

Abstract
Dichloroacetate (DCA) is a naturally occurring xenobiotic that has been used as an investigational drug for over 50 years. Originally found to lower blood glucose levels and alter fat metabolism in diabetic rats, this small molecule was found to serve primarily as a pyruvate dehydrogenase kinase inhibitor. Pyruvate dehydrogenase kinase inhibits pyruvate dehydrogenase complex, the catalyst for oxidative decarboxylation of pyruvate to produce acetyl coenzyme A. Several congenital and acquired disease states share a similar pathobiology with respect to glucose homeostasis under distress that leads to a preferential shift from the more efficient oxidative phosphorylation to glycolysis. By reversing this process, DCA can increase available energy and reduce lactic acidosis. The purpose of this review is to examine the literature surrounding this metabolic messenger as it presents exciting opportunities for future investigation and clinical application in therapy including cancer, metabolic disorders, cerebral ischemia, trauma, and sepsis.
Keywords: Dichloroacetate, Mitochondria, Lactic acidosis, Glycolysis, Energetics
1. Introduction
Dichloroacetate (DCA) is a naturally occurring investigational drug that first demonstrated potential for clinical use over 50 years ago by lowering blood glucose, first in animals, and subsequently in humans [1,2]. It is a small, halogenated carboxylic acid with a very limited number of important sites of action that have demonstrated increasingly diverse clinical applications [3]. Its ability to modulate a key process in cellular respiration has made it an intriguing target that continues to be investigated for application in the treatment of tumors, metabolic diseases, cerebral ischemia, and trauma.
2. Pyruvate dehydrogenase complex: the gatekeeper of mitochondria
The primary mechanism of interest for DCA has been centered around its direct modulation of the mitochondrial pyruvate dehydrogenase complex (PDC)/pyruvate dehydrogenase kinase (PDK) axis (Fig. 1). It has also been shown to act as an inhibitor of HMG-CoA-reductase and an irreversible inactivator of glutathione S-transferase zeta 1 (GSTZ1) [4–6].
Fig. 1.
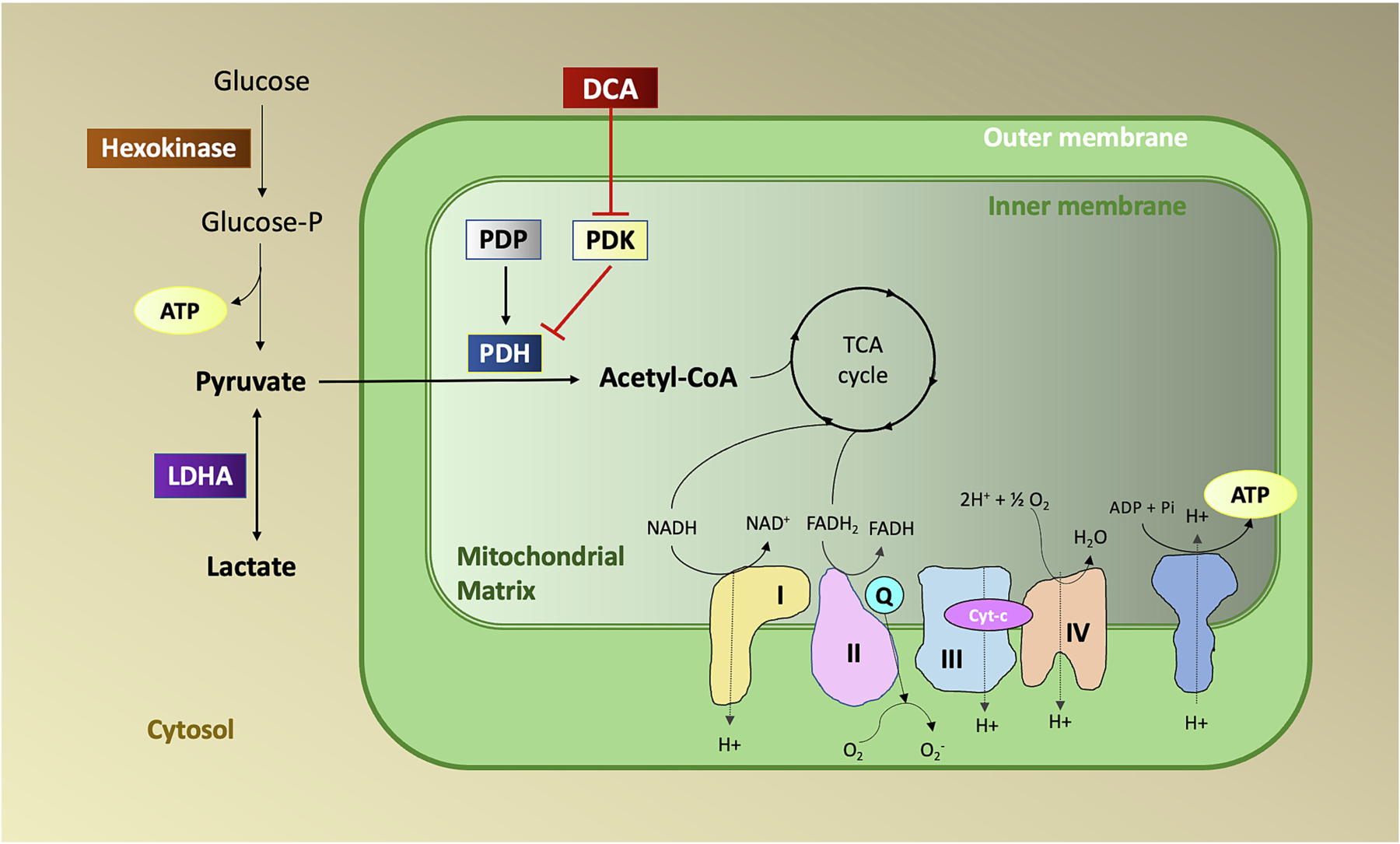
Mechanism of action of dichloroacetate (DCA). PDK phosphorylates and inhibits the activity of PDH while PDP dephosphorylates and activates PDH. Under hypoxic conditions activation of PDK results in phosphorylation and inactivation of PDH and reduced conversion of pyruvate to acetyl CoA. DCA inactivates PDK resulting in the activation of PDH.
PDC is a mitochondrial multienzyme complex that plays the paramount role as a gatekeeper in connecting cytoplasmic glycolysis with the tricarboxylic acid cycle and eventual oxidative phosphorylation. The rate-limiting step in this process involves oxidative decarboxylation of pyruvate leading to the production of acetyl-CoA, NADH, and CO2 [7,8]. PDC is a tightly regulated composite of three different catalytic enzymes: pyruvate dehydrogenase (E1), dihydrolipoamide acetyltransferase (E2), and dihydrolipoamide dehydrogenase (E3). These constituents act in concert as a primary regulator in mitochondria linking glucose metabolism, fatty acid metabolism, and the Krebs’ cycle.
Regulation of PDC is achieved through a cycle of phosphorylation and dephosphorylation which takes place at three serine residues, Ser293, Ser300, and Ser232, of the alpha-subunit of pyruvate dehydrogenase (PDH) [9]. The reversible phosphorylation of PDH by PDK leads to inactivation, and dephosphorylation by pyruvate dehydrogenase phosphatase (PDP) reactivates the complex [8,10,11]. PDK is activated by increased levels of acetyl-CoA and NADH and inactivated by the presence of pyruvate. Importantly, PDK activity is also increased by hypoxia.
Four isoforms of PDK have been identified in humans: PDK1, PDK2, PDK3, and PDK4. Under normal physiological conditions, the four isoenzymes are predictable in both distribution and expression: PDK1 is predominantly expressed in the heart, pancreatic islets, and skeletal muscles. PDK2 is broadly distributed in many tissues, except for the lung and spleen. PDK3 is found in the testicle, kidney, and brain. PDK4 is distributed in the heart, skeletal muscle, liver, kidney, and pancreatic islets [12]. However, both distribution and expression are altered under various pathologic states. Abnormal regulation of PDK2 and PDK4 has been associated with diabetes mellitus, whereas all four isoenzymes have been found to have a varied expression in different malignant tumors [12].
