

Abstract
To determine whether adenosine A3 receptors participate in adenosine-induced changes in coronary flow, isolated hearts from wild-type (WT) and A3 receptor knockout (A3KO) mice were perfused under constant pressure and effects of nonselective and selective agonists were examined. Adenosine and the selective A2A agonist 2-[p-(2-carboxyethyl)]phenylethylamino-5′-N-ethylcarboxam-idoadenosine (CGS-21680) produced augmented maximal coronary vasodilation in A3KO hearts compared with WT hearts. Selective activation of A3 receptors with 2-chloro-N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide (Cl-IB-MECA) at nanomolar concentrations did not effect coronary flow, but at higher concentrations it produced coronary vasodilation both in WT and A3KO hearts. Cl-IB-MECA-induced increases in coronary flow were susceptible to both pharmacological blockade and genetic deletion of A2A receptors. Because deletion or blockade of adenosine A3 receptors augmented coronary flow induced by nonselective adenosine and the selective A2A receptor agonist CGS-21680, we speculate that this is due to removal of an inhibitory influence associated with the A3 receptor subtype. These data indicate that the presence of adenosine A3 receptors may either inhibit or negatively modulate coronary flow mediated by other adenosine receptor subtypes.
Keywords: coronary vasodilation, knockout mice, A2A receptor knockout mice
adenosine produces potent coronary vasodilation in different mammalian species including bovine, canine, porcine, rat, guinea pig, mouse, and humans (1, 2, 8, 18, 19, 27, 34). The cardiovascular effects of adenosine are mediated by activation of four known cell surface adenosine receptors (A1, A2A, A2B, and A3); however, the relative contribution of each adenosine receptor subtype in modulating coronary flow is not yet fully understood (11, 21, 23, 32). Whereas it is well established that coronary vasodilation is primarily mediated through A2A receptor activation, it has been demonstrated that A2B receptor activation plays a role in coronary flow regulation in humans and mice (14, 20, 34). The physiological significance of A3 receptors in vascular responses is not yet characterized, although its role in myocardial ischemia and reperfusion is beginning to be understood (4, 7, 12). Recently, it has been reported (4, 7) that A3 receptors play an injurious role during myocardial ischemia-reperfusion, because targeted deletion of the A3 receptor confers resistance to myocardial ischemic injury. In isolated rat and rabbit hearts, selective activation of A3 receptors did not change coronary flow (16), yet infusion of adenosine in A3KO mice has been shown to cause a significant decrease in blood pressure compared with wild-type (WT) mice (36), suggesting that A3 receptors affect vascular tone in this species. However, there are no reports demonstrating whether and to what extent A3 receptors are involved in the modulation of coronary flow in mice.
With recent reports (20, 34) in murine hearts indicating a predominant role of A2A over A2B receptor activation in the regulation of coronary flow, the primary focus of the present study was to determine whether A3 receptors modulate this effect by comparing the coronary vascular responses to adenosine agonists in isolated hearts from WT and A3 receptor knockout (A3KO) mice. The strategy of combining receptor knockout technology with a traditional pharmacological approach has proven useful in determining the relative contribution of adenosine receptor subtypes in the complex regulation of coronary flow (20). This allows for a more precise and direct examination of specific adenosine receptor subtypes than previously possible through agonist-antagonist studies alone.
Thus, to determine whether A3 receptor activation participates in the regulation of coronary flow, coronary vascular responses to nonselective and selective adenosine receptor agonists were examined in hearts from both WT and A3KO mice. We hypothesized that targeted deletion of A3 receptors would modulate coronary flow mediated by other adenosine receptor subtypes.
MATERIALS AND METHODS
All of the experimental protocols were performed according to the guidelines of Animal Care and Use Committee at East Carolina University.
Source of mice.
Two sets of mice were used in the current study. A3KO and WT control mice were kindly provided by Merck Research Laboratories. Both populations of mice were on the mixed strain of Sv129J/C57Bl/6/D2. Details of generation and initial characterization of the A3KO mice have been described previously (28). A2A receptor knockout mice (A2AKO) and their WT littermate controls were raised at the Institute of Experimental Medicine, Brussels, Belgium, and kindly provided for the current study. Details of generation and initial characterization of the A2AKO mice have been described previously by Ledent et al. (17).
Langendorff-perfused heart preparation.
Hearts were isolated from age-matched mice of both WT and KO groups as previously described (9, 20, 33, 34). Briefly, mice were deeply anesthetized with pentobarbital sodium (100 mg/kg ip), and hearts were quickly excised and placed in heparinized ice-cold buffer to arrest cardiac contraction. After all extracardiac tissues were removed, the aorta was carefully tied to an aortic cannula made from a 20-gauge blunted needle. Hearts were retrogradely perfused at a constant pressure of 80 mmHg with warmed Krebs-Henseleit buffer in standard Langendorff fashion and allowed to beat spontaneously. The composition of the modified Krebs-Henseleit buffer was (in mM) 118 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 2.5 CaCl2, 0.5 Na2EDTA, 11 glucose, and 2.0 pyruvate. The buffer was prefiltered to particle size of <0.22 μm and bubbled continuously with 95% O2-5% CO2 at 37°C (pH 7.4). A water-filled balloon made of plastic wrap was inserted into the left ventricle across the mitral valve for continuous measurement of left ventricular developed pressure (LVDP) by a fluid-filled pressure transducer. Hearts were then immersed in perfusate maintained at 37°C and the ventricular balloon was inflated to yield a left ventricular end-diastolic pressure of 2–5 mmHg. Coronary flow was continuously measured using an ultrasonic flow probe (model T106, Transonic Systems; Ithaca, NY) placed in the aortic perfusion line, and aortic pressure was recorded via a pressure transducer attached to the side arm of the aortic cannula. All of the transducers and ultrasonic flow meter were coupled to a PowerLab/4sp data acquisition system (ADInstruments; Castle Hill, Australia) and functional data were recorded on a G4 Power Mac computer (Apple Computer) using PowerLab Chart version 3.5.6 software (ADInstruments). Baseline coronary flow, LVDP, and heart rate (derived from the ventricular pressure tracing) were monitored for an initial 30-min equilibration period. Hearts with persistent arrhythmias or poor LVDP (<50 mmHg) during equilibration were excluded from the study.
Protocol for isolated heart experiments.
When hearts reached a steady-state coronary flow, increasing concentrations of adenosine and its analogs were infused by a Harvard infusion pump (Harvard Apparatus) into the aortic cannula immediately above the heart at a rate of 1% of the basal flow to achieve the desired concentration in the perfusate. All agonist concentration-response curves (CRCs) were constructed noncumulatively and one CRC was performed on each heart. The concentration of agonist was tested in steps of 0.5 log units. Infusion of each agonist concentration was maintained until coronary flow demonstrated a new steady state, and a washout period of at least 5 min, unless otherwise indicated, was allowed before administration of next (higher) concentration. Changes in coronary flow, heart rate, and LVDP were expressed as the percent change from predrug baseline value.
