

Abstract
INTRODUCTION
Establishing valid diagnostic strategies is a precondition for successful therapeutic intervention in Alzheimer's disease (AD).
METHODS
One hundred forty‐four healthy 75‐year‐old participants from the Vienna‐Transdanube‐Aging longitudinal cohort study were tested for neuroaxonal damage by single molecular array (Simoa) plasma neurofilament light chain (NfL) levels at baseline, 30, 60, and 90 months, and onset of AD dementia. Individual risk for sporadic AD was estimated by continuous shrinkage polygenic risk score (PRS‐CS, genome‐wide association study).
RESULTS
Nineteen participants developed AD after a median of 60 months (interquartile range 30). In participants with AD, baseline NfL plasma levels correlated with PRS‐CS (r = 0.75, p < 0.001; difference to controls: Fisher's r‐to‐z: z = 3.89, p < 0.001). PRS‐CS combined with baseline plasma NfL predicted onset of AD (p < 0.01).
DISCUSSION
Our data suggest that polygenic risk for AD and plasma NfL closely interact years before onset of clinical symptoms. Peripheral NfL may serve as a diagnostic measure supporting early therapeutic intervention and secondary prevention in AD.
Keywords: Alzheimer's disease, genome‐wide association studies, neurofilament light chain, plasma biomarker, polygenic risk score, single molecular array
1. INTRODUCTION
Neurofilament light chain (NfL) is a correlate of neuroaxonal damage and considered an unspecific biomarker of neurodegeneration that can be measured in cerebrospinal fluid (CSF), but also in plasma and serum. 1 , 2 Levels of NfL in peripheral blood and CSF are strongly correlated and can be precisely inferred with single molecule array (Simoa) technology. 3 , 4 , 5 , 6
Plasma NfL has been reported previously to differentiate among persons with Alzheimer's disease (AD), mild cognitive impairment (MCI), and cognitively unimpaired participants in a cross‐sectional design. 7 A recent study confirmed a distinguishing capacity of plasma NfL between AD and MCI or persons with subjective cognitive decline in a memory clinic setting. 8 With regard to potential future disease‐modifying therapies, the identification of AD at‐risk subjects is of even higher interest. 9 In this context, the study by Gerards et al. did not find a distinguishing capacity of plasma NfL among MCI, an AD at‐risk group, and persons with subjective cognitive decline in this cross‐sectional approach. 8 Intriguingly, though, two recent longitudinal studies have suggested a predictive value of baseline plasma NfL for future cognitive decline or imaging parameters of neurodegeneration in cognitively impaired persons or a mixed sample of MCI and healthy control subjects. 1 , 10 Outcome parameters in the latter study such as global cognition, attention, and cortical thickness are non‐AD specific, though, and might be explained by other neurodegenerative processes. 10 A longitudinal study with familial AD mutation carriers found a predictive value of peripheral NfL values for neurodegeneration and clinical deterioration in cognitively unimpaired participants. 6 As age of onset is significantly younger in familial than in sporadic AD, which might impact NfL levels also in the control group, results cannot readily be translated to sporadic AD.
Taken together, no study has evaluated the predictive value of baseline peripheral NfL levels for the clinical manifestation of sporadic AD over time in non‐demented old‐aged persons.
Another emerging marker for individual risk stratification of AD development is the AD polygenic risk score (PRS). An advantage of PRS is that, although it might not be completely independent from it, an effect of age on the score is presumably rather low. Therefore, PRS can be calculated prior to disease onset at any point in an individual's life. Yet, due to the score being sample and methodology dependent, PRS in its current application is not recommended to be applied to determine AD risk as a single marker. 11 , 12 , 13
Based on the outlined findings we hypothesized that NfL in plasma might not be of predictive value for the future development of sporadic AD in non‐demented old‐aged participants as a single marker. This might be related to NfL being a neuron specific but etiologically unspecific marker of neurodegeneration.
We hypothesized that baseline plasma NfL combined with AD‐PRS, a biomarker with higher specificity for AD, might be associated with AD diagnosis over 90 months in a “community‐dwelling” cohort of non‐demented old‐aged persons, in which all were 75 years of age at baseline.