3. Dichloroacetate metabolism
In vivo, DCA is primarily metabolized via biotransformation executed by the zeta-1 family isoform of glutathione transferase (GSTZ1), also known as maleylacetoacetate isomerase (MAAI) (Fig. 2) [13]. This enzyme primarily resides in hepatocytes and proximal tubule cells of the kidney where it also serves to perform the final steps of tyrosine and phenylalanine metabolism [14]. Initially, DCA is transformed into glyoxylate, a highly reactive molecule that can cause oxidative stress to tissues and requires further metabolization [15].
Fig. 2.
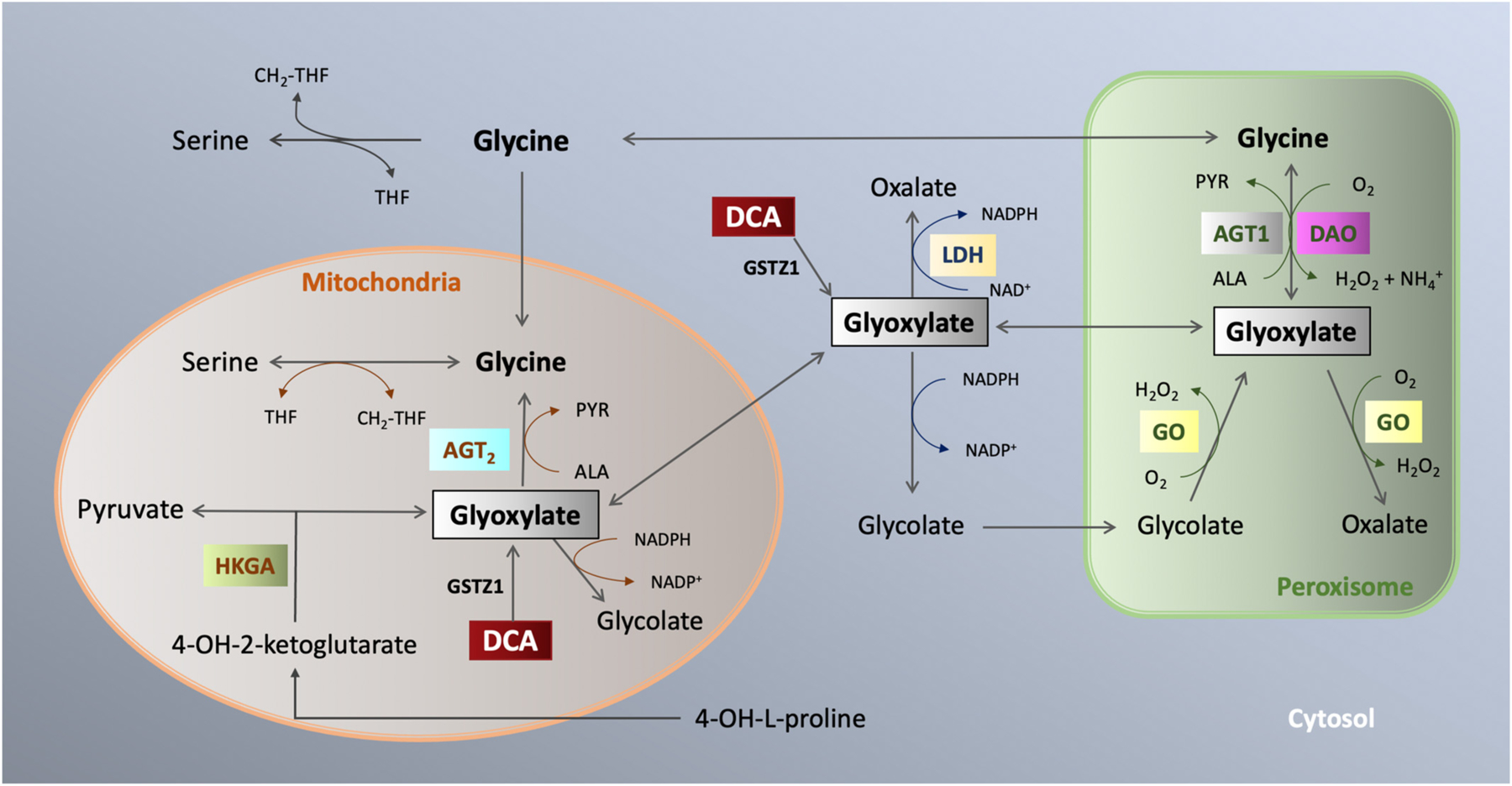
Dichloroacetate metabolism. GSTZ1 metabolizes DCA to glyoxylate which metabolizes to glycine, pyruvate, or oxalate in the mitochondria or peroxisome.
Glyoxylate is readily oxidized to oxalate via lactate dehydrogenase [16,17]. The oxalate is further metabolized via glyoxylate carboligase in a thiamine-dependent reaction to α-hydroxy-β-ketoadipate, a highly soluble molecule that is renally eliminated [18]. It was posited that the reversible polyneuropathy seen with DCA may be due to thiamine deficiency as thiamine stores are depleted through this reaction [19]. Case studies have also reported peripheral neuropathy from oxalate deposition in peripheral nerves and other tissues in primary hyperoxalurias, highlighting the metabolism to oxalate as a clinically significant event [20–22]. Through an alternative pathway, glyoxylate is also metabolized in peroxisomes and mitochondria (Fig. 2). Hepatocytes are rich in alanine glyoxylate aminotransferase which allows for abundant peroxisomal transformation to glycolate and subsequent breakdown to glycine which is used in several intracellular pathways [16].
DCA appears to restrict its own metabolism via inhibition of GSTZ1/MAAI and thus as DCA concentrations rise in the body via repeated administration or higher dose usage, the plasma clearance of the drug decreases [23,24]. Clearance of DCA from the plasma has been shown to be age-dependent, with older patients demonstrating decreased DCA clearance [25]. Both animal and human evidence have highlighted the importance of GSTZ1 genetic polymorphisms and their consequent influence on the metabolism of DCA, suggesting that individualized dosing regimens may be required to adequately address issues related to treatment effectiveness and adverse effects [24,26,27].
4. Lactic acidosis
Lactic acidosis is manifested when the production of lactic acid exceeds its clearance. Congenital lactic acidosis is generally a result of mitochondrial dysfunction due to deleterious mutations in genes coding for key proteins involved in mitochondrial function. As a result, cells are forced to rely on glycolysis as a primary means of energy production and suffer subsequent organ damage. Most cases prove fatal in the long term. DCA was proposed as a possible solution through enabling oxidative phosphorylation and consequent lowering of lactate.
Stacpoole and colleagues showed that DCA lowers blood, cerebrospinal fluid, and intracellular lactate in a dose dependent but not time dependent manner [28]. Barshop et al. performed a small open-label study in which 37 patients with “various mitochondrial disorders” were treated with DCA for a mean of 3.25 years though the treatment course ranged 3 weeks to 7 years. A subjective impression of overall disease showed improvement in nearly half of the patients (48.6 %) though the variability of underlying genetic disease, the open-label nature, and the discrepancies in treatment length made these findings difficult to generalize [29]. In a previous study using DCA as a potential therapeutic for all comers with lactic acidosis, there was a statistically significant decrease in the lactate level in arterial blood, but this did not translate to improved hemodynamic parameters or overall survival in a larger cohort of patients [30]. In another study, DCA was tested in congenital lactic acidosis. It was well tolerated orally and blunted post-prandial lactate but did not improve neurologic function or any other measured clinical outcome. [31] Thus, despite its success in lowering lactate, there does not as of yet appear to be a benefit in mortality from DCA use in congenital lactic acidosis [30]. This is likely due to the devastating nature of the pathophysiology and the site of mitochondrial gene mutations [32].