In our previous study (34), tachyphylaxis to repeat administration of a single concentration of agonist in the same heart was not observed; therefore, antagonist effects were investigated in a paired manner on the same hearts using only one agonist concentration. When examining responses to antagonists, the effect of agonist was first determined in the absence of antagonist (control). After complete washout of control response (when coronary flow returned to baseline value), antagonist was infused into the perfusion line and allowed to equilibrate for at least 10 min before adding the same dose of agonist in the perfusion. This time of incubation for the antagonist was chosen based on our previous studies of mouse hearts (20, 34). At 10–15 min into the antagonist infusion, data were sampled and normalized as a new “baseline” and agonist was added to the coronary perfusate at 1% of coronary flow. The antagonist remained present during agonist administration until steady-state response was achieved. Data were sampled at the end of this two-drug infusion for comparison with data resulting from infusion of agonist alone.
Data analysis.
Experimental values are presented as means ± SE. For each CRC to adenosine and CGS-21680, the concentration required to produce a 50% response (EC50) in coronary flow was obtained by graphic analysis of an individual curve. Significant differences were estimated by two-tailed Student’s t-test for paired data from the same experiment and unpaired data from different experiments. Differences in dose response between WT and A3KO groups at individual agonist concentrations were analyzed by ANOVA, followed by Student’s t-test. A P value of <0.05 was considered significant.
Chemicals.
Adenosine and CGS-21680 were purchased from RBI-Sigma (St. Louis, MO). Cl-IB-MECA was obtained by SRI International from the National Institute of Mental Health Chemical Synthesis and Drug Supply Program. MRS-1220 was obtained from National Institute of Diabetes, Digestive and Kidney Diseases (Bethesda, MD). All other chemicals were of the highest grade available and were purchased from Sigma. CGS-21680, Cl-IB-MECA, and adenosine antagonists were dissolved in 100% dimethyl sulfoxide (DMSO) as a 10 mM stock solution, followed by serial dilutions in 50% DMSO and distilled water. All other chemicals were dissolved in distilled water.
RESULTS
General characteristics and baseline functional parameters of isolated mouse hearts.
Baseline functional parameters of isolated murine hearts were recorded at the end of the 30-min equilibration period before beginning of the experimental protocol. Summarized data of coronary flow, heart rate, and LVDPs at equilibrium in WT and A3KO hearts are presented in Table 1.
Table 1.
General characteristics and baseline functional parameters of isolated hearts from both wild-type and adenosine A3 receptor knock-out mice
| Wild Type (n = 34) | A3 Receptor Knockout (n = 26) | |
|---|---|---|
| General characteristics | ||
| Age, wk | 16.6 ± 0.7 | 19.1 ± 0.5* |
| Body weight, g | 29.1 ± 0.6 | 32.3 ± 1.4* |
| Heart weight, mg | 117.6 ± 3.6 | 123.6 ± 3.1 |
| Baseline functional parameters | ||
| Coronary flow, ml/min | 1.08 ± 0.05 | 1.02 ± 0.04 |
| Heart rate, beats/min | 337 ± 5.5 | 337 ± 4.6 |
| Ventricular developed pressure, mmHg | 66.8 ± 2.05 | 69.8 ± 2.32 |
All values are means ± SE; n, number of animals.
P < 0.05 vs. wild-type mice.
Coronary vascular effects of adenosine and its analogues on isolated hearts from WT and A3KO mice.
Adenosine and its analogs CGS-21680 and Cl-IB-MECA produced concentration-dependent increases in coronary flow (vasodilation) in isolated hearts from both WT and A3KO mice (Fig. 1). The maximal coronary vasodilation elicited by adenosine in WT and A3KO hearts were 396.89 ± 32.59% (n = 6) and 554.94 ± 35.09% of baseline (n = 8), respectively (Fig. 1, P < 0.05 WT vs. A3KO). CGS-21680 induced maximal coronary vasodilation in WT and A3KO hearts were 415.49 ± 14.33% (n = 7) and 584.38 ± 30.10% of baseline (n = 6), respectively (Fig. 1, P < 0.05 WT vs. A3KO). Cl-IB-MECA is a highly potent A3 receptor agonist with inhibitor constant (Ki) values of 820, 470, and 0.33 nM at A1, A2A, and A3 receptors, respectively (12). Figure 1C shows that Cl-IB-MECA did not affect coronary flow even at 100 nM, but it increased coronary flow at concentrations ≥1 μM both in WT and A3KO hearts (Fig. 1). The increases in coronary flow with 5 μM Cl-IB-MECA in WT and A3KO hearts were 391.78 ± 38.08% (n = 5) and 534.77 ± 29.87% of baseline (n = 7), respectively (P < 0.05, WT vs. A3KO). The maximal response to Cl-IB-MECA could not be reached because of difficulty in washout of the drug even after 45-min drug-free perfusion; therefore, EC50 values for Cl-IB-MECA-induced increases in coronary flow were not determined. EC50 values for adenosine-induced increases in coronary flow in WT and A3KO hearts were 0.34 ± 0.05 and 0.76 ± 0.08 μM, respectively (P < 0.05 WT vs. A3KO), and those for CGS-21680 in WT and A3KO hearts were 17.2 ± 2.49 and 23.1 ± 0.64 nM, respectively. All agonists displayed an augmented maximal coronary flow in A3KO hearts compared with WT hearts (Fig. 1), suggesting that selective deletion of A3 receptors may have removed an inhibitory influence facilitating a greater maximal response mediated by other adenosine receptor subtypes.
Fig. 1.
