

Abstract
CARM1 is amplified or overexpressed in many cancer types and its overexpression correlates with poor prognosis. Potent small molecule inhibitors for CARM1 have been developed, but the cellular efficacy of the CARM1 inhibitors is limited. We herein report the development of the proteolysis targeting chimera (PROTAC) for CARM1 which contains a CARM1 ligand TP-064, a linker, and a VHL E3 ligase ligand. Compound 3b elicited potent cellular degradation activity (DC50 = 8 nM and Dmax > 95%) in a few hours. Compound 3b degraded CARM1 in VHL- and proteasome-dependent manner and was highly selective for CARM1 over other protein arginine methyltransferases. CARM1 degradation by 3b resulted in potent downregulation of CARM1 substrate methylation and inhibition of cancer cell migration in cell-based assays. Thus, CARM1 PROTACs can be used to interrogate CARM1’s cellular functions and potentially be developed as therapeutic agents for targeting CARM1-driven cancers.
Keywords: PROTAC, TPD, CARM1, PRMT4, SAR
Graphical Abstract
Introduction
Methylation is one of the major post-translational modifications catalyzed by nine protein arginine methyltransferases (PRMTs) in mammalian cells.1 PRMTs catalyze methyl group transfer from the S-adenosyl-L-methionine (SAM) to the two terminal nitrogen atoms of a protein arginine. They were classified into three types based on their enzymatic activities. The largest class Type I enzymes including PRMT1–4, 6, and 8, form asymmetric dimethylarginine (ADMA); Type II enzymes including PRMT 5 and 9 form symmetric dimethylarginine (SDMA); and Type III enzyme PRMT7 catalyzes mono-methylation of arginine. PRMT4, also known as coactivator-associated arginine methyltransferase 1 (CARM1), was first discovered as a nuclear hormone receptor coactivator through its association with p160 coactivators.2 Through methylating histone H3R17 and H3R26 and numerous non-histone substrates, CARM1 is involved in various cellular processes.3
CARM1 is amplified or overexpressed in many cancer types including breast cancer.4 Among breast cancer subtypes, CARM1 is overexpressed in triple-negative breast cancer, which is typically associated with poor outcome.5,6 The oncogenic functions of CARM1 are strongly associated with its methylation of non-histone substrates. For example, methylation of BAF155, a subunit in the SWI/SNF complex, not only turns on massive oncogene expression but also promotes tumor metastasis via an inhibiting antitumor immune response.7 Overexpressing CARM1 in transgenic mouse models augmented mammary gland tumorigenesis in MMTV-Neu mice.8 Using a genetically engineered CARM1 knockout (KO) cell line model, we demonstrated that CARM1 KO significantly decreased the growth and invasion of breast cancer cells in vitro and in vivo, reinforcing that CARM1 is an important oncogene during breast cancer progression.7,9
Since 2008, potent small-molecule CARM1 inhibitors have been reported but none has moved to clinical investigation. While most of them are designed as substrates competitive inhibitors,10–22 two of them are SAM competitors.23,24 Although these inhibitors elicit low IC50 in in vitro assays, high doses of the inhibitors are needed to inhibit the enzymatic activity of CARM1 in cell-based assays.19 Moreover, single-cell transcriptome analysis revealed the remarkable difference between inhibiting CARM1 and genetic knockout of CARM1 in cancer cells.23 In breast cancer cells, CARM1 knockout, but not CARM1 inhibition, decrease cancer cell growth.7,25 These studies suggest that CARM1 has non-enzymatic roles in driving cancer progression which necessitates the development of small molecule degraders of CARM1.
Proteolysis targeting chimera (PROTAC) is a transformative therapeutic paradigm to engage our body’s natural protein disposal system for targeting disease-causing proteins with small molecules.26–28 PROTACs consist of heterobifunctional small molecules bonded together via an appropriate linker.29 The bifunctional nature in this case is defined by one end of the PROTAC binding with high selectivity to an E3 ubiquitin ligase while the other end simultaneously engages the target protein. As the binding event occurs, the exposed lysine residues of the target protein are brought in close proximity to the E3 ligase complex. As a result, the protein is poly-ubiquitinated and degraded by the proteasome. The PROTAC is released to continue its catalytic activity for targeted protein degradation. The recent discovery of potent small molecule E3 ligase ligands, including thalidomide analogs for cereblon (CRBN) and hydroxyproline derivatives for Von Hippel–Lindau (VHL), led to the surge of small-molecule PROTACs with high potency in cells since 2015.30–33 Degraders of disease-causing proteins have a number of potential advantages over inhibitors of oncogenic proteins. The occupancy-driven pharmacology of inhibitors requires stoichiometric drug binding to the oncogenic protein to inhibit the function of the protein. In contrast, induced protein degradation by PROTACs is event-driven and catalytic, providing favorable pharmacology.16 For example, it has been reported that a number of PROTAC degraders induce sustained tumor regression even after cessation of the PROTAC treatment.34–38 Degraders based on inhibitors may still be effective for cancers that are resistant to the corresponding inhibitors in the case of target protein mutations. Mutations could reduce the binding affinity of inhibitors thus rendering drug resistance. Protein degrader is less sensitive to the decreased binding affinity as compared with the occupancy-driven inhibitors.39–42
Recently, Jin’s group reported a PROTAC for Type II enzyme PRMT5 as the first chemical degrader for PRMT enzymes43. To the best of our knowledge, CARM1 degrader has not been reported to date. Herein we describe the design, synthesis, and characterization of the first CARM1 PROTAC. We showed that CARM1 PROTAC 3b could promote rapid CARM1 degradation in two hours with a DC50 around 8 nM and a Dmax > 95%. Western blotting and proteomics analyses reveal that 3b does not induce other PRMT degradation and potently inhibits CARM1 substrate methylation in breast cancer cells. As a result, 3b inhibits breast cancer cell migration. CARM1 PROTACs may offer pharmacological advantages over inhibitors for the treatment of CARM1-driven cancers. The CARM1 degraders can also be used as chemical probes for studying the non-enzymatic functions of CARM1.
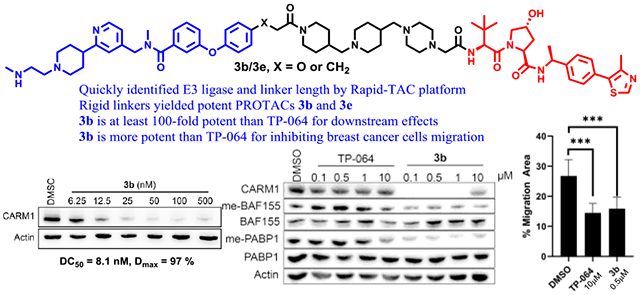
Results and Discussion
We designed the CARM1 PROTAC based on TP-064, a CARM1 small molecule inhibitor because TP-064 has a strong binding affinity and high selectivity for CARM1.21 To quickly identify the appropriate E3 ubiquitin ligase and the approximate linker length, we decided to utilize our Rapid-TAC platform to generate the first set of PROTACs by coupling a hydrazide-containing CARM1 inhibitor with the aldehyde group in our pre-assembled PROTAC library.44–46. After examining the crystal structure of CARM1 in complex with TP-064, the hydrazide moiety was placed on the para position of the terminal benzene, which is one of the solvent-exposed regions (Figure 1). Compound 1, TP-064 hydrazide derivative, was then prepared. Following our published procedures, six VHL ligand-based and six CRBN ligand-based PROTACs were quickly prepared in DMSO under miniaturized conditions. These PROTACs were tested in MCF7 cells at single concentrations (10 μM) for their ability to degrade CARM1. Western blot analysis revealed that VHL ligand based PROTACs induced obvious degradation of CARM1 in MCF7 cells (Figure S1A). A dose-response curve was generated for TPVC3 (VHL ligand and n=3), the most potent degrader. Significant CARM1 degradation was observed at 1.0 and 10 μM (Figure S1B).
Figure 1.
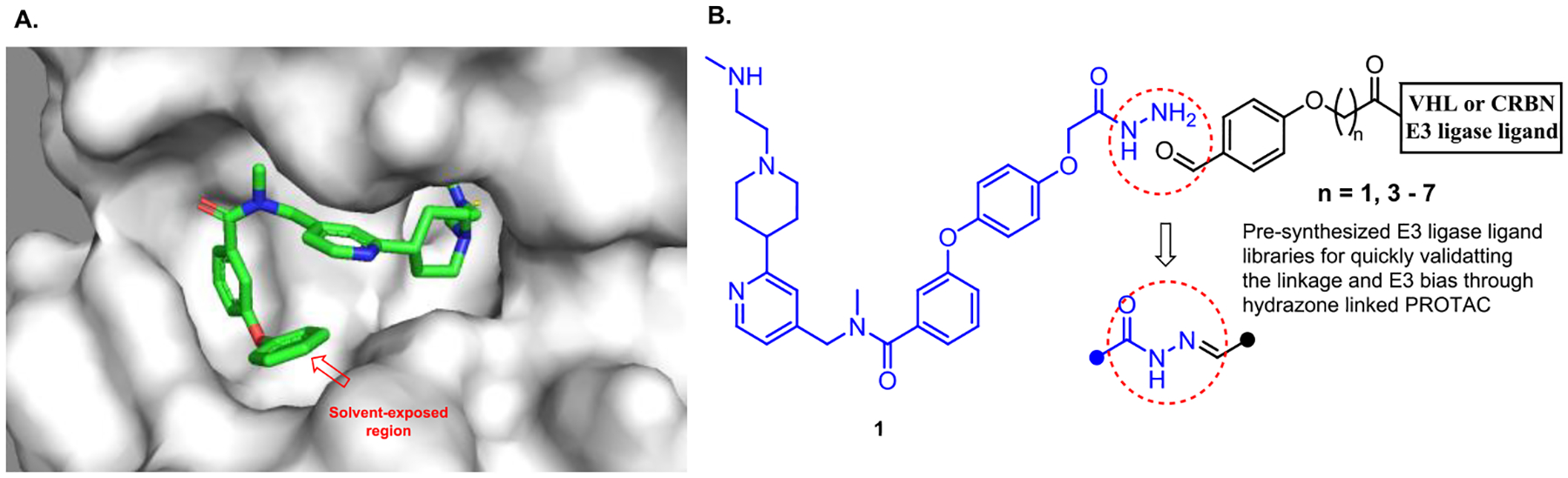
Quick identification of the linker length and E3 ubiquitin ligase for CARM1 PROTAC. (A) Crystal structure of CARM1 (gray) in complex with TP-064 (multicolor) (PDB: 5U4X). The highlighted phenyl ring of TP-064 is solvent-exposed. (B) The initial PROTAC library was derived from the coupling of the CARM1 ligand with a hydrazide functional group and a partial PROTAC library with a benzaldehyde functional group.
Based on the structure of TPVC3, CARM1 degraders with stable and flexible alkyl linkages 2a-e were designed and synthesized. Degrader 2b bears the same linker length as TPVC3, while 2a has one less carbon. However, we also changed the hydrolytic labile C=N double bond, which is part of the acylhydrazone motif, in TPVC3 to a C-C bond in compound 2b. This change may influence the required optimal linker length. To cover a wider range of linkers, we next prepared degraders 2c, 2d, and 2e, which possess one, two, and three more carbon atoms, respectively, than compound 2b (Figure 2A). Western blot assays were performed to evaluate the CARM1 degradation activity of the compounds after treating MCF7 cells with these degraders at two concentrations (0.5 and 1.0 μM) for 24 h. Compounds 2d and 2e showed stronger CARM1 degradation activities than 2a, 2b, and 2c (Figure 2B). Based on the structure-activity relationship (SAR) of these CARM1 degraders with alkyl linkages, we next designed and prepared a series of degraders with more rigid linkers to further improve the potency and potentially other pharmacological properties. Compound 3a has two heterocycles including a piperazine ring and a piperidine ring. It has one less carbon atom than 2a in terms of length. Compound 3b has one piperazine ring and two piperidine rings. It has the same linker length as 2d. MCF7 cells were then treated with 3a and 3b at four concentrations (0.1, 0.5, 1.0, and 10.0 μM) for 24 hours and compared with 2e, the best compound with a flexible linker, at 0.5 μM. Western blot results showed that 3b possessed significantly improved CARM1 degradation activity (Figure 2C). To fine-tune the linker length and to avoid the potential metabolic liability of having two para-substituted oxygen atoms on one benzene ring, compounds 3c, 3d, and 3e were designed and synthesized. The terminal phenyl group in TP-064 was conjugated with piperidine by benzamide, benzeneacetamide, and 3-phenylpropanamide groups, respectively (Figure 2A). After treating MCF7 cells with 0.5 μM of degraders 3b, 3c, 3d, or 3e, for 24 h, compounds 3b and 3e showed similar CARM1 degradation activity as expected since they have the same linker length. Interestingly, compounds 3d and 3c, which bear one and two less carbon atoms, respectively, than 3e, displayed significantly lower CARM1 degradation activities. The sharp change of CARM1 degradation activity with just two atoms difference between compounds 3c and 3b or compounds 3c and 3e (Figure 3D) is striking, indicating that the linker length is critical for positioning CARM1 to the proteasome for degradation.
Figure 2.
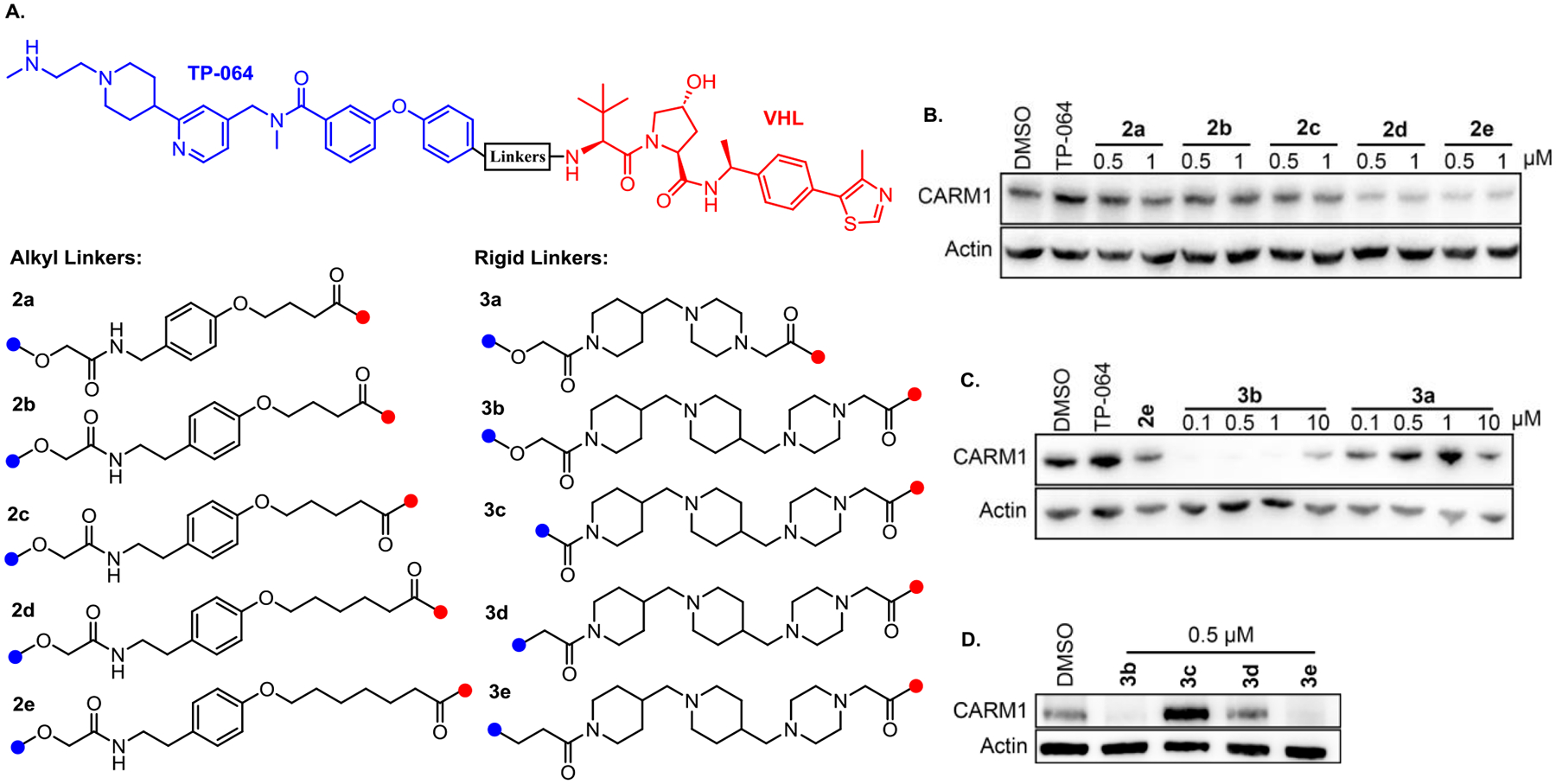
Structural activity relationship of CARM1 degraders. (A) Chemical structures of CARM1 degraders, 2a-2e bearing a flexible alkyl linker, 3a-3e bearing a rigid linker that contains the combination of piperidine and piperazine rings. (B) and (C) MCF7 cells were treated with the compounds at the indicated concentrations for 24 h. Cell lysates were collected and the CARM1 levels were detected by western blots. The results are representative of three independent experiments. (D) MCF7 cells were treated with DMSO and 0.5 μM of 3b, 3c, 3d, and 3e for 24 h.
