

Abstract
Environmental exposure to metals is known to cause a number of human toxicities including cancer. Metal-responsive transcription factor 1 (MTF-1) is an important component of metal regulation systems in mammalian cells. Here, we describe a novel method to identify chemicals that activate MTF-1 based on microarray profiling data. MTF-1 biomarker genes were identified that exhibited consistent, robust expression across 10 microarray comparisons examining the effects of metals (zinc, nickel, lead, arsenic, mercury, and silver) on gene expression in human cells. A subset of the resulting 81 biomarker genes was shown to be altered by knockdown of the MTF1 gene including metallothionein family members and a zinc transporter. The ability to correctly identify treatment conditions that activate MTF-1 was determined by comparing the biomarker to microarray comparisons from cells exposed to reference metal activators of MTF-1 using the rank-based Running Fisher algorithm. The balanced accuracy for prediction was 93%. The biomarker was then used to identify organic chemicals that activate MTF-1 from a compendium of 11 725 human gene expression comparisons representing 2582 chemicals. There were 700 chemicals identified that included those known to interact with cellular metals, such as clioquinol and disulfiram, as well as a set of novel chemicals. All nine of the novel chemicals selected for validation were confirmed to activate MTF-1 biomarker genes in MCF-7 cells and to lesser extents in MTF1-null cells by qPCR and targeted RNA-Seq. Overall, our work demonstrates that the biomarker for MTF-1 coupled with the Running Fisher test is a reliable strategy to identify novel chemical modulators of metal homeostasis using gene expression profiling.
Introduction
Homeostasis of metal ions is tightly regulated in biological systems due to the toxicity of many metals.1–3 At high concentrations, even essential metals like copper, zinc, iron, and manganese, which have important structural and catalytic functions in proteins, can cause toxicity. Human exposures to metals have been increasing as a result of increased use in a number of applications.4 Sources of heavy metals in the environment include geogenic, industrial, agricultural, pharmaceutical, domestic effluents, and atmospheric sources.5 Environmental pollution is significant from point sources involving mining and in foundries, smelters, and other metal-based industrial operations.5,6 Heavy metals, especially nonessential metals such as arsenic, cadmium, chromium, lead, and mercury are of public health significance due to their ability to act as systemic toxicants that induce damage in multiple organs, even at low levels of exposure. Many of these metals are classified as known or probable human carcinogens according to the US Environmental Protection Agency (EPA) and the International Agency for Research on Cancer.
One of the most important biological mechanisms used by cells to regulate and protect against toxic metal exposure is sequestration by metallothioneins (MTs) which are small, cysteine-rich proteins that bind excess metal ions under conditions of metal overload.7 During metal overload, MT-bound zinc ions are displaced, increasing levels of cellular zinc ions that activate the metal responsive transcription factor 1 (MTF-1), the major transcriptional regulator of responses to toxic metal overload. MTF-1 contains six highly conserved DNA binding zinc finger domains, which are sensitive to changes in zinc concentration. Excess zinc binds to the zinc fingers, increasing the binding affinity of MTF-1 with metal responsive elements (MREs) in the regulatory regions of target genes including MT family members allowing zinc-inducible gene regulation. The MTF-1-MT regulon forms a negative feedback loop in which activation of MTF-1 leads to increases in the expression of MT family members, sequestration of excess zinc, and subsequent decreases in MTF-1 activation.8–10 MTF-1 also increases the expression of a zinc transporter (ZnT1) which reduces intracellular zinc availability by promoting zinc efflux from the cytoplasm or into intracellular vesicles.11
There is evidence that increases in oxidative stress can activate MTF-1 through effects on MT family members. During conditions of oxidative stress or hypoxia, MTs become oxidized resulting in structural alterations that lead to release of zinc.12–14 Like metal overload, the increase in free zinc in the cell leads to activation of MTF-1. The universality of oxidative stress-induced MTF-1 activation under different chemical exposure conditions has not been previously explored. Nuclear factor erythroid 2-related factor 2 (Nrf2) is another transcription factor that responds to cellular oxidative stress; however, the relationship between the activation of MTF-1 and Nrf2 pathways is not fully defined but may include inactivation of the negative regulator of Nrf2 called Keap1, which has a phylogenetically conserved Zn binding site.15
Because of the relevance of metal exposure to human health and disease, high-throughput screening (HTS) methods to identify conditions in which metal homeostasis is disturbed would be useful to predict cellular responses to toxic metal overload and oxidative stress. The only known HTS assay that potentially measures alterations in metal homeostasis through MTF-1 activation is part of the EPA ToxCast screening program (http://epa.gov/ncct/toxcast/; Attagene, “ATG_MRE_CIS_up”) which measures the ability of endogenously expressed MTF-1 to activate a reporter gene in HepG2 cells. There are ~700 HTS assays representing ~350 molecular targets that have been used to screen more than 1800 chemicals.16 High-throughput transcriptomic (HTTr) technologies have now been added to the ToxCast screening battery17 in which targeted sequencing techniques are used to assess the expression of the human genome in chemically treated cells.18 HTTr is being used as a global assay to potentially identify any pathway that is perturbed upon chemical exposure, not just the ones examined by HTS assays. Targeted HTS assays could then be used to confirm the putative modulators.17 Approaches to assess MTF-1 activation in HTTr data have not been described but would be potentially useful to identify environmentally relevant chemicals that perturb metal homeostasis.
In the present study, we describe procedures for assessing MTF-1 activation using computational analysis of gene expression data. The large quantity of microarray data that already exists in public repositories and in commercial databases allows for in silico HTS prediction of chemical agents that activate or suppress a wide range of human molecular targets including MTF-1. As proof of principle, our lab has built and characterized gene expression biomarkers that predict modulation of the estrogen receptor and androgen receptor in human cell lines.19,20 Using similar approaches, we constructed a gene expression biomarker that accurately detects MTF-1 activation by different metals. We used the biomarker to screen a library of microarray profiles from cells treated with ~2600 organic chemicals to identify novel activators of MTF-1. All chemicals selected for validation were found to exhibit the properties of MTF-1 activators in MCF-7 cells.
Experimental
Use of a gene expression microarray experiment compendium
As described previously,21 data from BaseSpace Correlation Engine (BSCE) (https://www.illumina.com/products/by-type/informatics-products/basespace-correlationengine.html; formerly NextBio) were used to build an annotated spreadsheet of statistically filtered gene expression comparisons (biosets) from experiments carried out using human cell lines and tissues. This compendium included bioset name, cell line, tissue, chemical name, chemical concentration, treatment time, and study ID. Of the biosets included, 11725 biosets were derived from experiments in which cells were treated with one or more chemicals. Methods used to derive the statistically filtered gene lists are described in detail in Kuperschmidt et al.22
Construction of the MTF-1 biomarker
Biosets from the BSCE database were selected to represent a variety of human cell lines and metal treatment conditions expected to activate MTF-1, including treatments with silver, arsenic, mercury, zinc, and nickel. From these biosets, 10 were selected whose fold-changes were consistent across treatments and included genes which exhibited opposite regulation after MTF-1 was knocked down using siRNA.23 Raw expression microarray data for each of the 10 biosets was downloaded from the NCBI GEO database and processed using Partek Genomics Suite to generate gene lists with expression foldchange values for each bioset. Gene lists were filtered to include only genes which had an absolute fold change magnitude of ≥1.2 and derived using a one-way ANOVA. The gene lists were uploaded to BSCE and the meta-analysis function was used to identify genes with the greatest degree of overlap between the 10 biosets. To construct the biomarker, genes were selected that were differentially expressed in the same direction in at least 5 of the 10 biosets and had an average fold change across those biosets which showed significant expression of at least 1.5 in either direction. Genes in the biomarker that overlapped with those in the Nrf2 biomarker (SLC7A11, GCLM, ADM, SRXN1, TXNRD1, SLC3A2, LARP6)24 were excluded from the MTF-1 biomarker. The final biomarker consisted of 81 genes and average fold-change levels. The biomarker was uploaded to BSCE without any further filtering.
Comparison of the biomarker to microarray compendium biosets
The MTF-1 biomarker was uploaded to BSCE and compared with all biosets in the database using the Running Fisher test. This method provides an assessment of the statistical significance of the correlation of the overlapping genes between the biomarker and each bioset providing a summary -value. A complete description of the Running Fisher test is provided in ref. 22. The results were exported and each -value was converted to a, with negative values used to indicate negative correlation between the biomarker and the bioset. Biosets with or ≤ −4 were considered significant based on prior studies using this threshold.20,25 A column in the human gene expression spreadsheet was populated with the for each bioset. Biosets that were positively correlated with the biomarker were predicted to activate MTF-1, either directly or indirectly, such as by inducing oxidative stress or increasing labile cellular Zn; biosets that were negatively correlated were predicted to suppress MTF-1. The minor number of biosets identified here that exhibited negative correlation were not considered further.