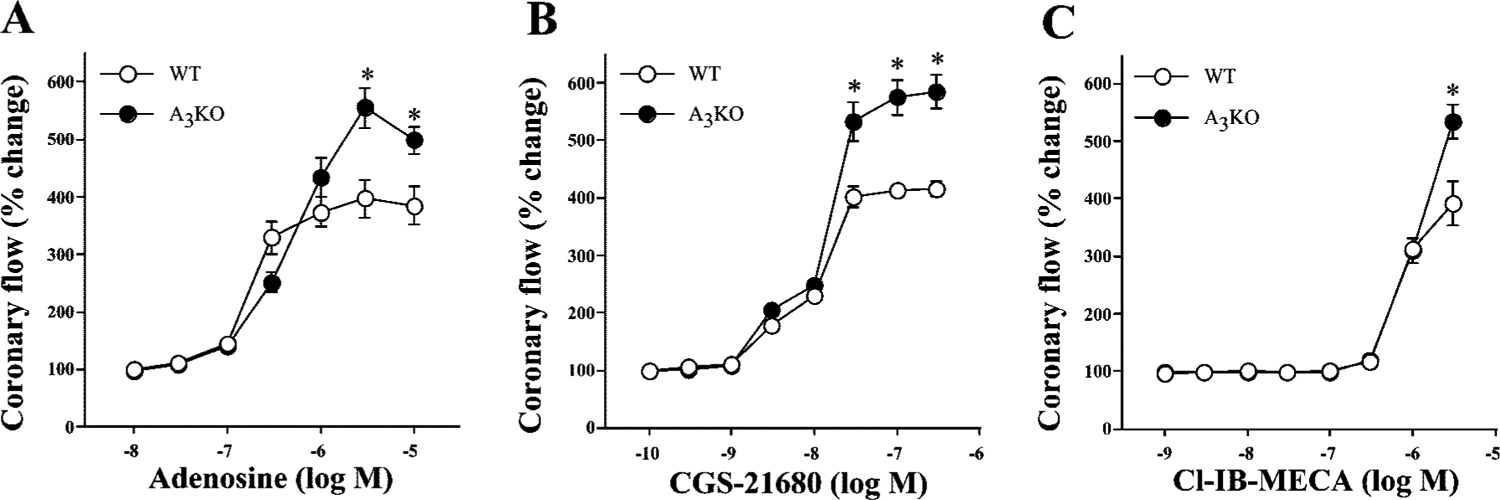
Concentration-dependent coronary vascular effects of adenosine (A), 2-[p-(2-carboxyethyl)]phenylethylamino-5′-N-ethylcarboxyamido-adenosine (CGS-21680) (B), and 2-chloro-N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide (Cl-IB-MECA) (C) in isolated hearts from both wild-type (WT) and A3 receptor knockout (A3KO) mice. Symbols represent means ± SE of 5–8 experiments. Concentration-response curve for coronary flow was constructed noncumulatively. y-Axis, changes in the coronary flow are expressed as %change from immediate baseline value that was assigned as 100%; x-axis, the molar concentration of agonists on a logarithmic scale. *P < 0.05 vs. corresponding WT value.
Influence of A3 receptor blockade on CGS-21680-induced increases in coronary flow in isolated mouse hearts.
With the observation that targeted deletion of A3 receptors augments the maximal coronary flow induced by adenosine receptor agonists (Fig. 1), the question arises whether this observation can be mimicked by acute pharmacological blockade of A3 receptors. To investigate this possibility, the influence of an A3 receptor antagonist 9-chloro-2-(2-furyl)–5-phenylacetylamino(1,2,4) triazolo(1,5-c)quinazoline (MRS-1220) on coronary vasodilation induced by the selective A2A receptor agonist CGS-21680 was examined in isolated WT hearts.
MRS-1220 is a potent A3 receptor antagonist with reported Ki values of 305, 52, and 0.65 nM at rat A1, A2A, and human A3 receptors, respectively (13). Species differences in the activity of MRS-1220 have been reported between rat and human. MRS-1220 is reported to have an affinity at rat A3 receptor at >1 μM (15), whereas it is >2,000-fold more potent at the human A3 receptor than at the rat receptor (13). CGS-21680 is the most potent, highly selective agonist at A2A receptors and is virtually ineffective at A2B receptors (6, 22), and it is the ligand of choice for the characterization of A2A receptors. CGS-21680 does not change coronary flow in A2AKO hearts even at micromolar concentrations (20). The Ki values for CGS-21680 at A1, A2A, and A2B receptors are >350 nM, 15 nM, and >100 μM, respectively (6). The reproducibility of responses induced by CGS-21680 (100 nM) on the coronary flow has been previously demonstrated in mice (34) and rats (16). A 5 nM concentration of MRS-1220 was chosen for A3 antagonist experiments to favor A3 selectivity. MRS-1220 (5 nM) decreased coronary flow to 93.63 ± 1.88% of baseline value (n = 6; P < 0.05 vs. baseline). After the control response to CGS-21680 (100 nM) in the absence of MRS-1220 (followed by subsequent 10-min agonist-free perfusion) was obtained, MRS-1220 (5 nM) was infused for at least 10 min before the second administration of CGS-21680 (100 nM) and was present during 5-min agonist infusion. Figure 2 shows that acute pharmacological blockade of A3 receptors with MRS-1220 can also augment agonist-induced coronary flow in isolated WT hearts. Infusion of 100 nM CGS-21680 in the absence of antagonist (control) increased coronary flow to 444.80 ± 22.18% (n = 6) of baseline, whereas, in the presence of MRS-1220, infusion of 100 nM CGS-21680 increased coronary flow to 502.13 ± 34.48% of the predrug baseline value (P < 0.05 vs. respective control value in the absence of MRS-1220). In these experiments, all hearts exhibited an augmented response to CGS-21680 in the presence of MRS-1220, although they had variable magnitude. Here it is also noteworthy that the magnitude of CGS-21680 (100 nM)-induced increases in coronary flow in WT hearts with MRS-1220 was quantitatively smaller (502.13 ± 34.48% of predrug baseline value, n = 6, Fig. 2) than that observed in A3KO hearts (574.36 ± 31.01% of predrug baseline value, n = 6, Fig. 1B). One possible reason for this quantitative difference of CGS-21680-induced response between pharmacological inhibition and genetic deletion of A3 receptors may be due to ineffective blockade of A3 receptors with MRS-1220 compared with total absence of A3 receptors in A3KO hearts. In an additional experiment, we tested a higher concentration of MRS-1220 on CGS-21680-induced coronary flow. However, a 10-fold increase in MRS-1220 (50 nM) did not augment but rather markedly reduced A2A receptor-mediated increases in coronary flow of CGS-21680 in WT hearts, thus illustrating its nonselective nature at a higher concentration (data not shown). Taken together, these findings indicate that the response to CGS-21680 with pharmacological blockade of A3 receptors in WT hearts (Fig. 2) is at least qualitatively similar to that observed in hearts with targeted deletion of A3 receptors (Fig. 1B).
Fig. 2.
