in turn, affect GSC biology, thus generating a GSC–immune symbiosis in a context-dependent manner, such as under specific genetic backgrounds5,53. RTK/RAS/PI3K/PTEN is one of the core signaling pathways that is altered in GBM54, and alteration of this pathway has been linked with GBM–immune cell crosstalk55. We have recently demonstrated that PTEN deficiency in GBM cells and GSCs promotes macrophage infiltration into the TME56. In addition, we have identified a CLOCK–OLFML3–LGMN pathway in GSCs that promotes tumor infiltration of microglia9,10. In this study, we found that the amplification patterns of CLOCK and TFPI2 are mutually exclusive in patients with GBM, suggesting that these two genes may share similar functions36. Results from the current study indicate that TFPI2-associated signaling is working to promote microglia infiltration into the GBM TME. Further studies are needed to characterize the relationship between CLOCK and TFPI2 in GSCs and their combined effects on microglia. Collectively, our studies indicate that GSCs utilize multiple mechanisms to modulate microglia biology, thus activating supportive rather than antitumor immune responses in GBM.
Myeloid cell infiltration is a hallmark of GBM, wherein microglia and macrophages are considered as prominent immune cell populations that act to suppress antitumor immunity9,10. Our findings of TFPI2-associated effects on microglia biology are consistent with previous reports that TFPI2 plays an important role in regulating innate immunity during pathogenic infection57. Through unbiased profiling followed by functional validation, we identified STAT6 as a key downstream signaling mediator responsible for TFPI2-induced microglia infiltration and immunosuppressive polarization. In addition, we show that TFPI2-associated STAT6 activation in microglia supports GBM progression in mouse models. In addition to highlighting STAT6 as a downstream signaling of TFPI2 in microglia, our work reinforces the importance of STAT6 signaling in TAM immunosuppressive polarization58,59 and identifies STAT6 as a potential target for TFPI2-high responder population for such intervention. Emerging evidence supports the existence of specific receptors (for example, CSF-1R and CD29) on TAMs as being crucial in determining their response to GSC-secreted factors53,56,60. Our data reveal that TFPI2 specifically binds to CD51 on microglia, which results in STAT6 activation and microglia immunosuppression. Extending the findings of a pancreatic cancer study showing that CD51 as being responsible for a macrophage-cancer stem cell interaction61, our studies identify an additional ligand, TFPI2, as a key mediator from GSCs that triggers microglia infiltration and polarization through CD51 signaling. Together, our findings highlight that CD51 serves as a functional TFPI2 receptor on microglia, thus providing biological insights to immunosuppression and GBM intervention.
The identification of the TFPI2–STAT6 axis as a driver of microglia immunosuppressive polarization in GBM, combined with our demonstration of antitumor effects resulting from TFPI2 and STAT6 inhibition in mouse models, indicates an effective therapeutic intervention strategy targeting microglia reprogramming. This strategy is supported by increasing evidence showing that inhibiting TAM immunosuppressive reprogramming shows robust antitumor effects in multiple types of cancer53,60,62, including GBM (this study). Clinical trial results involving the use of anti-PD1 therapy in treating GBM have been mostly disappointing63–65. However, the results of these trials have led to an increased recognition of the mechanisms used by immunosuppressive myeloid cells that influence outcomes of clinical trials in which immunotherapies are being investigated 66,67. The importance of TAMs as a determinant of tumor response to immunotherapy is reinforced by results from our recent study showing that inhibition of CLOCK-regulated microglia biology can synergize with anti-PD1 therapy in GBM mouse model9. Study results presented here further encourage the testing of combination regimens inhibiting microglia immunosuppressive reprogramming (for example, inhibiting the TFPI2–CD51–STAT6 signaling) while simultaneously targeting immune checkpoint molecules (for example, PD1) in the treatment of GBM.
Methods
Mice and intracranial xenograft tumor models
All animal experiments were performed with the approval of the Institutional Animal Care and Use Committee at Northwestern University (protocol numbers IS00017931, IS00020802, IS00016006 and IS00015772). Female C57BL/6 (#0000664), nude mice (#007850) and B6.129S2(C)-Stat6tm1Gru/J mice (STAT6-KO; #005977) at 6 weeks of age were purchased from the Jackson Laboratory. Animals were housed in temperature- (21–23 °C) and humidity- (30–70%) controlled rooms with 12:12 light/dark cycles. The intracranial xenograft tumor models were established as we described previously56. After tumor implantation, mice were monitored for recording survival. Mice with neurological deficits or moribund appearance were killed.
Cell culture
HMC3 microglia (ATCC, #CRL-3304), 293T cells (ATCC, #CRL-3216) and CT2A cells68, SIM-A9 microglia (ATCC, #CRL-3265), and SB28 (from Dr. Hideho Okada at UCSF) and GL261 cells (the National Cancer Institute) were cultured in Eagle’s Minimum Essential Medium (ATCC, #30–2003), Dulbecco’s Modified Eagle Medium (Gibco, #11995–065), Dulbecco’s Modified Eagle Medium–Ham’s F12 medium (Gibco, #10565–018) and RPMI 1640 medium (RPMI; Gibco, #11875093), respectively, containing 10% fetal bovine serum (FBS, Fisher Scientific, #16140071) and 1:100 antibiotic–antimycotic (Gibco, #15140–122). For stemness maintenance, CT2A cells were cultured in neural stem cell proliferation media (Millipore, #SCM005) containing 20 ng ml−1 epidermal growth factor (PeproTech, #AF-100–15) and basic fibroblast growth factor (PeproTech, #100–18B). Similarly, human and mouse GSCs were cultured in neural stem cell proliferation media containing 20 ng ml−1 epidermal growth factor and basic fibroblast growth factor. Human GSCs were gifted by Dr. Frederick F. Lang from the Brain Tumor Center (The University of Texas MD Anderson Cancer Center). QPP7 and 005 GSCs were provided by Dr. Jian Hu (The University of Texas MD Anderson Cancer Center) and Dr. Samuel D. Rabkin (Massachusetts General Hospital), respectively. JURKAT cells (from Dr. Colby Thaxton at Northwestern) were cultured in human plasma-like medium (Thermo Fisher Scientific, #A4899101) containing 10% FBS and 1:100 antibiotic–antimycotic. All cells were confirmed to be mycoplasma-free and were maintained at 37 °C and 5% CO2. CM was collected from number-matched control, TFPI2 shRNA knockdown or TFPI2-overexpressing cells after culturing for another 24 h in FBS- and growth factor-free culture medium.
Isolation of primary CD8+ T cells and microglia
Primary mouse CD8+ T cells were isolated from the spleens of C57BL/6 mice using a CD8+ T-cell isolation kit (Miltenyi Biotec, #130-104-075) according to the manufacturer’s instructions. Cells were expanded and activated using 30 U ml−1 mouse IL-2 (Miltenyi Biotec, #130-120-662) and Dynabeads mouse T-activator CD3/CD28 (Gibco, #11456D) before analysis and coculture studies. Adult human primary microglia (PrhMG) were isolated as we described previously37. Cells were labeled with CD11b magnetic beads, and MACS method was used to select CD11b+ cells. For isolation of mouse microglia, brains of neonate mice were homogenized in 10 ml HBSS on ice. Brain-derived microglia were then purified using 30/70 Percoll (GE Healthcare, #17-0891-01) with gradient separation for 30 min at 4 °C. The purity of microglia was determined using flow cytometry analysis for detecting CX3CR1-positive cells.
Plasmids, viral transfections and cloning
shRNAs targeting human TFPI2, mouse Tfpi2 and mouse Itgav in the pLKO.1 vector (Sigma, #SHC001) were used. Lentiviral particles were generated as we described previously56. The following mouse and human shRNA sequences (TFPI2: #1 TRCN0000373908, #2 TRCN0000373822 and #3 TRCN0000072725; Tfpi2: #2 TRCN0000271824 and #3 TRCN0000271717; and Itgav: #2 TRCN0000066588, #5 TRCN0000066590 and #6 TRCN0000066591) were selected following validation. For rescue experiments, TFPI2 shRNA knockdown QPP7 cells were transfected with a human TFPI2 construct that is resistant to TFPI2 shRNAs.
CRISPR KO of TFPI2 in human GSC272 cells was produced from the transfection of sgRNA (GenScript, #SC1969) targeting the TFPI2 sequence (TTCTCCGTTACTACTACGAC) and Cas9 Nuclease (GenScript, #Z03621–0.5). In total, 50 pmol of Cas9 Nuclease and 100 pmol of sgRNA were incubated in Opti-MEM medium (Gibco, #31985088) for 10 min at room temperature before combining with additional Opti-MEM medium containing Lipofectamine CRISPRMAX (Invitrogen, #CMAX00001). GSC272 cells were incubated with the cocktail of sgRNA, Cas9 nuclease and transfection reagent for 48 h. Transfected cells were expanded in culture, and GFP+ cells were sorted using FACS ARIA 4-Laser Sorter. CRISPR KO efficiency was verified using immunoblot to assess TFPI2 expression.
For overexpression, GSCs were transfected with a TFPI2 overexpression plasmid generated via a cloning methodology. In brief, a pCMV6-Entry vector containing the TFPI2 (Myc-DDK-tagged) sequence (Origene, #RC202760) and a pLenti-C-mGFP Lentiviral Gene Expression Vector (Origene, #PS100071) were digested using restriction enzymes corresponding to the open reading frames. Restriction enzymes Mlul (Promega, # R6381) and Sgfl (Promega, #R7103) were mixed with Restriction Digest Buffer C to enhance digestion efficiency. Following digestion, each mixture was incubated with antarctic phosphatase for 3 h to prevent self-ligation. T4 DNA ligase (Thermo Scientific, #B69) was included in a ligation mixture to induce the ligation of the TFPI2 insert into the pLenti-C-mGFP Lentiviral Gene Expression Vector. Ligation mixture was transformed into high-efficiency chemically competent Escherichia coli cells (Thermo Scientific, #C737303) and recovered in Lysogenia broth (LB, Fisher BioReagents, # BP9723). Post-recovery, LB containing E. coli transformants were plated on LB selection plates containing 34 μg/ml chloramphenicol (Fisher BioReagents, #BP904) for selection of clones containing the TFPI2 ORF in the pLenti-C-mGFP Lentiviral Gene Expression Vector. After 16 h of incubation at 37 °C, three colonies were picked from the selection plates for inoculation in LB broth supplemented with 34 μg ml−1 chloramphenicol to maintain selection, and then further purified for plasmid DNA using a QIAprep Spin Miniprep Kit (Qiagen, #27106). Purified plasmids were then transfected into GSCs using lentiviral transfection methodology as previously described10.