Selection of positive and negative controls and calculation of biomarker accuracy
In the database of human gene expression comparisons, 57 biosets were selected as true positives as they were expected to activate MTF-1, based on the fact that they came from human cells treated with known activators of MTF-1 such as zinc, arsenic, iron, gold, or silver. The 10 biosets used to create the biomarker (the training set) were not included in this list. Negative controls were identified using data from the EPA ToxCast program. In the EPA ToxCast online dashboard (https://comptox.epa.gov/dashboard), the ATG_MRE_CIS_up assay (carried out by contract with the EPA by Attagene, Inc.) was selected, and all chemical data was downloaded. Tested chemicals that were examined in both the HTS assay and the human microarray compendium were identified. This list was filtered to include only (1) chemicals with no activity in the ATG_MRE_CIS_up assay and (2) microarray biosets from chemical treatments that used 75–100% of the maximum concentration tested in the ToxCast assay. Using these criteria, there were 23 chemicals classified as true negatives. The values for predictive accuracy were calculated as follows: sensitivity (true positive rate) = true positive (TP)/(TP + false negative (FN)); specificity (true negative rate) = true negative (TN)/(false positive (FP) + TN); positive predictive value; negative predictive value; balanced accuracy = (sensitivity + specificity)/2.
Virtual screen for potential MTF-1 activators using the biomarker
Biosets were ranked by, and a list was generated of the top organic chemicals predicted to activate MTF-1 (Supplemental File 1, ESI†). Chemicals were excluded if they were not activated in the majority (more than half) of available biosets in which the chemical was tested in human cells. Of the top predicted MTF-1-activating chemicals remaining, nine commercially-available chemicals were selected for in vitro validation in our study.
Ingenuity pathway analysis
The MTF-1 biomarker genes were analyzed using the canonical pathway and upstream analysis functions of Ingenuity Pathway Analysis (IPA, Qiagen Bioinformatics, Redwood City, California). IPA calculates significance using a right-tailed Fisher’s Exact test. The -value is the probability of the overlap between the MTF-1 biomarker gene list and the IPA pathway gene list. Upstream analysis used the number of differentially expressed genes to predict upstream regulators of the biomarker genes.
Creation of a MTF1 knockout cell pool
The Knockout Cell Pool, MTF-1 Knockout MCF7, was created by Synthego, Inc. The MCF-7 cells were engineered by CRISPR-Cas9 to knock out the MTF-1 gene. The guide RNA was designed to bind to the antisense strand, in exon 2, of MTF1 gene to create a double stranded break at location 37857591 on chromosome 1. The knockout efficiency was determined by Synthego’s Inference of CRISPR Edits (ICE) analysis; 96% of the cell pool exhibited a disruption of the MTF-1 open reading frame.
Culture and treatment of wild-type and MTF-1-null MCF-7 cells
Chemicals were purchased from Sigma-Aldrich. All chemicals were ≥95% purity except for thiostrepton and sulforaphane (90%). All chemical stock solutions were made in DMSO. MCF-7 cells were cultured in DMEM media (GIBCO) supplemented with 10% FBS (Omega Scientific, Australia) and 1× penicillin/streptomycin/glutamine. An initial range finding experiment (data not shown) was conducted to determine the optimal concentration of the tested chemicals. Based on these preliminary results, the highest concentration that was clearly non-cytotoxic was chosen as the test concentration for each chemical. Cells were plated at 15 × 105 cells per well in 12-well plates. After 48 hours, media was replaced with dosing solutions containing DMSO (0.05%) or 1–100 μM of each chemical (final concentration of DMSO = 0.05%). After 6 hours of exposure, media was removed; cells were lysed in 0.5 mL Trizol, followed by RNA extraction.
Evaluation of gene expression using TempO-Seq
Gene expression in wild-type and MTF1-null cells were evaluated for gene expression changes after chemical exposure in a focused study using the human 1500+ Tempo-Seq platform (BioSpyder, Inc., Carlsbad, CA). After RNA extraction, samples were sent to BioSpyder for analysis. Raw read counts were normalized and statistically-filtered gene lists (-value <0.05 with no multiple test correction) were generated using the DESeq2 module in Partek Flow. The data is publicly available at Gene Expression Omnibus, accession number GSE152703.
Measurement of oxidative stress
MCF-7 cells were treated with 10 μM of the reactive oxygen species probe 2′,7′-dichlorofluorescin diacetate (DCFDA, Sigma-Aldrich) or no probe in Hank’s balanced salt solution (HBSS) for 30 min. This solution was removed and cells were washed twice before treatment with each compound or DMSO control for two hours. Fluorescence was measured (excitation 490 nm, emission 530 nm) with a SpectraMax i3X plate reader (Molecular Devices, San Jose, CA). The fluorescence for the no-probe controls was subtracted from the fluorescence of DCFDA conditions and normalized to the DMSO condition. The data was subjected to Student’s -test with significance criteria of.
Measurement of free zinc
MCF-7 cells were treated with each compound or DMSO control in DMEM and incubated for one hour. Media was removed and cells were washed twice with HBSS. New media containing 5 μM of the fluorescent zinc probe Zinpyr-1 (Santa Cruz Biotechnology) or no probe was added and cells were incubated for 30 minutes. Media was removed and cells were washed twice again before reading fluorescence (excitation 490 nm, emission 530 nm) with a SpectraMax i3X, Molecular Devices plate reader (San Jose, CA). The fluorescence for the no-probe controls was subtracted from the fluorescence of Zinpyr-1 conditions and normalized to the DMSO condition. The analysis was conducted on 4–5 independent experiments. The data was subjected to Student’s t-test with significance criteria of.
RT-qPCR analysis of MTF-1 biomarker genes
Wild-type MCF-7 cells and MTF-1 null cells were treated with 10 selected MTF-1 activators at concentrations indicated in Fig. 6 for 6 h in 3–4 independent experiments. Total RNA was isolated in Trizol, reverse transcribed (Bioline, UK), and subjected to qPCR analysis with SYBR Green master mix (BioRad, CA) on Bio-Rad CFX real-time PCR system. The primers were designed with Primer3 and listed in Supplemental Table S1 (ESI†). The Ct values were used to calculate the relative expression by the method and normalized with b-actin, setting WT control as 100%. The Student t-text was used to compare the difference between WT and MTF-1 null cells, with significance criteria of.
Fig. 6.

Expression of MTF-1 biomarker genes in wild-type and MTF-1-null cells. Wild-type and MTF-1-null cells were treated with the indicated chemicals for 6 h at the indicated concentrations. Expression of the four MTF-1 biomarker genes were examined by RT-qPCR. Expression levels are shown as mean and standard error of the mean. independent experiments. * Indicates significant difference between wild-type and MTF-1-null cells;.
Preparation of chemicals for clustering and association rule mining
Chemicals from the CMAP 2.0 collection of chemicals with defined structural information and observed MTF1 activity in at least 2 of the 3 cell lines were used to perform the clustering and association rule mining algorithms. Structures in the form of simplified molecular-input line entry system (SMILES) notation were taken from the EPA CompTox Chemicals dashboard (https://comptox.epa.gov/dashboard/, accessed November 19th 2018).26 Substances which did not meet the condition of activating MTF-1 in 2 or 3 of the 3 cell lines were removed from the cheminformatic analysis. A chemical fingerprint file (a matrix of binary values [1,0] for each chemical-feature pair) was derived using the publicly available ToxPrint feature set (https://toxprint.org/) generated within the associated Chemo-typer application (https://chemotyper.org/ accessed November 19th 2018), developed by Altamira [Altamira, Columbus, OH] and Molecular Networks [Molecular Networks, Erlangen, GmbH] under contract from FDA. The ToxPrint set (V2.0_r711) consists of 729 uniquely defined features designed to provide broad feature coverage of inventories consisting of tens of thousands of environmental and industrial chemicals, including pesticides, cosmetics ingredients, food additives, and drugs.27
Chemical clustering
To generate chemical clusters, the ChemmineR package (v.3.32.1) was used to calculate a distance matrix of the Tanimoto similarity index between each pair of ToxPrint fingerprints and, subsequently, chemicals were binned together using the single-linkage (nearest neighbor) clustering function.28 A range of different similarity thresholds from 0.6 (60%) to 0.9 (90%) in 5% increments was used to cluster the chemicals. The final threshold was chosen based upon a visual inspection of the clusters generated by each similarity threshold.
Identification of association rules
Association rule mining (ARM), as implemented within the arules package in R,29,30 was employed to investigate whether certain (sets of) ToxPrints were associated with a chemical having a positive designation. It should be noted that if a chemical was tested at two different concentrations and given a different designation at each concentration, only the concentration with the positive designation was retained. Briefly, ARM is a data mining method that can identify highly correlated (groups of) variables and has been utilized in a variety of application domains including bioinformatics, and chemoinformatics.31–37 The ARM workflow comprises two main steps: frequent itemset mining (FIM) and generation of rules. More information on FIM and ARM can be found in studies by Agrawal, Bagelt and colleagues.29,38,39
The first step of FIM aims to identify items in a dataset that frequently co-occur. Within FIM, there are 3 components to consider; a set of items, where, a set of transactions, where and a transaction t which contains a subset of the items in (also called an itemset). In this analysis, the individual ToxPrints and the MTF-1 activity call (positive or negative) represent the items, whereas a given chemical represents a transaction .