5. Diabetes mellitus
DCA has been studied in various metabolic diseases, including diabetes mellitus (DM). In animal models, DCA lowered the serum blood glucose of diabetic mice though no change was observed in healthy mice [1]. It was observed that DCA reduces the plasma glucose concentration in starved and diabetic rats, but not in normal, fed rats [4,33]. Later, Stacpoole and Greene found that DCA decreased hepatic gluconeogenesis and average glucose levels without an increase in insulin secretion in diabetic BB Wistar rats. The authors concluded that this decrease in glucose is likely due to reduced production of lactic acid and alanine, and subsequent disruption of the Cori cycle for gluconeogenesis [34]. A recent study in ob/ob mice also attributed the effect of DCA on blood glucose to the suppression of hepatic gluconeogenesis [35]. DCA improved diabetic outcomes in mice treated with streptozotocin (STZ), a selective toxin for pancreatic beta cells. Furthermore, di-isopropylammonium dichloroacetate (DIPA), a derivative of DCA, was also shown to improve survival in STZ-diabetic rats [36].
DCA exhibits cardioprotective effects in the setting of diabetic cardiomyopathy and it has been shown to increase peripheral glucose oxidation which may ameliorate microvascular ischemic changes within cardiac muscle [37]. Additionally, DCA improved cardiac contractility in decompensated congestive heart failure, increased cardiac function and overall survival, as well as decreased oxidative stress and prevented cell death in cultured cardiomyocytes [38]. In ischemic cardiac cells, DCA was able to reduce ischemia/reperfusion damage via an AMPK-mediated mechanisms [39,40]. There is a paucity of studies that tested DCA’s ability to counteract heart failure or improve cardiac contractility in diabetic mice. Outside of cardiac damage, diabetes has been associated with improper endometrial function and ovarian damage. The administration of DCA improved endocrine and reproductive function in rat models [41]. Despite all this, concerns about the long-term safety of DCA continue to limit its use as a chronic diabetic therapy [3].
6. Cancer
DCA has also been studied more recently in the treatment of cancer. The oxygen and energy demands of rapidly growing malignant cells are generally not met by the preexisting vascular supply [42]. The reduced intracellular oxygen leads to the stabilization of HIF-1α which regulates the release of vascular endothelial growth factor alpha (VEGF-A) [43]. Cancer cells generate new blood vessels in response to hypoxia through a VEGF-A-dependent process termed pathological angiogenesis [44]. VEGF-A also promotes lymphogenesis which potentiates lymphatic metastasis of cancer [45]. DCA has been shown to limit HIF-1α production by tumor cells, upregulate P53 and subsequent apoptosis, and reduce VEGF-A activity in brain glioblastoma multiforme [46].
A hallmark of cancer cells is the use of the anaerobic glycolytic pathway despite the presence of oxygen and glucose [47]. This aerobic glycolysis was later termed “The Warburg Effect.” Liberti et al. posited that the Warburg effect conferred a benefit to cancer cells through the rapid availability of energy provided by glycolysis, the shunting of anaerobic byproducts to other biosynthetic pathways, aiding in immune cell evasion, and cell signaling through reactive oxygen species (ROS) [48]. Aerobic glycolysis is preferentially used by tumor cells through TGF-β1 signaling, a process also observed in endometriosis [49]. It was suggested that DCA inhibits cancer progression through a reversal of the Warburg effect [50]. By forcing pyruvate through oxidative phosphorylation, the mitochondrial suppression exhibited by cancer cells is reversed; ultimately promoting apoptosis [50]. Cancer cells also use VEGF and other HIF-1-mediated actors to increase angiogenesis in an acidotic environment. [43] This enables cancer cells to gain access to blood vessels and promote metastasis. DCA is able to mitigate this by reducing lactic acidosis in the tumor microenvironment [51]. Finally, fatty acid oxidation is required for new nucleotides to be formed in order for cancer cells to replicate [52]. DCA can suppress mitochondrial betaoxidation, at least in mammalian muscle and liver, making it more difficult for cancer cells to generate the nucleotides necessary to replicate and stimulate endothelial growth [51,52].
DCA is also able to boost host cell immunity through indirect means. An elevated serum lactate can blunt T-cell proliferation and signaling via cytokines [53]. It was also observed to decrease glutathione levels in T-cells, making them more sensitive to ROS and subsequent T-cell apoptosis. It was found that in vitro treatment of cancer cells in the setting of DCA improved T-cell function, signaling, and survival which led to decreased cancer growth [53]. Thus, DCA appears to reinforce the host immune system to better identify and respond to tumor cells.
Research in the last decade has shown that DCA increases certain cancer cells’ sensitivity to radiotherapies and chemotherapies while decreasing the side effects of common chemotherapy agents. In colorectal cancer cells that were previously resistant to 5-fluorouracil (5-FU) treatment, the in vitro addition of DCA lead to increased chemosensitivity to 5-fluorouracil while increasing the intracellular level of intrinsic tumor suppressor p53. This vastly reduced tumorigenesis [54]. DCA has been used in trials with several other agents, such as cisplatin and arsenic trioxide, showing improved apoptosis, anti-proliferative effects, and increased treatment effectiveness in both lung and breast cancer, as well as playing a nephroprotective role in the treatment of breast cancer. [55–57]. Galgamuwa et al. were able to show that DCA played a nephroprotective role in the management of breast cancer with cisplatin without limiting the overall effectiveness of treatment [58]. It has also been linked to increased radiosensitization of tumors in breast cancer and gliomas [59,60]. One case report showed remission in a man with relapsed non-Hodgkin’s lymphoma after DCA treatment [61]. Furthermore, when DCA was added to chemoradiotherapy for locally advanced head and neck squamous cell carcinoma, though survival rates were not significantly different between groups, the DCA group had significantly reduced pyruvate and lactate [62].
One of the notable limitations of chronic DCA use (discussed further below) has been the peripheral neuropathy [5,13,26,63]. This adverse effect may be mitigated using antioxidant medications or even muscarinic antagonists and proves to be reversible as well [5]. Despite this side effect, recent data show DCA may mitigate the toxic effects of other chemotherapy agents. A common side effect of the chemotherapeutic agent bleomycin is pulmonary fibrosis mediated by lung myofibroblasts. This is posited to occur due to HIF-1α induction in the lungs leading to myofibroblast proliferation. DCA can downregulate the HIF-1α response leading to attenuation of myofibroblast proliferation and subsequent decrease in pulmonary fibrosis [64]. Doxorubicin, when co-administered with DCA, was found to be more tumor-selective than doxorubicin alone with similar chemotherapeutic potential [65]. Furthermore, it was found that DCA successfully decreases doxorubicin’s characteristic cardiotoxicity [66]. The pre-clinical and clinical studies show that the induction of glycolytic-mitochondrial shift and its anticancer effects have the potential to alter cancer metabolism and cancer progression and offer promise for further studies.
7. Ischemia and reperfusion
7.1. Trauma
For decades trauma has been cited as the leading cause of death in people under the age of 45, with hemorrhagic shock noted as the most common preventable cause of death in this subgroup [67–69]. Hemorrhagic shock leads to global tissue hypoxia, decreased coronary perfusion pressure, and subsequent depressed cardiac function, all of which led to an inevitable downward spiral of worsening hypoperfusion and dysfunction [70]. The treatment of hemorrhagic shock has evolved from large volume replacement with crystalloid in order to maintain a degree of tissue reperfusion and continued metabolic activity to a more straightforward “replace blood with blood” strategy, although this approach is limited by the logistical challenges of acquisition and storage [71]. This is particularly true for the far-forward management of patients in combat zones and rural emergency departments. Although more advanced tourniquets, kaolin-impregnated gauze (“combat gauze”), and other adjuncts have become standard measures, the number of trauma-related victims remains high, and other means of significantly improving survival are lacking.