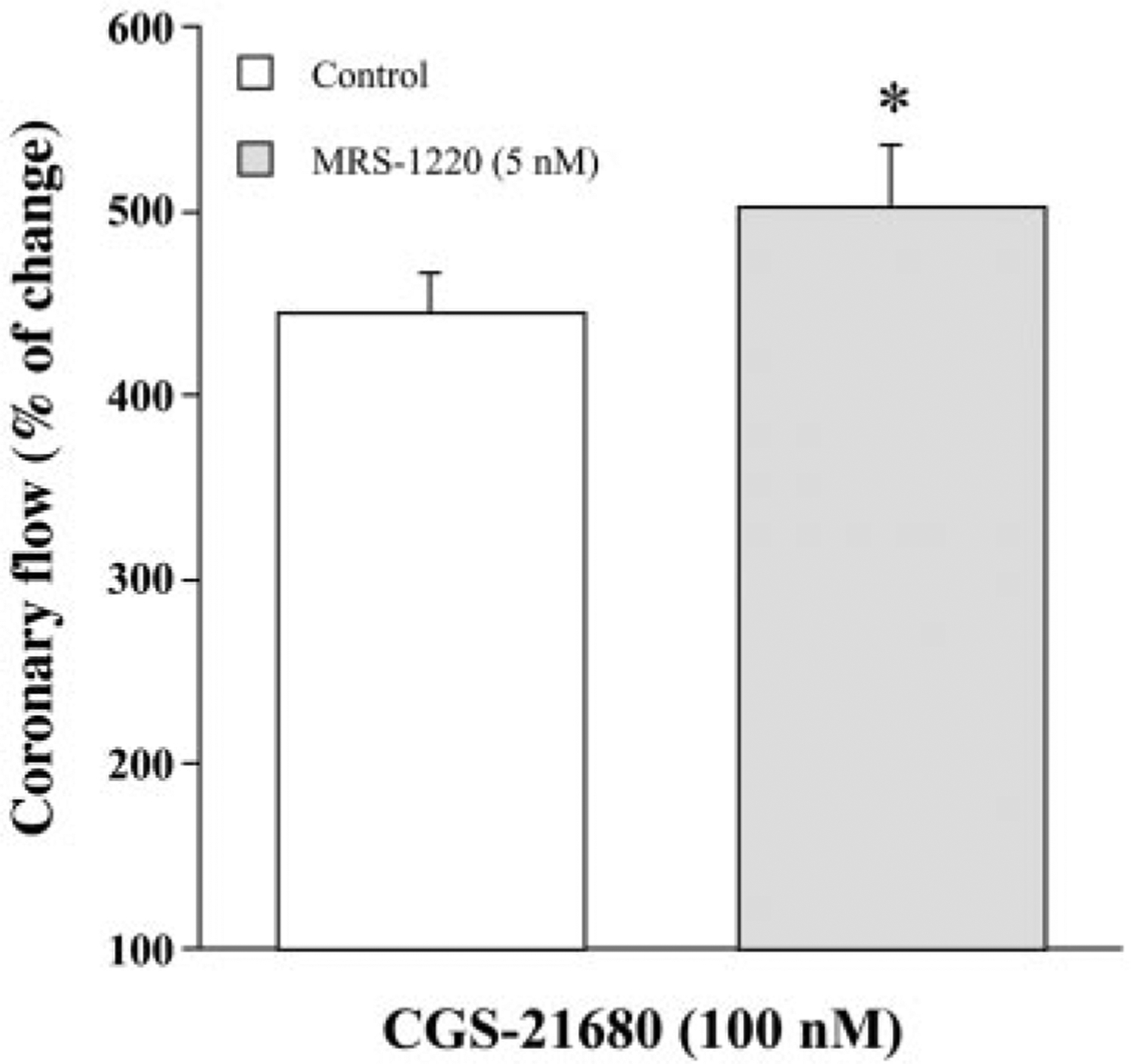
Influence of selective blockade of adenosine A3 receptor with 9-chloro-2-(2-furyl)-5-phenylacetylamino(1,2,4)-triazolo(1,5-c)quinazoline (MRS-1220) on CGS-21680-induced increases in coronary flow in WT mice. Control response to CGS-21680 (100 nM) was first determined in the absence of MRS-1220. After 10-min drug-free washout, hearts were pretreated with MRS-1220 (5 nM) for 15 min before second exposure to CGS-21680. Bars represent means ± SE of 6 experiments from different hearts in a paired manner. Vehicle control experiments were done in four hearts. *P < 0.05 vs. respective control value.
Influence of A2A receptor blockade on Cl-IB-MECA-induced increases in coronary flow in isolated mouse hearts.
Coronary flow was unaffected by Cl-IB-MECA in both WT and A3KO hearts at nanomolar concentrations (Fig. 1) where it is selective for A3 receptors. Yet in the micromolar range where Cl-IB-MECA becomes nonselective for A2A receptors, coronary flow was increased in both WT and A3KO hearts (Fig. 1). That Cl-IB-MECA-induced coronary vasodilation was observed in hearts with and without A3 receptors suggests that this results from nonselective activation of A2A receptors at micromolar concentrations. To characterize this effect, coronary vascular responses to Cl-IB-MECA were examined in A3KO hearts in the presence of a selective A2A receptor antagonist 7-(2-phenylethyl)-5-amino-2-(2-furyl)-pyrazolo-[4,3-e]-1,2,4-triazolo-[1,5-c]pyrimidine (SCH-58261).
Because it has been reported that successive activation of A3 receptors with Cl-IB-MECA demonstrates tachyphylaxis on the hemodynamic effects of conscious animal (29), it was necessary to assess the reproducibility of coronary flow responses induced by Cl-IB-MECA. Because Cl-IB-MECA elicited the same coronary vasodilation in WT and A3KO hearts at 1 μM (Fig. 1C), the coronary vascular effects at this concentration were assessed in a subset of three WT hearts (Fig. 3A). Each heart was exposed to Cl-IB-MECA (1 μM) until a steady response was observed (typically at 8–10 min). Two infusions of Cl-IB-MECA separated by at least 30 min of agonist-free perfusion were performed. The increases in coronary flow for the first and second Cl-IB-MECA infusions were 305.41 ± 15.13 and 296.23 ± 18.27% of baseline, respectively (Fig. 3A), and were significantly different from vehicle controls (P < 0.05). Thus Cl-IB-MECA-induced coronary vascular responses in mouse hearts did not exhibit tachyphylaxis with successive dosing as has been reported in isolated rat hearts (16).
Fig. 3.