Once identified, frequent itemsets can then be examined and expressed as a collection of probabilistic if/then statements called association rules. They take the form of where and are itemsets. Each association rule comprises two parts: (1) the “if” or antecedent (i.e. the left-hand side, LHS) and (2) the “then” or consequent (i.e. the right-hand side, RHS). The basic premise behind the generation of association rules is splitting a frequent itemset into two disjoint subsets and such that items present in the LHS are not present in the RHS (i.e.). In this study, the ToxPrints served as the antecedent, whereas the MTF-1 activity call served as the consequent. It should be noted that for the association rules generated the antecedent could be a set of up to three separate ToxPrints. Additionally, we filtered for only those rules containing a positive designation as the consequent.
Several interest measures were calculated to help identify a pragmatic and practical number of frequent itemsets and their resulting association rules for use. These measures themselves are defined as follows:
- • Support: the support of an association rule () describes the proportion of transactions that contain all items in the rule (i.e. the union of and ).
- • Confidence: the confidence of a rule meanwhile describes the proportion of all transactions containing that also contain . Alternatively, confidence is the conditional probability of the transaction containing given the presence of .
- • Lift: the lift of a rule describes the factor by which the observed co-occurrence of and exceeds what would be expected if and were independent.
- • Odds ratio: the odds ratio of a rule describes the odds of occurring given the presence of compared against occurring in the absence of .
A combination of the support and confidence interest measures were used to limit the number of association rules generated, specifically, a rule had to have a minimum support of 4 (i.e., ToxPrints and positive activity outcome needed to appear in at least 4 chemicals within the dataset) and a minimum confidence of 75% (i.e., the ToxPrints needed to be associated with a positive activity call at least 75% of the time). The derived rules were subsequently applied to the set of 284 unique chemicals from the list of CMAP chemicals where (1) no overall designation had been determined in the MTF-1 assays, and (2) structural information (in terms of SMILES notation) were present in the EPA’s CompTox Chemicals Dashboard (https://www.epa.gov/chemical-research/comptox-chemicalsdashboard; accessed March 17, 2020). This was carried out to identify other chemicals that contain the structural features present in at least one of the rules derived from the ARM algorithm.
Inventory comparison
As the ToxPrint chemotypes are a predefined set of structural features that cover a broad cross section of the available chemical space, they were used to compare the structural diversity of the chemicals studied within this analysis to those tested in the ATG_MRE_CIS_up assay as part of the ToxCast program. The comparison would provide an indication of whether the rules derived were generalizable. To do this the ToxPrint fingerprints were generated for those chemicals tested in the ATG_MRE_CIS_up assay. Subsequently, the fingerprints for both sets of chemicals were folded to reduce their length from 729 features to 70 features so the comparison could be more easily visualized. This folding took advantage of the hierarchical nature of the ToxPrints such that more specific ToxPrints were collapsed into their more generalised form. For example, the “bond:C#N_cyano_acylcyanide”, “bond:C#N_cyano_cyanamide”, and “bond:C#N_nitrile_ab-unsaturated” chemotypes would all fall under the more general “bond:C#N” chemotype. The relative quantities of each of these more general chemotypes were then calculated and compared between the two data sets.
Results and discussion
Construction of an MTF-1 biomarker
A set of biomarker genes was identified to predict modulation of MTF-1 activity. Genes were identified from 10 microarray comparisons (biosets) representing diverse metal treatment conditions and human cell types (Table 1). Raw gene expression data from the studies was used to generate the gene lists using Partek Genomics Suite. These statistically-filtered gene lists were compared to the corresponding gene lists found in BSCE (Fig. S1, ESI†); strong correlations were found between the two computationally-derived bioset gene lists. The Partek-generated gene lists were used to identify biomarker genes.
Table 1.
Biosets used to construct the MTF-1 biomarker
| Bioset name | Factor | Study |
|---|---|---|
|
| ||
| Jurkat T-cells 6 h post 3 μM arsenic trioxide treatment vs. untreated | Arsenic trioxide | GSE46909 |
| Colorectal cancer Caco-2 cells 6 h treated with 60 nM AgNPs vs. untreated controls | Silver nanoparticles | GSE84982 |
| Lymphoblastoid B cells treated 12 h with 5 μM sodium arsenite vs. untreated controls | Sodium arsenite | GSE51454 |
| PBMCs + 1 d 10 μM lead acetate + 1 d fresh media vs. 2 d untreated donor-matched controls | Lead acetate | GSE37567 |
| HUVEC cells + 1.5 mM nickel chloride for 5 h vs. control | Nickel chloride | GSE4852 |
| Neuroepithelial progenitors from ESCs in 6 d + mercury(II) bromide (HgBr2) vs. culture medium only | Mercuric bromide | GSE71127 |
| Macrophages derived from monocytes treated 6 h with 10 μg mL−1 ZnO particles vs. 6 h untreated | Zinc oxide nanoparticles | GSE39444 |
| Breast adenocarcinoma MCF-7 cells 6 h treated with 60 nM AgNPs vs. untreated controls | Silver nanoparticles | GSE84982 |
| Hepatic stellate cells 30 μg mL−1 ZnSO4 treated 24 h vs. untreated | Zinc sulfate | GSE60159 |
| Breast adenocarcinoma MCF-7 cells 24 h treated with 1.5 μg mL−1 AgNO3 vs. untreated controls | Silver nitrate | GSE84982 |
Genes that exhibited relatively robust and consistent expression changes across the 10 biosets were selected as candidate biomarker genes (absolute average fold-change ≥1.5-fold in either direction). Given that metal exposure can lead to oxidative stress that activates Nrf2,8 we filtered out any genes that overlapped with a previously characterized biomarker for Nrf2.24 The behavior of the resulting 81 genes across the 10 biosets is shown in Fig. 1A. The gene list and fold-change values were used as the MTF-1 biomarker. The biomarker consisted of 48 upregulated genes and 33 downregulated genes. The genes with the highest fold-change values included seven MT family members (MT1E, MT1F, MT1G, MT1H, MT1M, MT1X, MT2A) and the zinc transporter SLC30A1, all well known to be regulated by MTF-1.11 Several of the other top ranked genes in the biomarker are not known MTF-1 targets but have indirect connections to the oxidative stress response or metal detoxification and homeostasis. For example, a top ranked gene in the biomarker was AKR1C2 (aldo-keto reductase family 1 member C2), which has known antioxidant effects in cancer cells.40 Other highly ranked genes in the biomarker were ATF3 (activating transcription factor 3) and CLU (clusterin), known to be activated under conditions of oxidative stress.41–43
Fig. 1.

Building and characterization of a MTF-1 biomarker. (A) Gene expression profiles of the 10 biosets used to construct the MTF-1 biomarker. The expression of the statistically significant genes are shown across the 81 genes in biomarker. The biomarker consists of fold-change values averaged across the treatments with significant changes. (B) Ingenuity pathways analysis of biomarker genes. (top) Canonical pathways that significantly overlap with the biomarker genes. Biomarker genes were compared to the genes in the canonical pathway lists from IPA. (bottom) Predicted upstream transcriptional regulators. (C) The of the 10 Partek-generated biosets used to construct the biomarker. The bioset comparisons are in the same order as those in (A). (D) MTF-1-dependence of biomarker genes. Biomarker genes were compared to a microarray comparison between cells treated with siRNAs against MTF-1 vs. cells treated with control siRNA with both groups treated with 150 μM zinc.
To identify pathways enriched in biomarker genes, the canonical pathway and upstream analysis functions in Ingenuity Pathway Analysis were used (Fig. 1B). The analysis identified the Nrf2-mediated oxidative stress response pathway and the iron homeostasis signaling pathways as the top overlapping pathways, with 3.5% () and 2.9% () of the genes overlapping, respectively. Several of the top upstream regulators predicted by the analysis were transcription factors and included MTF-1 ().
Comparison of the biomarker to individual biosets used to make the biomarker
BSCE was used to determine the correlation between the biomarker and each of the 10 Partek-generated biosets used in the construction of the biomarker. BSCE uses the rank-based Running Fisher algorithm to calculate the statistical significance of each pair-wise correlation. Statistically significant correlations were defined as those with a or ≤ −4, where negative values indicate a negative correlation. Chemical treatment conditions that resulted in positively correlated biosets were predicted to be activators of MTF-1, while those conditions that resulted in negatively correlated biosets were predicted to be suppressors of MTF-1. (Suppressors of MTF-1 were not examined in this study.) Of the 10 biosets used to create the biomarker, nine were found to be significantly correlated with the biomarker (Fig. 1C). The one bioset not significantly correlated () had only two significantly altered genes overlapping with the biomarker. We also compared the biomarker to the same 10 BSCE-derived biosets in the human compendium. Fig. S2 (ESI†) shows that the biomarker correlations were similar independent of the method used to derive the bioset, although most of the biosets generated by the Partek approach had higher .
MTF-1 dependence of biomarker genes
To determine if any of the biomarker genes exhibited MTF-1 dependence in their expression profiles, we examined expression of the genes after MTF-1 siRNA knockdown from a recent microarray study using colorectal cancer cells.23 For this analysis, the data from this bioset was processed in Partek to generate a gene list using a fold-change cutoff of 1.2 in either direction and a significance cutoff of. There were 20 significantly altered genes that overlapped between the biomarker and a comparison in which MTF-1 knockdown was compared to cells treated with control siRNA in which both groups were treated with 150 μM zinc. Out of 20 genes, 9 of the biomarker upregulated genes exhibited decreased expression by MTF-1 siRNA (Fig. 1D), and 5 of the biomarker downregulated genes exhibited increased expression by MTF-1 siRNA. This bioset was significantly negatively correlated with the biomarker (). These results indicated that at least a subset of the biomarker genes were dependent on MTF-1 for metal-induced expression. We also show MTF-1-dependence of the biomarker gene responses in experiments described below.