Migration assays
For transwell migration assay, HMC3 microglia, SIM-A9 microglia and PrhMG were suspended in serum-free culture medium and seeded into transwell 24-well plates with permeable polycarbonate membrane inserts (5.0 μm, Corning, #07-200-149). GSC CM or basal cell culture medium with TFPI2 recombinant protein (BioVision, #7481–10) was added to the remaining receiver wells. The STAT6 inhibitor AS1517499 (Selleck Chemicals, #S8685) was used to study the role of STAT6 in microglia migration. The CD51 inhibitor MK-0429 (MedChemExpress, #HY-15102) was used to study the role of CD51 in TFPI2-induced microglia migration. After 10 h, the migrated microglia were fixed and stained with crystal violet (Sigma, #C-3886). The number of transferred cells was counted using ImageJ Version 1.53t (NIH).
The scratch-wound assay was performed as we reported previously69. For tracking single cell movement, microglia were cultured in 96-well plates (Corning, #3599) with different treatments. Incucyte Live Cell Analysis System was used to record cell movement over time. The time-lapse images were reconstructed and analyzed using TrackMate70,71. The speed of each individual cell was calculated by processing images with LoG detector and Linear Assignment Problem tracker.
Immunoblotting
The protein expression in cells was determined by western blotting analysis as we described previously56. Primary antibodies (1:1,000 dilution) against the following proteins were used: TFPI2 (Abcam, #ab186747), CD133 (Abcam, #ab19898), SOX2 (Abcam, #ab97959), cleaved caspase-3 (Cell Signaling Technology, #9661), P-STAT6 (Abcam, #ab28829), STAT6 (Cell Signaling Technology, #9362), P-STAT3 (Cell Signaling Technology, #9145), STAT3 (Cell Signaling Technology, #9139), P-AKT (Cell Signaling Technology, #4058S), AKT (Bio-Rad, #VMA00253), P-JNK (Cell Signaling Technology, #4668S), JNK (Cell Signaling Technology, #9252S), PLCγ1 (Cell Signaling Technology, #5690S), P-PLCγ1 (Cell Signaling Technology, #8713S), PKCζ (Cell Signaling Technology, #9368S), P-PKCζ (Cell Signaling Technology, #9378S), Integrin αV/CD51 (Cell Signaling Technology, #4711S) and Actin (Cell Signaling Technology, #3700). HRP-linked, anti-mouse (Cell Signaling Technology, #7076) or anti-rabbit (Cell Signaling Technology, #7074S) secondary antibodies (1:5,000 dilution) were used. The signaling was imaged under ChemiDoc Touch Imaging System (Bio-Rad). Each assay was repeated at least three times.
Co-IP
Co-immunoprecipitation (co-IP) was performed using Invitrogen Dynabeads Protein G Immunoprecipitation Kit (Thermo Fisher Scientific, #10007D). Briefly, microglia were treated with 20 ng ml−1 TFPI2 recombinant protein for 1 h, and then RIPA lysis buffer was used to extract protein. Magnetic beads (1.5 mg) were separated from the solution using a magnetic stand. Ten micrograms TFPI2 antibody (Abcam, #ab186747), CD51 antibody (Cell Signaling Technology, #4711S) or rabbit IgG (isotype control, Cell Signaling Technology, #2729) was prepared in 200 μl of antibody binding and washing buffer. Antibody or IgG was incubated with magnetic beads for 10 min at room temperature with rotation. After removing the supernatant, antibody/IgG-conjugated magnetic beads were incubated with cell lysis buffer for 2 h at room temperature with rotation to immunoprecipitate TFPI2- or CD51-interacting antigens. The magnetic bead–antibody–antigen complex was washed three times and then eluted with the elution buffer. The complex was loaded into 4–12% SurePAGE gels to evaluate target protein expression after co-IP.
IP–MS assay
PrhMG were incubated with 20 ng ml−1 TFPI2 recombinant protein for 1 h to allow the binding between TFPI2 and its potential receptors. After incubation, cell membrane-associated proteins were extracted using the Mem-PER Plus Membrane Protein Extraction Kit (Thermo Fisher Scientific, #89842). A total of 50 μg cell membrane lysate was incubated with 10 μl TFPI2 antibody (Abcam, #ab186747) or IgG control (Cell Signaling Technology, #2729) for 12 h with gentle rotation at 4 °C. A total of 100 μl Protein A/G PLUS-Agarose (#sc-2003, Santa Cruz Biotechnology) was mixed with each sample on ice. The lysate bead mixture was incubated at 4 °C under rotary agitation for 4 h. The tubes were centrifuged to remove supernatant. The beads were then washed in lysis buffer three times to remove nonspecific binding. After removing wash buffer, the complex was washed with pre-urea wash buffer (50 mM Tris pH 8.5, 1 mM egtazic acid and 75 mM KCl) to remove all residual supernatant. The protein was eluted with the elution buffer (20 mM Tris pH 7.5, and 100 mM NaCl) and sent to the Northwestern Proteomics Core for protein identification. Samples were in-gel digested (trypsin) and subject to mass spectrometry (MS) analysis. IP pulled-down samples were analyzed by liquid chromatography (LS)–tandem mass spectrometry (MS/MS) using a Dionex UltiMate 3000 Rapid Separation nanoLC coupled to the Orbitrap Elite Mass Spectrometer (Thermo Fisher Scientific). Raw files were searched against a human database and submitted to Mascot and Scaffold searches. Protein probabilities were assigned by the Protein Prophet algorithm. Proteins that contained similar peptides and could not be differentiated on the basis of tandem MS analysis alone were grouped to satisfy the principles of parsimony.
Calcium mobilization assay
Calcium mobilization in microglia was determined using the Fluo-4 Direct Calcium Assay Kit (Thermo Fisher Scientific, #F10472) according to the manufacturer’s instructions. Fluorescence kinetic was measured in a microplate reader using instrument settings for excitation at 494 nm and emission at 516 nm.
RT–qPCR
Cells were pelleted, and RNA was isolated with the RNeasy Mini Kit (Qiagen, #74106). RNA was quantified by NanoDrop spectrophotometers and then reverse-transcribed into cDNA by using the All-In-One 5X RT MasterMix (Applied Biological Materials, #G592) in T100 Thermal Cycler (Bio-Rad). RT–qPCR was performed using SYBR Green PCR Master Mix (Bio-Rad, #1725275) in CFX Connect Real-Time PCR Detection System (Bio-Rad). The expression of each gene was normalized to housekeeping genes. RT–qPCR primers are listed in Supplementary Table 2.
Immunohistochemistry and immunofluorescence
Immunohistochemistry and immunofluorescence were performed using a standard protocol as we previously described56,72. Sections were incubated with primary antibody (1:100 dilution) for 1 h at room temperature and then overnight at 4 °C. Sections were subjected to corresponding secondary antibodies (1:100 dilution) or rabbit-on-rodent HRP-polymer (Biocare, #RMR 622) for 1 h at room temperature. For immunofluorescence, cell nucleus was counterstained with 4′,6-diamidino-2-phenylindole/anti-fade mounting medium (Vector Laboratories, #H-1200–10). Protein signal was captured using Nikon AX/AX R Confocal Microscope System in the Center for Advanced Microscopy at Northwestern University. The relative intensity of protein was determined by ImageJ. For immunohistochemistry, tissues were incubated with DAB Quanto (Epredia, #TA125QHDX). The nucleus was then stained by hematoxylin. After dehydration, the images were captured by using EVOS Cell Imaging System. ImageJ Version 1.53t with IHC profiler plug-in was used for scoring positive signals73.
Hematoxylin and eosin staining
Hematoxylin and eosin staining was performed using the staining kit (Abcam, #ab245880) following a standard protocol. Briefly, the paraffin sections were baked at 65 °C for 2 h before staining. Sections were subjected to xylene and ethanol for deparaffinization and rehydration and then incubated with hematoxylin, Mayer’s (Lillie’s Modification) for 5 min. Slides were then incubated with the Bluing Reagent and Eosin Y Solution (Modified Alcoholic) for 15 s and 3 min, respectively. After washing, slides were dehydrated in three changes of absolute alcohol. The images of tissue sections were captured by TissueFAXS in the Center for Advanced Microscopy at Northwestern University.
Human phospho-kinase array
After starvation for 24 h followed by TFPI2 recombinant protein treatment for 1 h, HMC3 cell lysate was prepared using a lysis buffer with 10 μg ml−1 aprotinin, 10 μg ml−1 leupeptin and 10 μg ml−1 pepstatin. Phosphorylation profiles of kinases were determined using Proteome Profiler Human Phospho-Kinase Array Kit (Bio-Techne, #ARY003C) as the instruction manual. Signal was detected under ChemiDoc Touch Imaging System (Bio-Rad).
Brain tumor and spleen cell isolation
At the endpoint of experiments, mice were killed to collect their brains and spleens. Immune cells in the brain and spleen were isolated as we previously described9. Brain/spleen-derived immune cells were purified using 30/70 Percoll (GE Healthcare, #17–0891-01) with gradient separation for 30 min at 4 °C. Spleen cells were treated with 1 ml ACK buffer (Thermo Fisher, #A1049201) for 5 min and then filtered through 70 μm strainers (Thermo Fisher, #08–771-2).
T-cell-mediated cytotoxicity assay
T-cell-mediated cytotoxicity was determined by Cytotox96 nonradioactive cytotoxicity assay kit (Promega, #G1780) based on the release of lactate dehydrogenase. Briefly, after expansion and activation, mouse primary CD8+ T cells or human JURKAT T cells were collected. About 105 T cells were cocultured with SIM-A9 microglia or PrhMG (1:1 ratio) treated with TFPI2 recombinant protein in the presence or absence of STAT6 inhibitor AS1517499 for 24 h. T cells (effector cells) were then collected for further coculturing with CT2A cells or GSC272 cells (target cells) at different ratios for 5 h. T-cell-mediated cytotoxicity on GSCs was assessed according to the manufacturer’s instructions. Optical density (OD) was measured by a Biotek Synergy 2 SL Microplate Reader. The cytotoxicity was calculated by the following formula: (experimental OD value – effector spontaneous OD value – target spontaneous OD value)/(target maximum OD value – target spontaneous OD value).