2. MATERIALS AND METHODS
2.1. Participants
The present study is based on data collected in a subsample of the Vienna Transdanube Aging (VITA) study. The VITA study is a community‐based cohort study of 606 participants that were all 75 years old at study inclusion and originated from the 21st and 22nd district of Vienna, Austria. The study started in May 2000. At study inclusion (“baseline”), all 606 participants were healthy and had normal cognitive performance. The main scientific aim of the VITA study is the prediction of dementia in old‐aged persons. 14
The VITA study protocol includes a thorough neurologic and psychiatric examination, neuropsychological testing including the CERAD (Consortium to Establish a Registry for Alzheimer's Disease) test protocol, cerebral magnetic resonance imaging (cMRI), and blood sampling. Detailed descriptions of the study procedures have been published previously. 14 , 15 Participants were invited for follow‐up visits and examinations after 30, 60, and 90 months. Fulfillment of the National Institute for Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association criteria for AD were evaluated at each visit. 16 , 17 The data used for the present study is based on 169 persons, from whom genome‐wide association study (GWAS) data (n = 159) or plasma NfL levels (n = 160) from all time points (0, 30, 60, 90 months) were available. From 151 persons both NfL at baseline and GWAS data was available. Of these 144 passed GWAS quality control (QC) procedures.
2.2. Analysis of NfL in plasma
Levels of NfL were analyzed in plasma samples at baseline (0 months), 30, 60, and 90 months from all 160 participants. All frozen plasma samples were analyzed at the University Hospital of Basel using a Simoa assay (Quanterix NfL Advantage Kit). A detailed description of the assay has been published previously. 4 , 18 All samples and calibrators were measured in duplicate with follow‐up samples from the same participants analyzed in the same run. Interassay variability of the measurements was assessed with three native serum controls (sample 1: 7.2 pg/mL, sample 2: 20.7 pg/mL, sample 3: 38.5 pg/mL). Interassay mean coefficients of variation of duplicate measures were (sample 1, 2, and 3): 4.2%, 4.7%, and 1.3%, respectively.
2.3. Genome‐wide genotyping of VITA participants
159 VITA DNA samples were genotyped on the Infinium Global Screening Array (Illumina), as previously described. 19 GWAS data collection was part of the European Alzheimer's Disease DNA Biobank dataset (EADB) study. 20
2.4. Genotypes QC and imputation
The genotype QC of 159 VITA participants was performed using Ricopili Pipeline 21 using following standard parameters for retaining subjects and single nucleotide polymorphisms (SNPs): SNP missingness < 0.05 (before sample removal); subject missingness < 0.02; autosomal heterozygosity deviation (| Fhet | < 0.2); SNP missingness < 0.02 (after sample removal); difference in SNP missingness between cases and controls < 0.02; SNP Hardy–Weinberg equilibrium (p > 10−6 in controls or p > 10−10 in cases); and male subjects with heterozygosity rate for chromosome X > 0.5 and female subjects < 0.5. Three participants were excluded due to SNP missing > 0.02. Four participants were excluded as population outliers in a principal component analysis (PCA; two participants with principal component [PC] 1 > 0.2 and two with PC 2 > 0.2).
The genotype imputation was performed using the pre‐phasing/imputation stepwise approach implemented in EAGLE 22 / MINIMAC3 23 , 24 (with a variable chunk size of 132 genomic chunks and default parameters). The imputation reference set consisted of 54,330 phased haplotypes with 36,678,882 variants from the publicly available Haplotype Reference Consortium reference. 25
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional sources (e.g., PubMed). An ongoing major focus of the field is the identification of blood biomarkers valid for estimating risk for developing sporadic Alzheimer's disease (AD). We identified a knowledge gap regarding the predictive value of baseline peripheral neurofilament light chain (NfL) levels for the manifestation of sporadic AD in non‐demented old‐aged persons as a single marker or combined with individual polygenic risk scores (PRS).
Interpretation: Our data align well with the notion that peripheral NfL is a powerful biomarker of neurodegeneration. Our findings substantiate a role of peripheral NfL in early AD diagnostics combined with more disease‐specific markers.