Correlation between MTF-1 and Nrf2 activation
Because metal toxicity is known to induce oxidative stress, we examined the relationship between activation of MTF-1 and Nrf2 by comparing biomarker responses across the biosets in the human compendium. Fig. S3 (ESI†) compares the for each of the 11725 biosets for both biomarkers. The correlation coefficient was calculated to be with , indicating that predicted activation of the two transcription factors is correlated in a positive manner. This correlation does not change meaningfully when the biosets were filtered for time (e.g. r = 0.432, >6 h treatment) or cell line (e.g., >6 h treatment, MCF7 cells). This analysis indicates a tight relationship between activation of MTF-1 and Nrf2. The inclusion of a number of stress response genes in the MTF-1 biomarker including those that may not be directly regulated by MTF-1 indicates that the biomarker represents the gene expression profile of a general cellular response to metal-induced stress, not just the direct induction of metal regulatory genes by MTF-1. Using this biomarker as an indicator of the metal stress response may therefore be more useful as a measure of systems changes due to metal-induced toxicity than simply measuring changes in MT gene expression.
Predictive accuracy of the biomarker
In the compendium of human gene expression biosets, there were many examples of cells treated with metals under conditions expected to activate MTF-1 (excluding those used to make the biomarker). We randomly selected 57 to use as those that activate MTF-1. Using the Running Fisher test, correlation between each of these biosets and the biomarker was determined and 52 of the 57 were predicted to activate MTF-1 resulting in a sensitivity of 91%. Results from the EPA’s ToxCast chemical screening program (MTF-1 activation assay run by Attagene, Inc.) were used to identify 9 chemicals represented by 23 biosets in the database which were classified as true negatives for MTF-1 activation. Of these, only one of the 23 was predicted by the biomarker to activate MTF-1, demonstrating a specificity of 96%. Combining these results, the biomarker gave a balanced accuracy of 93%. Fig. 2 shows the distribution of the for the true positives and true negatives, demonstrating that the biomarker reliably identifies MTF-1 activators from non-activators.
Fig. 2.

Assessment of the predictive accuracy of the MTF-1 biomarker. The for biosets using expected MTF-1 activators (true positives, blue) and biosets using non-MTF-1-activators identified by ToxCast (true negatives, red). The cutoff for statistical significance was .
Virtual screen for chemical activators of MTF-1
Given the high predictive accuracy of the biomarker, we performed an in silico screen to identify novel chemicals that activate MTF-1. In the human compendium, there were 11725 biosets representing human cell lines exposed to 2582 chemicals. Excluding the biosets used to create the biomarker, the analysis identified 1547 biosets representing 700 chemicals that were positively correlated and 122 biosets representing 90 chemicals that were negatively correlated with the MTF-1 biomarker. The remaining 10066 biosets were not significantly correlated with the biomarker in either direction. The statistically filtered fold-change values of the biomarker genes for all 11725 biosets ranked by are shown in Fig. 3, top, and the for individual biosets are shown in Fig. 3, bottom. The top most correlated chemical treatments are shown. For those biosets that were significantly positively correlated with the biomarker, the expression of the biomarker genes was remarkably similar to the fold-change values of the biomarker genes. For those biosets that were significantly negatively correlated, the expression of the biomarker genes exhibited opposite expression compared to the biomarker genes.
Fig. 3.
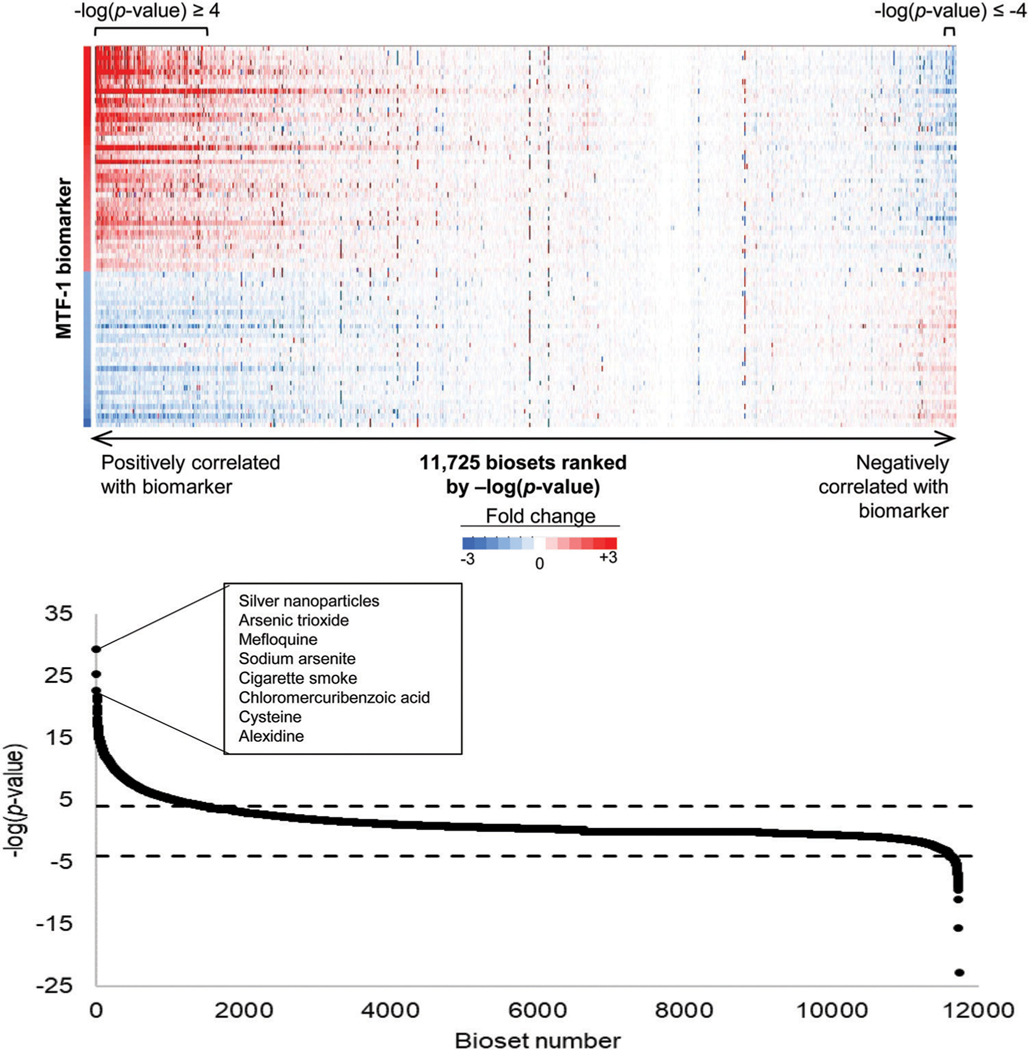
Screening a human microarray compendium for MTF-1 activators. (Top) Expression changes of the MTF-1 biomarker genes across 11 725 biosets evaluating chemical-induced changes in gene expression, excluding those used to make the biomarker. The MTF-1 biomarker fold change values are shown on the left. Biosets were ranked by of the correlation between the biomarker and the bioset. (Bottom) The for correlations between the MTF-1 biomarker and each of the 11 725 biosets, excluding those used to construct the biomarker. The cutoffs for statistical significance are shown with dashed lines. The biosets with were considered positively correlated while the biosets with were considered negatively correlated with the biomarker. Top chemical hits that led to activation of MTF-1 are indicated.
Characterization of putative MTF-1 activators
In addition to the chemicals used in the prediction analysis, there were additional metal treatments in the database. We compared those to the biomarker to examine which metal treatment conditions were correlated with the MTF-1 biomarker. There were a total of 89 metal treatment biosets identified (including the 58 true positives) discussed above, representing 10 different metals. For this analysis, all metal-containing compounds of the same metal were grouped together. The metals with the greatest number of database biosets were zinc, silver, and arsenic. The MTF-1 biomarker predicted that the majority of zinc treatments (87.1%), silver treatments (95.7%), and arsenic treatments (100%) activated MTF-1 (Table 2). All other metals in the database either have very few representative biosets in the database, or they are most often not predicted to activate MTF-1. Most surprisingly, cadmium, which is represented by seven biosets and known to activate MTF-1, was only predicted to activate MTF-1 in two of seven biosets. However, the majority of the cadmium biosets not predicted to activate MTF-1 were derived from one study using one cell line. The lack of MTF-1 activation in these experiments may be particular to this cell line or due to other experimental conditions used in this study. Comparison of the biomarker to a larger and more diverse sample of metal treatment experiments would be necessary to confidently assess the differences in MTF-1 activation by different metals as predicted by the biomarker.
Table 2.