Figure 3.
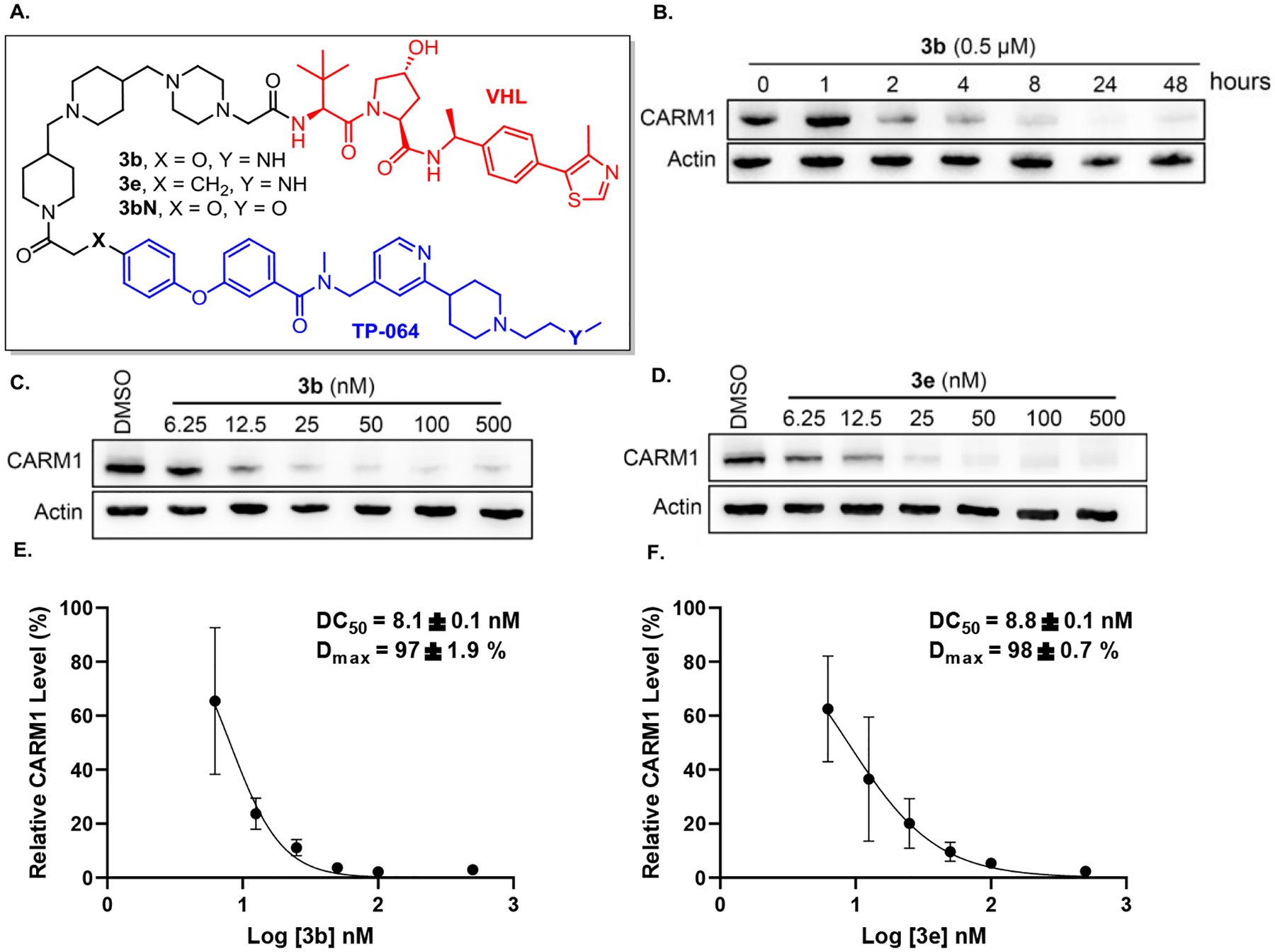
Degradation activities of compounds 3b and 3e. (A) Chemical structure of 3b, 3e and 3bN. (B) MCF7 cells were treated with the compounds at 0.5 μM for indicated time. (C) and (D) MCF7 cells were treated with the compounds at the indicated concentrations for 24 h. Cell lysates were collected and the CARM1 levels were detected by western blots. The results are representative of three independent experiments. (E) and (F) DC50 and Dmax were plotted and calculated based on three independent biological triplicate western blot assays.
The most potent degraders 3b and 3e were then selected for further evaluation. We performed a time course study for CARM1 degrader 3b. CARM1 degradation was observed as soon as 2 h after exposing to 3b, and degradation sustained and further maximized over the course of 48 h (Figure 3B). Next, dose-response curves were measured for degraders 3b and 3e. In MCF7 cells, 3b and 3e showed high potency for CARM1 degradation, with DC50 and Dmax of 8.1 ± 0.1 nM, 97 ± 1.9% and 8.8 ± 0.1 nM, 98 ± 0.7%, respectively (Figure 3C–F). Compound 3b also showed high potency for CARM1 degradation in MCF10A cells, a normal human mammary epithelial cell model (Figure S2). We then validated the mechanisms of action of these degraders using competitive inhibitors of CRL2VHL ligase complex and the 26S proteasome (Figure 4A). Pretreatment of the MCF7 cells with CARM1 inhibitor TP-064, VHL ligand VH-032, the proteasome inhibitor MG132, or neddylation inhibitor MLN492437 all abrogated 3b-induced CARM1 degradation, confirming that CARM1 degradation by 3b engages CARM1, VHL, the proteasome, and the Cullin-RING E3 ligase complex. We also prepared 3bN, a negative control compound, which cannot bind to CARM1 due to the lack of secondary amine (Y = O instead of NH) as reported in the literature.21 As expected, no CARM1 degradation was observed for 3bN in MCF7 cells (Figure 4B). To evaluate the selectivity of degrader 3b, PRMT1, PRMT6, and PRMT5 levels were measured by Western blot. In contrast to 3b induced CARM1 degradation with an 8.1 nM of DC50 and 97% of Dmax (Figure 3C), PRMT1, PRMT6, and PRMT5 degradation were not detected in MCF7 cells under the same conditions (Figure 4C–D). In order to systematically assess the alterations in the proteome caused by 3b, we performed a global quantitative proteomic analysis of MCF7 cells using mass spectrometry. The cells were treated with DMSO, 25 nM 3b or 3bN for 4 h. The treatment dose and time were determined based on time-course and dose titration experiments (Figure 3B–C). In total, 3491 proteins were quantified among three cohorts. Only 45 and 25 significantly changed proteins were identified in the 3bN and 3b groups as compared to the DMSO group with two-fold cutoff, respectively (p-value ≤ 0.05). As expected, the protein CARM1 demonstrated significant downregulation specifically in the 3b group and not in the 3bN group (Figure 4D). Two other PRMTs, PRMT1, and PRMT5, were detected in proteomics analyses but their levels were not significantly changed by 3b, reinforcing the selectivity of 3b to CARM1. Notably, several proteins (e.g. FAM129B) were also found to be significantly downregulated, which aligns with previous findings in CARM1 KO cells.9 This suggests that these proteins are likely associated with the functions regulated by CARM1. Relevant mechanistic investigations are ongoing and will be reported in due course.
Figure 4.
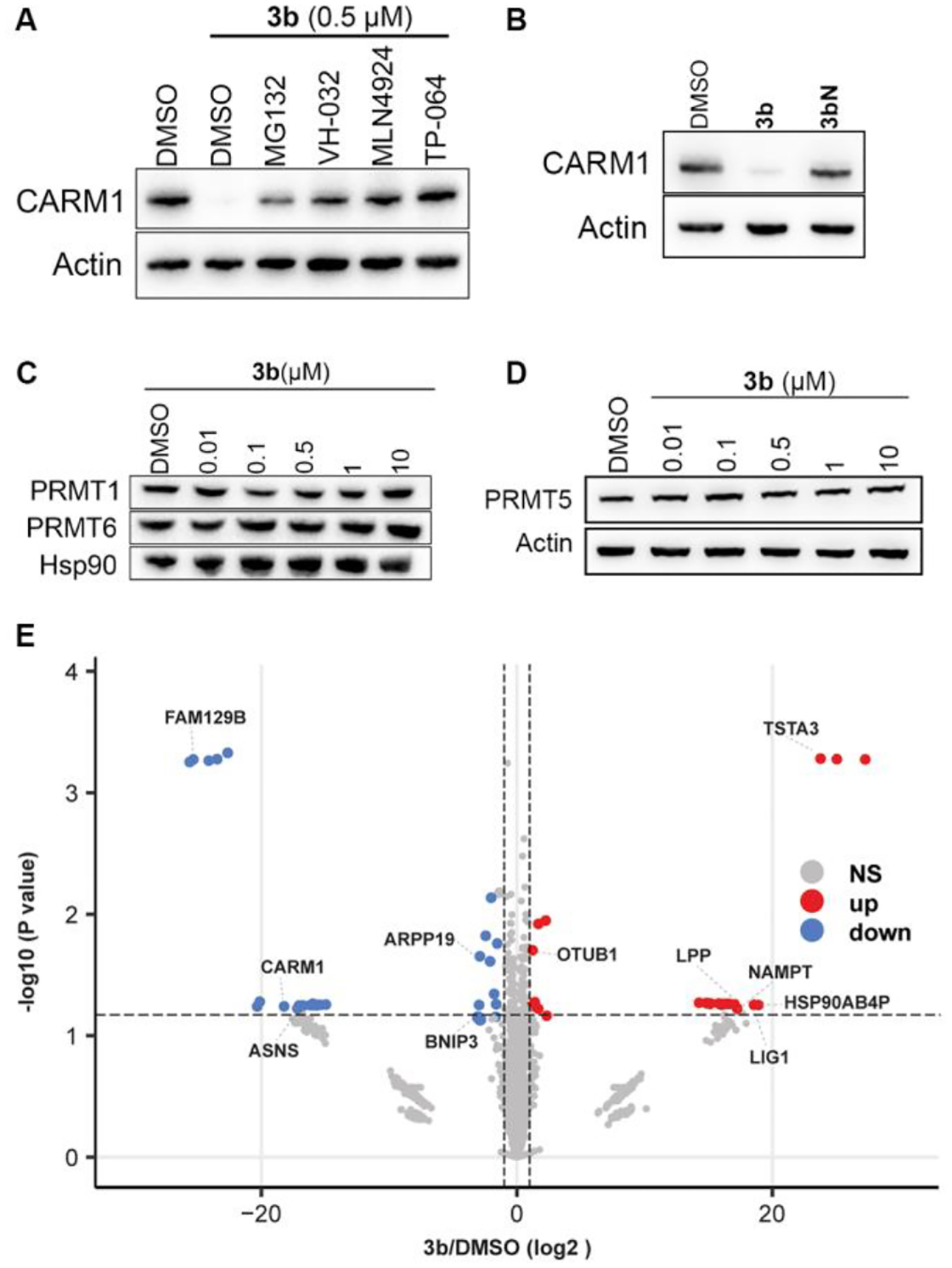
Mechanism and selectivity of degrader 3b. (A) MCF7 cells were co-treated with 0.5 μM of 3b and DMSO, 7.5 μM of MG132, 20 μM of VH-032, 2 μM of MLN4924, and 10 μM of TP-064 for 24 h. Cell lysates were collected and CARM1 levels were detected by Western blot. (B) MCF7 cells were treated with DMSO, 3b, or 3bN. Cell lysates were collected and CARM1 levels detected by Western blot. (C,D) MCF7 cells were treated with the indicated concentration of 3b for 24 h. Cell lysates were collected and PRMT1, PRMT6 and PRMT5 levels were detected by Western blot. (E) Volcano plots showed protein expression level changes for 3b versus DMSO group. Log2 protein fold changes are plotted against the negative log10 p-values. Proteins exhibiting significant alterations (p-value < 0.05, Student’s t-test) are represented by points above the non-axial horizontal line. Significantly down-regulated proteins are depicted in blue, while up-regulated proteins are depicted in red (protein |fold change| > 2).
To demonstrate the effectiveness of 3b to inhibit cellular protein methylation by CARM1, we selected CARM1-specific substrates Polyadenylate-binding protein 1 (PABP1) and BRG1-associated factor 155 (BAF155) for analyses because their methyl-specific antibodies are available.47,48 In fact, asymmetric dimethylation (me2a) of these proteins are routinely investigated for CARM1 inhibitors. Herein, the PABP1 and BAF155 di-methylation inhibitory effects in MCF7 cells by CARM1 inhibitor TP-064 and degrader 3b were compared side-by-side. As shown in Figure 5A, 3b is at least 100-fold more potent than TP-064 in terms of functional outcome. The levels of PABP1 and BAF155 me2a inhibition with 0.1 μM 3b are at least as equivalent to that of 10 μM of TP-064.49 The CARM1 degradation activity of 3b is not restricted to MCF7 cells, as CARM1 was also effectively degraded by 3b after 24 or 48 h in ER+/HER2+ BT474 and triple-negative MDA-MB-231 breast cancer cell lines. The decrease of BAF155 me2a level was observed after 48 h in both cell lines in TP-064 or 3b treated groups (Figure 5B, C).
Figure 5.