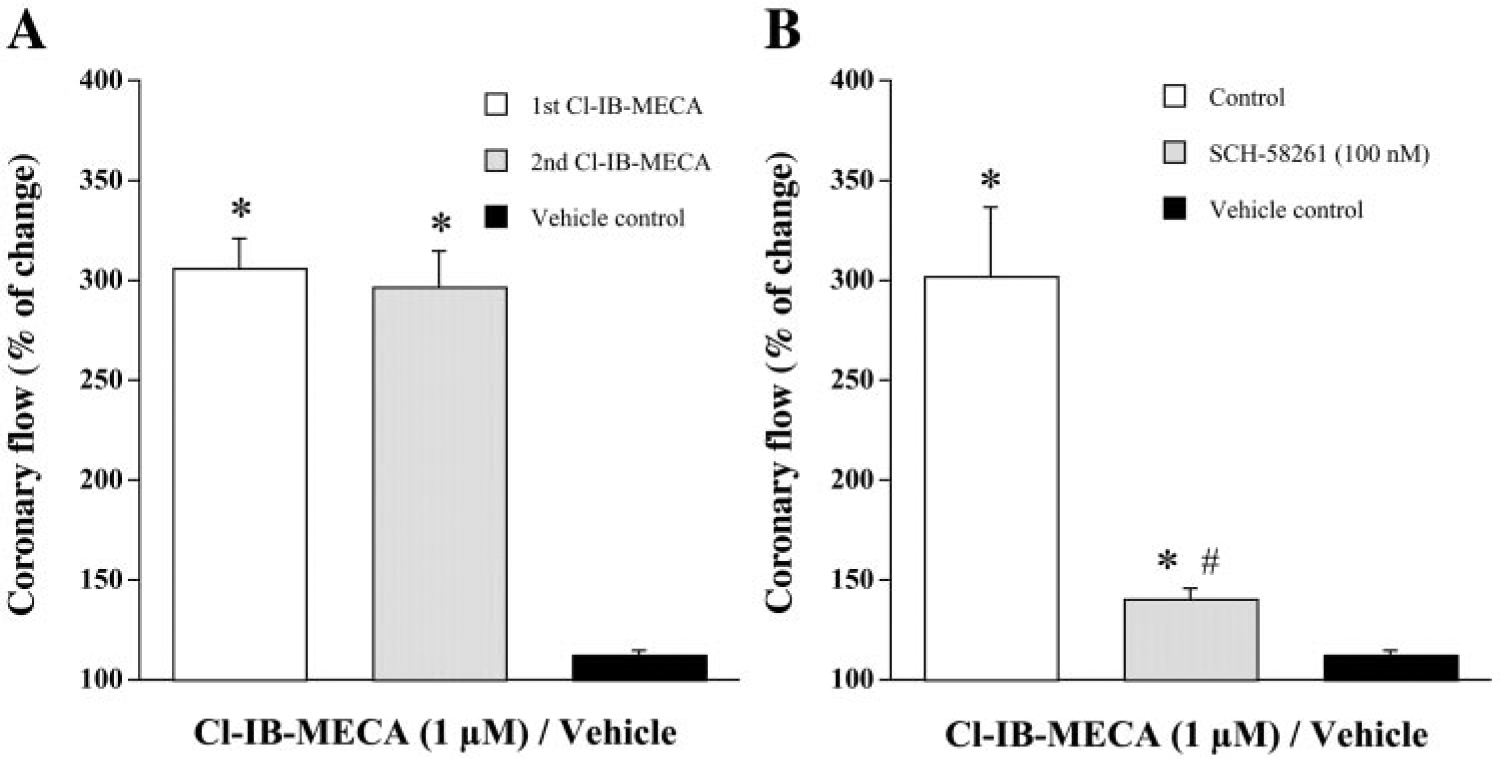
A: reproducibility of the effects of Cl-IB-MECA on coronary flow of isolated mouse hearts. Cl-IB-MECA (1 μM) was infused for 10 min, followed by 30-min drug-free wash before the next exposure. Bars represent means ± SE of three hearts in a paired manner. B: influence of selective blockade of adenosine A2A receptor with 7-(2-phenylethyl)-5-amino-2-(2-furyl)-pyrazolo-[4,3-e]-1,2,4-triazolo-[1,5-c]pyrimidine (SCH-58261) on Cl-IB-MECA-induced increases in coronary flow in A3KO hearts. Control responses to Cl-IB-MECA (1 μM) were first determined in the absence of SCH-58261. After 30-min washout, hearts were pretreated with SCH-58261 (100 nM) for 10 min before second exposure to Cl-IB-MECA. Bars represent means ± SE of 5 experiments from different hearts in a paired manner. Vehicle control experiments were done in four hearts. *P < 0.05 vs. vehicle control; #P < 0.05 vs. respective control.
The selective A2A receptor antagonist SCH-58261 was used to determine whether A2A receptor activation accounts for the Cl-IB-MECA-induced coronary vasodilation in isolated A3KO hearts. SCH-58261 is a potent and highly selective A2A receptor antagonist both in vivo and in vitro, and it has little or no affinity up to the micromolar range for A2B and A3 receptors (25, 26, 37). Because 100 nM SCH-58261 had been used to selectively characterize A2A receptor-mediated effects in related experiments (2, 25, 34, 37), this concentration was used in A3KO hearts. SCH-58261 alone decreased the coronary flow to 74.88 ± 3.91% of the baseline value (n = 5; P < 0.05 vs. baseline). After the control response to Cl-IB-MECA in the absence of SCH-58261 (followed by subsequent 30-min agonist-free perfusion) was obtained, SCH-58261 was infused for 10 min before the second administration of Cl-IB-MECA and was present during the 10-min agonist infusion. Infusion of Cl-IB-MECA (1 μM) in the absence of antagonist increased coronary flow to 301.68 ± 34.09% of baseline (n = 5, Fig. 3B). In the presence of SCH-58261, Cl-IB-MECA increased coronary flow to only 140.59 ± 4.49% of the predrug baseline value (P < 0.05 vs. respective control), which was markedly lower than that produced by Cl-IB-MECA alone (Fig. 3B). Cl-IB-MECA-induced coronary responses both in the absence and presence of SCH-58261 were significantly different from vehicle controls (P < 0.05). This inhibition of Cl-IB-MECA-induced coronary vasodilation in A3KO hearts with selective A2A receptor blockade supports the conclusion that the observed Cl-IB-MECA-induced increase in coronary flow is mediated in part by A2A receptor activation.
Effect of genetic deletion of A2A receptor on Cl-IB-MECA-induced coronary flow in isolated mouse hearts.
To confirm that the Cl-IB-MECA-induced increase in coronary flow results from activation of A2A receptors, coronary vascular responses to Cl-IB-MECA were examined in hearts from both WT and A2AKO mice (Fig. 4). In WT hearts, Cl-IB-MECA increased coronary flow to 268.57 ± 10.09% of baseline, whereas this response was limited to only 150.89 ± 4.95% of baseline in A2AKO hearts (n = 5, P < 0.05 WT vs. A2AKO). This increase in coronary flow in both WT and A2AKO hearts was significantly different from vehicle control. Thus inhibition of Cl-IB-MECA-induced coronary vasodilation in A3KO hearts by pharmacological blockade of A2A receptors (Fig. 3B) was mimicked by targeted deletion of A2A receptors (Fig. 4), suggesting that this response is mediated in part by A2A receptors.
Fig. 4.