Future directions: Relevance for developing AD needs to be confirmed in larger longitudinal cohorts. Further on, benefit for early therapeutic interventions could be assessed.
The relatedness was tested using a subset of 77,177 SNPs obtained with a stringent quality criterion (INFO > 0.8, missingness < 1%, minor allele frequency > 0.05) and by applying linkage disequilibrium (LD) pruning (r 2 > 0.02). The analysis did not find any related or overlapping individuals within VITA participants. A second PCA was performed using the same set of high‐quality SNPs to generate a set of PCs. PCs 1 to 4 were used to control for population stratification in downstream analysis.
2.5. Calculation of PRS for late onset AD
SNP data of 144 VITA participants, from whom both NfL at all time points and GWAS data were available, was used for estimating individual PRS for sporadic AD. We downloaded publicly available summary statistics of AD GWAS (cases/controls: 71,880/ 383,378) 26 from a complex trait genetics lab. 27 We used the PRS continuous shrinkage (CS) method, 28 which uses Bayesian regression, to calculate an updated posterior effect size by applying continuous shrinkage to the initial (prior) effect sizes from the AD GWAS. The posterior effect size accounts for the LD between SNPs using LD reference panels constructed from the 1000 Genomes Project Phase 3 European samples. 29 In total, 810,402 SNPs with a valid posterior effect size were used as weights to perform PRS on our sample by multiplying the updated effect size of each variant by its imputation probability for each participant. The resulting values were summed over each participant, to obtain a whole‐genome PRS.
2.6. Statistical analysis
Significance of differences in descriptive data, NfL values (pg/mL and z‐scores, and PRS (z‐scores) between the AD at 90 months and the control group were assessed with Wilcoxon rank sum test.
To investigate a possible association between the two groups AD/control (dependent variables) and PRS (independent variable), a logistic regression analysis was conducted adjusted for population stratification using PCs 1 to 4 as covariates. The explained variance for the logistic regression analyses was estimated with Nagelkerke R 2 using likelihood of reduced model (covariates only) and full model (containing covariates and PRS) and the p value was obtained by comparing the reduced and full model. The predictive value of PRS and baseline NfL (independent variables) for the diagnosis (AD/control group) was assessed with logistic regression with baseline body mass index (BMI) and sex as additional covariates.
The Spearman's correlation coefficient was used to describe the correlation of PRS and baseline NfL values in the AD and in the control group. Difference between the slopes of the correlation coefficients was calculated with Fisher's r‐to‐z transformation. 30 , 31
To further characterize the effect of baseline NfL, AD diagnosis, and time on NfL levels a linear mixed effects model was calculated with time as the independent variable, NfL as the dependent variable, and AD diagnosis at 90 months as well as baseline NfL and sex as covariates (MATLAB 2022b, version 9.13.0, Statistics and Machine Learning Toolbox Version 12.4).
For validity control NfL levels were controlled for the influence of BMI and age using a large reference database of healthy controls that was built with a generalized additive model for location, scale, and shape. 32 From this statistical model NfL z‐scores and percentiles were derived as age‐ and BMI‐adjusted measures (Figure S1 in supporting information). NfL z‐scores were used for control analysis of the data.
As a further validity control, analyses were repeated after excluding participants with a baseline NfL z‐score > 1.5 and concurrent vascular pathology (total n = 6, all control subjects), defined as magnetic resonance imaging evidence of infarction (diameter > 1.5 cm), lacunar infarcts (diameter < 1.5 cm), periventricular hyperintensities at baseline (periventricular bands and irregular with extension into deep white matter), or deep white matter hyperintensities (beginning confluent and confluent).
Multiple testing bias (false discovery rate [FDR]) was allowed for using the Benjamini–Hochberg procedure. 33
GraphPad Prism software (version 9.4.1) was used for data visualization.