Several studies have demonstrated diminished mitochondrial function in hemorrhagic shock [72–74]. On a metabolic level, hemorrhagic shock leads to hypoxia, and this scarcity of cellular oxygen suppresses mitochondrial oxidation through increased activity of glycolytic enzymes and PDK [75,76]. The subsequent inactivation of PDC by PDK may be a mechanism to reduce excess production of ROS, but at the cost of the more economic production of ATP [77]. Investigations using animal models have demonstrated the profound effect of DCA to enhance mitochondrial function within the context of hemorrhagic shock injury (HI) both as monotherapy and as part of a combinatorial strategy [74,78]. Rats subjected to HI using validated hemorrhagic shock models exhibited prolonged survival independent of the addition of volume resuscitation when treated with DCA. The elevated levels of PDK in rats subjected to HI that received DCA were restored to normal. DCA was shown to be superior to another mitochondria potentiating agent, resveratrol, by almost doubling the mean survival time of subjects (200 vs 110 min) [78,79]. Some of the earlier studies using higher doses of DCA in HI had mixed results [80,81]. It is worth noting that the method of inducing HI and the relatively high doses of DCA (150 mg/kg vs 10–25 mg/kg) used in the previous studies may have acted as confounding factors [80,81]. Although future studies will need to investigate both the efficacy and the safety of the drug in human patients, the single-dose regimen could be valuable in austere situations and avoids the well-known peripheral neuropathy observed with chronic use [5,39].
7.2. Sepsis and inflammation
Initial lactate build-up as a response to an acute disease state contributes to a heightened inflammatory and immune response. When the body’s regulatory mechanisms are overwhelmed, this initially favorable response can lead to shock, increased morbidity, and death [82–84]. Lactic acidosis is a key marker of sepsis and septic shock with elevated levels associated with worse outcomes [84,85]. Lactic acidosis in sepsis primarily originates from the byproducts of anaerobic respiration due to tissue hypoxia and the mitochondrial-glycolytic shift during periods of shock due to reduced microcirculation [84] The effect of DCA on the PDC limiting the transition to anaerobic respiration makes it an interesting adjunct therapy in the setting of sepsis and septic shock.
In 1992 a clinical trial in patients with lactic acidosis showed no improvement of morbidity or mortality in a randomized controlled study of 252 patients using DCA vs. placebo. In the study over half (~58 %) of the patients had sepsis as the cause of their lactic acidosis, however sub-group analysis was not reported [30]. Studies have shown clearance of lactic acidosis does not correlate directly to improved outcomes in sepsis, making the results difficult to interpret given trial participants had proven lactic acidosis >5.0 mmol/L prior to initiation of DCA therapy [30,82,84]. Notably, this trial was completed prior to the Surviving Sepsis Campaign in 2002 and treatment for the disease state has shifted dramatically.
Research continued in this sphere, using in vivo and in vitro models, to show how DCA’s effect on PDK lead to increased survival in septic mice [86]. They postulated that cellular divergence to less efficient energy production during times of oxidative stress can contribute to increased mortality by creating an environment favorable to endotoxins while limiting the function of the immune system [86].
In 2020, Bakalov et al. used Drosophila models to show that DCA in combination with antibiotics lead to longer overall survival in septic flies with no effect on sham control groups [82]. This study further reported that septic flies who received DCA had significantly lower levels of lactic acid, α-ketoglutarate, and pyruvate levels. These levels were unchanged in flies without infection regardless of DCA therapy leading us to conclude that the effect of DCA was disease-state specific [82].
Numerous studies have suggested that organisms with sepsis not only undergo cellular hypoperfusion but experience a cascade of metabolic shifts favoring anerobic respiration causing negative downstream effects. In these studies, manipulation of PDC has shown a reversal of these rearrangements by reliance on aerobic respiration through the TCA cycle [84,86–90]. Mainali et al. showed this metabolic shift specifically led to the inactivation of mitochondrial respiration through PDK1 inhibition of PDC causing significant prolonged liver injury limiting our internal sepsis recovery mechanisms [88]. Their findings also suggest a role for stress hormone pathway in sepsis-induced PDK activation. DCA has shown improved homeostasis, energy balance, and reduction of the inflammatory cascade with modulation of PDC in animal models, however no randomized controlled clinical trial in the setting of sepsis has been done.
7.3. Cerebral ischemia
DCA has been shown to have a significant role in pre-clinical studies as a potentially neuro-protective agent during cerebral ischemic and reperfusion events by modulating PDC activity [91–96]. Local tissue hypoxemia as the result of vessel occlusion leads to cell death, dysfunction, inflammation, and eventual breakdown of the blood-brain barrier (BBB) [91,92,94,97]. Like other pathologic states, inhibition of PDC leads to decreased oxidative phosphorylation [92,93]. In 1994 Dimlich used DCA in gerbil models to show evidence of reduced neurological damage after ischemia [95]. High dose DCA was used in a clinical trial for patients presenting 1–5 days after acute stroke. After a single high dose there was a lactate to N-acetyl compound ratio trend suggesting possible therapeutic benefit [98].
Further research has shown that the accumulation of NADPH and ROS species during the initial cerebral ischemic insult can cause worsening neuronal tissue damage after reperfusion. ROS disrupts the BBB leading to pro-inflammatory infiltration and edema. [94,97,99] The ability of DCA to mitigate the production of ROS in addition to increased energy availability at the site of injury provides a multifactorial advantage in the setting of cerebral ischemic pathology. Hong et al. showed in 2018 that an infusion of DCA and pyruvate in rat models reduced neuronal cell death and limited the downstream effects of neuroinflammatory response. However, they did not show improvement with DCA alone [97]. Zhao et al. in 2021 showed that DCA administration during cerebral ischemia and reperfusion was able to reduce infarct size, cerebral edema, and improve the integrity of the BBB in a murine model [94].
Data suggests that DCA may have a role in mitigating cerebral ischemic changes and negative downstream effects in the setting of cerebrovascular accident, however, animal models were dosed at the time of injury bringing into question the clinical applications of the data [94,95,97] Clinical trials using DCA in the setting of cerebral ischemia are warranted to take this orphan drug from bench to bedside.
8. Limitations of use
Dichloroacetate has been studied in clinical trials for several decades and used with limited FDA approval as an orphan drug for metabolic disorders. These trials have established a robust safety profile. DCA has been associated with neuropathic toxicity and sub-clinical hepatic enzyme elevation, with neuropathy the most frequently reported significant finding [13,26,29,31,63,100–106]. Adverse events trend towards transient, even with reported cases of mild to severe peripheral neuropathy [26,29,101,105]. In a trial presented by Kaufmann et al., peripheral neuropathy was the cause of prematurely stopping therapy for 19 of 22 patients treated with high doses of DCA (25 mg/kg/day) for a rare but progressive mitochondrial disease affecting the neurologic system termed mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). However, symptoms resolved completely in 17 of the 19 patients with the remaining two patients lost to follow-up [63]. Other trials with chronic DCA use in similar settings did not show similar neuropathic toxicity [29,31,101–104,106]. The three trials that used limited dosing did not report neuropathy or any other clinically significant side effects [30,98,107].
A deeper look into the dosing of DCA investigated biomechanical modifiers and genetic predispositions to drug DCA metabolism suggesting that sex, age, and a testable gene (GSTZ1) have a significant influence and should be considered when dosing [26,63,100]. Published studies have not shown severe life-threatening or chronically disabling toxicity associated with the use of DCA in pre-clinical or clinical studies. DCA continues to be investigated in both acute and chronic settings with variable dosing structures. The possibility of short-term therapy, particularly in severe acute illness, should not be limited by its published side-effect profile.