Metal treatment biosets in the human compendium and their rate of predicted MTF-1 activation
| Metal | % of biosets with | |
|---|---|---|
|
| ||
| Zinc | 87.1 | 31 |
| Silver | 95.7 | 23 |
| Arsenic | 100.0 | 8 |
| Cadmium | 28.6 | 7 |
| Nickel | 14.3 | 7 |
| Cobalt | 100.0 | 5 |
| Mercury | 100.0 | 3 |
| Iron | 50.0 | 2 |
| Lead | 100.0 | 2 |
| Titanium | 100.0 | 1 |
Identification of structural features associated with MTF-1 activation
We next asked whether there were any structural features in the organic compounds associated with MTF-1 activation. (Metals were not considered in this analysis.) To facilitate the analysis, we focused on a set of 616 unique CMAP chemicals that had been examined in three cell lines (HL60, MCF-7, PC3), had an overall activity call for MTF-1 consistent across the cell lines, and for which chemical structures could be readily identified. There was activation of MTF-1 by 105 unique chemicals in two or more cell lines, and of these there were 81 in which chemical structures could be identified. Two approaches were undertaken to address whether there were structural features associated with MTF-1 activation: (1) clustering based upon structural similarity, enabling assessment of how similar the chemicals were to one another and (2) association rule mining to specifically examine which sets of structural features discriminated for MTF-1 activation.
ChemmineR’s agglomerative clustering using a Tanimoto threshold of 80% was found to result in reasonable clusters of all 616 active and inactive chemicals in terms of total number of clusters, their membership size, and chemical makeup. At this similarity threshold, 21 clusters were extracted that had at least 3 or more members. Six clusters contained at least 1 active chemical whereas 5 of these 6 clusters contained multiple active chemicals. The 5 clusters with multiple active chemicals covered 32 unique chemicals, of which 18 chemicals were active. The remaining active chemicals were captured in “clusters” of either 1 or 2 chemicals. Fig. 4A represents a dendrogram of the clustering highlighting two specific clusters (cluster ID 25 and 125) for illustrative purposes. The full set of clusters and their membership are provided in Supplemental File 1 (ESI†). Fig. 4B shows the consistency in the structures identified in clusters 25 and 125.
Fig. 4.

Clustering of MTF-1 activating chemicals based on structural features. (A) Dendrogram focusing on those chemicals that were clustered into groups with at least three members. The dashed vertical line represents the similarity cut off used by the ChemmineR package to generate the chemical clusters, i.e. any splits that appear to the right of the dashed line are ignored when generating the clusters. (B) The structures of the chemicals found in Cluster 25 and Cluster 125 (indicated in red).
Utilizing the ToxPrint fingerprints for the 616 unique chemicals and their corresponding activity calls (i.e., positive or negative), the apriori algorithm identified 235 rules associated with a chemical being given an overall activity call of “positive”. The majority of these rules identified the same or similar sets of chemicals. These rules can be grouped into 4 larger bins. The reason behind this grouping is the hierarchical nature of the ToxPrints features themselves. For example, if a chemical contains one of the more specific ToxPrint features, then its molecular fingerprint will contain an “on” bit for both this more specific ToxPrint feature and the corresponding more general ToxPrint feature. Thus, both the more specific and more general ToxPrint features may be used in the generation of association rules, provided they meet the inclusion criteria (i.e., a minimum support of 4 and minimum confidence of 75%). All of the association rules generated by the apriori algorithm are available in Supplemental File 1 (ESI†). Fig. 5 illustrates one of the association rules generated in this study. A description of the support and confidence of this and other rules are found in Supplemental File 1 (ESI†). The association rules predicted 70 unique chemicals out of the 284 chemicals from the CMAP set with no activity designation (either positive or negative) as MTF-1 actives.
Fig. 5.

ToxPrint features and chemicals associated with MTF-1 activation. Illustration of the two ToxPrint features that comprise the left hand side of the rule {ring:fused_steroid_generic_[5_6_6_6], bond:C(=O)O_carboxylicEster_alkenyl} (A) and example chemicals that contain these two ToxPrints (B).
The structural diversity of the chemicals tested within this study were compared to those chemicals tested in the ATG_MRE_CIS_up assay. Fig. S4 (ESI†) shows the chemicals in this study contain a larger proportion of chemicals with aromatic and hetero ring systems. Additionally, the chemicals in this study contain a larger proportion of chemicals with amine, alcohol, ether, and amide bonds. However, the ATG_MRE_CIS_up data set contained more metal containing chemicals; although, it is likely that the majority of these metal-containing chemicals are salts rather than organometallics.
The initial clustering approach used in this study could identify the closest chemical neighbors, thereby facilitating predictive inferences between chemicals within a given cluster. This approach could also be expanded to categorizing novel chemicals and making inferences as to the likely (in)activity towards MTF-1 using a data gap filling approach. Additionally, we employed ARM to identify substructural features associated with chemicals being assigned a positive designation: a total of 253 rules were identified. These rules were applied prospectively to a set of 284 chemicals from the CMAP data set for which incomplete activity data was available. Confirmatory testing would help evaluate the performance of the rules inferred. To facilitate the generalization of the rules derived, the ToxPrint feature set was utilized which are a fixed set of defined features that are not specific to the dataset being analyzed. To explore the general ability of the rules, the set of chemicals of interest were compared to a larger inventory. The enrichments across the feature set was very similar between the CMAP chemicals and the chemicals tested in the ATG_MRE_CIS_up assay suggesting that the rules should be broadly applicable.
Confirmation of the MTF-1 activating chemicals
Nine organic chemicals predicted to activate MTF-1 by our virtual screen were selected for testing of MTF-1 activation in vitro (Table 3). Most of these came from the CMAP 2.0 dataset (GSE5258) in which ~1300 mostly pharmaceutical chemicals were examined in three cell lines;44 they had some of the highest activation scores of the putative positive chemicals. Fig. S5A (ESI†) shows the average for significantly correlated biosets for each of these positive chemicals. The chemical structures of the chemicals are shown in Fig. S5B (ESI†). Of these nine chemicals, two are known to interact with cellular metals. Clioquinol is an antibiotic that is known to act as a zinc and copper ionophore and has been shown to have metal-dependent activity against cancer and Alzheimer’s disease,45–47 and disulfiram is an alcohol aversion therapeutic that has also been shown to act as a copper and zinc ionophore with anticancer activity.48–50 The other seven chemicals, however, have no known direct relationships to metal homeostasis. Some of these chemicals, such as mefloquine,51 thiostrepton,52 and deoxycholic acid,53 are known to induce oxidative stress, which may explain why they activate MTF-1.
Table 3.
Predicted activators of MTF-1 identified using the biomarker and examined for further study
| Predicted MTF-1 activator | Description | Cell line(s) | Treatment s) (h) | Treatment concentration(s) (μM) |
Data source |
|---|---|---|---|---|---|
|
| |||||
| Mefloquine | Antimalarial | PC3, MCF7, HL60 | 6 | 9.6 | GSE5258 |
| Alexidine | Antimicrobial | PC3, MCF7, HL60 | 6 | 6.8 | GSE5258 |
| Phenoxybenzamine | Alpha blocker | PC3, MCF7 | 6 | 11.8 | GSE5258 |
| Astemizole | Antihistamine | PC3, MCF7, HL60 | 6 | 8.8 | GSE5258 |
| Disulfiram | Alcohol aversion drug | PC3, MCF7, HL60, hepatocytes | 6, 8 | 13.4, 60 | GSE5258, TG-GATES |
| Clioquinol | Antifungal | PC3, MCF7, HL60 | 6 | 13 | GSE5258 |
| Suloctidil | Vasodilator | PC3, MCF7, HL60 | 6 | 11.8 | GSE5258 |
| Thiostrepton | Antibiotic | PC3, MCF7 | 6 | 2.4, 10 | GSE40766, GSE5258 |
| Deoxycholic acid | Bile acid | SKGT4, HET1A | 4, 8, 12, 24 | 100, 300 | GSE13400 |
The ability of the nine chemicals to regulate MTF-1-dependent genes in MCF-7 cells was tested by treating MCF7 wild-type and MTF-1-null cells with each chemical and measuring gene expression by RT-qPCR. We chose four genes from the MTF-1 biomarker to measure (MT1M, MT2A, HMOX1, ATF3). Zinc activated three of the genes in a MTF-1-dependent manner. Exposure to all of the chemicals led to increases in expression of one or more of the four genes (Fig. 6). For all but two of the chemicals (chloroquine, phenoxybenzamine), the increases were abolished or decreased in MTF-1-null cells compared to wild-type cells demonstrating dependence of the increases on MTF-1. While ATF3 appeared to be regulated by the majority of metals in the microarray studies, there may be cell line differences in responses between those used to create the biomarker and MCF-7 cells used for validation.
Assessment of changes in intracellular zinc and oxidative stress by the nine chemicals
Having confirmed that most of the chemicals do activate MTF-1, we sought to determine the mechanism by which MTF-1 is being activated. Possible mechanisms of activation include induction of zinc influx, generation of reactive oxygen species that induce release of zinc from MTs, or direct binding to MTF-1 or another protein necessary for MTF-1 transcriptional activation. We used fluorescent probes Zinpyr-1 and 2′,7′-dichlorofluorescin diacetate (DCFDA) to detect intracellular amounts of labile zinc and reactive oxygen species (ROS), respectively. Our results indicate that in addition to Zn itself six of the nine chemicals cause an increase in cellular labile zinc (Fig. 7A). The positive control tert-butyl hydroperoxide (TBHP) caused increases in ROS production (Fig. 7B). Two of the nine chemicals caused increases in ROS while one chemical decreased ROS. Overall, the results suggest that increased labile zinc may play an important role in the mechanism of activation of MTF-1 by many of the predicted MTF-1-activating chemicals.