3. RESULTS
3.1. Study sample: 19 out of 144 initially healthy participants were diagnosed with AD at 90 months
Of the 144 participants included in data analysis, 19 persons developed AD while 125 participants did not develop AD over the study course of 90 months (Table 1). This corresponds to 1.76 AD cases per 100 person years. At study end, AD prevalence was 13.2%. Median time to AD diagnosis was 60 months (interquartile range [IQR] 30). Median age at study entry was 75.6 years with a low IQR of 0.7, due to inclusion criteria. Median age at study end was 83.2 years (IQR 0.7). Median score on the Mini‐Mental State Examination (MMSE) at study beginning was 28 (IQR 2) and participants had a high level of education of 11 (IQR 1) years. Between the AD at 90 months and the control group we found significant differences for the MMSE at 0‐ and 90‐month intervals as well as apolipoprotein E (APOE) ε4 carrier status, but for no other descriptive data such as age, years of education, or BMI.
TABLE 1.
Demographic, descriptive, plasma, and calculated PRS and correlation data. Medians with IQR.
| All | AD at 90 months | Controls | Wilcoxon (p) | |
|---|---|---|---|---|
| N | 144 | 19 | 125 | – |
| Females/males | 55/89 | 7/12 | 48/77 | 0.90 |
| Age at start | 75.6 (0.7) | 75.5 (0.7) | 75.7 (0.7) | 0.21 |
| Age at end | 83.2 (0.7) | 83.0 (0.6) | 83.2 (0.6) | 0.13 |
| Years of education | 11 (1) | 11 (1) | 11 (1) | 0.92 |
| MMSE at start | 28 (2) | 27 (1.5) | 28 (2) | * 0.015 |
| MMSE at end | 28 (2) | 25 (3.5) | 28 (2) | *** < 0.001 |
| BMI at baseline | 26.9 (3.6) | 26.49 (2.1) | 27.0 (4.1) | 0.24 |
| APOE ε4 carriers (ε4/ε4, ε4/ε3, ε4/ε2) | 38/144 | 10/19 | 28/125 | *** < 0.001 |
| Plasma NfL baseline (pg/mL) | 14.8 (8.2) | 17.9 (7.4) | 14.7 (7.8) | 0.19 |
| Plasma NfL 30 months (pg/mL) | 19.8 (11.1) | 23.6 (10.0) | 19.4 (10.7) | 0.21 |
| Plasma NfL 60 months (pg/mL) | 22.3 (10.9) | 21.0 (9.9) | 22.4 (11.0) | 0.49 |
| Plasma NfL 90 months (pg/mL) | 24.7 (12.6) | 27.7 (11.2) | 24.3 (12.8) | 0.35 |
| Delta NfL (pg/mL) 0 to 90 months | 9.3 (10.1) | 9.2 (9.6) | 9.3 (10.2) | 0.91 |
| PRS‐CS AD | −0.06 (1.5) | 0.35 (1.8) | −0.08 (1.4) | 0.33 |
| Correlation (r) PRS‐CS/baseline NfL (z‐score) | 0.05 | 0.75 | ‐0.06 | ***<0.001 a |
Abbreviations: AD, Alzheimer's disease; APOE, apolipoprotein E; BMI, body mass index; IQR, interquartile range; MMSE, Mini‐Mental State Examination; NfL, neurofilament light chain; PRS, polygenic risk score; PRS‐CS, polygenic risk score continuous shrinkage.
Fisher's r‐to‐z transformation.
p < 0.05.
p < 0.001.
3.2. At 90 months, plasma NfL was significantly increased in the entire study population, compared to baseline
Median NfL at baseline (0 months) was 17.9 pg/mL (IQR 7.4) in the AD at 90 months and 14.7 pg/mL (IQR 7.8) in the control group. At 30 months mean NfL values were 23.6 (IQR 10.0) and 19.4 (IQR 10.7) and at 60 months 21.0 (IQR 9.6) and 22.4 (11.0), respectively. In the AD at 90 months group plasma NfL was 27.7 (IQR 11.2) at 90 months and 24.3 (IQR 12.8) in the control group. In both groups there was a significant increase of NfL over time (0 vs. 90 months: AD: 17.9 pg/mL [IQR 7.4] vs. 27.7 [IQR 11.2]; controls: 14.7 pg/mL [IQR 7.8] vs. 24.3 [IQR 12.8], AD: p = 1.2 × 10−4, controls: p = 6.0 × 10−18). Yet, NfL levels between the AD at 90 months and the control group were not significantly different at any time point (Figure 1). Neither was the mean increase of NfL from baseline to 90 months significantly different between the AD at 90 months and the control group (AD: 9.2 [IQR 9.6], controls: 9.3 [IQR 10.2], p = 0.91).