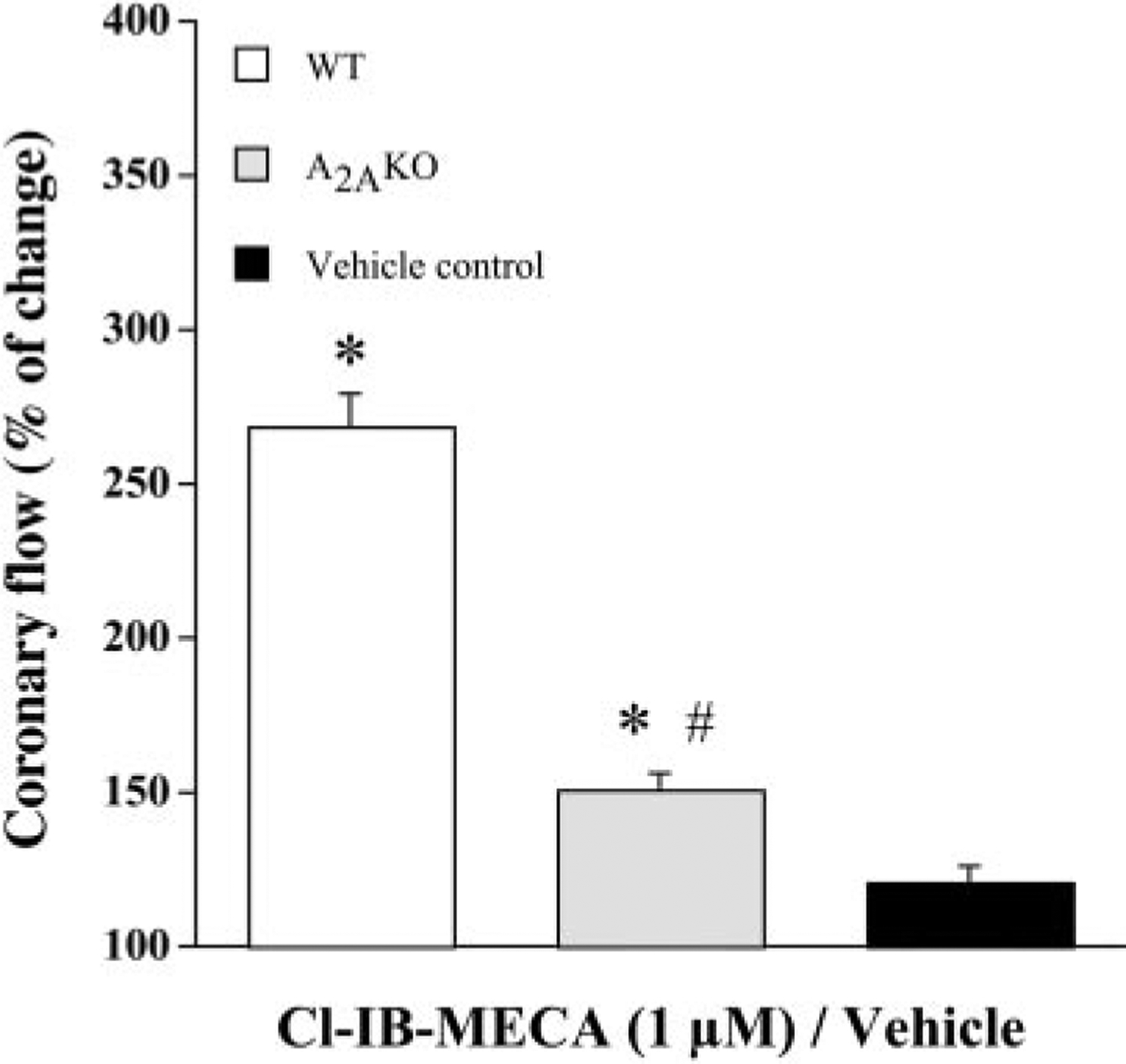
Influence of genetic deletion of adenosine A2A receptors on Cl-IB-MECA-induced increases in coronary flow. Cl-IB-MECA (1 μM)-induced increases in the coronary flow were determined in both WT and A2AKO hearts. Bars represent mean ± SE of 5 experiments from different hearts in each group. Vehicle control experiments were done in five hearts. *P < 0.05 vs. vehicle control; #P < 0.05 vs. respective WT hearts. For other details, see Fig. 3.
DISCUSSION
The primary intent of this study was to determine whether A3 receptor activation participates in adenosine-mediated changes in coronary flow in isolated murine hearts. Hearts with targeted deletion of A3 receptors demonstrate increased maximal coronary vasodilation in response to all agonists tested (adenosine, CGS-21680, and Cl-IB-MECA). Acute pharmacological blockade of either A3 or A2A receptors mimics the coronary vascular effect of genetic deletion of each of these respective receptor subtypes. Importantly, high concentrations of the A3 receptor agonist Cl-IB-MECA produce coronary vasodilation in A3KO hearts and the majority of this response can be inhibited by A2A receptor blockade. Taken together, these findings support the conclusion that adenosine A3 receptors participate in coronary flow regulation of isolated murine hearts via inhibition or negative modulation of A2A receptor-mediated coronary vasodilation.
We (34) have recently shown that adenosine-induced coronary vasodilation in isolated mouse hearts is predominantly mediated by activation of the A2A receptor subtype where a role for A2B receptors was suggested. Subsequent studies in A2AKO mice have confirmed that the A2B receptor subtype contributes to adenosine-induced coronary vasodilation (20). The physiological significance of A3 receptors in coronary vasculature is still unknown, although it has been reported to cause a peripheral vasoconstriction in hamster cheek arterioles (30).
Until recently, characterization of the physiological significance of A3 receptors has been hindered mainly due to the unavailability of selective A3 receptor antagonists (7). In the present study, we combined receptor knockout technology with the traditional pharmacological approach to determine whether A3 receptors modulate coronary flow. Because activation of A3 receptors by endogenous adenosine requires a high concentration (Ki values at A1, A2A, and A3 receptors are 3–30 nM, 1–20 nM, and >1 μM, respectively) (6), a highly potent A3 receptor agonist, Cl-IB-MECA, was chosen to isolate the effect of A3 receptors in WT and A3KO hearts. At concentrations selective for A3 receptors (nanomolar range), Cl-IB-MECA did not effect coronary flow in WT hearts (Fig. 1C). Instead, it increased coronary flow at micromolar concentrations where selectivity favors A2A receptors (12). A similar response was observed in A3KO hearts where Cl-IB-MECA increased coronary flow at micromolar concentrations (Fig. 1C). However, the maximal coronary vasodilation with Cl-IB-MECA was greater in A3KO hearts than in WT hearts (Fig. 1C). These findings suggest that selective activation of A3 receptors with Cl-IB-MECA (nanomolar range) has no effect on coronary flow of isolated mouse hearts as has been reported in isolated rabbit and rat hearts (16). Rather, it indicates that at higher concentrations, Cl-IB-MECA elicits coronary vasodilation by nonselective activation of a receptor subtype other than A3, possibly the A2A and/or A2B receptor subtype.