Apoptosis analysis
Cell apoptosis was evaluated using Apotracker Green (BioLegend, #427402) as described previously74. Briefly, cells were collected and stained with Apotracker (1:10 dilution). Propidium iodide solution (BioLegend, # 421301) was added for labeling late apoptotic and necrotic cells. Fluorescein isothiocyanate (FITC) and propidium iodide signals were analyzed in BD FACSymphony flow cytometer. The gating strategy for apoptosis analysis is shown in Supplementary Fig. 8.
Flow cytometry
For membrane protein staining, single-cell suspensions were incubated with fixable viability dye (Invitrogen, #5211229035) on ice for 10 min. Following washing with phosphate-buffered saline (PBS), cells were incubated with the anti-CD16/CD32 cocktail (BioLegend, #103132) in 2% bovine serum albumin PBS to block Fc receptors for 30 min on ice. Different antibody combinations, including Percp/Cy5.5 anti-mouse CD45 (BioLegend, #103132), AF488 anti-mouse CD3 (BioLegend, #100210), BV421 anti-mouse CX3CR1 (BD Bioscience, #567531), BUV395 anti-mouse CD4 (BD Bioscience, #740208), BV711 anti-mouse CD8 (BioLegend, #100747), PE/Cy7 anti-mouse/human CD11b (BioLegend, #101216), PE anti-mouse CD68 (BD Bioscience, #566386), PE anti-mouse PD1 (BioLegend, #135205), AF647 anti-mouse CD206 (BD Bioscience, #565250), AF647 anti-human CD206 (BioLegend, #321116), PE anti-human CD69 (BioLegend, #310906), PE/Cy7 anti-mouse CD69 (BioLegend, #104512), and BV650 anti-human HLA-DR (BioLegend, #307650) were added to cell suspensions (1:100 dilution) for 30 min on ice. CD51 (Cell Signaling Technology, #4711S) was added to cell suspension with corresponding antibody combinations for 30 min on ice, followed by incubation of goat anti-rabbit IgG cross-adsorbed secondary antibody (AF594) for 30 min. After washing with PBS, cells were fixed in fixation buffer (BioLegend, #420801) overnight. Samples were read in BD FACSymphony flow cytometer and analyzed in FlowJo v10.8.1.
Intracellular protein staining was performed separately or followed by cell surface marker staining. Cells were fixed in fixation buffer at room temperature for 20 min. After washing in permeabilization buffer twice, cells were incubated with AF700 anti-human IFNγ (BioLegend, #502520) or FITC anti-human/mouse ARG1 (BioLegend, #IC5868F) for 20 min at room temperature. For staining with Phospho-STAT6, samples were fixed in a fixation buffer at room temperature for 15 min. Cells were then stained with a fixable viability dye and permeabilized in 90% cold methanol for 15 min on ice. After blocking Fc receptors, cells were resuspended with P-STAT6 primary antibody for 1 h on ice, and then incubated with goat anti-rabbit IgG cross-adsorbed secondary antibody (AF594). Cell suspensions were then rewashed and read in BD FACSymphony flow cytometer. The gating strategies for flow cytometry analyses are shown in Supplementary Figs. 9–13.
Proliferation analysis
Cell proliferation was assessed using the CellTrace carboxyfluorescein succinimidyl ester (CFSE) Cell Proliferation Kit (Invitrogen, #C34554). Briefly, 106 cells were collected and incubated with CFSE working solution (1:1,000) for 20 min at 37 °C. The staining was stopped by adding complete cell culture media. After washing, cells were continuously cultured for 5 days in the dark and used for flow cytometry analysis. The percentage of CFSE positive peaks over the undivided peak (generation 0) was analyzed using the ImageJ software.
Tumorsphere formation assay
The tumorsphere formation assay was performed as previously described75. Briefly, GSCs were treated with different concentrations of recombinant TFPI2 protein for 24 h with or without inhibiton of JNK or STAT3, or modified with TFPI21 KO, knockdown or overexpression. Cells were seeded into a 96-well plate at 100 cells per well and tumorsphere numbers in each well were imaged and quantified after 10 days.
Enzyme-linked immunoassay
The levels of TFPI2 in human plasma were measured by enzyme-linked immunoassay (ELISA) using the commercial TFPI2 kit (MyBioSource, #MBS2507217) following the manufacturer’s instructions. OD value was measured by a Biotek Synergy 2 SL Microplate Reader.
Bulk RNA-seq and scRNA-seq data analysis
Extracted RNA from shC and shTFPI2 GSC272 cells were processed to Oligo-dT-based library and analyzed using a NovaSeq 6000 instrument to generate RNA-seq dataset at The University of Chicago Functional Genomics (RRID: SCR_019196). Raw FASTQ data from two flowcells per sample was uploaded to Galaxy server76. Trimmomatic was performed to read and trim paired-end reads. HISAT2 program was used to align the data where hg38 was selected as the reference genome. Samtools merge was performed on BAM files to merge data from different flowcells. Gene expression was then determined by FeatureCounts. Limma was used to calculate differential expression (DE) genes and generate DE table. The upregulated and downregulated pathways were determined by GSEA analysis. scRNA-seq data of GSE131928 (ref. 29) was used for analyzing the expression of TFPI2 and stemness signature in malignant cells, and the scRNA-seq data of EGAS00001004422 (ref. 77) and GSE131928 (ref. 29) was used for analyzing microglia signature in GBM patient tumors with TFPI2 high versus low in GBM cells.
Computational analysis of human GBM datasets
For bioinformatics analyses of human GBM patient tumor data, we downloaded the microarray gene expression and survival data of TCGA dataset or other available datasets from GlioVis (http://gliovis.bioinfo.cnio.es/). The analyses of gene expression, signature expression, correlation, survival and the GSEA of interesting gene signatures in patients with IDH-WT GBM were performed as we reported previously9,10,56.
Patient samples
GBM patient peripheral blood plasma samples (n = 55) were collected from the Northwestern Central Nervous System Tissue Bank. Patient tumor samples (n = 60) from surgically resected IDH-WT GBMs were collected at the Northwestern Central Nervous System Tissue Bank under the institutional review board protocol STU00095863. All patients were diagnosed according to the World Health Organization diagnostic criteria by neuropathologist Dr. Craig Horbinski. Detailed patient information is provided in Supplementary Table 3. Control plasma samples (n = 6) from healthy human blood were purchased from Solomonpark (#4345), which are commercially available and de-identified. According to The George Washington University Institutional Review Board and based on the guidelines from the Office of Human Research Protection, the conducted research meets the criteria for exemption #4 (45 CFR 46.101(b) Categories of Exempt Human Subjects Research) and does not constitute human research.
Statistical analysis
Statistical analysis was conducted using GraphPad Prism 9 (GraphPad Software). Data distribution was assumed to be normal, but this was not formally tested. Data collection and analysis were not performed blind to the conditions of the experiments. We did not perform randomization of study participants or samples within each group because not relevant/needed for this study. No statistical methods were used to predetermine sample sizes but our sample sizes are similar to those reported in previous publications9,10,56. In vitro and in vivo measurement data were presented as mean ± standard error of the mean (s.e.m.) or standard deviation (s.d.). Comparison between two groups was evaluated using Student’s t-test, and multiple comparisons among groups were evaluated using a one-way analysis of variance (ANOVA) test in Tukey’s method. The survival analysis for animal models was performed using log-rank (Mantel–Cox) test. Correlation analysis was performed using the Pearson test to determine the R and P values. Differences with a minimum of P < 0.05 were indicated as statistically significant. No data were excluded from the analysis.
Extended Data
Extended Data Fig. 1. TFPI2 promotes GSC self-renewal.
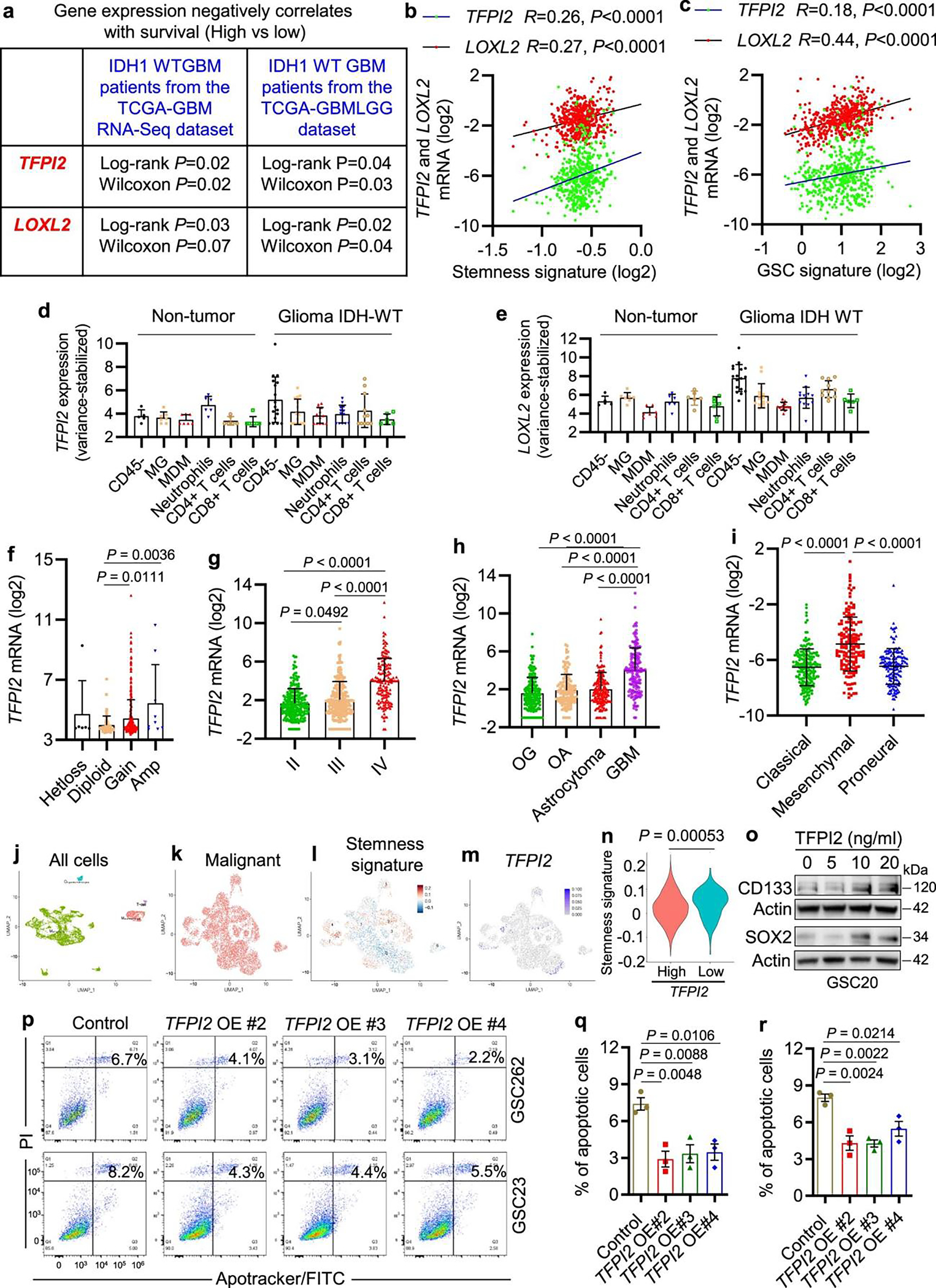
a, Correlation between TFPI2 or LOXL2 expression and IDH1-WT GBM survival in TCGA datasets. P values are shown. b, c, Correlation between stemness (b) or GSC signature (c) and TFPI2 or LOXL2 expression in TCGA GBM dataset. R and P values were determined by Pearson correlation. d, e, Expression of TFPI2 (d) and LOXL2 (e) in CD45− cells and immune cells of non-tumor tissues and GBM tumors. n = 5 and 17 for CD45− cells, 8 and 15 for microglia (MG), 7 and 13 for macrophages (MDM), 6 and 13 for neutrophils, 6 and 11 for CD4+ T cells, as well as 7 and 6 for CD8+ T cells of non-tumor tissues and GBM, respectively. f, TFPI2 copy number correlates with TFPI2 in HG-U133A TCGA GBM tumors. n = 6, 88, 405 and 9 for Hetloss, Diploid, Gain and Amp, respectively. g, TFPI2 expression in glioma tumors at different stages. n = 256, 244, and 150 for stage II, III and IV, respectively. h, TFPI2 expression in different types of brain tumors. n = 191, 130, 194 and 152 for oligodendroglioma (OG), oligoastrocytoma (OA), astrocytoma and GBM, respectively. i, TFPI2 expression in different subtypes of GBM tumors. n = 182, 156 and 151 for classical, mesenchymal and proneural, respectively. j, k, High-resolution UMAP dimensional reduction of all cells (j) and malignant cells (k) from GBM patient tumors. l, m, UMAP dimensional reduction of the basis of the expression pattern of hallmark stemness signature (l) and TFPI2 (m) in malignant cells. n, Quantification of hallmark stemness signature in TFPI2-low and TFPI2-high tumors. o, Immunoblots for CD133 and SOX2 in cell lysates from GSC20 treated with TFPI2 protein at indicated concentrations. p–r, Representative (p) and quantification (q,r) of flow cytometry analysis of apoptosis in GSC262 and GSC23. n = 3 biological replicates. Error bars indicate mean ± SD (f–i) or SEM (q, r). One-way ANOVA test (f–i, q, r) and two-tailed Student’s t-test (n).