9. Conclusion
DCA represents a potentially limitless opportunity for investigation. The PDC/PDK axis can be modulated by DCA in such a way to negate the body’s adaptive preference for glycolysis under pathologic states and instead dramatically increase the efficiency of energy production through aerobic respiration. Despite over fifty years of use as an investigational drug with numerous potential clinical applications, there are still a dearth of clinical trials focused on the efficacy of this metabolic modifier in humans. Although many inquiries surrounding the use of DCA have centered on its ability to reverse the Warburg effect in the setting of malignancy, recent work has also shown promise in the acute setting of cerebral ischemia-reperfusion injuries, hemorrhagic shock, and sepsis. These areas of clinical interest are particularly exciting given that the most often cited and well-studied adverse effect of DCA is its potential to cause reversible peripheral neuropathy in the setting of chronic administration [5]. These acute disease processes are generally short in duration; therefore, drug administration requirements would likely be limited to a single dose or brief regimen [74,78,90]. Future studies should seek to prove the efficacy of DCA in the aforementioned clinical contexts and validate its safety suggested by studies already reviewed. It is possible that this orphan drug and its successors will find several homes within the house of medicine.
Acknowledgments
We would like to thank Efrain M. Castillo, M.D. for the design of our figures. This work was supported by the National Institutes of Health (R01GM122059 and R01AG073338) and the United States Department of Defense (W81XWH-22-1-0903) to RR.
Abbreviations:
- DCA
Dichloroacetate
- PDK
Pyruvate dehydrogenase kinase
- PDH
Pyruvate dehydrogenase
- PDC
Pyruvate dehydrogenase complex
Footnotes
CRediT authorship contribution statement
Nick Schoenmann: Conceptualization, Writing, Editing; Nicholas Tannenbaum: Writing, Editing; Ryan M. Hodgeman: Writing, Editing; Raghavan Pillai Raju: Conceptualization, Writing, Supervision.
Declaration of competing interest
Authors have no interest to declare.
Data availability
No data was used for the research described in the article.
References
- [1].Stacpoole PW, Felts JM, Diisopropylammonium dichloroacetate (DIPA) and sodium dichloracetate (DCA): effect on glucose and fat metabolism in normal and diabetic tissue, Metabolism 19 (1) (1970) 71–78. [DOI] [PubMed] [Google Scholar]
- [2].Stacpoole PW, Moore GW, Kornhauser DM, Metabolic effects of dichloroacetate in patients with diabetes mellitus and hyperlipoproteinemia, N. Engl. J. Med 298 (10) (1978) 526–530. [DOI] [PubMed] [Google Scholar]
- [3].Constantin-Teodosiu D, Regulation of muscle pyruvate dehydrogenase complex in insulin resistance: effects of exercise and dichloroacetate, Diabetes Metab. J 37 (5) (2013) 301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Whitehouse S, Randle PJ, Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate (short communication), Biochem. J 134 (2) (1973) 651–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Stacpoole PW, Martyniuk CJ, James MO, Calcutt NA, Dichloroacetate-induced peripheral neuropathy, Int. Rev. Neurobiol 145 (2019) 211–238. [DOI] [PubMed] [Google Scholar]
- [6].Zhong G, Li W, Gu Y, Langaee T, Stacpoole PW, James MO, Chloride and other anions inhibit dichloroacetate-induced inactivation of human liver GSTZ1 in a haplotype-dependent manner, Chem. Biol. Interact 215 (2014) 33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].James MO, Jahn SC, Zhong G, Smeltz MG, Hu Z, Stacpoole PW, Therapeutic applications of dichloroacetate and the role of glutathione transferase zeta-1, Pharmacol. Ther 170 (2017) 166–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Patel MS, Nemeria NS, Furey W, Jordan F, The pyruvate dehydrogenase complexes: structure-based function and regulation, J. Biol. Chem 289 (24) (2014) 16615–16623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Anwar S, Shamsi A, Mohammad T, Islam A, Hassan MI, Targeting pyruvate dehydrogenase kinase signaling in the development of effective cancer therapy, Biochim. Biophys. Acta Rev. Cancer 1876 (1) (2021), 188568. [DOI] [PubMed] [Google Scholar]
- [10].Sugden MC, Holness MJ, Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs, Am. J. Physiol. Endocrinol. Metab 284 (5) (2003) E855–E862. [DOI] [PubMed] [Google Scholar]
- [11].Zimmer AD, Walbrecq G, Kozar I, Behrmann I, Haan C, Phosphorylation of the pyruvate dehydrogenase complex precedes HIF-1-mediated effects and pyruvate dehydrogenase kinase 1 upregulation during the first hours of hypoxic treatment in hepatocellular carcinoma cells, Hypoxia (Auckl.) 4 (2016) 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang X, Shen X, Yan Y, Li H, Pyruvate dehydrogenase kinases (PDKs): an overview toward clinical applications, Biosci. Rep 41 (4) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, Theriaque DW, Shuster JJ, Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate, Pediatrics 121 (5) (2008) e1223–e1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fernandez-Canon JM, Baetscher MW, Finegold M, Burlingame T, Gibson KM, Grompe M, Maleylacetoacetate isomerase (MAAI/GSTZ)-deficient mice reveal a glutathione-dependent nonenzymatic bypass in tyrosine catabolism, Mol. Cell. Biol 22 (13) (2002) 4943–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Stacpoole PW, Henderson GN, Yan Z, James MO, Clinical pharmacology and toxicology of dichloroacetate, Environ. Health Perspect 106 (Suppl. 4) (1998) 989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Salido E, Pey AL, Rodriguez R, Lorenzo V, Primary hyperoxalurias: disorders of glyoxylate detoxification, Biochim. Biophys. Acta 1822 (9) (2012) 1453–1464. [DOI] [PubMed] [Google Scholar]
- [17].Baker PR, Cramer SD, Kennedy M, Assimos DG, Holmes RP, Glycolate and glyoxylate metabolism in HepG2 cells, Am. J. Physiol. Cell Physiol 287 (5) (2004) C1359–C1365. [DOI] [PubMed] [Google Scholar]
- [18].Salyer WR, Salyer DC, Thiamine deficiency and oxalosis, J. Clin. Pathol 27 (7) (1974) 558–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stacpoole PW, Harwood HJ Jr., Cameron DF, Curry SH, Samuelson DA, Cornwell PE, Sauberlich HE, Chronic toxicity of dichloroacetate: possible relation to thiamine deficiency in rats, Fundam. Appl. Toxicol 14 (2) (1990) 327–337. [DOI] [PubMed] [Google Scholar]
- [20].Bilbao JM, Berry H, Marotta J, Ross RC, Peripheral neuropathy in oxalosis. A case report with electron microscopic observations, Can. J. Neurol. Sci 3 (1) (1976) 63–67. [DOI] [PubMed] [Google Scholar]
- [21].Berini SE, Tracy JA, Engelstad JK, Lorenz EC, Milliner DS, Dyck PJ, Progressive polyradiculoneuropathy due to intraneural oxalate deposition in type 1 primary hyperoxaluria, Muscle Nerve 51 (3) (2015) 449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Darras BT, ScienceDirect, Neuromuscular Disorders of Infancy, Childhood, and Adolescence: A clinician’s Approach, Second edition ed, Academic Press, London, 2015. [Google Scholar]