It is well documented that A1 receptor activation results in negative inotropic and antiadrenergic effects in hearts (6, 11, 32), and, recently, Headrick et al. (10) reported that adenosine-induced coronary vascular responses in mouse hearts remained the same both in WT and transgenic hearts overexpressing A1 receptor. Thus the A1 receptor has little or no influence on adenosine agonist-induced coronary vascular response in isolated mouse heart. Here, referring to our earlier report (19), after examining adenosine-induced coronary vascular responses in A2AKO hearts and blocking A2B receptors in A2AKO hearts, only A3 receptors remained as a possible candidate site for adenosine receptor agonists to modulate coronary vascular response in murine hearts. In the present study, the A3 receptor agonist Cl-IB-MECA did not affect coronary flow even at 100 nM, but at higher concentrations it increased coronary flow both in WT and A3KO hearts (Fig. 1). Because A2A receptor activation is predominantly responsible for coronary vasodilation across most species, including mice (20, 34), we examined in parallel the influence of pharmacological blockade and genetic deletion of A2A receptors on Cl-IB-MECA-induced coronary vasodilation. Pharmacological blockade of A2A-receptors in A3KO hearts (Fig. 3B) and genetic deletion of A2A receptors (Fig. 4) resulted in similar inhibition of Cl-IB-MECA-induced coronary vasodilation. This suggests that Cl-IB-MECA-induced increases in coronary flow in murine hearts (at higher concentrations) are mediated primarily by activation of A2A receptors, as has been reported in isolated rat hearts (16).
The most remarkable finding in the present study was that targeted deletion of A3 receptors resulted in an augmented maximal coronary flow in isolated hearts by all adenosine agonists (Fig. 1). The mechanisms by which deletion of A3 receptors augment this maximal response remain to be elucidated. Zhao et al. (35) have demonstrated that A3 receptors are functionally inhibitory through attenuation of adenosine-induced increases in cAMP in rat vascular smooth muscle cells. Recently, Zhao et al. (36) observed that steady-state levels of cAMP were elevated in aortas and hearts of A3KO mice compared with WT mice. Therefore, it is possible that the current finding of augmented coronary vasodilation in A3KO hearts results from removal of an inhibitory influence at the level of subcellular signaling pathway.
Several observations in the present study suggest that A3 receptor activation does not increase coronary flow; rather, it inhibits or negatively modulates A2A receptor-mediated coronary vasodilation. First, low concentrations of Cl-IB-MECA, the most selective and potent for A3 agonist, did not affect coronary flow in WT hearts even at a concentration of 100 nM (Fig. 1C). Second, Cl-IB-MECA increased coronary flow at micromolar (high) concentration in A3KO hearts where the maximal response was greater than in WT hearts (Fig. 1C). Third, pharmacological blockade and targeted deletion of A2A receptors equally blocked Cl-IB-MECA-induced increases in the coronary flow (Figs. 3B and 4). Finally, pharmacological blockade (Fig. 2) and targeted deletion of A3 receptors (Fig. 1B) significantly augmented the maximal coronary vasodilation-induced by the A2A receptor agonist CGS-21680, suggesting that A3 receptors indirectly participate in the regulation of coronary flow in isolated mouse heart.
In summary, the present study provides the first evidence that A3 receptors participate in the regulation of coronary flow in the isolated mouse heart. Selective activation of A3 receptors does not affect coronary flow, whereas targeted deletion of A3 receptors increases the maximal coronary flow mediated by adenosine receptor agonists. These findings suggest that A3 receptors either inhibit or negatively modulate coronary vasodilation mediated by other adenosine receptor subtypes.
Acknowledgments
The authors gratefully acknowledge the generous gift of SCH-58261 from Dr. A. Monopoli (Shearing Plough; Milan, Italy).
This work is supported by National Heart, Lung, and Blood Institute Grant HL-27339.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Abebe W, Makujina SR, and Mustafa SJ. Adenosine receptor-mediated relaxation of porcine coronary artery in presence and absence of endothelium. Am J Physiol Heart Circ Physiol 266: H2018–H2025, 1994. [DOI] [PubMed] [Google Scholar]
- 2.Belardinelli L, Shryock JC, Snowdy S, Zhang Y, Monopoli A, Lozza G, Ongini E, Olsson RA, and Dennis DM. The A2A adenosine receptor mediates coronary vasodilation. J Pharmacol Exp Ther 284: 1066–1073, 1998. [PubMed] [Google Scholar]
- 3.Brackett LE and Daly JW. Functional characterization of the A2B adenosine receptor in NIH 3T3 fibroblasts. Biochem Pharmacol 47: 801–814, 1994. [DOI] [PubMed] [Google Scholar]
- 4.Cerniway RJ, Yang Z, Jacobson MA, Linden J, and Matherne GP. Targeted deletion of A3 adenosine receptors improves tolerance to ischemia-reperfusion injury in mouse myocardium. Am J Physiol Heart Circ Physiol 281: H1751–H1758, 2001. [DOI] [PubMed] [Google Scholar]
- 5.Feoktistov I and Biaggioni I. Adenosine A2B receptors. Pharmacol Rev 49: 381–402, 1997. [PubMed] [Google Scholar]
- 6.Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, and Williams M. Nomenclature and classification of purinoceptors. Pharmacol Rev 46: 143–156, 1994. [PMC free article] [PubMed] [Google Scholar]
- 7.Guo Y, Bolli R, Bao W, Wu WJ, Black RG Jr, Murphree SS, Salvatore CA, Jacobson MA, and Auchampach JA. Targeted deletion of the adenosine A3 receptor confers resistance to myocardial ischemic injury and does not prevent early preconditioning. J Mol Cell Cardiol 33: 825–830, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gurden MF, Coates J, Ellis F, Evans B, Foster M, Hornby E, Kennedy I, Martin DP, Strong P, Vardey CJ, and Wheeldon A. Functional characterization of three adenosine receptor types. Br J Pharmacol 109: 693–698, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Headrick JP, Gauthier NS, Morrison RR, and Matherne GP. Cardioprotection by KATP channels in wild-type hearts and hearts overexpressing A1 adenosine receptors. Am J Physiol Heart Circ Physiol 279: H1690–H1697, 2000. [DOI] [PubMed] [Google Scholar]