Extended Data Fig. 2. TFPI2 regulates GSC self-renewal, proliferation, and apoptosis.
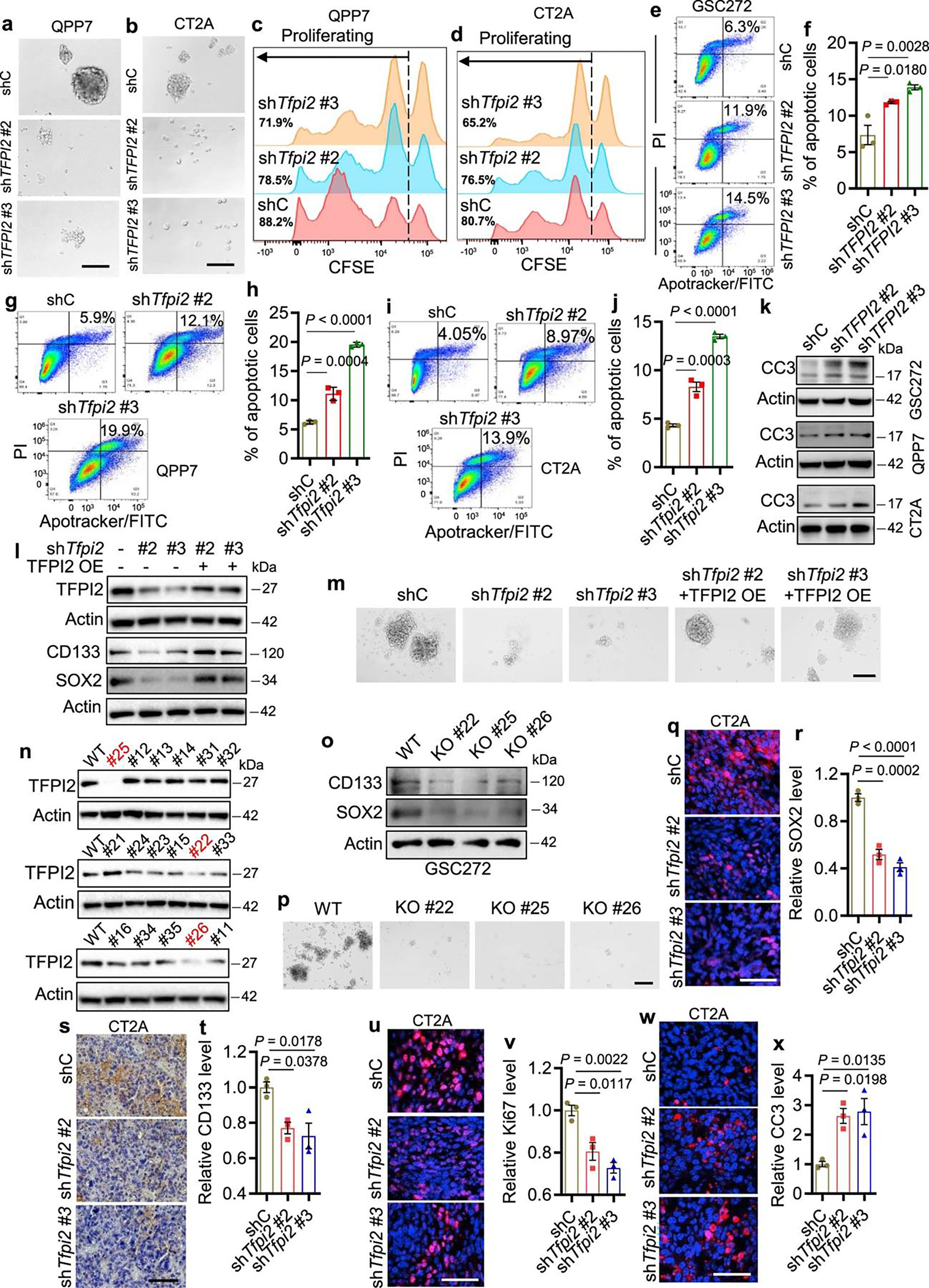
a, b, Representative of tumorspheres of QPP7 (a) and CT2A (b) cells expressing control shRNA (shC) and Tfpi2 shRNAs (shTfpi2). Scale bar, 200 μm. c, d, Representative of proliferation in shC and shTfpi2 QPP7 (c) and CT2A (d) cells. e–j, Representative and quantification of flow cytometry for apoptosis in GSC272 (e, f), QPP7 GSCs (g, h) and CT2A cells (i, j) expressing shC and shTFPI2. k, Immunoblots for cleaved caspase 3 (CC3) in cell lysates from GSC272, QPP7, and CT2A cells expressing shC and shTFPI2. l, Immunoblots for CD133 and SOX2 in cell lysates from QPP7 GSCs expressing shTfpi2 with or without reexpression of shRNA-resistant TFPI2 cDNA (OE). m, Representative of tumorspheres in QPP7 GSCs expressing shTfpi2 with or without TFPI2 OE. Scale bar, 400 μm. n, Immunoblots for TFPI2 in cell lysates from TFPI2 wild-type (WT) and different clones of CRISPR knockout (KO) GSC272. o, Immunoblots for CD133 and SOX2 in cell lysates from TFPI2-WT and TFPI2-GSC272. p, Representative of tumorspheres in TFPI2-WT and TFPI2-KO GSC272. Scale bar, 400 μm. q, r, Representative (q) and quantification (r) of immunofluorescence staining of SOX2 in size-matched tumors from C57BL/6 mice intracranially implanted with shC and shTfpi2 CT2A cells. Scale bar, 50 μm. s, t, Representative (s) and quantification (t) of immunochemistry staining of CD133 in tumors from C57BL/6 mice intracranially implanted with shC and shTfpi2 CT2A cells. Scale bar, 200 μm. u–x, Representative and quantification of immunofluorescence staining of Ki67 (u,v) and CC3 (w,x) in tumors from C57BL/6 mice intracranially implanted with shC and shTfpi2 CT2A cells. Scale bar, 50 μm. In (f, h, j, r, t, v, x), n = 3 biological replicates. Error bars indicate mean ± SEM. One-way ANOVA test.
Extended Data Fig. 3. TFPI2 regulates GSC proliferation and apoptosis via JNK and STAT3 pathways.