- [23].Stacpoole PW, The dichloroacetate dilemma: environmental hazard versus therapeutic goldmine—both or neither? Environ. Health Perspect 119 (2) (2011) 155–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tzeng HF, Blackburn AC, Board PG, Anders MW, Polymorphism- and species-dependent inactivation of glutathione transferase zeta by dichloroacetate, Chem. Res. Toxicol 13 (4) (2000) 231–236. [DOI] [PubMed] [Google Scholar]
- [25].Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW, Age-dependent kinetics and metabolism of dichloroacetate: possible relevance to toxicity, J. Pharmacol. Exp. Ther 324 (3) (2008) 1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tian DD, Bennett SK, Coupland LA, Forwood K, Lwin Y, Pooryousef N, Tea I, Truong TT, Neeman T, Crispin P, D’Rozario J, Blackburn AC, GSTZ1 genotypes correlate with dichloroacetate pharmacokinetics and chronic side effects in multiple myeloma patients in a pilot phase 2 clinical trial, Pharmacol. Res. Perspect 7 (6) (2019), e00526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Blackburn AC, Tzeng HF, Anders MW, Board PG, Discovery of a functional polymorphism in human glutathione transferase zeta by expressed sequence tag database analysis, Pharmacogenetics 10 (1) (2000) 49–57. [DOI] [PubMed] [Google Scholar]
- [28].Stacpoole PW, Nagaraja NV, Hutson AD, Efficacy of dichloroacetate as a lactate-lowering drug, J. Clin. Pharmacol 43 (7) (2003) 683–691. [PubMed] [Google Scholar]
- [29].Barshop BA, Naviaux RK, McGowan KA, Levine F, Nyhan WL, Loupis-Geller A, Haas RH, Chronic treatment of mitochondrial disease patients with dichloroacetate, Mol. Genet. Metab 83 (1–2) (2004) 138–149. [DOI] [PubMed] [Google Scholar]
- [30].Stacpoole PW, Wright EC, Baumgartner TG, Bersin RM, Buchalter S, Curry SH, Duncan CA, Harman EM, Henderson GN, Jenkinson S, et al. , A controlled clinical trial of dichloroacetate for treatment of lactic acidosis in adults. The Dichloroacetate-Lactic Acidosis Study Group, N. Engl. J. Med 327 (22) (1992) 1564–1569. [DOI] [PubMed] [Google Scholar]
- [31].Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore RL, Greer M, Henderson GN, Hutson AD, Neiberger RE, O’Brien RG, Perkins LA, Quisling RG, Shroads AL, Shuster JJ, Silverstein JH, Theriaque DW, Valenstein E, Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children, Pediatrics 117 (5) (2006) 1519–1531. [DOI] [PubMed] [Google Scholar]
- [32].Stacpoole PW, Harman EM, Curry SH, Baumgartner TG, Misbin RI, Treatment of lactic acidosis with dichloroacetate, N. Engl. J. Med 309 (7) (1983) 390–396. [DOI] [PubMed] [Google Scholar]
- [33].Backshear PJ, Holloway PA, Alberti KG, Metabolic interactions of dichloroacetate and insulin in experimental diabetic ketoacidosis, Biochem. J 146 (2) (1975) 447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Stacpoole PW, Greene YJ, Dichloroacetate, Diabetes Care 15 (6) (1992) 785–791. [DOI] [PubMed] [Google Scholar]
- [35].Katayama Y, Kawata Y, Moritoh Y, Watanabe M, Dichloroacetate, a pyruvate dehydrogenase kinase inhibitor, ameliorates type 2 diabetes via reduced gluconeogenesis, Heliyon 8 (2) (2022), e08889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Eichner HL, Stacpoole PW, Forsham PH, Treatment of streptozotocin diabetes with di-isopropylammonium dichloroacetate (DIPA), Diabetes 23 (3) (1974) 179–182. [DOI] [PubMed] [Google Scholar]
- [37].Le Page LM, Rider OJ, Lewis AJ, Ball V, Clarke K, Johansson E, Carr CA, Heather LC, Tyler DJ, Increasing pyruvate dehydrogenase flux as a treatment for diabetic cardiomyopathy: a combined 13C hyperpolarized magnetic resonance and echocardiography study, Diabetes 64 (8) (2015) 2735–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, Iwanaga Y, Narazaki M, Matsuda T, Soga T, Kita T, Kimura T, Shioi T, Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure, Circ. Heart Fail 3 (3) (2010) 420–430. [DOI] [PubMed] [Google Scholar]
- [39].Li X, Liu J, Hu H, Lu S, Lu Q, Quan N, Rousselle T, Patel MS, Li J, Dichloroacetate ameliorates cardiac dysfunction caused by ischemic insults through AMPK signal pathway-not only shifts metabolism, Toxicol. Sci 167 (2) (2019) 604–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Matsiukevich D, Piraino G, Klingbeil LR, Hake PW, Wolfe V, O’Connor M, Zingarelli B, The AMPK activator Aicar ameliorates age-dependent myocardial injury in murine hemorrhagic shock, Shock 47 (1) (2017) 70–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pala HG, Pala EE, Artunc Ulkumen B, Erbas O, Protective effects of dichloroacetic acid on endometrial injury and ovarian reserve in an experimental rat model of diabetes mellitus, J. Obstet. Gynaecol. Res 47 (12) (2021) 4319–4328. [DOI] [PubMed] [Google Scholar]
- [42].Nagy JA, Chang SH, Dvorak AM, Dvorak HF, Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 100 (6) (2009) 865–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ramakrishnan S, Anand V, Roy S, Vascular endothelial growth factor signaling in hypoxia and inflammation, J. NeuroImmune Pharmacol 9 (2) (2014) 142–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Nagy JA, Dvorak AM, Dvorak HF, VEGF-A and the induction of pathological angiogenesis, Annu. Rev. Pathol 2 (2007) 251–275. [DOI] [PubMed] [Google Scholar]
- [45].Bjorndahl MA, Cao R, Burton JB, Brakenhielm E, Religa P, Galter D, Wu L, Cao Y, Vascular endothelial growth factor-a promotes peritumoral lymphangiogenesis and lymphatic metastasis, Cancer Res. 65 (20) (2005) 9261–9268. [DOI] [PubMed] [Google Scholar]
- [46].Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC, Metabolic modulation of glioblastoma with dichloroacetate, Sci. Transl. Med 2 (31) (2010) 31ra34. [DOI] [PubMed] [Google Scholar]
- [47].Warburg O, Wind F, Negelein E, The metabolism of tumors in the body, J. Gen. Physiol 8 (6) (1927) 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liberti MV, Locasale JW, The Warburg effect: how does it benefit cancer cells? Trends Biochem. Sci 41 (3) (2016) 211–218.26778478 [Google Scholar]
- [49].Horne AW, Ahmad SF, Carter R, Simitsidellis I, Greaves E, Hogg C, Morton NM, Saunders PTK, Repurposing dichloroacetate for the treatment of women with endometriosis, Proc. Natl. Acad. Sci. U. S. A 116 (51) (2019) 25389–25391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kankotia S, Stacpoole PW, Dichloroacetate and cancer: new home for an orphan drug? Biochim. Biophys. Acta 1846 (2) (2014) 617–629. [DOI] [PubMed] [Google Scholar]
- [51].Stacpoole PW, Therapeutic targeting of the pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer, J. Natl. Cancer Inst 109 (11) (2017). [DOI] [PubMed] [Google Scholar]