- 10.Headrick JP, Gauthier NS, Morrison RR, and Matherne GP. Chronotropic and vasodilatory responses to adenosine and isoproterenol in mouse heart: effects of adenosine A1 receptor overexpression. Clin Exp Pharmacol Physiol 27: 185–190, 2000. [DOI] [PubMed] [Google Scholar]
- 11.Hori M and Kitakaze M. Adenosine, the heart, and coronary circulation. Hypertension 18: 565–574, 1991. [DOI] [PubMed] [Google Scholar]
- 12.Jacobson KA. Adenosine A3 receptors: novel ligands and paradoxical effects. Trends Pharmacol Sci 19: 184–191, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacobson KA, Park KS, Jiang JL, Kim YC, Olah ME, Stiles GL, and Ji XD. Pharmacological characterization of novel A3 adenosine receptor-selective antagonists. Neuropharmacology 36: 1157–1165, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kemp BK and Cocks TM. Adenosine mediates relaxation of human small resistance-like coronary arteries via A2B receptors. Br J Pharmacol 126: 1796–1800, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim YC, Ji XD, and Jacobson KA. Derivatives of the triazoloquinazoline adenosine antagonist (CGS15943) are selective for the human A3 receptor subtype. J Med Chem 39: 4142–4148, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lasley RD, Narayan P, Jahania MS, Partin EL, Kraft KR, and Mentzer RM Jr. Species-dependent hemodynamic effects of adenosine A3 receptor agonists IB-MECA and Cl-IB-MECA. Am J Physiol Heart Circ Physiol 276: H2076–H2084, 1999. [DOI] [PubMed] [Google Scholar]
- 17.Ledent C, Vaugeois JM, Schiffman SN, Pedrazzii T, El Yacoubi M, Vanderhaeghen JJ, Costentin J, Heath JK, Vassart G, and Parmentier M. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2A receptor. Nature 388: 674–678, 1997. [DOI] [PubMed] [Google Scholar]
- 18.Lewis CD and Hourani SMO. Involvement of functional antagonism in the effects of adenosine antagonists and l-NAME in the rat isolated heart. Gen Pharmacol 29: 421–427, 1997. [DOI] [PubMed] [Google Scholar]
- 19.Makujina SR, Sabouni MH, Bhatia S, Douglas FL, and Mustafa SJ. Vasodilatory effects of adenosine A2 receptor agonists CGS 21680 and CGS 22492 in human vasculature. Eur J Pharmacol 221: 243–247, 1992. [DOI] [PubMed] [Google Scholar]
- 20.Morrison RR, Talukder MAH, Ledent C, and Mustafa SJ. The cardiac effects of adenosine in A2A receptor knockout hearts: uncovering A2B receptors. Am J Physiol Heart Circ Physiol 282: H437–H444, 2002. [DOI] [PubMed] [Google Scholar]
- 21.Mubagwa K, Mullane K, and Flameng W. Role of adenosine in the heart and circulation. Cardiovasc Res 32: 797–813, 1996. [PubMed] [Google Scholar]
- 22.Müller CE and Stein B. Adenosine receptor antagonist: structures and potential therapeutic applications. Curr Pharm Des 2: 501–530, 1996. [Google Scholar]
- 23.Mustafa SJ and Abebe W. Coronary vasodilation by adenosine: receptor subtypes and mechanism(s) of action. Drug Dev Res 39: 308–313, 1996. [Google Scholar]
- 24.Mustafa SJ and Askar AO. Evidence suggesting an Ra-type adenosine receptor in bovine coronary arteries. J Pharmacol Exp Ther 232: 49–56, 1985. [PubMed] [Google Scholar]
- 25.Ongini E. SCH 58261: a selective A2A adenosine receptor antagonist. Drug Dev Res 42: 63–70, 1997. [Google Scholar]
- 26.Ongini E, Dionisotti S, Gessi S, Irenius E, and Fredholm BB. Comparison of CGS 15943, ZM 241385 and SCH 58261 as antagonists at human adenosine receptors. Arch Pharm (Weinheim) 359: 7–10, 1999. [DOI] [PubMed] [Google Scholar]
- 27.Ramagopal MV, Chitwood RWJ, and Mustafa SJ. Evidence for an A2 adenosine receptor in human coronary arteries. Eur J Pharmacol 151: 483–486, 1988. [DOI] [PubMed] [Google Scholar]
- 28.Salvatore CA, Tilley SL, Latour AM, Fletcher DS, Koller BH, and Jacobson MA. Disruption of the A3 adenosine receptor gene in mice and its effect on stimulated inflammatory cells. J Biol Chem 275: 4429–4434, 2000. [DOI] [PubMed] [Google Scholar]
- 29.Schaick EAV, Jacobson KA, Kim HO, Ijzerman AP, and Danhof M. Hemodynamic effects and histamine release elicited by the selective adenosine A3 receptor agonist 2-Cl-IB-MECA in conscious rats. Eur J Pharmacol 308: 311–314, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shepherd RK, Linden J, and Duling BR. Adenosine-induced vasoconstriction in vivo. Role of the mast cell and A3 adenosine receptor. Circ Res 78: 627–634, 1996. [DOI] [PubMed] [Google Scholar]
- 31.Shin HK, Shin YW, and Hong KW. Role of adenosine A2B receptors in vasodilation of rat pial artery and cerebral blood flow autoregulation. Am J Physiol Heart Circ Physiol 278: H339–H344, 2000. [DOI] [PubMed] [Google Scholar]
- 32.Shryock JC and Belardinelli L. Adenosine and adenosine receptors in the cardiovascular system: biochemistry, physiology, and pharmacology. Am J Cardiol 79: 2–10, 1997. [DOI] [PubMed] [Google Scholar]
- 33.Sutherland FJ and Hearse DJ. The isolated blood and perfusion fluid perfused heart. Pharm Res 41: 613–627, 2000. [DOI] [PubMed] [Google Scholar]
- 34.Talukder MAH, Morrison RR, and Mustafa SJ. Comparison of the vascular effects of adenosine in isolated mouse heart and aorta. Am J Physiol Heart Circ Physiol 282: H49–H57, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Zhao Z, Francis CE, and Ravid K. An A3-subtype adenosine receptor in highly expressed in rat vascular smooth muscle cells: its role in attenuating adenosine-induced increas in cAMP. Microvasc Res 54: 243–252, 1997. [DOI] [PubMed] [Google Scholar]
- 36.Zhao Z, Makaritsis K, Francis CE, Gavras H, and Ravid K. A role for A3 adenosine receptor in determining tissue levels of cAMP and blood pressure: studies in knock-out mice. Biochem Biophys Acta 1500: 280–290, 2000. [DOI] [PubMed] [Google Scholar]
- 37.Zocchi C, Ongini E, Conti A, Monopoli A, Negretti A, Baraldi PG, and Dionisotti S. The non-xanthine heterocyclic compound SCH 58261 is a new potent and selective A2A adenosine receptor antagonist. J Pharmacol Exp Ther 276: 398–404, 1996. [PubMed] [Google Scholar]