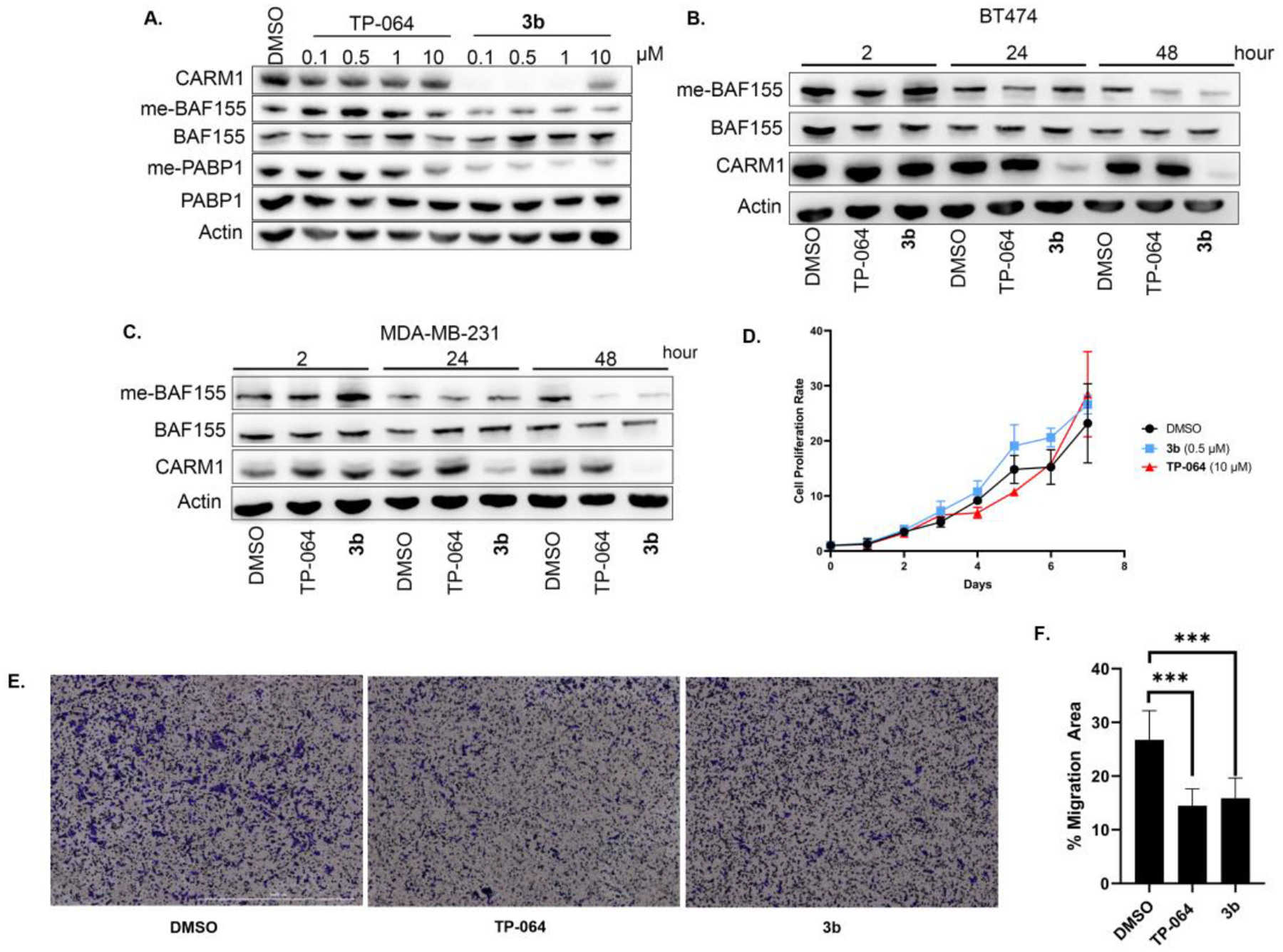
Biological effect comparison of inhibitor and degrader 3b in cell lines. (A) MCF7 cells were treated with indicated concentrations of TP-064 or 3b for 24 h. Cell lysates were collected and CARM1, methyl-BAF155, total BAF155, methyl-PABP1, and total PABP1 levels were detected by Western blot. (B) BT474 and (C) MDA-MB-231 cells were treated with DMSO, 10 μM of TP-064, and 0.5 μM of 3b for the indicated times. Cell lysates were collected and CARM1, methyl-BAF155, total BAF155 levels were detected by Western blot. (D) MDA-MB-231 cells were treated with DMSO, 0.5 μM of 3b, or 10 μM of TP-064 for 7 days. Cell proliferation rate normalized to day 0 was plotted. (E) and (F) Transwell cell migration assays after MDA-MB-231 cells were treated with DMSO, 0.5 μM 3b, or 10 μM TP-064. Migrated cells were stained with 1% crystal violet and the percentage of migrated cells were plotted. Data mean ± s.d. ***. P ≤ 0.001.
After confirming that 3b induces CARM1 degradation and inhibits CARM1 downstream enzymatic effect in different breast cancer cell lines representing three breast cancer subtypes, the effects of 3b on cell proliferation and migration were determined. CARM1 inhibitors were reported to have low cytotoxic effects in breast cancer cells, yet they significantly inhibited cell migration, an indicator of metastasis.25 Similarly, 3b was found not to inhibit cell proliferation in normal mammary epithelial cell line MCF10A and breast cancer cell lines MCF7 and MDA-MB-231 cells (Figure S3 and 5D). Because ER-positive breast cancer cells have low migratory effects and CARM1 is highly expressed in triple-negative breast cancer cells, we measured the effects of 3b on cell migration using a transwell migration assay for MDA-MB-231 cells. We found that 10 μM of TP-064 and 0.5 μM of 3b treatment inhibited cell migration at a similar level (Figure 5E, 5F). Compound 3b can induce the same anti-migratory effects with a 20-fold lower dose compared to the corresponding inhibitor, indicating that CARM1 degrader can be much more potent than the corresponding inhibitor in cell-based assays for eliciting the same biological effects.
The synthesis of Boc-protected CARM1 binders S2–S5 and a negative control S6 is summarized in Scheme 1. Secondary amine S1b is a common buidling block for the preparation of the right half of compounds S2–S6. The left half of compounds S2–S6 are dervied from carboxylic acid derivatives S2c, S3a, S4c, and S5c. First, cross-coupling of boronic ester with a bromo pyridine derivative afforded intermediate S1a, which can then undergo reductive amination to yield amine S1b. Carboxyclic acid S2c was prepared from p-iodophenol in four steps through standard alkylation, cross-coupling, saponification followed by acidification. Acid S3a was prepared by nucleophilic aromatic substitution (SNAr) reaction. Acid S4c was prepared in four steps through the sequence of SNAr, reduction, saponification followed by esterification. Finally, acid S5c was prepared by SNAr, olefination, saponification followed by esterification. The coupling of carboxylic acids S2c, S3a, S4c, and S5c with amine S1b afforded the corresponding amides, which can then undergo de-protection followed by reductive amination to yield key building blocks S2–S6.
Scheme 1.
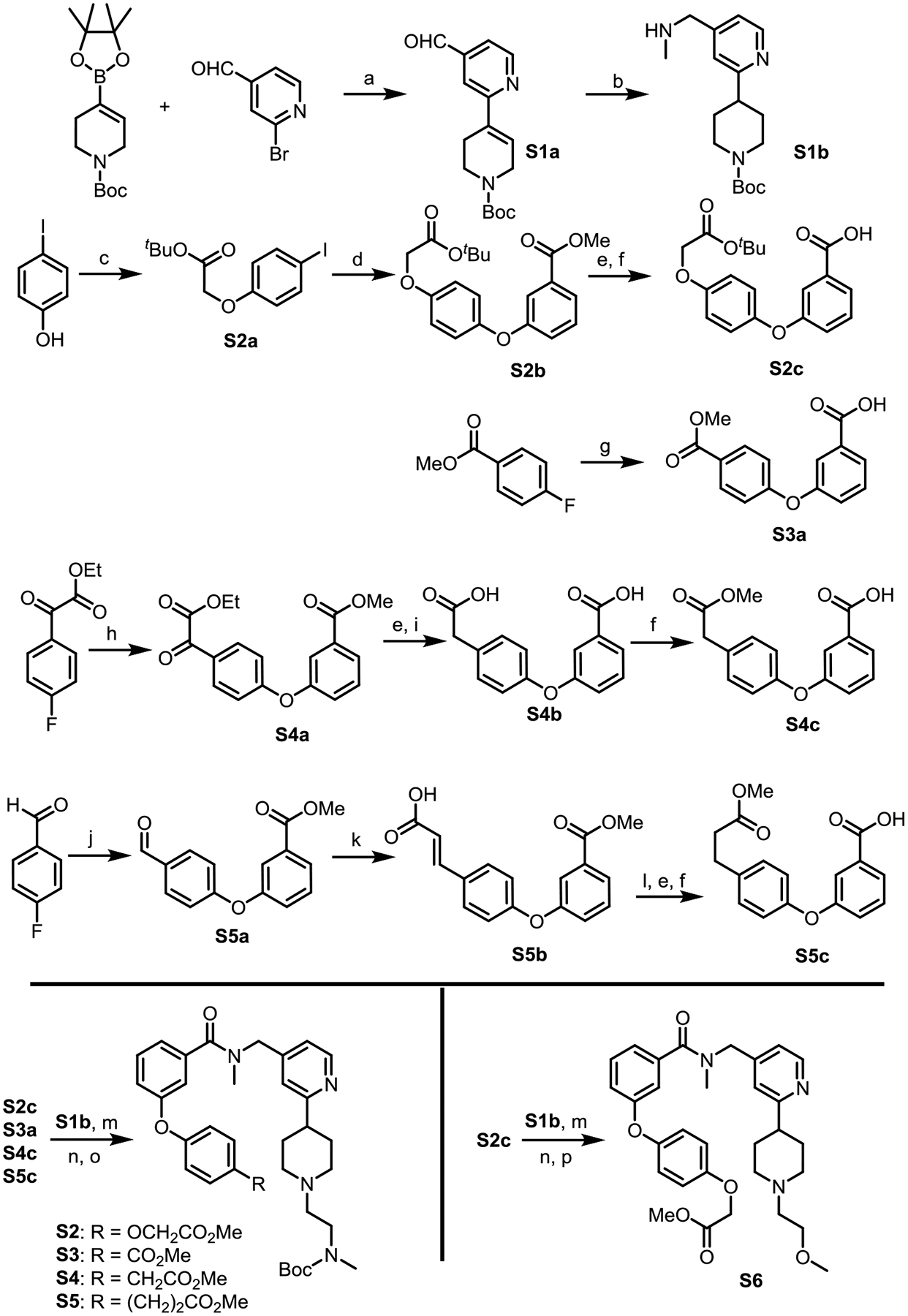
Synthesis of the functionalized CARM1 binder
Reagents and conditions: (a) Pd(dppf)Cl2, Na2CO3, DME/H2O (5/1), 80 °C, yield 59%; (b) MeNH2, Pd/C, H2 (1.0 atm), MeOH, rt; (c) tert-Butyl bromoacetate, K2CO3, KI, Acetone, 60 °C; (d) Methyl 3-hydroxybenzoic acid, CuI, dimethylglycine, Cs2CO3, dioxane, 90 °C, yield 56%; (e) NaOH, THF/MeOH/H2O, rt; (f) Amberlyst-15, MeOH, rt; (g) 3-Hydroxybenzoic acid, NaH (2.1 equiv), DMSO, 70 °C; (h) K2CO3 (2.1 equiv), DMSO, 70 °C; (i) Hydrazine hydrate, NaOH, Ethylene glycol, 160 °C; (j) Methyl 3-hydroxybenzoate, K2CO3 (2.1 equiv), DMSO, 130 °C; (k) Malonic acid, piperidine (10%), pyridine, reflux; (l) Pd/C, H2 (1.0 atm), MeOH, rt; (m) HATU, DIPEA, DMF, rt; (n) TFA/DCM, rt; (o) N-Boc-(methylamino)acetaldehyde, Pd/C, H2 (1.0 atm), MeOH, rt; (p) 2-Bromoethyl methyl ether, Cs2CO3, acetonitrile, rt.
The synthesis of CARM1 PROTACs is summarized in Scheme 2. First, linkers AL1+3 to AL2+6 were prepared by Boc-protection and alkylation from phenols with a remote amino group linked by different number of methylenes. Next, piperidine- and piperazine-containing rigid linkers RL1 and RL2 were prepared by alkylation followed by deprotection and reductive amination. Commercially available ligand Boc-VHL, linker AL1+3, and key intermediate S2 were linked together to form CARM1 PROTAC 2a via amide formation reactions followed by de-protection of the Boc-group. All other CARM1 PROTACs were prepared similarly by simply coupling the three pieces through amide bond formation.
Scheme 2.
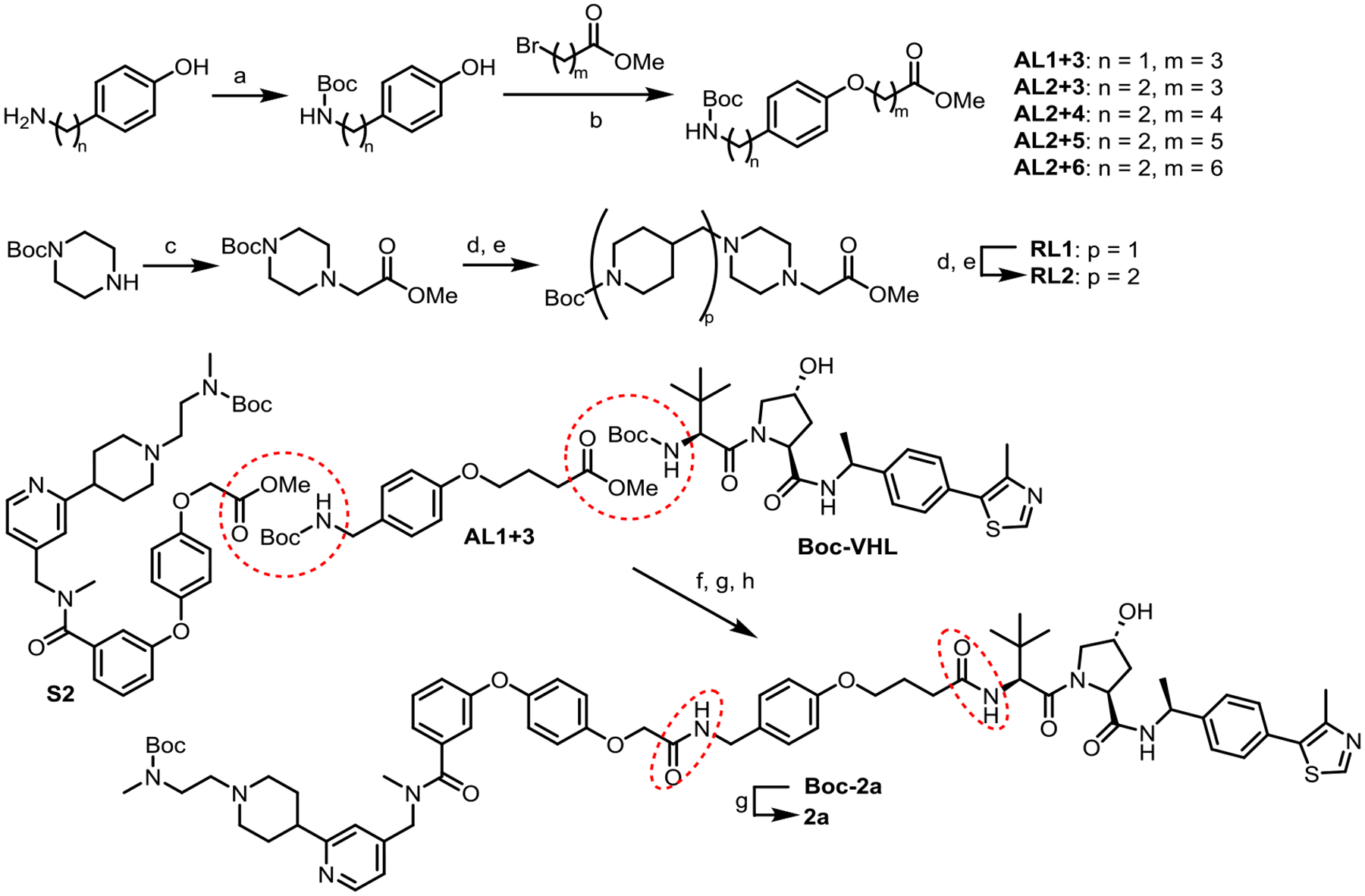
Synthesis of CARM1 PROTAC 2a (All other PROTACs were prepared similarly.)
Reagents and conditions: (a) Boc2O, Et3N, DCM, rt; (b) K2CO3, acetonitrile, 80 °C; (c) Methyl bromoacetate, DIPEA, acetonitrile, rt; (d) TFA/DCM, rt; (e) 1-Boc-piperidine-4-carboxaldehyde, Pd/C, DIPEA, H2 (1.0 atm), MeOH, rt; (f) NaOH, MeOH/THF/H2O, rt; (g) TFA/DCM, rt; (h) PyAOP, DIPEA, DMF, rt.
Conclusions
In summary, potent and selective CARM1 PROTACs were developed for the first time. Appropriate E3 ubiquitin ligase and linker length were quickly identified using our Rapid-TAC platform. Further optimization of the linker by replacing the flexible alkyl group with rigid piperidine and piperazine rings yielded potent PROTACs 3b and 3e with DC50 around 8 nM and Dmax over 95%. We are able to achieve more significant downstream effects (i.e. methylation of the substrates) using 100 times less concentrated degrader than the inhibitor. Functional studies indicated that CARM1 PROTACs could potently inhibit the migration of breast cancer cells. Further investigation of the non-enzymatic effect of CARM1 by comparing the CARM1 degrader and inhibitor is underway, and results will be reported in due course.