Fig. 7.

Measurement of changes in free zinc and oxidative stress after exposure to MTF-1 activators. Measurement of free zinc (A) and oxidative stress (B) were carried out as described in the Methods. Levels are shown as mean and standard error of the mean. independent experiments. * Indicates significant difference between treatment and control cells;. ZnSO4 was used as the positive control for zinc release and tert-butyl hydroperoxide (TBHP) was used as the positive control to measure oxidative stress.
Transcript profiling of chemicals in wild-type and MTF-1-null cells
In future HTTr studies, we propose that the use of cell lines nullizygous for important targets of environmental chemicals will greatly facilitate the interpretation of regulated genes. To provide proof of this concept, we generated transcript profiles of Zn and 6 putative activators of MTF-1 in wild-type and MTF-1-null cells generated by TempO-Seq targeted sequencing of ~3000 human genes. In the absence of chemical exposure, knockout of MTF-1 resulted in effects on 44 genes (Fig. S6, ESI†) indicating that MTF-1 has some effect on gene expression in the absence of overt activation.
Fig. 8A shows the heat maps of the filtered changes altered by each chemical in the two cell lines. Fig. 8B shows the number of genes altered by each chemical in wild-type cells and the number of those genes that are altered in the MTF-1-null cells. Almost all of the genes regulated by Zn and phenoxybenzamine were no longer regulated in the MTF-1-null cells. For the rest of the chemicals, more than half of the genes regulated in the wild-type cells were no longer regulated in the MTF-1-null cells.
Fig. 8.

Transcript profiling of chemicals in wild-type and MTF-1-null cells. The treatments described in Fig. 6 legend were analyzed by TempO-Seq human 1500+ platform examining the expression changes in ~3000 genes. Significant expression changes were identified as described in the Methods. (A) The heat maps show the genes altered by the indicated chemical in wild-type MCF-7 cells and their expression after treatment in the MTF-1-null cells. (B) Number of genes significantly altered in each treatment described in (A) are shown.
The profiles derived from the wild-type cells were each compared to those found in the CMAP 2.0 collection to determine if the same chemical would exhibit high correlation despite differences in transcriptomic platform and number of genes evaluated. Each profile was compared to 3442 biosets in the CMAP collection. The chemicals ranked highest to lowest to the same chemical examined in MCF-7 cells were astemizole and disulfiram (both 1st), suloctidil (29th), alexidine (41st) and phenoxybenzamine (816th). Zn and deoxycholic acid were not examined in the CMAP collection. The top hit for Zn was the comparison “Lung epithelial BEAS-2B cells + 3 μM zinc sulfate heptahydrate for 4 h vs. 24 h untreated” from GSE80733 (). The top hit for deoxycholic acid was the comparison “Hepatic progenitor HepaRG cells differentiated 96 h – 200037.13 nM chenodeoxycholic acid vs. DMSO” () examined on the 1500+ platform.54 Except for phenoxybenzamine, these rankings indicate that the 1500+ gene set allows for a high correlation to the same chemical within an independent dataset.
These studies demonstrate that many of the changes in gene expression after exposure to the putative MTF-1 activators were MTF-1-dependent. We also found that the chemical-induced profiles generated in wild-type cells exhibited correlation to the same chemicals profiled in various cell lines in our compendium. The wild-type vs. null cell line comparisons can unequivocally identify targets of environmental chemicals. To our knowledge, this is the first study to compare profiles of chemicals between wild-type and nullizygous cell lines. Although the study was limited in scope, it provides support for the use of nullizygous cell lines in HTTr screening, especially in targeted screening to confirm predicted targets from primary HTTr screens.
Summary
Gene expression biomarkers have been shown to be useful for identifying chemical modulators of transcription factors in previous work by our group.19,20 In the present study, we used a similar approach to identify activators of MTF-1 in a compendium of gene expression profiles derived from chemically-treated human cell lines. The biomarker was built by identifying genes with consistent expression behavior across comparisons from metal-treated cells under conditions known to activate MTF-1. The resulting biomarker consisted of 48 up-regulated and 33 down-regulated genes. The genes in the biomarker with the highest fold-change values included seven MT family members and SLC30A1 encoding the zinc transporter ZnT1 that are well-established targets of MTF-1-mediated gene regulation.11 A subset of the biomarker genes was shown to be MTF-1-dependent based on a previously published MTF-1 siRNA knockdown experiment and transcript profile comparisons between wild-type and MTF-1-null cell lines. We tested the biomarker for predictive accuracy using a set of biosets with known MTF-1 responses including a number of metal activators of MTF-1 and found that the biomarker was very accurate (balanced accuracy = 93%). The biomarker was used to identify chemical activators of MTF-1 in our human microarray compendium. We identified 700 chemicals in 1547 biosets that were putative activators, and they included a large number of metals, metal complexes, and chemicals known to modulate metal homeostasis. Nine of the positive chemicals were examined further and we found that in RT-qPCR and transcript profile experiments most of the chemicals were confirmed to be MTF-1 activators. We performed a chemoinformatic analysis to identify structural features associated with MTF-1 activity and used these features to reprofile chemicals with an equivocal result to demonstrate their utility in chemical prioritization. The results indicate that our methods can readily identify MTF-1 activators. Our approach will not only be useful in analyzing HTTr data of environmental chemicals in ToxCast screens to identify those that disrupt metal homeostasis but will allow identification of environmental samples with contamination by metals.
We envision that batteries of validated biomarkers (including the MTF-1 biomarker described here) with known accuracies and context of use55 could be systematically applied to HTTr data to identify the modulation of tens if not hundreds of factors important in chemical toxicity. There are a number of examples in which characterized biomarkers have been used to screen large sets of chemicals for those that modulate the factor similar to the screening strategy used in this study. A set of 1152 chemicals was screened for androgen receptor (AR) agonism/antagonism in AR+ human prostate cancer cell lines.19 A set of ~1950 chemicals was screened for those that potentially cause DNA damage using the TGx-DDI biomarker identifying ~10 novel compounds not previously known to cause DNA damage.56 The same set of chemicals was screened using a biomarker for Nrf2 and identified both known and novel activators.24 Finally, an estrogen receptor (ER) biomarker was characterized that could be used to screen chemicals in ER+ human breast cancer cell lines.20 Prioritization of identified chemicals for further analysis could then be carried out using the network of adverse outcome pathways (AOP)s as a starting point,57,58 especially if strategies are in place that stratify the AOPs in terms of human relevance, number of key events perturbed, and overlap with AOPs of special concern including developmental or reproductive toxicity. This approach would then allow targeted follow-up studies for confirmation of phenotypic perturbation in lower throughput organotypic-based assays, zebrafish, or short-term rodent studies. Benchmark dose analysis will be used to identify transcriptional points of departure for the genes in the biomarkers to estimate minimal concentrations of chemical that lead to activation or suppression of the factor similar to methods carried out to determine transcriptional points of departure for pathways.59 These concentration values could then be used for in vitro to in vivo extrapolation (IVIVE) to predict tissue concentrations of the chemical in humans followed by use of exposure models to determine the margin between estimated human exposures and those that induce responses in cell-based assays.60 These efforts would allow a preliminary assessment of “safe” levels of exposure.60 Our efforts to characterize a biomarker that predicts MTF-1 modulation contributes to a broader effort within EPA to comprehensively and systematically identify targets of chemicals17 and to move towards reduction of the use of animals in toxicity testing.61
Supplementary Material
Significance to metallomics.
This work describes the use of a novel computational method to identify chemical exposure conditions that activate the metal-responsive transcription factor 1 (MTF-1) in vitro. Our method determines the significance in the correlation between a biomarker of 81 metal-regulated genes and statistically-derived gene lists from chemically-treated human cells. The method has a predictive accuracy of 93% and can readily identify metals, metal complexes, and novel nonmetal organic chemicals whose exposure conditions lead to activation of MTF-1. The methods have applications to not only identify environmental water samples contaminated with metals but organic chemicals that perturb metal homeostasis.
Acknowledgements
We wish to thank Drs James Samet and Keith Houck for critical review of this manuscript and Molly Windsor for assistance with creating the figures. Funding for this study came from the U.S. Environmental Protection Agency Office of Research and Development. A. C. J. acknowledges support from the NSF Graduate Research Fellowship Program and Graduate Research Internship Program (DGE 1644868). This work was carried out as part of the US-EPA Chemical Safety for Sustainability (CSS) research program 12.01: Adverse Outcome Pathways, Project 1.1 g (Cancer AOPs).
Footnotes
Conflicts of interest
The authors have no conflicts of interest.