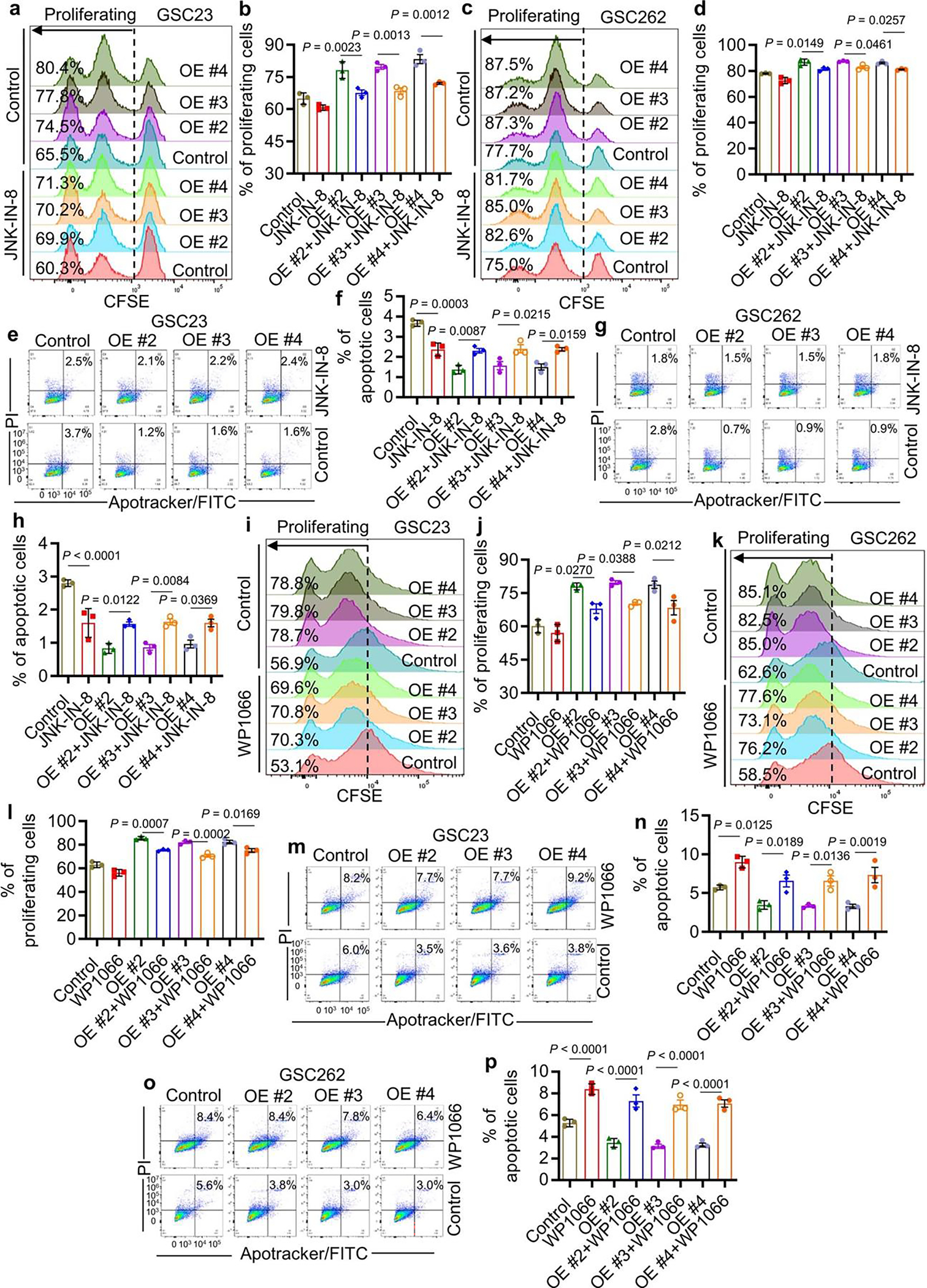
a, b, Representative (a) and quantification (b) of proliferation in GSC23 expressing control and TFPI2 overexpression (OE) plasmid treated with or without JNK inhibitor JNK-IN-8 (10 nM). n = 3 biological replicates. c, d, Representative (c) and quantification (d) of proliferation in GSC262 expressing control and TFPI2 OE plasmid treated with or without JNK-IN-8 (10 nM). n = 3 biological replicates. e, f, Representative (e) and quantification (f) of apoptosis in GSC23 expressing control and TFPI2 OE plasmid treated with or without JNK-IN-8 (10 nM). n = 3 biological replicates. g, h, Representative (g) and quantification (h) of apoptosis in GSC262 expressing control and TFPI2 OE plasmid treated with or without JNK-IN-8 (10 nM). n = 3 biological replicates. i, j, Representative (i) and quantification (j) of proliferation in GSC23 expressing control and TFPI2 OE plasmid treated with or without STAT3 inhibitor WP1066 (20 nM). n = 3 biological replicates. k, l, Representative (k) and quantification (l) of proliferation in GSC262 expressing control and TFPI2 OE plasmid treated with or without WP1066 (20 nM). n = 3 biological replicates. m, n, Representative (m) and quantification (n) of apoptosis in GSC23 expressing control and TFPI2 OE plasmid treated with or without WP1066 (20 nM). n = 3 biological replicates. o, p, Representative (o) and quantification (p) of apoptosis in GSC262 expressing control and TFPI2 OE plasmid treated with or without WP1066 (20 nM). n = 3 biological replicates. Error bars indicate mean ± SEM. One-way ANOVA test.
Extended Data Fig. 4. TFPI2 promotes microglia migration via activating STAT6 signaling.

a, Amplification pattern of TFPI2 and CLOCK in GBM and low-grade glioma (LGG) patient tumors. b, GSEA analysis shows the top 10 enriched hallmark pathways in TFPI2-high GBM patient tumors compared to TFPI2-low tumors. Blue highlighted pathways relate to immune response. c, Representative of transwell migration of PrhMG following stimulation with conditioned media (CM) from GSC262 or GSC23 expressing control and TFPI2 overexpression (OE) plasmid. Scale bar, 400 μm. d, Representative of transwell migration of PrhMG, HMC3 and SIM-A9 microglia following stimulation with CM from GSC272 (for PrhMG and HMC3) or QPP7 cells (for SIM-A9) expressing control shRNA (shC) and TFPI2 shRNAs (shTFPI2). Scale bars, 400 μm for PrhMG; 200 μm for HMC3 and SIM-A9. e, f, Representative (e) and quantification (f) of flow cytometry for the percentage of CD45lowCD11b+ and CD45highCD11b+ cells in size matched tumors from C57BL/6 mice intracranially implanted with shC and shTfpi2 CT2A cells. n = 4 and 3 biological replicates for shC and shTfpi2, respectively. g, h, Representative (g) and quantification (h) of flow cytometry for the percentage of CD45highCD11b+CD68+ macrophages in size matched tumors from C57BL/6 mice intracranially implanted with shC and shTfpi2 CT2A cells. n = 4 and 3 biological replicates for shC and shTfpi2, respectively. i, Correlation between microglia signature and STAT6, AKT1, AKT2 or AKT3 in TCGA GBM dataset. R and P values were determined by Pearson correlation. j, Representative of transwell migration of mouse primary microglia isolated from wild-type (WT) and STAT6 knockout (KO) mice treated with or without TFPI2 recombinant protein (20 ng/ml). Scale bar, 200 μm. k, Representative of transwell migration of PrhMG, HMC3 and SIM-A9 microglia following stimulation with TFPI2 protein (20 ng/ml) in the presence or absence of STAT6 inhibitor AS1517499 at indicated concentrations. Scale bars, 400 μm for PrhMG; 200 μm for HMC3 and SIM-A9. Error bars indicate mean ± SEM. One-way ANOVA test.
Extended Data Fig. 5. TFPI2-STAT6 axis regulates microglia polarization.

a, Representative of flow cytometry for CD206 in PrhMG treated with conditioned media (CM) from GSC23 expressing control and TFPI2 overexpression (OE) plasmid. b, Representative of flow cytometry for CD206 in HMC3 microglia treated with CM from GSC272 expressing shRNA control (shC) and TFPI2 shRNAs (shTFPI2). c, Representative of flow cytometry for CD206 in SIM-A9 microglia treated with CM from QPP7 GSCs expressing shC and shTfpi2 with or without reexpression of shRNA-resistant TFPI2 cDNA (OE). d, e, Representative (d) and quantification (e) of flow cytometry for the percentage of CD45hiCD11b+CD68+CD206+ macrophages in size matched tumors from C57BL/6 mice intracranially implanted with shC and shTfpi2 CT2A cells. n = 4 and 3 for shC and shTfpi2, respectively. f–i, Immunofluorescence and quantification of CX3CR1+CD206+ (f,g) and CX3CR1+CD163+ (h,i) cells in tumors from C57BL/6 mice intracranially implanted with shC and shTfpi2 CT2A cells. Scale bar, 25 μm. n = 3 (g) or 6 (i) biological replicates. j–m, Immunofluorescence and quantification of CX3CR1+CD206+ (j,k) and CX3CR1+CD163+ (l,m) cells in tumors from nude mice intracranially implanted with shC and shTFPI2 GSC272. Scale bar, 25 μm. n = 6 biological replicates. n, Representative of CD206 positive microglia isolated from wild-type (WT) and STAT6 knockout (KO) mice and treated with or without TFPI2 recombinant protein (20 ng/ml). o, p, Representative of flow cytometry for CD206 (o) and ARG1 (p) in SIM-A9 microglia treated with or without TFPI2 protein (20 ng/ml) and STAT6 inhibitor (STAT6i) AS1517499 (25 nM). q, r, Representative of flow cytometry for CD206 (q) and ARG1 (r) in HMC3 microglia treated with or without TFPI2 protein (20 ng/ml) and AS1517499 (25 nM). s, Relative mRNA expression of IL6, TNFA, LFNG, and HLA-DR in HMC3 microglia treated with or without TFPI2 protein (20 ng/ml) and AS1517499 (25 nM). n = 3 biological replicates. Error bars indicate mean ± SEM. A one-way ANOVA followed by a Tukey test was performed to compare more than two groups.
Extended Data Fig. 6. CD51 is a receptor for TFPI2 activities on microglia.