Experimental Sections
Western Blot Assay.
Cells were harvested and washed with Dulbecco’s phosphate-buffered saline (PBS), then lysed with 1X RIPA lysis buffer (50mM Tris pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) supplemented with 1mM phenylmethylsulfonyl fluoride, 10μg/mL aprotinin, 1μM leupeptin, and 10μg/mL pepstatin on ice for 20 min. The supernatant was collected after spinning down at 15000 RPM at 4 degrees Celsius for 10 min. The concentration of whole lysates was quantified using a Bradford assay. 50–75μg of cell lysates was boiled in 4x Laemmli Loading Dye and boiled at 100 degrees Celsius for 10min. 25 μg were run in 8% SDS-PAGE gels and transferred to nitrocellulose membranes. Blots were blocked in 5% skim milk in PBST [PBS and 0.1% Tween 20] for 45 min, then incubated with primary antibodies at 4 degrees Celsius overnight. Blots were washed in PBST for 30 min, then incubated with horseradish peroxidase-linked secondary antibodies (Jackson ImmunoResearch) for 1hr. Afterwards, blots were washed in PBST for 15–30 min, then treated with chemiluminescent ECL reagent and imaged with the ChemiDoc Imaging System (Bio-Rad) or Azure 600 (Azure Biosystems). Expression levels of the indicated proteins were probed by the following antibodies: anti-CARM1, anti-me-BAF155, anti-me-PABP1, anti-PABP1,7 anti-BAF155 (D7F8S, Cell Signaling Technology), anti-PRMT1 (Bethyl), anti-PRMT6 (D-5, Santa Cruz Biotechnology), anti-β-Actin (ABclonal Technology, Sigma-Aldrich), anti-Hsp90 (H-114, Santa Cruz Biotechnology).
Sample preparation for global proteomics analysis.
Sample preparation is similar to the previous report.50 Briefly, treated MCF7 cells were harvested by washing with PBS buffer (Gibco, pH 7.4) and lysed in RIPA buffer supplemented with protease inhibitors for 20 min, then protein concentration was determined using a BCA Protein Assay Kit (Thermo Pierce, Rockford, IL) per manufacturer instructions. Samples were reduced with 5mM DTT for 1hr and alkylated with 10 mM IAA for 30 min in the dark before quenching with 5 mM DTT. Proteins were digested by trypsin at 37°C for 16 hours in a 50:1 (protein: enzyme) ratio. Digests were quenched by lowering the pH to < 3 with 10% TFA. Peptides were desalted with SepPak C18 solid-phase extraction (SPE) cartridges (Waters, Milford, MA). The concentrations of the peptide mixture were measured by peptide assay (Thermo Fisher Scientific). All samples were dried in vacuo and stored at −80°C until LC-ESI-MS/MS analysis.
LC-MS/MS data acquisition.
Samples were analyzed on an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific) coupled to a Dionex UltiMate 3000 UPLC system. Each sample was dissolved in 0.1% formic acid in water before being loaded onto a 75 μm inner diameter homemade microcapillary column that is packed with 15 cm of Bridged Ethylene Hybrid C18 particles (1.7 μm, 130 Å, Waters) and fabricated with an integrated emitter tip. Mobile phase A was composed of water and 0.1% formic acid, while mobile phase B was composed of ACN and 0.1% formic acid. LC separation was achieved across a 137-min gradient elution of 3–30% mobile phase B at a flow rate of 300 nL/min. Survey scans of peptide precursors from m/z 300 to 1500 were performed at a resolving power of 60k (at m/z 200) with an AGC target of 2 × 105 and maximum injection time of 100 ms. The top 20 precursors were then selected for higher energy collisional dissociation fragmentation with a normalized collision energy of 30, an isolation width of 1.0 Da, a resolving power of 15k, and an AGC target of 1 × 104. Precursors were subject to dynamic exclusion for 45 s with a 10 ppm tolerance. Each sample was acquired in technical triplicates.
Data analysis.
Protein identification and quantification by MaxQuant (version 1.5.3.8) based database searching, using the integrated Andromeda search engine with FDR < 1% at peptide and protein levels. The tandem mass spectra were searched against the Homo sapiens reviewed database (version updated December 2023). A reverse database for the decoy search was generated automatically in MaxQuant. Enzyme specificity was set to ‘Trypsin/p’, and a minimum number of seven amino acids were required for peptide identification. For label-free protein quantification (LFQ), the MaxQuant LFQ algorithm was used to quantitate the MS signals, and the proteins’ intensities were represented in LFQintesity.51 Cysteine carbamidomethylation was set as the fixed modification. The oxidation of M and acetylation of the protein N-terminal were set as variable modifications. The first search mass tolerance was 20 ppm, and the main search peptide tolerance was 6 ppm. The false discovery rates of the peptide-spectrum match and proteins were set to less than 1%. For peptide quantification, the intensities of all samples were extracted from the MaxQuant result peptide files. Then, the expression matrix was subjected to normalization followed by log2-transformed by Perseus.52 From three technical replicates, the mean protein intensities were calculated for each biological replicate and subjected to statistical analysis. Bioinformatic analyses were performed with R software environment.53
Cell Proliferation Assay.
Cell proliferation was measured by direct cell count via high contrast brightfield microscopy. MDA-MB-231 cells (3 × 103 / well) were seeded in 100μL of DMEM into the wells of a 96-well plate. Cells were treated with either DMSO, TP-064, or 3b and refreshed every two days. The plate was imaged daily for 7 days using the High-Contrast Brightfield Kit for Label-Free Cell Counting (BioTek) on the BioTek Imager.
Transwell Migration Assay.
Cells were harvested and washed in serum-free DMEM. The wells of a 24-well plate were filled with 800μL of DMEM with 10% FBS. Transwell inserts with 8.0μM pore size were then added to the wells with DMEM. Cells were resuspended in serum-free DMEM, then 1 × 105 cells in 200μL were added to the upper chambers. Drug treatments were added to the lower chambers, then incubated for 20 h at 37°C. The cells on the upper surface were removed with cotton swabs. Migratory cells were fixed in 3.7% formaldehyde at room temperature, followed by 100% methanol at −20°C. Fixed cells were stained with 1% crystal violet in 20% methanol for 30 min. The migration area was quantified from five independent fields of view of the lower surface of the membrane under a microscope.
General Information in Synthetic Chemistry
All reactions were conducted under a positive pressure of dry argon in glassware that had been oven-dried prior to use. Anhydrous solutions of reaction mixtures were transferred via an oven-dried syringe or cannula. All solvents were dried prior to use unless noted. Thin-layer chromatography (TLC) was performed using precoated silica gel plates. Flash column chromatography was performed with silica gel. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on Bruker 400, 500, 600 MHz and Varian 500 MHz spectrometers. 1H NMR spectra were reported in parts per million (ppm) referenced to 7.26 ppm of CDCl3 or referenced to the center line of a septet at 2.50 ppm of DMSO-d6. Signal splitting patterns were described as singlet (s), doublet (d), triplet (t), quartet (q), quintet (quint), or multiplet (m), with coupling constants (J) in hertz. High-resolution mass spectra (HRMS) were performed on an electron spray injection (ESI) TOF mass spectrometer.
The liquid chromatography–mass spectrometry (LC–MS) analysis of final products was processed on an Agilent 1290 Infinity II LC system using a Poroshell 120 EC-C18 column (5 cm × 2.1 mm, 1.9 μm) for chromatographic separation. Agilent 6120 Quadrupole LC/MS with multimode electrospray ionization plus atmospheric pressure chemical ionization was used for detection. The mobile phases were 5.0% methanol and 0.1% formic acid in purified water (A) and 0.1% formic acid in methanol (B). The gradient was held at 5% (0–0.2 min), increased to 100% at 2.5 min, then held at isocratic 100% B for 0.4 min, and then immediately stepped back down to 5% for 0.1 min re-equilibration. The flow rate was set at 0.8 mL/min. The column temperature was set at 40 °C. The purities of all of the final compounds were determined to be over 95% by LC–MS. See the Supporting Information for 1H and 13C NMR spectra and LC–MS purity analysis of all compounds.
Procedure for the preparation of compound S1b.
To a 250 mL flask with a magnetic stirring bar, commercially available N-Boc-1,2,3,6-tetrahydropyridine-4-boronic acid pinacol ester (6.0 g, 19.4 mmol), 2-bromopyridine-4-carboxaldehyde (2.4 g, 12.9 mmol), Pd(dppf)Cl2 (512 mg, 0.63 mmol), Na2CO3 (3.4 g, 31.5 mmol), were sequentially added, evacuated, and backfilled with argon. Dimethoxyethane (DME) (30 mL) and water (6 mL) were successively added using a syringe. The reaction mixture was heated to 80 °C until the starting material 2-bromopyridine-4-carboxaldehyde disappeared, as indicated by TLC (~4 h). The mixture was partitioned between ethyl acetate and a saturated solution of sodium bicarbonate; the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide S1a as colorless oil. (2.2 g, yield of 59%).
A 250 mL flask was charged with a magnetic stirring bar and S1a (2.0 g, 6.94 mmol). Next, methanol (70 mL), palladium on carbon (10 wt. %) (0.4 g) and methylamine (40 wt. % solution in water) (1.4 mL) were added. A hydrogen balloon was connected with the reaction mixture through a needle. The reaction mixture was stirred at room temperature until the starting material S1a disappeared, as indicated by TLC. The mixture was filtered through Celite and washed with methanol (3 × 5 mL); the mixture was then concentrated in vacuum. The residue was purified by column chromatography on silica to provide S1b as colorless oil. (2.0 g, yield of 95%).
Procedure for the preparation of compound S2c.
To a 100 mL flask with a magnetic stirring bar, 4-Iodophenol (4.4 g, 20.0 mmol), tert-butyl bromoacetate (3.25 mL, 22.0 mmol), K2CO3 (3.04 g, 22.0 mmol), acetone (20 mL) was sequentially added. The reaction mixture was heated to 60 °C, until the starting material disappeared, as indicated by TLC. The mixture was partitioned between ethyl acetate and a saturated solution of sodium bicarbonate; the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide S2a as light-yellow solid. (6.4 g, yield of 95%).
In a 100 mL flask with a magnetic stirring bar, S2a (3.0 g, 9.0 mmol), methyl 3-hydroxybenzoic acid (5.0 g, 32.9 mmol), CuI (0.8 g, 4.2 mmol), dimethylglycine (1.0 g, 9.7 mmol), Cs2CO3 (7.8 g, 23.9 mmol) were sequentially added, evacuated, and backfilled with argon. Dioxane (20 mL) was added using a syringe. The reaction mixture was heated to 90 °C, until the starting material S2a disappeared, as indicated by TLC. The mixture was partitioned between ethyl acetate and a saturated solution of sodium bicarbonate; the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide S2b as solid. (1.8 g, yield of 56%).
In a 250 mL flask with a magnetic stirring bar, S2b (1.5 g, 4.2 mmol), tetrahydrofuran (20 mL), methanol (20 mL), sodium hydroxide solution (2.0 M) (20 mL) was sequentially added. The reaction mixture was stirred at room temperature, until the starting material S2b disappeared, as indicated by TLC. The mixture was diluted by 80 mL water, concentrated in vacuum to remove methanol and tetrahydrofuran. The aqueous was adjusted to pH 1.0 by 1.0 N HCl solution, white solid was precipitated, filtrated and dried under vacuum. In a 250 mL flask with a magnetic stirring bar, the white solid was dissolved in methanol (40 mL). Amberlyst-15 (15 g) was added. The reaction mixture was stirred at room temperature, until the starting material disappeared, as indicated by TLC. Amberlyst-15 was removed by filtration and washed by methanol (20 mL × 3). The methanol solution was concentrated in vacuum. The residue was purified by column chromatography on silica to provide S2c as solid. (0.69 g, yield of 54%).
Procedure for the preparation of compound S3a.
In a 250 mL flask with a magnetic stirring bar, 3-hydroxybenzoic acid (3.05 g, 22.0 mmol) was dissolved in 60 mL of dimethylsulfoxide. Sodium hydride (1.6 g, 42 mmol) was added in batches for half an hour. Next, methyl 4-fluorobenzoate (3.24 g, 21.0 mmol) was added. The reaction mixture was heated to 70 °C, until the starting material disappeared, as indicated by TLC. The mixture was diluted by 140 mL water, then adjusted to pH 1.0 by 1.0 N HCl solution. The white solid was precipitated, filtrated and dried under vacuum, S3a (3.7 g, yield of 65%) was obtained as white solid.
Procedure for the preparation of compound S4c.
In a 100 mL flask with a magnetic stirring bar, methyl 3-hydroxybenzoic acid (3.0 g, 19.7 mmol), ethyl 2-(4-fluorophenyl)-2-oxoacetate (4.0 g, 20.4 mmol), K2CO3 (5.6 g, 40.5 mmol) and dimethylsulfoxide (60 mL) were sequentially added. The reaction mixture was heated to 70 °C, until the starting material disappeared, as indicated by TLC. The mixture was partitioned between ethyl acetate and a saturated solution of sodium bicarbonate; the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide S4a as oil. (5.8 g, yield of 90%). S4a (5.7 g) was hydrolyzed following the hydrolysis procedure of the S2b, the diacid intermediate (4.0 g, yield of 80%) was obtained as white solid. In a 250 mL flask with a magnetic stirring bar, the diacid intermediate (3.0 g, 10.5 mmol), ethylene glycol (50 mL) and hydrazine monohydrate (3.28 mL, 52.5 mmol) were sequentially added. The reaction mixture was heated to 60 °C for an hour. Next, sodium hydroxide (4.2 g, 105 mmol) was added to the reaction mixture and heated to 150 °C for five hours. The mixture was cooled to room temperature and diluted by 100 mL water, then adjusted to pH 1.0 by 1.0 N HCl solution. The white solid was precipitated, filtrated and dried under vacuum, S4b (2.6 g, yield of 91%) was obtained as white solid. In a 250 mL flask with a magnetic stirring bar, the white solid S4b (1.3 g, 4.8 mmol) was dissolved in methanol (50 mL). Amberlyst-15 (13 g) was added. The reaction mixture was stirred at room temperature, until the starting material disappeared, as indicated by TLC. Amberlyst-15 was removed by filtration and washed by methanol (20 mL × 3). The methanol solution was concentrated in vacuum. The residue was purified by column chromatography on silica to provide S4c as solid. (1.0 g, yield of 73%).
Procedure for the preparation of compound S5c.
In a 100 mL flask with a magnetic stirring bar, methyl 3-hydroxybenzoic acid (1.52 g, 10.0 mmol), 4-fluorobenzaldehyde (1.3 g, 10.5 mmol), K2CO3 (2.9 g, 21 mmol) and dimethylformamide (10 mL) were sequentially added. The reaction mixture was heated to 80 °C, until the starting material disappeared, as indicated by TLC. The mixture was partitioned between ethyl acetate and a saturated solution of sodium bicarbonate; the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide S5a as oil. (2.4 g, yield of 92%). In a 100 mL flask with a magnetic stirring bar, S5a (1.3 g, 5.0 mmol), malonic acid (780 mg, 7.5 mmol), piperidine (0.15 mL, 1.5 mmol) and pyridine (25mL) were sequentially added. The reaction mixture was heated to 110 °C, until the starting material disappeared, as indicated by TLC. The mixture was concentrated in vacuum. The residue was purified by column chromatography on silica to provide S5b (1.1 g, yield of 74%) as white solid. In a 100 mL flask with a magnetic stirring, S5b (1.1 g, 3.7 mmol), palladium on carbon (10 wt. %) (0.1 g) and methanol (20 mL) were added. A hydrogen balloon was connected with the reaction mixture through a needle. The reaction mixture was stirred at room temperature until the starting material S5b disappeared, as indicated by TLC. The mixture was filtered through celite and washed with methanol (3 × 5 mL); the mixture was then concentrated in vacuum. Following the procedure of preparation S2c, the reduced intermediate was hydrolyzed and monomethylated, S5c (0.8 g, total yield of 72%) was obtained as white solid.