Electronic supplementary information (ESI) available: Supplemental File 1. (Excel) Contains (1) genes in the MTF-1 biomarker, (2) biomarker for each pairwise comparison across the microarray compendium, (3) biosets used to determine predictive accuracy, (4) ToxPrint fingerprints for each of the chemicals identified as having either positive or negative activity across three MTF-1 assays, (5) cluster information generated by ChemmineR at the different similarity thresholds and the activity call for each chemical, (6) the full list of 235 association rules generated using the arules package in R, and (7) profiling results of the equivocal chemicals using the generated association rules. Supplemental File 2. (Word) Contains (1) Fig. S1. Comparison of gene lists from BSCE and those generated in Partek Genomics Suite for the 10 biosets used in the biomarker. (2) Fig. S2. Comparison of the derived from the Partek-derived vs. the BSCE-derived lists. (3) Fig. S3. MTF-1 and Nrf2 are simultaneously activated by chemical exposure. (4) Fig. S4. Comparison of the frequency of the 70 “level 2” ToxPrints for the chemicals in the CMAP dataset and the chemicals tested in the ToxCast ATG_MRE_CIS_up assay. (5) Fig. S5. Analysis of nine putative MTF-1 activators, and (6) Fig. S6. Identification of genes that exhibit expression changes in the MTF-1-null cells. See DOI: 10.1039/d0mt00071j
References
- 1.Rolfs A. and Hediger MA, Metal ion transporters in mammals: structure, function and pathological implications, J. Physiol, 1999, 518, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Halloran T, Transition metals in control of gene expression, Science, 1993, 261, 715–725. [DOI] [PubMed] [Google Scholar]
- 3.Choi SY and Bird AJ, Zinc’ing sensibly: controlling zinc homeostasis at the transcriptional level, Metallomics, 2014, 6, 1198–1215. [DOI] [PubMed] [Google Scholar]
- 4.Jaishankar M, Tseten T, Anbalagan N, Mathew BB and Beeregowda KN, Toxicity, mechanism and health effects of some heavy metals, Interdiscip. Toxicol, 2014, 7, 60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He ZL, Yang XE and Stoffella PJ, Trace elements in agroecosystems and impacts on the environment, J. Trace Elem. Med. Biol, 2005, 19, 125–140. [DOI] [PubMed] [Google Scholar]
- 6.Ferguson J, The Heavy Elements, Chemistry, Environmental Impact and Health Effects, Oxford, 1990, pp. 211–212. [Google Scholar]
- 7.Miles AT, Hawksworth GM, Beattie JH and Rodilla V, Induction, Regulation, Degradation, and Biological Significance of Mammalian Metallothioneins, Crit. Rev. Biochem. Mol. Biol, 2000, 35, 35–70. [DOI] [PubMed] [Google Scholar]
- 8.Gunther V, Lindert U and Schaffner W, The taste of heavy metals: gene regulation by MTF-1, Biochim. Biophys. Acta, 2012, 1823, 1416–1425. [DOI] [PubMed] [Google Scholar]
- 9.Dong G, Chen H, Qi M, Dou Y. and Wang Q, Balance between metallothionein and metal response element binding transcription factor 1 is mediated by zinc ions (review), Mol. Med. Rep, 2015, 11, 1582–1586. [DOI] [PubMed] [Google Scholar]
- 10.Lindert U, Cramer M, Meuli M, Georgiev O. and Schaffner W, Metal-Responsive Transcription Factor 1 (MTF-1) Activity Is Regulated by a Nonconventional Nuclear Localization Signal and a Metal-Responsive Transactivation Domain, Mol. Cell. Biol, 2009, 29, 6283–6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardyman JE, Tyson J, Jackson KA, Aldridge C, Cockell SJ, Wakeling LA, Valentine RA and Ford D, Zinc sensing by metal-responsive transcription factor 1 (MTF1) controls metallothionein and ZnT1 expression to buffer the sensitivity of the transcriptome response to zinc, Metallomics, 2016, 8, 337–343. [DOI] [PubMed] [Google Scholar]
- 12.Ruttkay-Nedecky B, Nejdl L, Gumulec J, Zitka O, Masarik M, Eckschlager T, Stiborova M, Adam V. and Kizek R, The Role of Metallothionein in Oxidative Stress, Int. J. Mol. Sci, 2013, 14, 6044–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maret W, Metallothionein/disulfide interactions, oxidative stress, and the mobilization of cellular zinc, Neurochem. Int, 1995, 27, 111–117. [DOI] [PubMed] [Google Scholar]
- 14.Maret W, Oxidative metal release from metallothionein via zinc-thiol/disulfide interchange, Proc. Natl. Acad. Sci. U. S. A, 1994, 91, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu W, Bromberg PA and Samet JM, Zinc ions as effectors of environmental oxidative lung injury, Free Radical Biol. Med, 2013, 65, 57–69. [DOI] [PubMed] [Google Scholar]
- 16.Judson R, Houck K, Martin M, Knudsen T, Thomas RS, Sipes N, Shah I, Wambaugh J. and Crofton K, In vitro and modelling approaches to risk assessment from the U.S. Environmental Protection Agency ToxCast programme, Basic Clin. Pharmacol. Toxicol, 2014, 115, 69–76. [DOI] [PubMed] [Google Scholar]
- 17.Thomas RS, Bahadori T, Buckley TJ, Cowden J, Deisenroth C, Dionisio KL, Frithsen JB, Grulke CM, Gwinn MR, Harrill JA, Higuchi M, Houck KA, Hughes MF, Hunter ES, Isaacs KK, Judson RS, Knudsen TB, Lambert JC, Linnenbrink M, Martin TM, Newton SR, Padilla S, Patlewicz G, Paul-Friedman K, Phillips KA, Richard AM, Sams R, Shafer TJ, Setzer RW, Shah I, Simmons JE, Simmons SO, Singh A, Sobus JR, Strynar M, Swank A, TorneroValez R, Ulrich EM, Villeneuve DL, Wambaugh JF, Wetmore BA and Williams AJ, The Next Generation Blueprint of Computational Toxicology at the U.S. Environmental Protection Agency, Toxicol. Sci, 2019, 169, 317–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeakley JM, Shepard PJ, Goyena DE, VanSteenhouse HC, McComb JD and Seligmann BE, A trichostatin A expression signature identified by TempO-Seq targeted whole transcriptome profiling, PLoS One, 2017, 12, e0178302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rooney JP, Chorley B, Kleinstreuer N. and Corton JC, Identification of Androgen Receptor Modulators in a Prostate Cancer Cell Line Microarray Compendium, Toxicol. Sci, 2018, 166, 146–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryan N, Chorley B, Tice RR, Judson R. and Corton JC, Moving Toward Integrating Gene Expression Profiling Into High-Throughput Testing: A Gene Expression Biomarker Accurately Predicts Estrogen Receptor alpha Modulation in a Microarray Compendium, Toxicol. Sci, 2016, 151, 88–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rooney J, Oshida K, Vasani N, Vallanat B, Ryan N, Chorley BN, Wang X, Bell DA, Wu KC, Aleksunes LM, Klaassen CD, Kensler TW and Corton JC, Activation of Nrf2 in the liver is associated with stress resistance mediated by suppression of the growth hormone-regulated STAT5b transcription factor, PLoS One, 2018, 13, e0200004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kupershmidt I, Su QJ, Grewal A, Sundaresh S, Halperin I, Flynn J, Shekar M, Wang H, Park J, Cui W, Wall GD, Wisotzkey R, Alag S, Akhtari S. and Ronaghi M, Ontology-based meta-analysis of global collections of high-throughput public data, PLoS One, 2010, 5, e13066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardyman JEJ, Tyson J, Jackson KA, Aldridge C, Cockell SJ, Wakeling LA, Valentine RA and Ford D, Zinc sensing by metal-responsive transcription factor 1 (MTF1) controls metallothionein and ZnT1 expression to buffer the sensitivity of the transcriptome response to zinc, Metallomics, 2016, 8, 337–343. [DOI] [PubMed] [Google Scholar]
- 24.Rooney J, Chorley B. and Corton JC, Mining a human transcriptome database for chemical modulators of Nrf2, 2020, submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oshida K, Vasani N, Thomas RS, Applegate D, Rosen M, Abbott B, Lau C, Guo G, Aleksunes LM, Klaassen C. and Corton JC, Identification of modulators of the nuclear receptor peroxisome proliferator-activated receptor alpha (PPARalpha) in a mouse liver gene expression compendium, PLoS One, 2015, 10, e0112655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams AJ, Grulke CM, Edwards J, McEachran AD, Mansouri K, Baker NC, Patlewicz G, Shah I, Wambaugh JF, Judson RS and Richard AM, The CompTox Chemistry Dashboard: a community data resource for environmental chemistry, J. Cheminf, 2017, 9, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang C, Tarkhov A, Marusczyk J, Bienfait B, Gasteiger J, Kleinoeder T, Magdziarz T, Sacher O, Schwab CH,Schwobel J, Terfloth L, Arvidson K, Richard A, Worth A. and Rathman J, New publically available chemical query language, CSRML, to support chemotype representations for application to data mining and modeling, J. Chem. Inf. Model, 2015, 55, 510–528. [DOI] [PubMed] [Google Scholar]