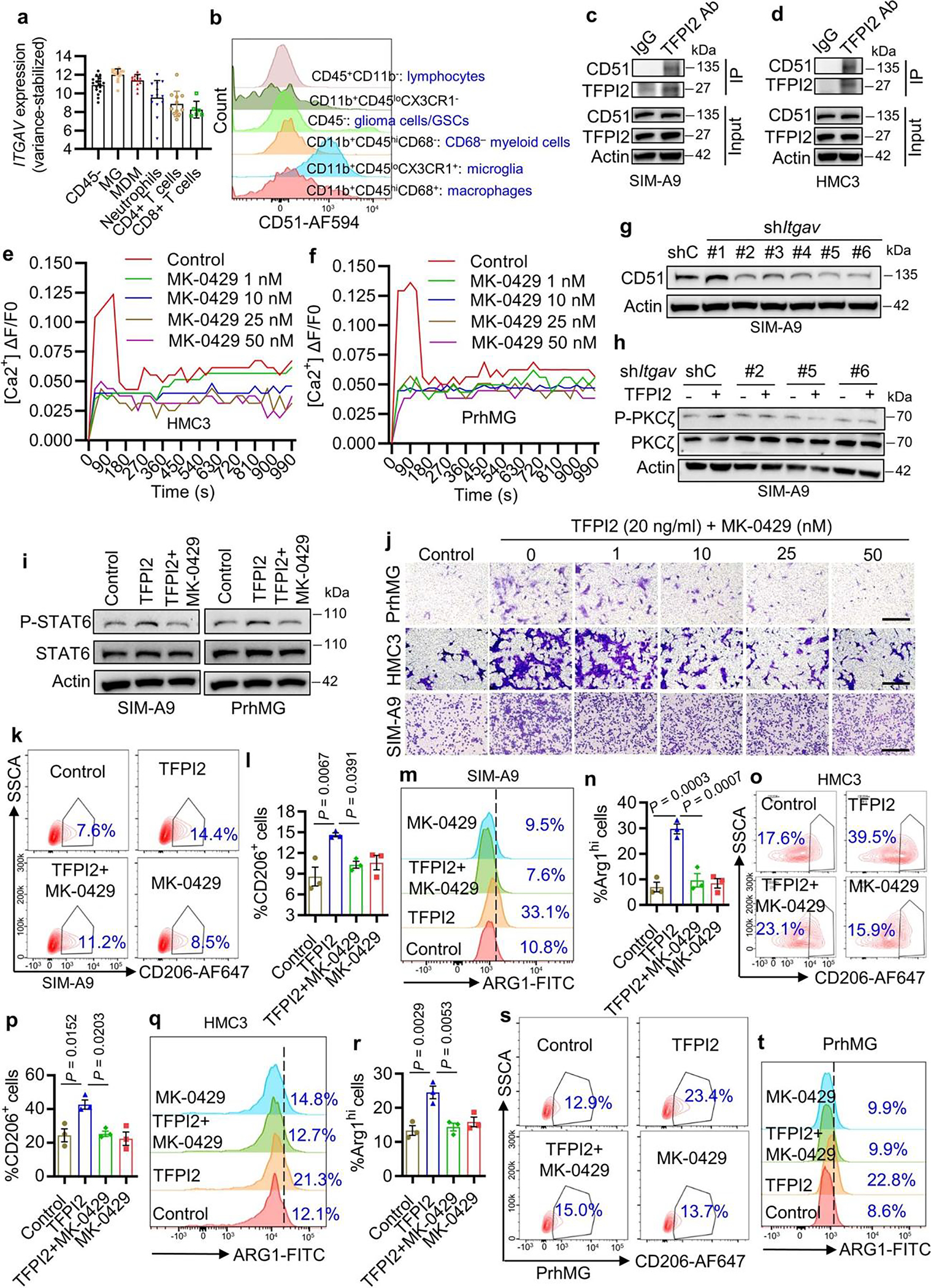
a, CD51 expression level in human GBM samples from the TIME dataset. n = 17, 15, 13, 13, 11, and 6 for CD45− cells, microglia (MG), macrophages (MDM), neutrophils, CD4+ T cells and CD8+ T cells, respectively. b, CD51 expression in different cell populations in mouse CT2A tumor tissues. c, d, Immunoprecipitation with TFPI2 antibody (TFPI2 Ab) and immunoblotting for the interaction between TFPI2 and CD51 in SIM-A9 (c) and HMC3 (d) microglia. e, f, Calcium mobilization triggered by TFPI2 protein in HMC3 microglia (e) and PrhMG (f) treated with or without CD51 inhibitor MK-0429 at indicated concentrations. g, Immunoblots for CD51 in cell lysates of SIM-A9 microglia expressing shRNA control (shC) and Itgav shRNAs (shItgav). h, Immunoblots for P-PKCζ and PKCζ in cell lysates of shC and shItgav SIM-A9 microglia treated with or without TFPI2 protein. i, Immunoblots for P-STAT6 and STAT6 in cell lysates from SIM-A9 microglia and PrhMG treated with TFPI2 protein and MK-0429. j, Representative of transwell migration of PrhMG human HMC3, and mouse SIM-A9 microglia treated with or without TFPI2 protein and MK-0429 at indicated concentrations. Scale bars, 200 μm for HMC3 and SIM-A9, and 400 μm for PrhMG. k–n, Representative and quantification of flow cytometry for the percentage of CD206+ (k,l) and ARG1+ (m,n) SIM-A9 microglia treated with or without TFPI2 protein and MK-0429. n = 3 biological replicates. o–r, Representative (o) and quantification (p) of flow cytometry for the percentage of CD206+ (o,p) and ARG1+ (q,r) HMC3 microglia treated with or without TFPI2 recombinant protein and MK-0429 (50 nM). n = 3 biological replicates. s, t, Representative of flow cytometry for the percentage of CD206+ (s) and ARG1+ (t) PrhMG treated with or without TFPI2 protein and MK-0429. TFPI2 protein at 20 ng/ml and MK-0429 at 50 nM were used unless indicated. Error bars indicate mean ± SEM. One-way ANOVA test.
Extended Data Fig. 7. TFPI2-STAT6 signaling mediates microglia-induced T cell suppression.

a, b, Survival curves of nude mice implanted with QPP7 cells (2×104 cells; a) or CT2A cells (2×104 cells; b) expressing control shRNA (shC) and Tfpi2 shRNAs (shTfpi2). n = 8 biological replicates. c, d, Immunofluorescence (c) and quantification (d) of CD8 in size matched tumors from C57BL/6 mice intracranially implanted with shC and shTfpi2 CT2A cells expressing. Scale bar, 50 μm. n = 3 biological replicates. e, f, Immunofluorescence (e) and quantification (f) of CD4 in size matched tumors from C57BL/6 mice intracranially implanted with shC and shTfpi2 QPP7 GSCs. Scale bar, 50 μm. n = 3 biological replicates. g, Representative of flow cytometry analysis for CD45+CD3+CD8+PD1high and CD45+CD3+CD4+PD1high T cells in the spleens of C57BL/6 mice bearing size matched shC and shTfpi2 CT2A tumors. h, Representative of flow cytometry for IFNγ+CD8+ T cells cocultured with SIM-A9 mouse microglia treated with or without TFPI2 recombinant protein (20 ng/ml) and STAT6 inhibitor (STAT6i) AS1517499 (25 nM). i, Representative of flow cytometry for fixable viability dye-labeled JURKAT T cells cocultured with PrhMG treated with or without TFPI2 protein (20 ng/ml) and AS1517499 (25 nM). Percentage of live cells and dead cells are indicated. j, k, Representative (j) and quantification (k) of flow cytometry for the percentage of HLA-DR+ cells out of total JURKAT T cells cocultured with PrhMG treated with TFPI2 protein (20 ng/ml) in the presence or absence of AS1517499 (25 nM). n = 3 biological replicates. l, Representative of flow cytometry for CD69+ JURKAT T cells cocultured with PrhMG treated with or without TFPI2 protein (20 ng/ml) and AS1517499 (25 nM). m, Representative of flow cytometry analysis for IFNγ+ JURKAT T cells cocultured with PrhMG treated with TFPI2 protein (20 ng/ml) in the presence or absence of AS1517499 (25 nM). Error bars indicate mean ± SEM. One-way ANOVA test. In (a,b), log-rank test.
Extended Data Fig. 8. TFPI2-CD51-STAT6 signaling mediates microglia-induced T cell suppression.

cer cell cytotoxicity induced by activated mouse primary CD8+ T cells cocultured with SIM-A9 microglia treated with TFPI2 recombinant protein (20 ng/ml) in the presence or absence of STAT6 inhibitor (STAT6i) AS1517499 (25 nM). The ratios between CD8+ T cells and CT2A cells are indicated. n = 10 biological replicates. c, GSC272 cytotoxicity induced by activated JURKAT T cells cocultured with PrhMG treated with TFPI2 protein (20 ng/ml) in the presence or absence of AS1517499 (25 nM). The ratios between CD8+ T cells and CT2A cells are indicated. n = 10 biological replicates. d, Correlation between CD274 (PD-L1) and TFPI2, STAT6, or ITGAV (CD51) in TCGA GBM dataset. e, Correlation between PDCD1LG2 (PD-L2) and TFPI2, STAT6, or ITGAV (CD51) in TCGA GBM dataset. In (d, e), R and P values were determined by Pearson correlation. Error bars indicate mean ± SEM. One-way ANOVA test.
Extended Data Fig. 9. TFPI2 correlates with proliferation and apoptosis markers in GBM patient tumors.
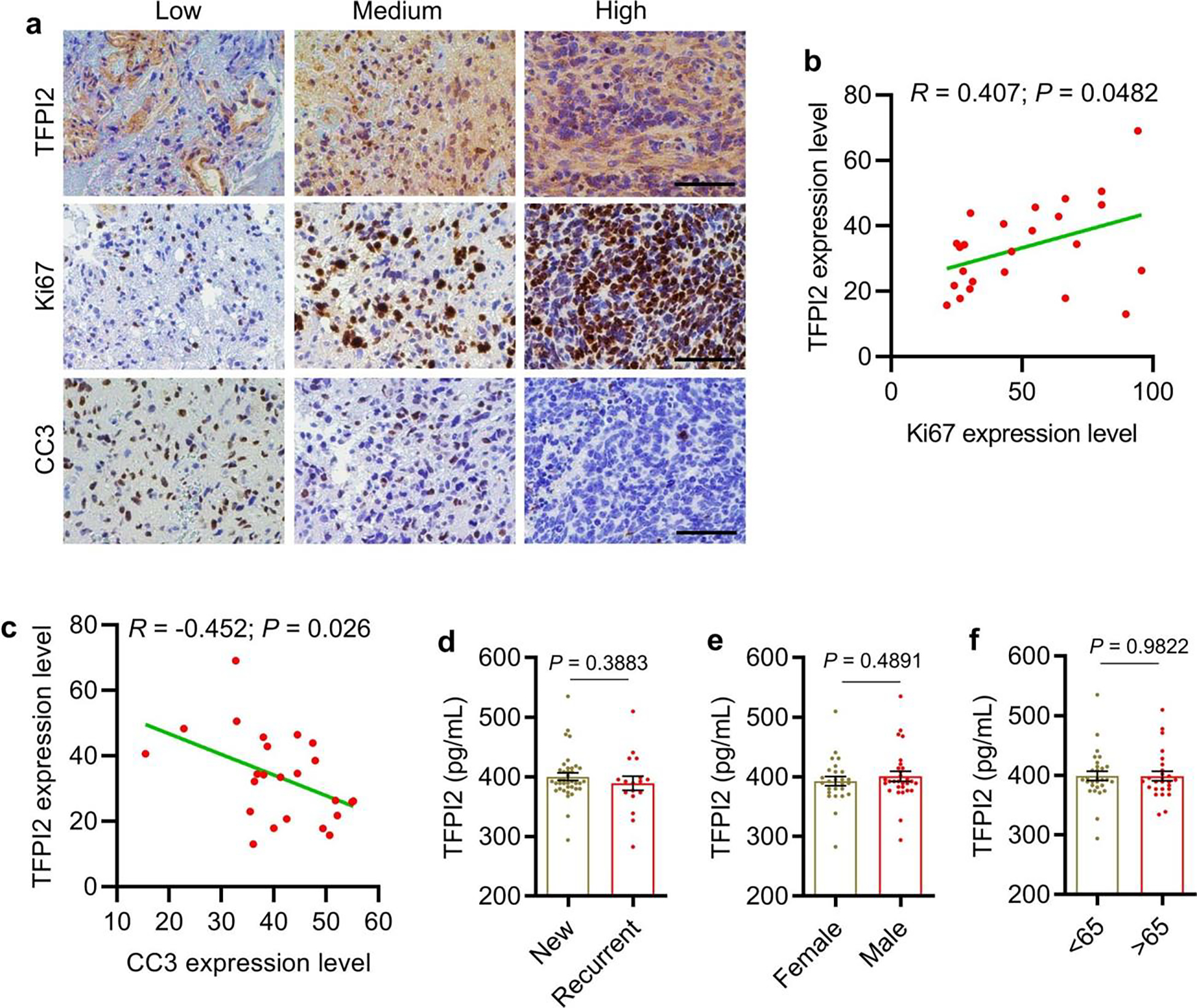
a, Representative images show low, medium, and high expression of TFPI2, Ki67, and cleaved caspase 3 (CC3) in human GBM tumor samples based on immunohistochemistry staining. Scale bar, 100 μm. b,c, Quantification of immunohistochemistry staining showing strong positive correlation between TFPI2 and Ki67 (b) and negative correlation between TFPI2 and CC3 (c) in human GBM tumor samples (n = 24). R and P values were determined by Pearson correlation. d-f, The plasma TFPI2 level in newly diagnosed (n = 38) and recurrent (n = 17) (d), female (n = 26) and male (n = 29) (e), as well as old (>65) (n = 29) and young (<65) (n = 26) GBM patients (f). Error bars indicate mean ± SEM. Student’s t-test.