General procedure for the preparation of compound S2–S6.
In a 20 mL flask with a magnetic stirring bar, S1b (305 mg, 1.0 mmol), S2c (303 mg, 1.0 mmol), dimethylformamide (5 mL), N,N-diisopropylethylamine (0.45 mL, 2.5 mmol) and Hexafluorophosphate Azabenzotriazole Tetramethyl Uronium (460 mg, 1.2 mmol) were sequentially added. The reaction mixture was stirred at room temperature, until the starting material disappeared, as indicated by TLC. The mixture was partitioned between ethyl acetate and a saturated solution of sodium bicarbonate; the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide intermediate (492 mg, yield of 83%) as oil. The intermediate (492 mg, 0.83 mmol) was dissolved in mixture solvent (6 mL, 50% (vol/vol) trifluoroacetic acid in dichloromethane) at room temperature for half an hour. The mixture was concentrated in vacuum. The residue was partitioned between ethyl acetate and a saturated solution of sodium carbonate; the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The deprotected intermediate residue was dissolved by 4 mL of methanol and transferred to a 20 mL flask with a magnetic stirring, N-Boc-(methylamino)acetaldehyde (0.16 mL, 0.92 mmol), palladium on carbon (10 wt. %) (0.1 g) were sequentially added. A hydrogen balloon was connected with the reaction mixture through a needle. The reaction mixture was stirred at room temperature until the starting material disappeared, as indicated by TLC. The mixture was filtered through celite and washed with washed with methanol (3 × 10 mL). The methanol solution was concentrated in vacuum. The residue was purified by column chromatography on silica to provide S2 as oil. (321 mg, total yield of 60%). S3, S4 and S5 were prepared by the same procedure as S2. The deprotected intermediate (116 mg, 0.23 mmol) was dissolved by 1.0 mL of acetonitrile and transferred to a 10 mL vial with a magnetic stirring, 2-bromoethyl methyl ether (22 μL, 0.23 mmol), Cs2CO3 (370 mg, 1.1 mmol) were sequentially added. The reaction mixture was stirred at room temperature until the starting material disappeared, as indicated by TLC. The mixture was filtered through celite and washed with washed with ethyl acetate (3 × 10 mL). The solution was concentrated in vacuum. The residue was purified by column chromatography on silica to provide S6 as oil. (89 mg, yield of 75%).
General procedure for the preparation of linker AL1+3, AL2+3 to AL2+6.
In a 100 mL flask with a magnetic stirring bar, tyramine (1.89 g, 13.8 mmol), Di-tert-butyl decarbonate (3.18 g, 14.6 mmol), dichloromethane (20 mL), triethylamine (2 mL, 14.6 mmol) was sequentially added. The reaction mixture was stirred at room temperature, until the starting material disappeared, as indicated by TLC. The mixture was concentrated in vacuum. The residue was dissolved in ethyl acetate (80 mL) and sequentially washed with 1N HCl (2 × 30 mL), water and brine, dried over Na2SO4, and concentrated in vacuum. The residue (3.26 g, yield of 100%) was used directly for next step reaction without purification. In a 25 mL flask with a magnetic stirring bar, the Boc protected tyramine (680 mg, 2.86 mmol), K2CO3 (0.95 g, 6.87 mmol), acetonitrile (8 mL), methyl 4-bromobutyrate (0.44 mL, 3.44 mmol) were sequentially added. The reaction mixture was heated to 80 °C, until the starting material disappeared, as indicated by TLC. The mixture was partitioned between ethyl acetate and a saturated solution of sodium bicarbonate; the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to obtained AL2+3 (736 mg, yield of 76%) as white solid.
General procedure for the preparation of linker RL1 and RL2.
In a 250 mL flask with a magnetic stirring bar, N-boc-piperazine (11.0 g, 59.0 mmol), methyl bromoacetate (5.9 mL, 62.0 mmol), acetonitrile (80 mL), triethylamine (16.5 mL, 118 mmol) was sequentially added. The reaction mixture was stirred at room temperature, until the starting material disappeared, as indicated by TLC. The mixture was concentrated in vacuum. The residue was dissolved in ethyl acetate (100 mL) and sequentially washed water, saturated sodium carbonate and brine, dried over Na2SO4, and concentrated in vacuum. The residue (14.5 g, yield of 95%) was used directly for next step reaction without purification. The residue (2.3 g, 9.0 mmol) was dissolved in mixture solvent (10 mL, 50% (vol/vol) trifluoroacetic acid in dichloromethane) at room temperature for half an hour. The mixture was concentrated in vacuum. The deprotected intermediate residue was dissolved with 18 mL of methanol and transferred to a 100 mL flask with a magnetic stirring, 1-boc-piperidine-4-carboxaldehyde (2.1 g, 9.9 mmol), palladium on carbon (10 wt. %) (0.2 g), N,N-diisopropylethylamine (4.5 mL, 27.0 mmol) were sequentially added. A hydrogen balloon was connected with the reaction mixture through a needle. The reaction mixture was stirred at room temperature until the starting material disappeared, as indicated by TLC. The mixture was filtered through celite and washed with washed with methanol (3 × 20 mL). The methanol solution was concentrated in vacuum. The residue was purified by column chromatography on silica to provide RL1 as oil. (2.4 g, total yield of 76%).
General procedure for the preparation of 2a-2e, 3a-3e and 3bN (3e was used as an example).
In a 20 mL vial with a magnetic stirring bar, RL2 (272 mg, 0.6 mmol), methanol (2 mL), sodium hydroxide solution (2.0 N) (2 mL) was sequentially added. The reaction mixture was stirred at room temperature, until the starting material RL2 disappeared, as indicated by TLC. The mixture was adjusted to pH 7.0 with HCl (2.0 N) (2 mL) and concentrated in high vacuum. Boc-VHL was synthesized based on previously published procedures.44 In a 20 mL vial with a magnetic stirring bar, Boc-VHL (327 mg, 0.6 mmol) was dissolved in mixture solvent (6 mL, 50% (vol/vol) trifluoroacetic acid in dichloromethane) and stirred at room temperature until the starting material disappeared, as indicated by TLC. The mixture was concentrated in high vacuum. This Boc deprotected intermediate was dissolved in 3 mL of dimethylformamide and transfer to the vial contained hydrolyzed RL2, a magnetic stirring bar, N,N-diisopropylethylamine (0.43 mL, 2.4 mmol) (Adjust to pH 7.0), (7-Azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyAOP) (376 mg, 0.72 mmol) were sequentially added. The reaction mixture was stirred at room temperature, until the starting material disappeared, as indicated by TLC. The mixture was partitioned between ethyl acetate and a saturated solution of sodium carbonate; the organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica to provide intermediate RL2-VHL (379 mg, yield of 73%). Following the same procedure, S5 (15 mg, 0.023 mmol) was hydrolyzed; intermediate RL2-VHL (20 mg, 0.023 mmol) was deprotected the Boc; then amide coupling by PyAOP (13 mg, 0.025 mmol) afford Boc-3e (20 mg, yield of 63%). In a 20 mL vial with a magnetic stirring bar, Boc-3e (20 mg, 0.0145 mmol) was dissolved in mixture solvent (2 mL, 50% (vol/vol) trifluoroacetic acid in dichloromethane) and stirred at room temperature until the starting material disappeared, as indicated by TLC. The mixture was concentrated in high vacuum. The residue was redissolved in acetonitrile (2 mL), 1N HCl methanol solution (44 μL, 0.044 mmol) was added, white solid precipitation was observed. The solvent was removed by high vacuum, 3e (19 mg, 0.0145 mmol) was obtained in x·HCl salts form.
Compound Characterization Data
tert-butyl 4-(4-((methylamino)methyl)pyridin-2-yl)piperidine-1-carboxylate (S1b)
1H NMR (400 MHz, CDCl3) δ 8.45 (d, J = 5.0 Hz, 1H), 7.12 (s, 1H), 7.08 (dd, J = 5.1, 1.6 Hz, 1H), 3.75 (s, 2H), 2.95 – 2.71 (m, 4H), 2.46 (s, 3H), 2.0 – 1.81 (m, 3H), 1.80 – 1.60 (m, 3H), 1.46 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 164.7, 154.9, 149.5, 149.4, 121.1, 120.3, 79.5, 54.9, 44.7, 44.1, 36.1, 31.8, 28.6.
tert-butyl 2-(4-iodophenoxy)acetate (S2a)
1H NMR (400 MHz, CDCl3) δ 7.61 – 7.52 (m, 2H), 6.72 – 6.64 (m, 2H), 4.47 (s, 2H), 1.48 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 167.8, 158.0, 138.4, 117.1, 83.9, 82.7, 65.8, 28.2.
methyl 3-(4-(2-(tert-butoxy)-2-oxoethoxy)phenoxy)benzoate (S2b)
1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 7.7 Hz, 1H), 7.62 – 7.56 (m, 1H), 7.36 (dd, J = 8.2, 7.7 Hz, 1H), 7.15 (dd, J = 8.2, 2.6 Hz, 1H), 7.02 – 6.94 (m, 2H), 6.93 – 6.86 (m, 2H), 4.51 (s, 2H), 3.88 (s, 3H), 1.49 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 168.2, 166.7, 158.5, 154.6, 150.6, 131.9, 129.8, 123.9, 122.5, 120.9, 118.6, 116.1, 82.6, 66.4, 52.3, 28.2.
3-(4-(2-methoxy-2-oxoethoxy)phenoxy)benzoic acid (S2c)
1H NMR (400 MHz, DMSO-d6) δ 7.64 (d, J = 7.7 Hz, 1H), 7.48 (dd, J = 8.1, 7.7 Hz, 1H), 7.38 – 7.30 (m, 1H), 7.23 (dd, J = 8.1, 2.7 Hz, 1H), 7.08 – 7.02 (m, 2H), 7.02 – 6.95 (m, 2H), 4.80 (s, 2H), 3.71 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 169.3, 166.8, 158.2, 154.4, 149.4, 132.5, 130.3, 123.5, 121.9, 121.2, 117.0, 116.1, 65.0, 51.9.
3-(4-(methoxycarbonyl)phenoxy)benzoic acid (S3a)
1H NMR (400 MHz, CDCl3) δ 8.08 – 8.00 (m, 2H), 7.93 (d, J = 7.8 Hz, 1H), 7.80 – 7.75 (m, 1H), 7.49 (dd, J = 8.1, 7.8 Hz, 1H), 7.32 (dd, J = 8.1, 2.6 Hz, 1H), 7.05 – 6.98 (m, 2H), 3.91 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.4, 166.7, 161.2, 156.2, 132.0, 131.5, 130.4, 126.2, 125.4, 125.3, 121.4, 117.8, 52.2.
methyl 3-(4-(2-ethoxy-2-oxoacetyl)phenoxy)benzoate (S4a)
1H NMR (400 MHz, CDCl3) δ 8.05 – 7.99 (m, 2H), 7.91 (ddd, J = 7.8, 2.6, 1.1 Hz, 1H), 7.74 (dd, J = 2.5, 1.5 Hz, 1H), 7.49 (dd, J = 8.1, 7.8 Hz, 1H), 7.29 (ddd, J = 8.1, 2.5, 1.1 Hz, 1H), 7.05 – 7.01 (m, 2H), 4.43 (q, J = 7.2 Hz, 2H), 3.91 (s, 3H), 1.42 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 184.9, 166.2, 163.9, 163.2, 155.2, 132.8, 132.5, 130.4, 127.6, 126.3, 125.1, 121.5, 117.7, 62.5, 52.5, 14.2.
3-(4-(carboxymethyl)phenoxy)benzoic acid (S4b)
1H NMR (400 MHz, DMSO-d6) δ 7.72 – 7.67 (m, 1H), 7.53 – 7.46 (m, 1H), 7.45 – 7.41 (m, 1H), 7.35 – 7.24 (m, 3H), 7.07 – 6.96 (m, 2H), 3.58 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.8, 166.8, 157.4, 154.7, 132.7, 131.3, 131.0, 130.5, 124.1, 122.8, 119.3, 118.1, 40.0.
3-(4-(2-methoxy-2-oxoethyl)phenoxy)benzoic acid (S4c)
1H NMR (400 MHz, CDCl3) δ 7.91 – 7.84 (m, 1H), 7.74 (dd, J = 2.6, 1.5 Hz, 1H), 7.45 (dd, J = 8.1, 7.9 Hz, 1H), 7.33 – 7.25 (m, 3H), 7.05 – 6.97 (m, 2H), 3.74 (s, 3H), 3.65 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 172.2, 171.7, 157.6, 155.8, 131.2, 131.0, 130.0, 129.6, 125.1, 124.3, 120.1, 119.4, 52.3, 40.5.
methyl 3-(4-formylphenoxy)benzoate (S5a)
1H NMR (400 MHz, CDCl3) δ 9.93 (s, 1H), 7.98 – 7.81 (m, 3H), 7.73 (dd, J = 2.5, 1.5 Hz, 1H), 7.48 (dd, J = 8.1, 7.9 Hz, 1H), 7.28 (ddd, J = 8.1, 2.5, 1.1 Hz, 1H), 7.12 – 7.02 (m, 2H), 3.91 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 190.9, 166.3, 162.7, 155.5, 132.5, 132.2, 131.8, 130.3, 126.1, 125.0, 121.3, 118.0, 52.5.
(E)-3-(4-(3-(methoxycarbonyl)phenoxy)phenyl)acrylic acid (S5b)
1H NMR (400 MHz, DMSO-d6) δ 7.79 – 7.72 (m, 3H), 7.63 – 7.55 (m, 2H), 7.50 (dd, J = 2.6, 1.5 Hz, 1H), 7.38 (ddd, J = 8.2, 2.6, 1.1 Hz, 1H), 7.11 – 7.02 (m, 2H), 6.47 (d, J = 16.0 Hz, 1H), 3.83 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.6, 165.5, 157.8, 156.3, 143.1, 131.6, 130.8, 130.4, 130.0, 124.6, 124.0, 119.0, 118.9, 118.5, 52.4.