- [52].Schoors S, Bruning U, Missiaen R, Queiroz KC, Borgers G, Elia I, Zecchin A, Cantelmo AR, Christen S, Goveia J, Heggermont W, Godde L, Vinckier S, Van Veldhoven PP, Eelen G, Schoonjans L, Gerhardt H, Dewerchin M, Baes M, De Bock K, Ghesquiere B, Lunt SY, Fendt SM, Carmeliet P, Fatty acid carbon is essential for dNTP synthesis in endothelial cells, Nature 520 (7546) (2015) 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Rostamian H, Khakpoor-Koosheh M, Jafarzadeh L, Masoumi E, Fallah-Mehrjardi K, Tavassolifar MJ, Jas MP, Mirzaei HR, Hadjati J, Restricting tumor lactic acid metabolism using dichloroacetate improves T cell functions, BMC Cancer 22 (1) (2022) 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Liang Y, Hou L, Li L, Li L, Zhu L, Wang Y, Huang X, Hou Y, Zhu D, Zou H, Gu Y, Weng X, Wang Y, Li Y, Wu T, Yao M, Gross I, Gaiddon C, Luo M, Wang J, Meng X, Dichloroacetate restores colorectal cancer chemosensitivity through the p53/miR-149–3p/PDK2-mediated glucose metabolic pathway, Oncogene 39 (2) (2020) 469–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Al-Azawi A, Sulaiman S, Arafat K, Yasin J, Nemmar A, Attoub S, Impact of sodium dichloroacetate alone and in combination therapies on lung tumor growth and metastasis, Int. J. Mol. Sci 22 (22) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sun RC, Board PG, Blackburn AC, Targeting metabolism with arsenic trioxide and dichloroacetate in breast cancer cells, Mol. Cancer 10 (2011) 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC, Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo, Breast Cancer Res. Treat 120 (1) (2010) 253–260. [DOI] [PubMed] [Google Scholar]
- [58].Galgamuwa R, Hardy K, Dahlstrom JE, Blackburn AC, Wium E, Rooke M, Cappello JY, Tummala P, Patel HR, Chuah A, Tian L, McMorrow L, Board PG, Theodoratos A, Dichloroacetate prevents cisplatin-induced nephrotoxicity without compromising cisplatin anticancer properties, J. Am. Soc. Nephrol 27 (11) (2016) 3331–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].de Mey S, Dufait I, Jiang H, Corbet C, Wang H, Van De Gucht M, Kerkhove L, Law KL, Vandenplas H, Gevaert T, Feron O, De Ridder M, Dichloroacetate radiosensitizes hypoxic breast cancer cells, Int. J. Mol. Sci 21 (24) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Cook KM, Shen H, McKelvey KJ, Gee HE, Hau E, Targeting glucose metabolism of cancer cells with dichloroacetate to radiosensitize high-grade gliomas, Int. J. Mol. Sci 22 (14) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Strum SB, Adalsteinsson O, Black RR, Segal D, Peress NL, Waldenfels J, Case report: sodium dichloroacetate (DCA) inhibition of the “Warburg effect” in a human cancer patient: complete response in non-Hodgkin’s lymphoma after disease progression with rituximab-CHOP, J. Bioenerg. Biomembr 45 (3) (2013) 307–315. [DOI] [PubMed] [Google Scholar]
- [62].Powell SF, Mazurczak M, Dib EG, Bleeker JS, Geeraerts LH, Tinguely M, Lohr MM, McGraw SC, Jensen AW, Ellison CA, Black LJ, Puumala SE, Reed VJ, Miskimins WK, Lee JH, Spanos WC, Phase II study of dichloroacetate, an inhibitor of pyruvate dehydrogenase, in combination with chemoradiotherapy for unresected, locally advanced head and neck squamous cell carcinoma, Investig. New Drugs 40 (3) (2022) 622–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, Stacpoole PW, DiMauro S, De Vivo DC, Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial, Neurology 66 (3) (2006) 324–330. [DOI] [PubMed] [Google Scholar]
- [64].Goodwin J, Choi H, Hsieh MH, Neugent ML, Ahn JM, Hayenga HN, Singh PK, Shackelford DB, Lee IK, Shulaev V, Dhar S, Takeda N, Kim JW, Targeting hypoxia-inducible factor-1alpha/pyruvate dehydrogenase kinase 1 Axis by dichloroacetate suppresses bleomycin-induced pulmonary fibrosis, Am. J. Respir. Cell Mol. Biol 58 (2) (2018) 216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sharma A, Chun J, Ji MS, Lee S, Kang C, Kim JS, Binary prodrug of dichloroacetic acid and doxorubicin with enhanced anticancer activity, ACS Appl. Bio Mater 4 (3) (2021) 2026–2032. [DOI] [PubMed] [Google Scholar]
- [66].Saleh MF, Elsayad ME, Goda AE, Mitigation of doxorubicin-induced cardiotoxicity by dichloroacetate: potential roles of restoration of PGC-1alpha/SIRT3 signaling and suppression of oxidative stress and apoptosis, Eur. Rev. Med. Pharmacol. Sci 25 (21) (2021) 6573–6584. [DOI] [PubMed] [Google Scholar]
- [67].Minino AM, Anderson RN, Fingerhut LA, Boudreault MA, Warner M, Deaths: injuries, 2002, Natl. Vital Stat. Rep 54 (10) (2006) 1–124. [PubMed] [Google Scholar]
- [68].Camazine MN, Hemmila MR, Leonard JC, Jacobs RA, Horst JA, Kozar RA, Bochicchio GV, Nathens AB, Cryer HM, Spinella PC, Massive transfusion policies at trauma centers participating in the American College of Surgeons Trauma Quality Improvement Program, J. Trauma Acute Care Surg 78 (6 Suppl 1) (2015) S48–S53. [DOI] [PubMed] [Google Scholar]
- [69].Black JA, Pierce VS, Juneja K, Holcomb JB, Complications of hemorrhagic shock and massive transfusion-a comparison before and after the damage control resuscitation era, Shock 56 (1) (2021) 42–51. [DOI] [PubMed] [Google Scholar]
- [70].Cannon JW, Hemorrhagic shock, N. Engl. J. Med 378 (4) (2018) 370–379. [DOI] [PubMed] [Google Scholar]
- [71].Black JA, Pierce VS, Kerby JD, Holcomb JB, The evolution of blood transfusion in the trauma patient: whole blood has come full circle, Semin. Thromb. Hemost 46 (2) (2020) 215–220. [DOI] [PubMed] [Google Scholar]
- [72].Gomez H, Kautza B, Escobar D, Nassour I, Luciano J, Botero AM, Gordon L, Martinez S, Holder A, Ogundele O, Loughran P, Rosengart MR, Pinsky M, Shiva S, Zuckerbraun BS, Inhaled carbon monoxide protects against the development of shock and mitochondrial injury following hemorrhage and resuscitation, PLoS One 10 (9) (2015), e0135032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Rao G, Xie J, Hedrick A, Awasthi V, Hemorrhagic shock-induced cerebral bioenergetic imbalance is corrected by pharmacologic treatment with EF24 in a rat model, Neuropharmacology 99 (2015) 318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Chu X, Schwartz R, Diamond MP, Raju RP, A combination treatment strategy for hemorrhagic shock in a rat model modulates autophagy, Front. Med. (Lausanne) 6 (2019) 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Semenza GL, Hypoxia-inducible factors in physiology and medicine, Cell 148 (3) (2012) 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Jian B, Wang D, Chen D, Voss J, Chaudry I, Raju R, Hypoxia-induced alteration of mitochondrial genes in cardiomyocytes: role of Bnip3 and Pdk1, Shock 34 (2) (2010) 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Kim JW, Tchernyshyov I, Semenza GL, Dang CV, HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia, Cell Metab. 3 (3) (2006) 177–185. [DOI] [PubMed] [Google Scholar]
- [78].Subramani K, Lu S, Warren M, Chu X, Toque HA, Caldwell RW, Diamond MP, Raju R, Mitochondrial targeting by dichloroacetate improves outcome following hemorrhagic shock, Sci. Rep 7 (1) (2017) 2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Ayub A, Poulose N, Raju R, Resveratrol improves survival and prolongs life following hemorrhagic shock, Mol. Med 21 (1) (2015) 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kline JA, Maiorano PC, Schroeder JD, Grattan RM, Vary TC, Watts JA, Activation of pyruvate dehydrogenase improves heart function and metabolism after hemorrhagic shock, J. Mol. Cell. Cardiol 29 (9) (1997) 2465–2474. [DOI] [PubMed] [Google Scholar]