- 28.Cao Y, Backman T, Wang Y. and Girke T, ChemmineR – V2: Analysis of small molecule and screening data, ChemMineR Manual, 2011. [Google Scholar]
- 29.Agrawal R. and Srikant R, Fast algorithms for mining association rules, VLDB, Santiago, Chile, 1994, vol. 1215, pp. 487–499. [Google Scholar]
- 30.Hahsler M, Grun B. and Hornik K, Arules – A Computational Environment for Mining Association Rules and Frequent Item Sets, J. Stat. Softw, 2005, 14(15), 1–25. [Google Scholar]
- 31.Oki NO and Edwards SW, An integrative data mining approach to identifying adverse outcome pathway signatures, Toxicology, 2016, 350–352, 49–61. [DOI] [PubMed] [Google Scholar]
- 32.Bell SM and Edwards SW, Building associations between markers of environmental stressors and adverse human health impacts using frequent itemset mining, Proceedings of the 2014 SIAM International Conference on Data Mining, Society for Industrial and Applied Mathematics, 2014, pp. 551–559. [Google Scholar]
- 33.Bell SM and Edwards SW, Identification and Prioritization of Relationships between Environmental Stressors and Adverse Human Health Impacts, Environ. Health Perspect, 2015, 123, 1193–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gardiner EJ and Gillet VJ, Perspectives on Knowledge Discovery Algorithms Recently Introduced in Chemoinformatics: Rough Set Theory, Association Rule Mining, Emerging Patterns, and Formal Concept Analysis, J. Chem. Inf. Model, 2015, 55, 1781–1803. [DOI] [PubMed] [Google Scholar]
- 35.Naulaerts S, Meysman P, Bittremieux W, Vu TN, Vanden Berghe W, Goethals B. and Laukens K, A primer to frequent itemset mining for bioinformatics, Briefings Bioinf., 2015, 16, 216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu P. and Wild DJ, Fast rule-based bioactivity prediction using associative classification mining, J. Cheminf, 2012, 4, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park SH, Jang SY, Kim H. and Lee SW, An association rule mining-based framework for understanding lifestyle risk behaviors, PLoS One, 2014, 9, e88859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agrawal R, Imielinski T. and Swami A, Mining association rules between sets of items in large databases, Proceedings of the 1993 ACM SIGMOD international conference on Management of data, 1993, pp. 207–216. [Google Scholar]
- 39.Borgelt C, Frequent item set mining, WIREs Data Min. Knowl. Discov, 2012, 2, 437–456. [Google Scholar]
- 40.Shirato A, Kikugawa T, Miura N, Tanji N, Takemori N, Higashiyama S. and Yokoyama M, Cisplatin resistance by induction of aldo-keto reductase family 1 member C2 in human bladder cancer cells, Oncol. Lett, 2014, 7, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hai T. and Hartman MG, The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis, Gene, 2001, 273, 1–11. [DOI] [PubMed] [Google Scholar]
- 42.Hai T, Wolfgang CD, Marsee DK, Allen AE and Sivaprasad U, ATF3 and Stress Responses, Gene Expression, 1999, 7, 321–335. [PMC free article] [PubMed] [Google Scholar]
- 43.Schwochau GB, Nath KA and Rosenberg ME, Clusterin protects against oxidative stress in vitro through aggregative and nonaggregative properties, Kidney Int., 1998, 53, 1647–1653. [DOI] [PubMed] [Google Scholar]
- 44.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES and Golub TR, The Connectivity Map: using geneexpression signatures to connect small molecules, genes, and disease, Science, 2006, 313, 1929–1935. [DOI] [PubMed] [Google Scholar]
- 45.Ding W-Q and Lind SE, Metal ionophores – An emerging class of anticancer drugs, IUBMB Life, 2009, 61, 1013–1018. [DOI] [PubMed] [Google Scholar]
- 46.Mao X, Li X, Sprangers R, Wang X, Venugopal A, Wood T, Zhang Y, Kuntz DA, Coe E, Trudel S,Rose D, Batey RA, Kay LE and Schimmer AD, Clioquinol inhibits the proteasome and displays preclinical activity in leukemia and myeloma, Leukemia, 2008, 23, 585. [DOI] [PubMed] [Google Scholar]
- 47.Regland B, Lehmann W, Abedini I, Blennow K, Jonsson M, Karlsson I, Sjogren M¨, Wallin A, Xilinas M. and Gottfries CG, Treatment of Alzheimer’s Disease with Clioquinol, Dementia Geriatr. Cognit. Disord, 2001, 12, 408–414. [DOI] [PubMed] [Google Scholar]
- 48.Allensworth JL, Evans MK, Bertucci F, Aldrich AJ, Festa RA, Finetti P, Ueno NT, Safi R, McDonnell DP, Thiele DJ, Van Laere S. and Devi GR, Disulfiram (DSF) acts as a copper ionophore to induce copper-dependent oxidative stress and mediate anti-tumor efficacy in inflammatory breast cancer, Mol. Oncol, 2015, 9, 1155–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wiggins HL, Wymant JM, Solfa F, Hiscox SE, Taylor KM, Westwell AD and Jones AT, Disulfiram-induced cytotoxicity and endo-lysosomal sequestration of zinc in breast cancer cells, Biochem. Pharmacol, 2015, 93, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Allensworth JL, Evans MK, Bertucci F, Aldrich AJ, Festa RA, Finetti P, Ueno NT, Safi R, McDonnell DP, Thiele DJ, Van Laere S. and Devi GR, Disulfiram (DSF) acts as a copper ionophore to induce copper-dependent oxidative stress and mediate anti-tumor efficacy in inflammatory breast cancer, Mol. Oncol, 2015, 9, 1155–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hood JE, Jenkins JW, Milatovic D, Rongzhu L. and Aschner M, Mefloquine induces oxidative stress and neurodegeneration in primary rat cortical neurons, Neuro-toxicology, 2010, 31, 518–523. [DOI] [PubMed] [Google Scholar]
- 52.Qiao S, Lamore SD, Cabello CM, Lesson JL, Muñoz-Rodriguez JL and Wondrak GT, Thiostrepton is an inducer of oxidative and proteotoxic stress that impairs viability of human melanoma cells but not primary melanocytes, Biochem. Pharmacol, 2012, 83, 1229–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lechner S, Muller-Ladner K, Fau-Schlottmann U, Schlottmann B, Fau-Jung K, Jung M, Fau-McClelland B, McClelland J, Fau-Ruschoff M, Ruschoff J, Fau-Welsh J, Welsh J, Fau-Scholmerich J, Scholmerich F, Fau-Kullmann J and Kullmann F, Bile acids mimic oxidative stress induced upregulation of thioredoxin reductase in colon cancer cell lines, Carcinogenesis, 2002, 23, 1281–1288. [DOI] [PubMed] [Google Scholar]
- 54.Ramaiahgari SC, Auerbach SS, Saddler TO, Rice JR, Dunlap PE, Sipes NS, DeVito MJ, Shah RR, Bushel PR, Merrick BA, Paules RS and Ferguson SS, The Power of Resolution: Contextualized Understanding of Biological Responses to Liver Injury Chemicals Using High-throughput Transcriptomics and Benchmark Concentration Modeling, Toxicol. Sci, 2019, 169, 553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Corton JC, Kleinstreuer NC and Judson RS, Identification of potential endocrine disrupting chemicals using gene expression biomarkers, Toxicol. Appl. Pharmacol, 2019, 114683, DOI: 10.1016/j.taap.2019.114683. [DOI] [PubMed] [Google Scholar]
- 56.Corton JC, Williams A. and Yauk CL, Using a gene expression biomarker to identify DNA damage-inducing agents in microarray profiles, Environ. Mol. Mutagen, 2018, 59, 772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Edwards SW, Tan YM, Villeneuve DL, Meek ME and McQueen CA, Adverse Outcome Pathways-Organizing Toxicological Information to Improve Decision Making, J. Pharmacol. Exp. Ther, 2016, 356, 170–181. [DOI] [PubMed] [Google Scholar]
- 58.Perkins EJ, Ashauer R, Burgoon L, Conolly R, Landesmann B, Mackay C, Murphy CA, Pollesch N, Wheeler JR, Zupanic A. and Scholz S, Building and Applying Quantitative Adverse Outcome Pathway Models for Chemical Hazard and Risk Assessment, Environ. Toxicol. Chem, 2019, 38, 1850–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Phillips JR, Svoboda DL, Tandon A, Patel S, Sedykh A, Mav D, Kuo B, Yauk CL, Yang L, Thomas RS, Gift JS, Davis JA, Olszyk L, Merrick BA, Paules RS, Parham F, Saddler T, Shah RR and Auerbach SS, BMDExpress 2: enhanced transcriptomic dose-response analysis workflow, Bioinformatics, 2019, 35, 1780–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bell SM, Chang X, Wambaugh JF, Allen DG, Bartels M, Brouwer KLR, Casey WM, Choksi N, Ferguson SS, Fraczkiewicz G, Jarabek AM, Ke A, Lumen A, Lynn SG, Paini A, Price PS, Ring C, Simon TW, Sipes NS, Sprankle CS, Strickland J, Troutman J, Wetmore BA and Kleinstreuer NC, In vitro to in vivo extrapolation for high throughput prioritization and decision making, Toxicol. In Vitro, 2018, 47, 213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.AR W, Directive to prioritize efforts to reduce animal testing, 2019, https://www.epa.gov/sites/production/files/2019-09/documents/image2019-09-09-231249.pdf.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.