3-(4-(3-methoxy-3-oxopropyl)phenoxy)benzoic acid (S5c)
1H NMR (400 MHz, CDCl3) δ 7.88 – 7.82 (m, 1H), 7.75 – 7.69 (m, 1H), 7.44 (dd, J = 8.2, 7.9 Hz, 1H), 7.26 (dd, J = 8.2, 2.6 Hz, 1H), 7.24 – 7.18 (m, 2H), 7.02 – 6.95 (m, 2H), 3.71 (s, 3H), 2.98 (t, J = 7.7 Hz, 2H), 2.67 (t, J = 7.7 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 173.5, 171.7, 157.9, 154.9, 136.3, 131.1, 130.0, 129.9, 124.8, 124.0, 119.7, 119.5, 51.8, 35.9, 30.3.
methyl 2-(4-(3-(((2-(1-(2-((tert-butoxycarbonyl)(methyl)amino)ethyl)piperidin-4-yl)pyridin-4-yl)methyl)(methyl)carbamoyl)phenoxy)phenoxy)acetate (S2)
1H NMR (400 MHz, CDCl3) δ 8.48 (d, J = 5.1 Hz, 1H), 7.42 – 7.27 (m, 1H), 7.15 – 6.77 (m, 9H), 4.76 – 4.36 (m, 4H), 3.81 (s, 3H), 3.54 – 3.31 (m, 2H), 3.31 – 2.83 (m, 8H), 2.82 – 2.50 (m, 3H), 2.49 – 1.65 (m, 6H), 1.45 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 171.2, 169.4, 158.6, 155.9, 154.5, 150.6, 149.7, 146.5, 137.3, 130.1, 121.1, 120.6, 119.9, 119.1, 118.5, 116.4, 116.1, 115.4, 79.6, 65.9, 56.2, 54.1, 52.4, 50.3, 46.4, 44.0, 37.6, 35.0, 31.6, 28.6.
methyl 4-(3-(((2-(1-(2-((tert-butoxycarbonyl)(methyl)amino)ethyl)piperidin-4-yl)pyridin-4-yl)methyl)(methyl)carbamoyl)phenoxy)benzoate (S3)
1H NMR (400 MHz, CDCl3) δ 8.49 (d, J = 5.1 Hz, 1H), 8.07 – 7.90 (m, 2H), 7.50 – 7.32 (m, 1H), 7.25 – 6.81 (m, 7H), 4.76 – 4.41 (m, 2H), 3.89 (s, 3H), 3.51 – 3.30 (m, 2H), 3.26 – 2.99 (m, 3H), 2.96 – 2.81 (m, 5H), 2.79 – 2.47 (m, 3H), 2.39 – 2.11 (m, 2H), 2.06 – 1.71 (m, 4H), 1.45 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 170.8, 166.5, 161.1, 156.0, 149.7, 146.4, 137.7, 131.9, 130.5, 125.3, 123.0, 122.3, 121.3, 120.6, 119.9, 119.3, 118.6, 117.9, 79.6, 56.3, 54.2, 52.2, 50.4, 46.4, 44.1, 37.6, 34.9, 31.7, 28.5.
methyl 2-(4-(3-(((2-(1-(2-((tert-butoxycarbonyl)(methyl)amino)ethyl)piperidin-4-yl)pyridin-4-yl)methyl)(methyl)carbamoyl)phenoxy)phenyl)acetate (S4)
1H NMR (400 MHz, CDCl3) δ 8.49 (d, J = 5.2 Hz, 1H), 7.44 – 7.15 (m, 3H), 7.13 – 6.70 (m, 7H), 4.77 – 4.38 (m, 2H), 3.70 (s, 3H), 3.60 (s, 2H), 3.43 – 3.28 (m, 2H), 3.13 – 2.85 (m, 8H), 2.76 – 2.65 (m, 1H), 2.63 – 2.42 (m, 2H), 2.27 – 2.05 (m, 2H), 2.02 – 1.70 (m, 4H), 1.45 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 172.1, 171.1, 157.5, 155.8, 149.7, 146.4, 137.4, 130.9, 130.2, 129.6, 121.8, 121.0, 120.4, 120.0, 119.4, 118.6, 117.3, 116.4, 79.5, 56.4, 54.3, 52.2, 50.4, 46.6, 44.3, 40.5, 37.6, 34.8, 31.9, 28.6.
methyl 3-(4-(3-(((2-(1-(2-((tert-butoxycarbonyl)(methyl)amino)ethyl)piperidin-4-yl)pyridin-4-yl)methyl)(methyl)carbamoyl)phenoxy)phenyl)propanoate (S5)
1H NMR (400 MHz, CDCl3) δ 8.48 (d, J = 5.0 Hz, 1H), 7.40 – 7.27 (m, 1H), 7.20 – 6.85 (m, 9H), 4.79 – 4.38 (m, 2H), 3.67 (s, 3H), 3.44 – 3.27 (m, 2H), 3.13 – 2.98 (m, 3H), 2.97 – 2.82 (m, 7H), 2.76 – 2.66 (m, 1H), 2.62 (t, J = 7.8 Hz, 2H), 2.58 – 2.40 (m, 2H), 2.24 – 2.08 (m, 2H), 2.02 – 1.69 (m, 4H), 1.45 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 173.3, 171.2, 157.8, 155.8, 149.7, 146.4, 137.4, 136.3, 130.1, 129.8, 121.6, 120.8, 120.4, 119.8, 119.5, 118.6, 117.1, 116.2, 79.5, 56.5, 54.3, 51.8, 50.4, 46.6, 44.3, 37.6, 35.8, 34.8, 31.9, 30.3, 28.6.
methyl 2-(4-(3-(((2-(1-(2-methoxyethyl)piperidin-4-yl)pyridin-4-yl)methyl)(methyl)carbamoyl)phenoxy)phenoxy)acetate (S6)
1H NMR (400 MHz, CDCl3) δ 8.47 (d, J = 5.1 Hz, 1H), 7.40 – 7.28 (m, 1H), 7.12 – 6.83 (m, 9H), 4.62 (s, 4H), 3.81 (s, 3H), 3.53 (t, J = 5.7 Hz, 2H), 3.35 (s, 3H), 3.16 – 3.04 (m, 2H), 3.03 – 2.86 (m, 3H), 2.79 – 2.49 (m, 3H), 2.24 – 2.04 (m, 2H), 2.10 – 1.70 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 171.1, 169.4, 158.5, 154.5, 150.6, 149.7, 146.4, 137.3, 130.1, 121.1, 120.4, 119.6, 119.1, 118.4, 116.4, 116.1, 115.4, 70.3, 65.9, 59.0, 58.2, 54.4, 52.4, 50.3, 44.2, 37.5, 31.7.
methyl 4-(4-(((tert-butoxycarbonyl)amino)methyl)phenoxy)butanoate (AL1+3)
1H NMR (400 MHz, CDCl3) δ 7.24 – 7.08 (m, 2H), 6.90 – 6.78 (m, 2H), 4.83 (s, 1H), 4.22 (d, J = 5.9 Hz, 2H), 3.97 (t, J = 6.1 Hz, 2H), 3.67 (s, 3H), 2.51 (t, J = 7.3 Hz, 2H), 2.09 (tt, J = 7.3, 6.1 Hz, 2H), 1.44 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 173.7, 158.2, 155.9, 131.3, 128.9, 114.6, 79.4, 66.8, 51.7, 44.2, 30.6, 28.5, 24.7.
methyl 4-(4-(2-((tert-butoxycarbonyl)amino)ethyl)phenoxy)butanoate (AL2+3)
1H NMR (400 MHz, CDCl3) δ 7.16 – 7.00 (m, 2H), 6.88 – 6.73 (m, 2H), 4.57 (s, 1H), 3.97 (t, J = 6.1 Hz, 2H), 3.67 (s, 3H), 3.41 – 3.16 (m, 2H), 2.71 (t, J = 7.1 Hz, 2H), 2.51 (t, J = 7.3 Hz, 2H), 2.21 – 2.01 (m, 2H), 1.42 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 173.7, 157.5, 156.0, 131.2, 129.8, 114.7, 79.2, 66.8, 51.7, 42.0, 35.4, 30.6, 28.5, 24.7.
methyl 5-(4-(2-((tert-butoxycarbonyl)amino)ethyl)phenoxy)pentanoate (AL2+4)
1H NMR (400 MHz, CDCl3) δ 7.15 – 6.98 (m, 2H), 6.89 – 6.71 (m, 2H), 4.57 (s, 1H), 4.01 – 3.87 (m, 2H), 3.66 (s, 3H), 3.44 – 3.16 (m, 2H), 2.71 (t, J = 7.1 Hz, 2H), 2.46 – 2.31 (m, 2H), 1.86 – 1.74 (m, 4H), 1.42 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 174.0, 157.7, 156.0, 131.1, 129.8, 114.6, 79.2, 67.4, 51.6, 42.1, 35.4, 33.8, 28.8, 28.5, 21.7.
methyl 6-(4-(2-((tert-butoxycarbonyl)amino)ethyl)phenoxy)hexanoate (AL2+5)
1H NMR (400 MHz, CDCl3) δ 7.15 – 7.01 (m, 2H), 6.85 – 6.74 (m, 2H), 4.59 (s, 1H), 3.91 (t, J = 6.4 Hz, 2H), 3.65 (s, 3H), 3.40 – 3.21 (m, 2H), 2.70 (t, J = 7.0 Hz, 2H), 2.33 (t, J = 7.5 Hz, 2H), 1.82 – 1.73 (m, 2H), 1.73 – 1.63 (m, 2H), 1.53 – 1.45 (m, 2H), 1.42 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 174.0, 157.7, 155.9, 130.9, 129.7, 114.6, 79.1, 67.6, 51.5, 42.0, 35.3, 34.0, 29.0, 28.4, 25.7, 24.7.
methyl 7-(4-(2-((tert-butoxycarbonyl)amino)ethyl)phenoxy)heptanoate (AL2+6)
1H NMR (400 MHz, CDCl3) δ 7.13 – 7.00 (m, 2H), 6.89 – 6.74 (m, 2H), 4.59 (s, 1H), 3.91 (t, J = 6.4 Hz, 2H), 3.65 (s, 3H), 3.39 – 3.17 (m, 2H), 2.71 (t, J = 7.1 Hz, 2H), 2.30 (t, J = 7.5 Hz, 2H), 1.81 – 1.71 (m, 2H), 1.69 – 1.60 (m, 2H), 1.51 – 1.33 (m, 13H). 13C NMR (101 MHz, CDCl3) δ 174.2, 157.8, 155.9, 130.9, 129.7, 114.6, 79.1, 67.8, 51.5, 42.0, 35.3, 34.0, 29.2, 28.9, 28.5, 25.8, 24.9.
tert-butyl 4-((4-(2-methoxy-2-oxoethyl)piperazin-1-yl)methyl)piperidine-1-carboxylate (RL1)
1H NMR (400 MHz, CDCl3) δ 3.71 (s, 3H), 3.20 (s, 2H), 2.73 – 2.34 (m, 10H), 2.18 (d, J = 7.0 Hz, 2H), 1.75 – 1.66 (m, 2H), 1.66 – 1.58 (m, 1H), 1.46 – 1.41 (m, 11H), 1.11 – 0.99 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 170.8, 155.0, 79.3, 64.4, 59.5, 53.4, 53.1, 51.8, 43.9, 33.6, 30.8, 28.5.
(2S,4R)-1-((S)-3,3-dimethyl-2-(4-(4-((2-(4-(3-(methyl((2-(1-(2-(methylamino)ethyl)piperidin-4-yl)pyridin-4-yl)methyl)carbamoyl)phenoxy)phenoxy)acetamido)methyl)phenoxy)butanamido)butanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (2a)
1H NMR (400 MHz, MeOD) δ 9.67 (s, 1H), 8.73 (d, J = 6.3 Hz, 1H), 8.01 – 7.73 (m, 2H), 7.58 – 7.37 (m, 5H), 7.34 – 6.82 (m, 11H), 5.05 – 4.95 (m, 3H), 4.69 – 4.51 (m, 4H), 4.50 – 4.32 (m, 3H), 4.08 – 3.81 (m, 5H), 3.74 (dd, J = 11.0, 4.0 Hz, 1H), 3.66 – 3.49 (m, 5H), 3.45 – 3.36 (m, 2H), 3.14 (s, 3H), 2.83 (s, 3H), 2.57 (s, 3H), 2.53 – 2.28 (m, 6H), 2.25 – 2.15 (m, 1H), 2.10 – 2.00 (m, 2H), 1.99 – 1.87 (m, 1H), 1.59 – 1.48 (m, 3H), 1.03 (s, 9H). HRMS (ESI/[M+H]+) Calcd for [C64H79N9O9S+H]+: 1150.5794, found: 1150.5804. HPLC purity: 99.1%.
(2S,4R)-1-((S)-3,3-dimethyl-2-(4-(4-(2-(2-(4-(3-(methyl((2-(1-(2-(methylamino)ethyl)piperidin-4-yl)pyridin-4-yl)methyl)carbamoyl)phenoxy)phenoxy)acetamido)ethyl)phenoxy)butanamido)butanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (2b)
1H NMR (400 MHz, MeOD) δ 9.81 (s, 1H), 8.74 (d, J = 6.1 Hz, 1H), 8.06 – 7.77 (m, 2H), 7.61 – 7.37 (m, 5H), 7.35 – 6.79 (m, 11H), 5.08 – 4.94 (m, 3H), 4.66 – 4.31 (m, 5H), 4.05 – 3.81 (m, 5H), 3.74 (dd, J = 11.1, 4.0 Hz, 1H), 3.65 – 3.36 (m, 9H), 3.14 (s, 3H), 2.83 (s, 3H), 2.77 (t, J = 7.4 Hz, 2H), 2.59 (s, 3H), 2.54 – 2.32 (m, 6H), 2.26 – 2.15 (m, 1H), 2.08 – 2.00 (m, 2H), 1.98 – 1.88 (m, 1H), 1.60 – 1.46 (m, 3H), 1.03 (s, 9H). HRMS (ESI/[M+H]+) Calcd for [C65H81N9O9S+H]+: 1164.5951, found: 1164.5938. HPLC purity: 99.6%.
(2S,4R)-1-((S)-3,3-dimethyl-2-(5-(4-(2-(2-(4-(3-(methyl((2-(1-(2-(methylamino)ethyl)piperidin-4-yl)pyridin-4-yl)methyl)carbamoyl)phenoxy)phenoxy)acetamido)ethyl)phenoxy)pentanamido)butanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (2c)
1H NMR (400 MHz, MeOD) δ 9.50 (s, 1H), 8.73 (d, J = 6.1 Hz, 1H), 8.01 – 7.70 (m, 2H), 7.61 – 7.36 (m, 5H), 7.35 – 6.78 (m, 11H), 5.05 – 4.95 (m, 3H), 4.65 – 4.33 (m, 5H), 4.13 – 3.80 (m, 5H), 3.75 (dd, J = 11.0, 4.0 Hz, 1H), 3.65 – 3.36 (m, 9H), 3.13 (s, 3H), 2.83 (s, 3H), 2.78 (t, J = 7.0 Hz, 2H), 2.55 (s, 3H), 2.48 – 2.26 (m, 6H), 2.24 – 2.14 (m, 1H), 1.99 – 1.90 (m, 1H), 1.87 – 1.70 (m, 4H), 1.59 – 1.47 (m, 3H), 1.04 (s, 9H). HRMS (ESI/[M+H]+) Calcd for [C66H83N9O9S+H]+: 1178.6107, found: 1178.6109. HPLC purity: 99.0%.
(2S,4R)-1-((S)-3,3-dimethyl-2-(6-(4-(2-(2-(4-(3-(methyl((2-(1-(2-(methylamino)ethyl)piperidin-4-yl)pyridin-4-yl)methyl)carbamoyl)phenoxy)phenoxy)acetamido)ethyl)phenoxy)hexanamido)butanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (2d)
1H NMR (400 MHz, MeOD) δ 9.73 (s, 1H), 8.74 (d, J = 6.0 Hz, 1H), 8.07 – 7.72 (m, 2H), 7.59 – 7.39 (m, 5H), 7.35 – 6.78 (m, 11H), 5.07 – 4.93 (m, 3H), 4.64 – 4.33 (m, 5H), 4.03 – 3.80 (m, 5H), 3.74 (dd, J = 11.0, 3.9 Hz, 1H), 3.66 – 3.36 (m, 9H), 3.14 (s, 3H), 2.83 (s, 3H), 2.77 (t, J = 7.4 Hz, 2H), 2.58 (s, 3H), 2.49 – 2.16 (m, 7H), 1.98 – 1.89 (m, 1H), 1.83 – 1.61 (m, 4H), 1.60 – 1.43 (m, 5H), 1.04 (s, 9H). 13C NMR (126 MHz, MeOD) δ 175.9, 173.7, 173.3, 172.3, 171.0, 161.4, 160.0, 159.2, 158.3, 155.9, 155.6, 151.8, 147.4, 144.2, 143.1, 137.8, 136.4, 132.1, 131.4, 130.8, 130.6, 128.9, 128.1, 127.8, 125.0, 124.8, 122.5, 122.2, 120.5, 117.3, 115.6, 71.0, 69.1, 68.8, 60.6, 59.1, 58.0, 53.7, 53.6, 52.3, 50.2, 44.3, 41.8, 39.2, 38.8, 37.6, 36.5, 36.4, 35.6, 33.9, 30.1, 29.2, 27.1, 26.8, 26.8, 22.4, 13.8. HRMS (ESI/[M+H]+) Calcd for [C67H85N9O9S+H]+: 1192.6264, found: 1192.6290. HPLC purity: 99.5%.