- [81].Barbee RW, Kline JA, Watts JA, Depletion of lactate by dichloroacetate reduces cardiac efficiency after hemorrhagic shock, Shock 14 (2) (2000) 208–214. [DOI] [PubMed] [Google Scholar]
- [82].Bakalov V, Reyes-Uribe L, Deshpande R, Maloy AL, Shapiro SD, Angus DC, Chang CH, Le Moyec L, Wendell SG, Kaynar AM, Dichloroacetate-induced metabolic reprogramming improves lifespan in a Drosophila model of surviving sepsis, PLoS One 15 (11) (2020), e0241122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, Cline GW, Phillips AJ, Medzhitov R, Functional polarization of tumour-associated macrophages by tumour-derived lactic acid, Nature 513 (7519) (2014) 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Suetrong B, Walley KR, Lactic acidosis in sepsis: it’s not all anaerobic: implications for diagnosis and management, Chest 149 (1) (2016) 252–261. [DOI] [PubMed] [Google Scholar]
- [85].Foucher CD, Tubben RE, Lactic Acidosis, StatPearls, Treasure Island (FL), 2022. [Google Scholar]
- [86].McCall CE, Zabalawi M, Liu T, Martin A, Long DL, Buechler NL, Arts RJW, Netea M, Yoza BK, Stacpoole PW, Vachharajani V, Pyruvate dehydrogenase complex stimulation promotes immunometabolic homeostasis and sepsis survival, JCI Insight 3 (15) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Oh TS, Zabalawi M, Jain S, Long D, Stacpoole PW, McCall CE, Quinn MA, Dichloroacetate improves systemic energy balance and feeding behavior during sepsis, JCI Insight 7 (12) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Mainali R, Zabalawi M, Long D, Buechler N, Quillen E, Key CC, Zhu X, Parks JS, Furdui C, Stacpoole PW, Martinez J, McCall CE, Quinn MA, Dichloroacetate reverses sepsis-induced hepatic metabolic dysfunction, Elife 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Langley RJ, Tsalik EL, van Velkinburgh JC, Glickman SW, Rice BJ, Wang C, Chen B, Carin L, Suarez A, Mohney RP, Freeman DH, Wang M, You J, Wulff J, Thompson JW, Moseley MA, Reisinger S, Edmonds BT, Grinnell B, Nelson DR, Dinwiddie DL, Miller NA, Saunders CJ, Soden SS, Rogers AJ, Gazourian L, Fredenburgh LE, Massaro AF, Baron RM, Choi AM, Corey GR, Ginsburg GS, Cairns CB, Otero RM, Fowler VG Jr., Rivers EP, Woods CW, Kingsmore SF, An integrated clinico-metabolomic model improves prediction of death in sepsis, Sci. Transl. Med 5 (195) (2013) 195ra95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Zeng Z, Huang Q, Mao L, Wu J, An S, Chen Z, Zhang W, The pyruvate dehydrogenase complex in sepsis: metabolic regulation and targeted therapy, Front. Nutr 8 (2021), 783164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Thibodeau A, Geng X, Previch LE, Ding Y, Pyruvate dehydrogenase complex in cerebral ischemia-reperfusion injury, Brain Circ. 2 (2) (2016) 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Zaidan E, Sims NR, Reduced activity of the pyruvate dehydrogenase complex but not cytochrome c oxidase is associated with neuronal loss in the striatum following short-term forebrain ischemia, Brain Res. 772 (1–2) (1997) 23–28. [DOI] [PubMed] [Google Scholar]
- [93].Geng X, Elmadhoun O, Peng C, Ji X, Hafeez A, Liu Z, Du H, Rafols JA, Ding Y, Ethanol and normobaric oxygen: novel approach in modulating pyruvate dehydrogenase complex after severe transient and permanent ischemic stroke, Stroke 46 (2) (2015) 492–499. [DOI] [PubMed] [Google Scholar]
- [94].Zhao X, Li S, Mo Y, Li R, Huang S, Zhang A, Ni X, Dai Q, Wang J, DCA protects against oxidation injury attributed to cerebral ischemia-reperfusion by regulating glycolysis through PDK2-PDH-Nrf2 Axis, Oxidative Med. Cell. Longev 2021 (2021) 5173035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Dimlich RV, Marangos PJ, Dichloroacetate attenuates neuronal damage in a gerbil model of brain ischemia, J. Mol. Neurosci 5 (2) (1994) 69–81. [DOI] [PubMed] [Google Scholar]
- [96].Peeling J, Sutherland G, Brown RA, Curry S, Protective effect of dichloroacetate in a rat model of forebrain ischemia, Neurosci. Lett 208 (1) (1996) 21–24. [DOI] [PubMed] [Google Scholar]
- [97].Hong DK, Kho AR, Choi BY, Lee SH, Jeong JH, Lee SH, Park KH, Park JB, Suh SW, Combined treatment with dichloroacetic acid and pyruvate reduces hippocampal neuronal death after transient cerebral ischemia, Front. Neurol 9 (2018) 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Graham GD, Barker PB, Brooks WM, Morris DC, Ahmed W, Bryniarski E, Hearshen DO, Sanders JA, Holshouser BA, Turkel CC, MR spectroscopy study of dichloroacetate treatment after ischemic stroke, Neurology 55 (9) (2000) 1376–1378. [DOI] [PubMed] [Google Scholar]
- [99].Suh SW, Shin BS, Ma H, Van Hoecke M, Brennan AM, Yenari MA, Swanson RA, Glucose and NADPH oxidase drive neuronal superoxide formation in stroke, Ann. Neurol 64 (6) (2008) 654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].James MO, Stacpoole PW, Pharmacogenetic considerations with dichloroacetate dosing, Pharmacogenomics 17 (7) (2016) 743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Dunbar EM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, Shuster JJ, Wagner DA, Stacpoole PW, Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors, Investig. New Drugs 32 (3) (2014) 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Chu QS, Sangha R, Spratlin J, Vos LJ, Mackey JR, McEwan AJ, Venner P, Michelakis ED, A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors, Investig. New Drugs 33 (3) (2015) 603–610. [DOI] [PubMed] [Google Scholar]
- [103].Spruijt L, Naviaux RK, McGowan KA, Nyhan WL, Sheean G, Haas RH, Barshop BA, Nerve conduction changes in patients with mitochondrial diseases treated with dichloroacetate, Muscle Nerve 24 (7) (2001) 916–924. [DOI] [PubMed] [Google Scholar]
- [104].Abdelmalak M, Lew A, Ramezani R, Shroads AL, Coats BS, Langaee T, Shankar MN, Neiberger RE, Subramony SH, Stacpoole PW, Long-term safety of dichloroacetate in congenital lactic acidosis, Mol. Genet. Metab 109 (2) (2013) 139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Michelakis ED, Gurtu V, Webster L, Barnes G, Watson G, Howard L, Cupitt J, Paterson I, Thompson RB, Chow K, O’Regan DP, Zhao L, Wharton J, Kiely DG, Kinnaird A, Boukouris AE, White C, Nagendran J, Freed DH, Wort SJ, Gibbs JSR, Wilkins MR, Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients, Sci. Transl. Med 9 (413) (2017). [DOI] [PubMed] [Google Scholar]
- [106].Mori M, Yamagata T, Goto T, Saito S, Momoi MY, Dichloroacetate treatment for mitochondrial cytopathy: long-term effects in MELAS, Brain and Development 26 (7) (2004) 453–458. [DOI] [PubMed] [Google Scholar]
- [107].Stacpoole PW, Pharmacotoxicology of Trichloroethylene Metabolites, ClinicalTrials.gov Identifier: NCT01128270, 2014. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.