Extended Data Fig. 10. Working model.
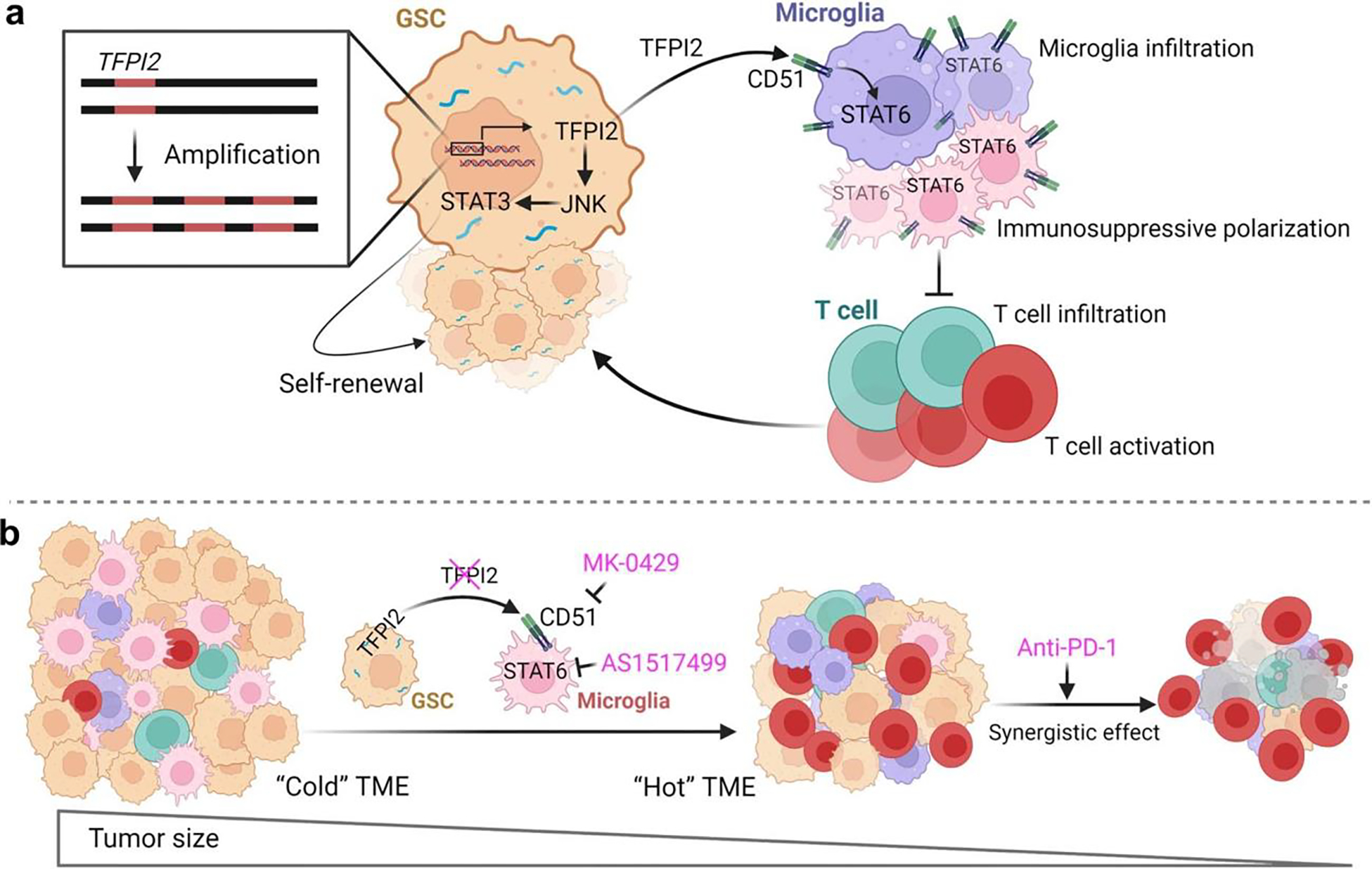
a, Illustration shows a proposed mechanism for the TFPI2 and its associated signaling axis in regulating self-renewal of glioma stem cells (GSCs) and GSC-microglia symbiosis in GBM. TFPI2 is amplified in GBM and essential for tumor progression. Upon secreting from GSCs, TFPI2 promotes GSC self-renewal by activating the JNK-STAT3 signaling pathway. Secreted TFPI2 binds to CD51 on microglia to promote microglia infiltration and immunosuppressive polarization via activation of STAT6 signaling. Consequently, these activated microglia inhibit the infiltration and activation of T cells and damage their tumor killing potential. b, Inhibition of TFPI2-CD51-STAT6 signaling axis impairs tumor progression by reducing immunosuppressive microglia and activating T cell-mediated antitumor immunity and synergizes with anti-PD1 therapy in GBM. This image was created with BioRender.com.
Supplementary Material
Acknowledgements
We thank S. D. Rabkin, F. Lang, J. Hu and H. Okada for providing 005 GSCs, patient-derived GSCs, QPP7 GSC and SB28, respectively. This work was supported in part by NIH R00 CA240896 (P.C.), NIH R01 NS124594 (P.C.), DoD Career Development Award W81XWH-21-1-0380 (P.C.), Cancer Research Foundation Young Investigator Award (P.C.), Lynn Sage Scholar Award (P.C.), American Cancer Society Institutional Research Grant IRG-21-144-27 (P.C.), NIH P50 CA221747 (to P.C., Brain Cancer SPORE CEP Award), philanthropic donation from Mindy Jacobson and the Bill Bass Foundation (P.C.), Northwestern University start-up funds (P.C.), the Northwestern Medicine Malnati Brain Tumor Insitute of the Robert H. Lurie Comprehensive Cancer Center (P.C.), NIH R24 NS104160 (C.G.) and NIH P30 AG072977 (C.G.). W.H.H. is funded by the CPRIT Training Program (RP210028). Imaging work was performed at the Northwestern University Center for Advanced Microscopy generously supported by NCI CCSG P30 CA060553. Proteomics experiments were performed at the Northwestern Proteomics Core Facility, which is generously supported by NCI CCSG P30 CA060553, instrumentation award (S10OD025194) from NIH Office of Director, and P41 GM108569. Incucyte analysis was performed in the Analytical bioNanoTechnology Core (ANTEC) Facility supported by the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633).
Footnotes
Ethics declarations
Competing interests
L.P. and P.C. are listed as inventors on a patent related to targeting the TFPI2–CD51–STAT6 signaling axis combining with or without anti-PD1 therapy. All other authors declare no competing interests.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Peer review information
Nature Immunology thanks the anonymous reviewers for their contribution to the peer review of this work. N. Bernard was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team. Peer reviewer reports are available.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements ofdata and code availability are available at https://doi.org/10.1038/s41590-023-01605-y.
Data availability
Proteomic data generated by IP–MS have been deposited in the Mass spectrometry Interactive Virtual Environment under dataset accession number of MSV000090624 (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?accession = MSV000090624). RNA-seq data have been desposited in the Gene Expression Omnibus under the accession code is GSE232486 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE232486). The TCGA GBM datasets, TCGA GBMLGG merged datasets, Brain Tumor Immune Micro Environment dataset (https://joycelab.shinyapps.io/braintime/) and published GEO datasets EGAS00001004422 (https://ega-archive.org/datasets/EGAD00001006206) and GSE131928 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131928) were also accessed. All other data are available within the article and supplementary information. Source data are provided with this paper.