(2S,4R)-1-((S)-3,3-dimethyl-2-(7-(4-(2-(2-(4-(3-(methyl((2-(1-(2-(methylamino)ethyl)piperidin-4-yl)pyridin-4-yl)methyl)carbamoyl)phenoxy)phenoxy)acetamido)ethyl)phenoxy)heptanamido)butanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (2e)
1H NMR (400 MHz, MeOD) δ 9.89 (s, 1H), 8.81 – 8.62 (m, 1H), 8.07 – 7.73 (m, 2H), 7.65 – 7.43 (m, 5H), 7.39 – 6.67 (m, 11H), 5.06 – 4.96 (m, 3H), 4.65 – 4.33 (m, 5H), 4.05 – 3.83 (m, 5H), 3.74 (dd, J = 10.9, 3.9 Hz, 1H), 3.64 – 3.38 (m, 9H), 3.14 (s, 3H), 2.83 (s, 3H), 2.78 (t, J = 7.4 Hz, 2H), 2.60 (s, 3H), 2.46 – 2.16 (m, 7H), 1.98 – 1.86 (m, 1H), 1.81 – 1.60 (m, 4H), 1.54 – 1.36 (m, 7H), 1.04 (s, 9H). HRMS (ESI/[M+H]+) Calcd for [C68H87N9O9S+H]+: 1206.6420, found: 1206.6421. HPLC purity: 99.9%.
(2S,4R)-1-((S)-3,3-dimethyl-2-(2-(4-((1-(2-(4-(3-(methyl((2-(1-(2-(methylamino)ethyl)piperidin-4-yl)pyridin-4-yl)methyl)carbamoyl)phenoxy)phenoxy)acetyl)piperidin-4-yl)methyl)piperazin-1-yl)acetamido)butanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (3a)
1H NMR (400 MHz, MeOD) δ 9.39 (s, 1H), 8.79 – 8.66 (m, 1H), 7.98 – 7.64 (m, 2H), 7.59 – 7.35 (m, 5H), 7.31 – 6.66 (m, 7H), 5.04 – 4.97 (m, 3H), 4.66 – 4.43 (m, 4H), 4.07 – 3.71 (m, 6H), 3.71 – 3.35 (m, 16H), 3.24 – 3.02 (m, 8H), 2.91 – 2.73 (m, 4H), 2.53 (s, 3H), 2.50 – 2.11 (m, 6H), 2.09 – 1.83 (m, 3H), 1.62 – 1.49 (m, 3H), 1.47 – 1.33 (m, 2H), 1.07 (s, 9H). HRMS (ESI/[M+H]+) Calcd for [C65H87N11O8S+H]+: 1182.6533, found: 1182.6534. HPLC purity: 99.3%.
(2S,4R)-1-((S)-3,3-dimethyl-2-(2-(4-((1-((1-(2-(4-(3-(methyl((2-(1-(2-(methylamino)ethyl)piperidin-4-yl)pyridin-4-yl)methyl)carbamoyl)phenoxy)phenoxy)acetyl)piperidin-4-yl)methyl)piperidin-4-yl)methyl)piperazin-1-yl)acetamido)butanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (3b)
1H NMR (400 MHz, MeOD) δ 9.76 (s, 1H), 8.75 (d, J = 6.3 Hz, 1H), 8.05 – 7.70 (m, 2H), 7.62 – 7.39 (m, 5H), 7.35 – 6.61 (m, 7H), 5.09 – 4.95 (m, 3H), 4.67 – 4.35 (m, 4H), 4.10 – 3.34 (m, 26H), 3.29 – 2.95 (m, 10H), 2.84 (s, 4H), 2.59 (s, 3H), 2.50 – 2.09 (m, 9H), 1.99 – 1.70 (m, 5H), 1.64 – 1.49 (m, 3H), 1.44 – 1.28 (m, 2H), 1.07 (s, 9H). 13C NMR (126 MHz, MeOD) δ 173.8, 173.2, 171.6, 168.7, 161.5, 160.1, 158.3, 156.3, 155.8, 147.5, 143.9, 143.1, 137.7, 136.6, 131.4, 130.7, 130.6, 128.7, 128.1, 127.9, 125.1, 124.8, 122.3, 122.1, 120.5, 117.3, 117.2, 71.0, 68.1, 63.4, 62.1, 60.5, 59.4, 58.0, 53.7, 52.3, 51.1, 50.6, 50.2, 45.5, 44.3, 42.6, 39.3, 39.0, 38.8, 36.6, 34.0, 32.4, 31.5, 30.7, 30.3, 29.2, 28.5, 27.0, 26.9, 22.4, 13.6. HRMS (ESI/[M+H]+) Calcd for [C71H98N12O8S+H]+: 1279.7424, found: 1279.7440. HPLC purity: 99.4%.
(2S,4R)-4-hydroxy-1-((S)-2-(2-(4-((1-((1-(2-(4-(3-(((2-(1-(2-methoxyethyl)piperidin-4-yl)pyridin-4-yl)methyl)(methyl)carbamoyl)phenoxy)phenoxy)acetyl)piperidin-4-yl)methyl)piperidin-4-yl)methyl)piperazin-1-yl)acetamido)-3,3-dimethylbutanoyl)-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (3bN)
1H NMR (400 MHz, MeOD) δ 9.85 (s, 1H), 8.75 (d, J = 5.9 Hz, 1H), 8.12 – 7.71 (m, 2H), 7.64 – 7.38 (m, 5H), 7.30 – 6.64 (m, 7H), 5.10 – 4.97 (m, 3H), 4.67 – 4.35 (m, 4H), 4.14 – 3.34 (m, 29H), 3.30 – 3.02 (m, 10H), 2.90 – 2.75 (m, 1H), 2.60 (s, 3H), 2.46 – 2.16 (m, 9H), 2.02 – 1.88 (m, 3H), 1.87 – 1.68 (m, 2H), 1.63 – 1.49 (m, 3H), 1.45 – 1.31 (m, 2H), 1.08 (s, 9H). HRMS (ESI/[M+H]+) Calcd for [C71H97N11O9S+H]+: 1280.7264, found: 1280.7287. HPLC purity: 99.4%.
(2S,4R)-1-((S)-3,3-dimethyl-2-(2-(4-((1-((1-(4-(3-(methyl((2-(1-(2-(methylamino)ethyl)piperidin-4-yl)pyridin-4-yl)methyl)carbamoyl)phenoxy)benzoyl)piperidin-4-yl)methyl)piperidin-4-yl)methyl)piperazin-1-yl)acetamido)butanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (3c)
1H NMR (400 MHz, MeOD) δ 9.73 – 9.46 (m, 1H), 8.75 (d, J = 6.1 Hz, 1H), 8.15 – 7.73 (m, 2H), 7.63 – 6.89 (m, 12H), 5.08 – 4.96 (m, 3H), 4.70 – 4.34 (m, 4H), 3.97 – 3.81 (m, 5H), 3.79 – 3.36 (m, 19H), 3.27 – 2.87 (m, 11H), 2.83 (s, 3H), 2.57 (s, 3H), 2.49 – 2.11 (m, 9H), 2.00 – 1.67 (m, 5H), 1.63 – 1.49 (m, 3H), 1.45 – 1.31 (m, 2H), 1.07 (s, 9H). HRMS (ESI/[M+H]+) Calcd for [C70H96N12O7S+H]+: 1249.7318, found: 1249.7345. HPLC purity: 99.6%.
(2S,4R)-1-((S)-3,3-dimethyl-2-(2-(4-((1-((1-(2-(4-(3-(methyl((2-(1-(2-(methylamino)ethyl)piperidin-4-yl)pyridin-4-yl)methyl)carbamoyl)phenoxy)phenyl)acetyl)piperidin-4-yl)methyl)piperidin-4-yl)methyl)piperazin-1-yl)acetamido)butanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (3d)
1H NMR (400 MHz, MeOD) δ 9.77 (s, 1H), 8.75 (d, J = 6.1 Hz, 1H), 8.13 – 7.74 (m, 2H), 7.65 – 7.40 (m, 5H), 7.39 – 6.78 (m, 7H), 5.11 – 4.95 (m, 3H), 4.69 – 4.35 (m, 4H), 4.15 – 3.86 (m, 6H), 3.85 – 3.36 (m, 21H), 3.27 – 3.02 (m, 9H), 2.83 (s, 3H), 2.80 – 2.67 (m, 1H), 2.59 (s, 3H), 2.52 – 2.10 (m, 9H), 1.99 – 1.67 (m, 5H), 1.64 – 1.49 (m, 3H), 1.26 – 1.02 (m, 11H). HRMS (ESI/[M+H]+) Calcd for [C71H98N12O7S+H]+: 1263.7475, found: 1263.7491. HPLC purity: 99.1%.
(2S,4R)-1-((S)-3,3-dimethyl-2-(2-(4-((1-((1-(3-(4-(3-(methyl((2-(1-(2-(methylamino)ethyl)piperidin-4-yl)pyridin-4-yl)methyl)carbamoyl)phenoxy)phenyl)propanoyl)piperidin-4-yl)methyl)piperidin-4-yl)methyl)piperazin-1-yl)acetamido)butanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (3e)
1H NMR (400 MHz, MeOD) δ 9.76 (s, 1H), 8.76 (d, J = 5.4 Hz, 1H), 8.11 – 7.77 (m, 2H), 7.67 – 7.43 (m, 5H), 7.38 – 6.82 (m, 7H), 5.09 – 4.97 (m, 3H), 4.70 – 4.34 (m, 4H), 4.11 – 3.85 (m, 6H), 3.82 – 3.36 (m, 19H), 3.28 – 3.00 (m, 9H), 2.93 (t, J = 7.2 Hz, 2H), 2.83 (s, 3H), 2.79 – 2.64 (m, 3H), 2.59 (s, 3H), 2.52 – 2.08 (m, 9H), 1.98 – 1.68 (m, 5H), 1.64 – 1.49 (m, 3H), 1.26 – 1.01 (m, 11H). 13C NMR (126 MHz, MeOD) δ 173.7, 173.2, 173.0, 171.6, 161.5, 159.5, 156.1, 155.8, 147.5, 143.9, 143.1, 138.3, 137.8, 136.6, 131.4, 131.3, 130.7, 130.6, 128.7, 128.1, 127.9, 125.0, 124.9, 122.8, 121.1, 120.6, 117.9, 71.0, 63.5, 62.1, 60.5, 59.4, 58.0, 53.8, 52.4, 51.0, 50.6, 50.2, 46.4, 44.3, 42.4, 39.3, 39.0, 38.8, 36.6, 35.6, 34.0, 32.3, 31.9, 31.6, 30.8, 30.2, 29.3, 28.5, 27.0, 26.9, 22.4, 13.7. HRMS (ESI/[M+H]+) Calcd for [C72H100N12O7S+H]+: 1277.7631, found: 1277.7608. HPLC purity: 98.1%.
Supplementary Material
ACKNOWLEDGMENT
This study made use of the Medicinal Chemistry Center (MCC) and Analytical Instrumentation Center (AIC) at UW-Madison, funded by the NIH shared instrument grants (NIH-NCRR S10RR029531 and S10OD028473), UW School of Pharmacy and the Lachman Institute of Pharmaceutical Development.
Funding Sources
W.T. thanks the financial support from the University of Wisconsin–Madison Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation (WARF) through a UW2020 award before June 30, 2022 and retention funds from the School of Pharmacy, UW-Madison. M.B. thanks the financial support from NRSA award T32 CA009135. W.X. thanks the financial support from National Institutes of Health R01 grants, R01CA236356, R01CA268183, R01CA281024, UWCCC Transdisciplinary Cancer Immunology-Immunotherapy Pilot grant, and Falk Medical Research Foundation Catalyst Award. L.L. acknowledges funding support of NIH grants R01DK071801, RF1AG052324, R01AG078794, and NIH shared instrument grants (NIH-NCRR S10RR029531, S10OD028473 and S10OD025084).
ABBREVIATIONS
- ADMA
asymmetric dimethylarginine
- BAF155
BRG1-associated factor 155
- CARM1
coactivator-associated Arginine Methyltransferase 1
- CRBN
cereblon
- PABP1
polyadenylate-binding protein 1
- PRMT
protein arginine methyltransferases
- PROTAC
proteolysis targeting chimera
- SAM
S-adenosyl-L-methionine
- SAR
structure activity relationship
- SDMA
symmetric dimethylarginine
- TLC
thin-layer chromatography
- TPD
target protein degradation
- VHL
von hippel–lindau
Footnotes
Supporting Information. Supporting Information is available free of charge.
Rapid-TAC CARM1 degradation (Figure S1A, S1B), CARM1 degradation activity in MCF10A cells (Figure S2), 3b cell proliferation assay in MCF7 and MCF10A cells (Figure S3), HPLC traces, NMR spectra (PDF).
Molecular formula strings (CSV).
The authors declare no competing financial interest.
REFERENCES
- (1).Yang Y; Bedford MT Protein Arginine Methyltransferases and Cancer. Nat. Rev. Cancer 2013, 13 (1), 37–50. 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- (2).Chen D; Ma H; Hong H; Koh SS; Huang S-M; Schurter BT; Aswad DW; Stallcup MR Regulation of Transcription by a Protein Methyltransferase. Science 1999, 284 (5423), 2174–2177. 10.1126/science.284.5423.2174. [DOI] [PubMed] [Google Scholar]
- (3).Suresh S; Huard S; Dubois T CARM1/PRMT4: Making Its Mark beyond Its Function as a Transcriptional Coactivator. Trends Cell Biol. 2021, 31 (5), 402–417. 10.1016/j.tcb.2020.12.010. [DOI] [PubMed] [Google Scholar]
- (4).Cheng H; Qin Y; Fan H; Su P; Zhang X; Zhang H; Zhou G Overexpression of CARM1 in Breast Cancer Is Correlated with Poorly Characterized Clinicopathologic Parameters and Molecular Subtypes. Diagn. Pathol 2013, 8 (1), 129. 10.1186/1746-1596-8-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Davis MB; Liu X; Wang S; Reeves J; Khramtsov A; Huo D; Olopade OI Expression and Sub-Cellular Localization of an Epigenetic Regulator, Co-Activator Arginine Methyltransferase 1 (CARM1), Is Associated with Specific Breast Cancer Subtypes and Ethnicity. Mol. Cancer 2013, 12 (1), 40. 10.1186/1476-4598-12-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Greenblatt SM; Man N; Hamard P-J; Asai T; Karl D; Martinez C; Bilbao D; Stathias V; Jermakowicz AM; Duffort S; Tadi M; Blumenthal E; Newman S; Vu L; Xu Y; Liu F; Schurer SC; McCabe MT; Kruger RG; Xu M; Yang F-C; Tenen DG; Watts J; Vega F; Nimer SD CARM1 Is Essential for Myeloid Leukemogenesis but Dispensable for Normal Hematopoiesis. Cancer Cell 2018, 33 (6), 1111–1127.e5. 10.1016/j.ccell.2018.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wang L; Zhao Z; Meyer MB; Saha S; Yu M; Guo A; Wisinski KB; Huang W; Cai W; Pike JW; Yuan M; Ahlquist P; Xu W CARM1 Methylates Chromatin Remodeling Factor BAF155 to Enhance Tumor Progression and Metastasis. Cancer Cell 2014, 25 (1), 21–36. 10.1016/j.ccr.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bao J; Di Lorenzo A; Lin K; Lu Y; Zhong Y; Sebastian MM; Muller WJ; Yang Y; Bedford MT Mouse Models of Overexpression Reveal Distinct Oncogenic Roles for Different Type I Protein Arginine Methyltransferases. Cancer Res. 2019, 79 (1), 21–32. 10.1158/0008-5472.CAN-18-1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Shishkova E; Zeng H; Liu F; Kwiecien NW; Hebert AS; Coon JJ; Xu W Global Mapping of CARM1 Substrates Defines Enzyme Specificity and Substrate Recognition. Nat. Commun 2017, 8 (1), 15571. 10.1038/ncomms15571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Purandare AV; Chen Z; Huynh T; Pang S; Geng J; Vaccaro W; Poss MA; Oconnell J; Nowak K; Jayaraman L Pyrazole Inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett 2008, 18 (15), 4438–4441. 10.1016/j.bmcl.2008.06.026. [DOI] [PubMed] [Google Scholar]
- (11).Allan M; Manku S; Therrien E; Nguyen N; Styhler S; Robert M-F; Goulet A-C; Petschner AJ; Rahil G; Robert MacLeod A; Déziel R; Besterman JM; Nguyen H; Wahhab A N-Benzyl-1-Heteroaryl-3-(Trifluoromethyl)-1H-Pyrazole-5-Carboxamides as Inhibitors of Co-Activator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett 2009, 19 (4), 1218–1223. 10.1016/j.bmcl.2008.12.075. [DOI] [PubMed] [Google Scholar]
- (12).Huynh T; Chen Z; Pang S; Geng J; Bandiera T; Bindi S; Vianello P; Roletto F; Thieffine S; Galvani A; Vaccaro W; Poss MA; Trainor GL; Lorenzi MV; Gottardis M; Jayaraman L; Purandare AV Optimization of Pyrazole Inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett 2009, 19 (11), 2924–2927. 10.1016/j.bmcl.2009.04.075. [DOI] [PubMed] [Google Scholar]
- (13).Therrien E; Larouche G; Manku S; Allan M; Nguyen N; Styhler S; Robert M-F; Goulet A-C; Besterman JM; Nguyen H; Wahhab A 1,2-Diamines as Inhibitors of Co-Activator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett 2009, 19 (23), 6725–6732. 10.1016/j.bmcl.2009.09.110. [DOI] [PubMed] [Google Scholar]
- (14).Wan H; Huynh T; Pang S; Geng J; Vaccaro W; Poss MA; Trainor GL; Lorenzi MV; Gottardis M; Jayaraman L; Purandare AV Benzo[d]Imidazole Inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1)—Hit to Lead Studies. Bioorg. Med. Chem. Lett 2009, 19 (17), 5063–5066. 10.1016/j.bmcl.2009.07.040. [DOI] [PubMed] [Google Scholar]
- (15).Sack JS; Thieffine S; Bandiera T; Fasolini M; Duke GJ; Jayaraman L; Kish KF; Klei HE; Purandare AV; Rosettani P; Troiani S; Xie D; Bertrand JA Structural Basis for CARM1 Inhibition by Indole and Pyrazole Inhibitors. Biochem. J 2011, 436 (2), 331–339. 10.1042/BJ20102161. [DOI] [PubMed] [Google Scholar]
- (16).Ferreira de Freitas R; Eram MS; Smil D; Szewczyk MM; Kennedy S; Brown PJ; Santhakumar V; Barsyte-Lovejoy D; Arrowsmith CH; Vedadi M; Schapira M Discovery of a Potent and Selective Coactivator Associated Arginine Methyltransferase 1 (CARM1) Inhibitor by Virtual Screening. J. Med. Chem 2016, 59 (14), 6838–6847. 10.1021/acs.jmedchem.6b00668. [DOI] [PubMed] [Google Scholar]
- (17).Shen Y; Szewczyk MM; Eram MS; Smil D; Kaniskan HÜ; Ferreira de Freitas R; Senisterra G; Li F; Schapira M; Brown PJ; Arrowsmith CH; Barsyte-Lovejoy D; Liu J; Vedadi M; Jin J Discovery of a Potent, Selective, and Cell-Active Dual Inhibitor of Protein Arginine Methyltransferase 4 and Protein Arginine Methyltransferase 6. J. Med. Chem 2016, 59 (19), 9124–9139. 10.1021/acs.jmedchem.6b01033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kaniskan HÜ; Eram MS; Liu J; Smil D; Martini ML; Shen Y; Santhakumar V; Brown PJ; Arrowsmith CH; Vedadi M; Jin J Design and Synthesis of Selective, Small Molecule Inhibitors of Coactivator-Associated Arginine Methyltransferase 1 (CARM1). MedChemComm 2016, 7 (9), 1793–1796. 10.1039/C6MD00342G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Drew AE; Moradei O; Jacques SL; Rioux N; Boriack-Sjodin AP; Allain C; Scott MP; Jin L; Raimondi A; Handler JL; Ott HM; Kruger RG; McCabe MT; Sneeringer C; Riera T; Shapiro G; Waters NJ; Mitchell LH; Duncan KW; Moyer MP; Copeland RA; Smith J; Chesworth R; Ribich SA Identification of a CARM1 Inhibitor with Potent In Vitro and In Vivo Activity in Preclinical Models of Multiple Myeloma. Sci. Rep 2017, 7 (1), 17993. 10.1038/s41598-017-18446-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kaniskan HÜ; Jin J Recent Progress in Developing Selective Inhibitors of Protein Methyltransferases. Curr. Opin. Chem. Biol 2017, 39, 100–108. 10.1016/j.cbpa.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Nakayama K; Szewczyk MM; dela Sena C; Wu H; Dong A; Zeng H; Li F; de Freitas RF; Eram MS; Schapira M; Baba Y; Kunitomo M; Cary DR; Tawada M; Ohashi A; Imaeda Y; Saikatendu KS; Grimshaw CE; Vedadi M; Arrowsmith CH; Barsyte-Lovejoy D; Kiba A; Tomita D; Brown PJ TP-064, a Potent and Selective Small Molecule Inhibitor of PRMT4 for Multiple Myeloma. Oncotarget 2018, 9 (26), 18480–18493. 10.18632/oncotarget.24883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhang Z; Guo Z; Xu X; Cao D; Yang H; Li Y; Shi Q; Du Z; Guo X; Wang X; Chen D; Zhang Y; Chen L; Zhou K; Li J; Geng M; Huang X; Xiong B Structure-Based Discovery of Potent CARM1 Inhibitors for Solid Tumor and Cancer Immunology Therapy. J. Med. Chem 2021, 64 (22), 16650–16674. 10.1021/acs.jmedchem.1c01308. [DOI] [PubMed] [Google Scholar]
- (23).Cai X-C; Zhang T; Kim E; Jiang M; Wang K; Wang J; Chen S; Zhang N; Wu H; Li F; dela Seña CC; Zeng H; Vivcharuk V; Niu X; Zheng W; Lee JP; Chen Y; Barsyte D; Szewczyk M; Hajian T; Ibáñez G; Dong A; Dombrovski L; Zhang Z; Deng H; Min J; Arrowsmith CH; Mazutis L; Shi L; Vedadi M; Brown PJ; Xiang J; Qin L-X; Xu W; Luo M A Chemical Probe of CARM1 Alters Epigenetic Plasticity against Breast Cancer Cell Invasion. eLife 2019, 8, e47110. 10.7554/eLife.47110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Iannelli G; Milite C; Marechal N; Cura V; Bonnefond L; Troffer-Charlier N; Feoli A; Rescigno D; Wang Y; Cipriano A; Viviano M; Bedford MT; Cavarelli J; Castellano S; Sbardella G Turning Nonselective Inhibitors of Type I Protein Arginine Methyltransferases into Potent and Selective Inhibitors of Protein Arginine Methyltransferase 4 through a Deconstruction–Reconstruction and Fragment-Growing Approach. J. Med. Chem 2022, 65 (17), 11574–11606. 10.1021/acs.jmedchem.2c00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kim E-J; Liu P; Zhang S; Donahue K; Wang Y; Schehr JL; Wolfe SK; Dickerson A; Lu L; Rui L; Zhong X; Wisinski KB; Yu M; Suzuki A; Lang JM; Ong IM; Xu W BAF155 Methylation Drives Metastasis by Hijacking Super-Enhancers and Subverting Anti-Tumor Immunity. Nucleic Acids Res. 2021, 49 (21), 12211–12233. 10.1093/nar/gkab1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lai AC; Crews CM Induced Protein Degradation: An Emerging Drug Discovery Paradigm. Nat. Rev. Drug Discov 2017, 16 (2), 101–114. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cromm PM; Crews CM Targeted Protein Degradation: From Chemical Biology to Drug Discovery. Cell Chem. Biol 2017, 24 (9), 1181–1190. 10.1016/j.chembiol.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Békés M; Langley DR; Crews CM PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov 2022, 21 (3), 181–200. 10.1038/s41573-021-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Sakamoto KM; Kim KB; Kumagai A; Mercurio F; Crews CM; Deshaies RJ Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (15), 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348 (6241), 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Bondeson DP; Mares A; Smith IED; Ko E; Campos S; Miah AH; Mulholland KE; Routly N; Buckley DL; Gustafson JL; Zinn N; Grandi P; Shimamura S; Bergamini G; Faelth-Savitski M; Bantscheff M; Cox C; Gordon DA; Willard RR; Flanagan JJ; Casillas LN; Votta BJ; den Besten W; Famm K; Kruidenier L; Carter PS; Harling JD; Churcher I; Crews CM Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol 2015, 11 (8), 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lu J; Qian Y; Altieri M; Dong H; Wang J; Raina K; Hines J; Winkler JD; Crew AP; Coleman K; Crews CM Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol 2015, 22 (6), 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zengerle M; Chan K-H; Ciulli A Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol 2015, 10 (8), 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Burslem GM; Smith BE; Lai AC; Jaime-Figueroa S; McQuaid DC; Bondeson DP; Toure M; Dong H; Qian Y; Wang J; Crew AP; Hines J; Crews CM The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol 2018, 25 (1), 67–77.e3. 10.1016/j.chembiol.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Qin C; Hu Y; Zhou B; Fernandez-Salas E; Yang C-Y; Liu L; McEachern D; Przybranowski S; Wang M; Stuckey J; Meagher J; Bai L; Chen Z; Lin M; Yang J; Ziazadeh DN; Xu F; Hu J; Xiang W; Huang L; Li S; Wen B; Sun D; Wang S Discovery of QCA570 as an Exceptionally Potent and Efficacious Proteolysis Targeting Chimera (PROTAC) Degrader of the Bromodomain and Extra-Terminal (BET) Proteins Capable of Inducing Complete and Durable Tumor Regression. J. Med. Chem 2018, 61 (15), 6685–6704. 10.1021/acs.jmedchem.8b00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Li Y; Yang J; Aguilar A; McEachern D; Przybranowski S; Liu L; Yang C-Y; Wang M; Han X; Wang S Discovery of MD-224 as a First-in-Class, Highly Potent, and Efficacious Proteolysis Targeting Chimera Murine Double Minute 2 Degrader Capable of Achieving Complete and Durable Tumor Regression. J. Med. Chem 2019, 62 (2), 448–466. 10.1021/acs.jmedchem.8b00909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Burslem GM; Schultz AR; Bondeson DP; Eide CA; Savage Stevens SL; Druker BJ; Crews CM Targeting BCR-ABL1 in Chronic Myeloid Leukemia by PROTAC-Mediated Targeted Protein Degradation. Cancer Res. 2019, 79 (18), 4744–4753. 10.1158/0008-5472.CAN-19-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Liao H; Li X; Zhao L; Wang Y; Wang X; Wu Y; Zhou X; Fu W; Liu L; Hu H-G; Chen Y-G A PROTAC Peptide Induces Durable β-Catenin Degradation and Suppresses Wnt-Dependent Intestinal Cancer. Cell Discov. 2020, 6 (1), 1–12. 10.1038/s41421-020-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Saenz DT; Fiskus W; Qian Y; Manshouri T; Rajapakshe K; Raina K; Coleman KG; Crew AP; Shen A; Mill CP; Sun B; Qiu P; Kadia TM; Pemmaraju N; DiNardo C; Kim M-S; Nowak AJ; Coarfa C; Crews CM; Verstovsek S; Bhalla KN Novel BET Protein Proteolysis-Targeting Chimera Exerts Superior Lethal Activity than Bromodomain Inhibitor (BETi) against Post-Myeloproliferative Neoplasm Secondary (s) AML Cells. Leukemia 2017, 31 (9), 1951–1961. 10.1038/leu.2016.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Buhimschi AD; Armstrong HA; Toure M; Jaime-Figueroa S; Chen TL; Lehman AM; Woyach JA; Johnson AJ; Byrd JC; Crews CM Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 2018, 57 (26), 3564–3575. 10.1021/acs.biochem.8b00391. [DOI] [PubMed] [Google Scholar]
- (41).Burslem GM; Song J; Chen X; Hines J; Crews CM Enhancing Antiproliferative Activity and Selectivity of a FLT-3 Inhibitor by Proteolysis Targeting Chimera Conversion. J. Am. Chem. Soc 2018, 140 (48), 16428–16432. 10.1021/jacs.8b10320. [DOI] [PubMed] [Google Scholar]
- (42).Zhang X; Lee HC; Shirazi F; Baladandayuthapani V; Lin H; Kuiatse I; Wang H; Jones RJ; Berkova Z; Singh RK; Lu J; Qian Y; Raina K; Coleman KG; Crews CM; Li B; Wang H; Hailemichael Y; Thomas SK; Wang Z; Davis RE; Orlowski RZ Protein Targeting Chimeric Molecules Specific for Bromodomain and Extra-Terminal Motif Family Proteins Are Active against Pre-Clinical Models of Multiple Myeloma. Leukemia 2018, 32 (10), 2224–2239. 10.1038/s41375-018-0044-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Shen Y; Gao G; Yu X; Kim H; Wang L; Xie L; Schwarz M; Chen X; Guccione E; Liu J; Bedford MT; Jin J Discovery of First-in-Class Protein Arginine Methyltransferase 5 (PRMT5) Degraders. J. Med. Chem 2020, 63 (17), 9977–9989. 10.1021/acs.jmedchem.0c01111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Roberts BL; Ma Z-X; Gao A; Leisten ED; Yin D; Xu W; Tang W Two-Stage Strategy for Development of Proteolysis Targeting Chimeras and Its Application for Estrogen Receptor Degraders. ACS Chem. Biol 2020, 15 (6), 1487–1496. 10.1021/acschembio.0c00140. [DOI] [PubMed] [Google Scholar]
- (45).Guo L; Zhou Y; Nie X; Zhang Z; Zhang Z; Li C; Wang T; Tang W A Platform for the Rapid Synthesis of Proteolysis Targeting Chimeras (Rapid-TAC) under Miniaturized Conditions. Eur. J. Med. Chem 2022, 236, 114317. 10.1016/j.ejmech.2022.114317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Guo L; Liu J; Nie X; Wang T; Ma Z; Yin D; Tang W Development of Selective FGFR1 Degraders Using a Rapid Synthesis of Proteolysis Targeting Chimera (Rapid-TAC) Platform. Bioorg. Med. Chem. Lett 2022, 75, 128982. 10.1016/j.bmcl.2022.128982. [DOI] [PubMed] [Google Scholar]
- (47).Wang L; Zhao Z; Meyer MB; Saha S; Yu M; Guo A; Wisinski KB; Huang W; Cai W; Pike JW; Yuan M; Ahlquist P; Xu W CARM1 Methylates Chromatin Remodeling Factor BAF155 to Enhance Tumor Progression and Metastasis. Cancer Cell 2014, 25 (1), 21–36. 10.1016/j.ccr.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Lee J; Bedford MT PABP1 Identified as an Arginine Methyltransferase Substrate Using High-Density Protein Arrays. EMBO Rep. 2002, 3 (3), 268–273. 10.1093/embo-reports/kvf052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kim E-J; Liu P; Zhang S; Donahue K; Wang Y; Schehr JL; Wolfe SK; Dickerson A; Lu L; Rui L; Zhong X; Wisinski KB; Yu M; Suzuki A; Lang JM; Ong IM; Xu W BAF155 Methylation Drives Metastasis by Hijacking Super-Enhancers and Subverting Anti-Tumor Immunity. Nucleic Acids Res. 2021, 49 (21), 12211–12233. 10.1093/nar/gkab1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Ma M; Zhao X; Chen S; Zhao Y; yang L; Feng Y; Qin W; Li L; Jia C Strategy Based on Deglycosylation, Multiprotease, and Hydrophilic Interaction Chromatography for Large-Scale Profiling of Protein Methylation. Anal. Chem 2017, 89 (23), 12909–12917. 10.1021/acs.analchem.7b03673. [DOI] [PubMed] [Google Scholar]
- (51).Schaab C; Geiger T; Stoehr G; Cox J; Mann M Analysis of High Accuracy, Quantitative Proteomics Data in the MaxQB Database*. Mol. Cell. Proteomics 2012, 11 (3), M111.014068. 10.1074/mcp.M111.014068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Tyanova S; Temu T; Sinitcyn P; Carlson A; Hein MY; Geiger T; Mann M; Cox J The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13 (9), 731–740. 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- (53).RStudio Team (2020). RStudio: Integrated Development for R. RStudio, PBC, Boston, MA: URL http://www.rstudio.com/. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.