References
- 1.Prager BC, Bhargava S, Mahadev V, Hubert CG & Rich JN Glioblastoma stem cells: driving resilience through chaos. Trends Cancer 6, 223–235 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Couturier CP et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 11, 3406 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guilhamon P et al. Single-cell chromatin accessibility profiling of glioblastoma identifies an invasive cancer stem cell population associated with lower survival. eLife 10, e64090 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen P, Hsu WH, Han J, Xia Y & DePinho RA Cancer stemness meets immunity: from mechanism to therapy. Cell Rep. 34, 108597 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xuan W, Lesniak MS, James CD, Heimberger AB & Chen P Context-dependent glioblastoma-macrophage/microglia symbiosis and associated mechanisms. Trends Immunol. 42, 280–292 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khan F et al. Macrophages and microglia in glioblastoma: heterogeneity, plasticity, and therapy. J. Clin. Invest. 133, e163446 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou W et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 17, 170–182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi Y et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat. Commun. 8, 15080 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xuan W et al. Circadian regulator CLOCK drives immunosuppression in glioblastoma. Cancer Immunol. Res. 10, 770–784 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen P et al. Circadian regulator CLOCK recruits immune-suppressive microglia into the GBM tumor microenvironment. Cancer Discov. 10, 371–381 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sprecher CA, Kisiel W, Mathewes S & Foster DC Molecular cloning, expression, and partial characterization of a second human tissue-factor-pathway inhibitor. Proc. Natl Acad. Sci. USA 91, 3353–3357 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herman MP et al. Tissue factor pathway inhibitor-2 is a novel inhibitor of matrix metalloproteinases with implications for atherosclerosis. J. Clin. Investig. 107, 1117–1126 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winkler J, Abisoye-Ogunniyan A, Metcalf KJ & Werb Z Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 11, 5120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rollin J et al. Expression and methylation status of tissue factor pathway inhibitor-2 gene in non-small-cell lung cancer. Br. J. Cancer 92, 775–783 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sato N et al. Epigenetic inactivation of TFPI-2 as a common mechanism associated with growth and invasion of pancreatic ductal adenocarcinoma. Oncogene 24, 850–858 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Wang GL et al. TFPI-2 suppresses breast cancer cell proliferation and invasion through regulation of ERK signaling and interaction with actinin-4 and myosin-9. Sci. Rep. 8, 14402 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Neaud V et al. Paradoxical pro-invasive effect of the serine proteinase inhibitor tissue factor pathway inhibitor-2 on human hepatocellular carcinoma cells. J. Biol. Chem. 275, 35565–35569 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Ruf W et al. Differential role of tissue factor pathway inhibitors 1 and 2 in melanoma vasculogenic mimicry. Cancer Res. 63, 5381–5389 (2003). [PubMed] [Google Scholar]
- 19.Mo J et al. TFPI2 promotes perivascular migration in an angiotropism model of melanoma. Front. Oncol. 11, 662434 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ota Y et al. Tissue factor pathway inhibitor-2 is specifically expressed in ovarian clear cell carcinoma tissues in the nucleus, cytoplasm and extracellular matrix. Oncol. Rep. 45, 1023–1032 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.George J, Gondi CS, Dinh DH, Gujrati M & Rao JS Restoration of tissue factor pathway inhibitor-2 in a human glioblastoma cell line triggers caspase-mediated pathway and apoptosis. Clin. Cancer Res. 13, 3507–3517 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gessler F, Voss V, Seifert V, Gerlach R & Kogel D Knockdown of TFPI-2 promotes migration and invasion of glioma cells. Neurosci. Lett. 497, 49–54 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Rao CN et al. Expression of tissue factor pathway inhibitor 2 inversely correlates during the progression of human gliomas. Clin. Cancer Res. 7, 570–576 (2001). [PubMed] [Google Scholar]
- 24.Miranda A et al. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl Acad. Sci. USA 116, 9020–9029 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshihara K et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chai R et al. A novel gene signature based on five glioblastoma stem-like cell relevant genes predicts the survival of primary glioblastoma. J. Cancer Res. Clin. Oncol. 144, 439–447 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uhlen M et al. The human secretome. Sci. Signal 12, eaaz0274 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Klemm F et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell 181, 1643–1660 e1617 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neftel C et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835–849 e821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shingu T et al. Qki deficiency maintains sternness of glioma stem cells in suboptimal environment by downregulating endolysosomal degradation. Nat. Genet. 49, 75–86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saha D, Martuza RL & Rabkin SD Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell 32, 253–267 e255 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoon CH et al. c-Jun N-terminal kinase has a pivotal role in the maintenance of self-renewal and tumorigenicity in glioma stem-like cells. Oncogene 31, 4655–4666 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Gu CY et al. Tumor-specific activation of the C-JUN/MELK pathway regulates glioma stem cell growth in a p53-dependent manner. Stem Cells 31, 870–881 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi Y et al. Ibrutinib inactivates BMX-STAT3 in glioma stem cells to impair malignant growth and radioresistance. Sci. Transl. Med. 10, eaah6816 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stechishin OD et al. On-target JAK2/STAT3 inhibition slows disease progression in orthotopic xenografts of human glioblastoma brain tumor stem cells. Neuro Oncol. 15, 198–207 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deng Y et al. Identifying mutual exclusivity across cancer genomes: computational approaches to discover genetic interaction and reveal tumor vulnerability. Brief. Bioinform. 20, 254–266 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Guo L et al. Postmortem adult human microglia proliferate in culture to high passage and maintain their response to amyloid-β. J. Alzheimers Dis. 54, 1157–1167 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagashima S et al.Synthesis and evaluation of 2-{[2-(4-hydroxyphenyl)-ethyl] amino} pyrimidine-5-carboxamide derivatives as novel STAT6 inhibitors. Bioorgan. Med. Chem 15, 1044–1055 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Uhlen M et al. Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Chen J et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell 19, 541–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen CH et al. Novel murine glioblastoma models that reflect the immunotherapy resistance profile of human disease. Neuro Oncol. 27, noad025 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bloch O et al. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 19, 3165–3175 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gimple RC, Yang KL, Halbert ME, Agnihotri S & Rich JN Brain cancer stem cells: resilience through adaptive plasticity and hierarchical heterogeneity. Nat. Rev. Cancer 22, 497–514 (2022). [DOI] [PubMed] [Google Scholar]
- 44.Wang X et al. Sequential fate-switches in stem-like cells drive the tumorigenic trajectory from human neural stem cells to malignant glioma. Cell Res 31, 684–702 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bachoo RM et al. Epidermal growth factor receptor and Ink4a/ Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 1, 269–277 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Dong Z et al. Targeting glioblastoma stem cells through disruption of the circadian clock. Cancer Discov. 9, 1556–1573 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dirkse A et al. Stem cell-associated heterogeneity in glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 10, 1787 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schaettler MO et al. Characterization of the genomic and immunologic diversity of malignant brain tumors through multisector analysis. Cancer Discov. 12, 154–171 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Konduri SD et al. A novel function of tissue factor pathway inhibitor-2 (TFPI-2) in human glioma invasion. Oncogene 20, 6938–6945 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Jacob F et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell 180, 188–204.e122 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.LeBlanc VG et al. Single-cell landscapes of primary glioblastomas and matched explants and cell lines show variable retention of inter- and intratumor heterogeneity. Cancer Cell 40, 379–392.e379 (2022). [DOI] [PubMed] [Google Scholar]
- 52.Bayik D & Lathia JD Cancer stem cell–immune cell crosstalk in tumour progression. Nat. Rev. Cancer 21, 526–536 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pang L, Khan F, Dunterman M & Chen P Pharmacological targeting of the tumor–immune symbiosis in glioblastoma. Trends Pharmacol. Sci. 43, 686–700 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brennan CW et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parsa AT et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 13, 84–88 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Chen P et al. Symbiotic macrophage–glioma cell interactions reveal synthetic lethality in PTEN-null glioma. Cancer Cell 35, 868–884 e866 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu H, Wang G, Hao D, Wang C & Zhang M Antimicrobial and immunoregulatory activities of TS40, a derived peptide of a TFPI-2 homologue from black rockfish (Sebastes schlegelii). Mar. Drugs 20, 353 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cai W et al. STAT6/Arg1 promotes microglia/macrophage efferocytosis and inflammation resolution in stroke mice. JCI Insight 4, e131355 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang N, Liang H & Zen K Molecular mechanisms that influence the macrophage m1–m2 polarization balance. Front. Immunol. 5, 614 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pang L, Khan F, Heimberger AB & Chen P Mechanism and therapeutic potential of tumor–immune symbiosis in glioblastoma. Trends Cancer 8, 839–854 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang B et al. Macrophage-expressed CD51 promotes cancer stem cell properties via the TGF-β1/smad2/3 axis in pancreatic cancer. Cancer Lett. 459, 204–215 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Chen P & Dey P Co-dependencies in the tumor immune microenvironment. Oncogene 41, 3821–3829 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chamberlain MC & Kim BT Nivolumab for patients with recurrent glioblastoma progressing on bevacizumab: a retrospective case series. J. Neurooncol. 133, 561–569 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Reiss S, Yerram P, Modelevsky L & Grommes C Retrospective review of safety and efficacy of checkpoint inhibition in refractory high-grade gliomas. J. Clin. Oncol. 35, no. 15_suppl. 2033 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reardona DA et al. Randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: Checkmate 143. Neuro Oncol. 19, 21 (2017). [Google Scholar]
- 66.Zhao J et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 25, 462–469 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arrieta VA et al. ERK1/2 phosphorylation predicts survival following anti-PD-1 immunotherapy in recurrent glioblastoma. Nat. Cancer 2, 1372–1386 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seyfried TN, El-Abbadi M, Ecsedy JA, Bai HW & Yohe HC Influence of host cell infiltration on the glycolipid content of mouse brain tumors. J. Neurochem. 66, 2026–2033 (1996). [DOI] [PubMed] [Google Scholar]
- 69.Chen P et al. Collagen VI regulates peripheral nerve regeneration by modulating macrophage recruitment and polarization. Acta Neuropathol. 129, 97–113 (2015). [DOI] [PubMed] [Google Scholar]
- 70.Ershov D et al. TrackMate 7: integrating state-of-the-art segmentation algorithms into tracking pipelines. Nat. Methods 19, 829–832 (2022). [DOI] [PubMed] [Google Scholar]
- 71.Tinevez JY et al. TrackMate: an open and extensible platform for single-particle tracking. Methods 115, 80–90 (2017). [DOI] [PubMed] [Google Scholar]
- 72.Chen P et al. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc. Natl Acad. Sci. USA 114, 580–585 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Varghese F, Bukhari AB, Malhotra R & De A IHC Profiler: an open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS ONE 9, e96801 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barth ND et al. A fluorogenic cyclic peptide for imaging and quantification of drug-induced apoptosis. Nat. Commun. 11, 4027 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ong Derrick Sek T et al. PAF promotes stemness and radioresistance of glioma stem cells. Proc. Natl Acad. Sci. USA 114, E9086–E9095 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Galaxy C The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 50, W345–W351 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Couturier CP et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 11, 3406 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Proteomic data generated by IP–MS have been deposited in the Mass spectrometry Interactive Virtual Environment under dataset accession number of MSV000090624 (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?accession = MSV000090624). RNA-seq data have been desposited in the Gene Expression Omnibus under the accession code is GSE232486 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE232486). The TCGA GBM datasets, TCGA GBMLGG merged datasets, Brain Tumor Immune Micro Environment dataset (https://joycelab.shinyapps.io/braintime/) and published GEO datasets EGAS00001004422 (https://ega-archive.org/datasets/EGAD00001006206) and GSE131928 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131928) were also accessed. All other data are available within the article and supplementary information. Source data are provided with this paper.