FIGURE 1.
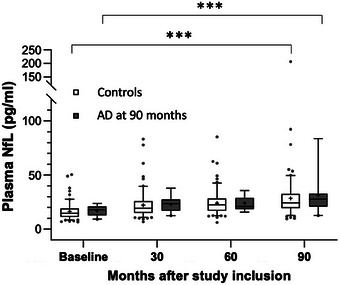
Plasma NfL at all time points in the AD group at 90 months and the control group. Box whisker plot of NfL (pg/mL) in plasma at 0, 30, 60, and 90 months in the AD at 90 months and the control group. Whiskers indicate 5% to 95% percentile, data points represent outliers. Differences between groups were not significant at any time point; in both groups NfL levels at 90 months were significantly different from baseline; *** p < 0.001. AD, Alzheimer's disease; NfL, neurofilament light chain.
p values of differences between groups at each time point as well as NfL increase from baseline to 90 months were comparable using the NfL z‐scores (Table S1 in supporting information). Likewise, the increase of NfL within each group from baseline to 90 months was also highly significant using NfL z‐scores (AD: p = 4.8 × 10−3, controls: p = 3.6 × 10−11).
Baseline NfL did not significantly predict diagnostic group (AD/controls) neither with raw values (pg/mL) nor z‐scores in a logistic regression model with BMI as a covariate.
A linear mixed effects model confirmed a significant increase of NfL levels as a function of time in all participants (p = 0.012), which was independent of AD diagnosis at 90 months in the observation period (p = 0.51, Figure 2). Adding sex as a covariate to the model had no relevant influence on the p values (p = 0.013 and p = 0.52, respectively).
FIGURE 2.
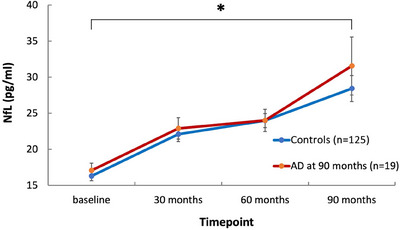
Plasma NfL evolution over time in the AD group at 90 months and the control group. Linear plot of NfL (pg/mL; means and SEM) per time point in the AD group at 90 months and the control group. NfL levels increased significantly as a function of time (p = 0.012), independent of AD diagnosis at 90 months (p = 0.51) according to linear effects modeling. AD, Alzheimer's disease; NfL, neurofilament light chain; SEM, standard error of the mean. * p < 0.05.
Results remained significant after correction for multiple testing using the Benjamini–Hochberg procedure.
3.3. Polygenic risk did not differ significantly between participants with and without AD diagnosis at 90 months
In the control group PRS‐CS z‐scores ranged from −2.53 to 2.21 with a median of −0.08 (IQR 1.3). In the AD at 90 months group PRS z‐scores ranged from −1.21 to 2.43 with a median of 0.35 (IQR 1.8). A Wilcoxon rank sum test showed no significant difference of PRS between the two groups. Also, PRS‐CS could not significantly predict diagnostic group (AD/controls). The variance (i.e., Nagelkerke R 2) explained was 2.2%, but not significant.
Clumping and thresholding (C+T) PRS results are presented in the supporting information.
3.4. Baseline plasma‐NfL correlated with polygenic risk in participants with AD at 90 months
Spearman's correlation coefficient showed a distinct correlation of NfL at study inclusion with PRS‐CS between participants that developed AD (Figure 3B) and the control group (Figure 3C; AD: r = 0.71, p = 8.5 × 10−4, control: r = −0.06, p = 0.48; all participants: Figure 3A). Comparison of the correlation coefficient's slopes with Fisher's r‐to‐z transformation revealed a significant difference between the slopes (z = 3.61, p = 3.1 × 10−4). Effects were more pronounced when using the NfL z‐scores values (AD: r = 0.75, p = 2.2 × 10−4, control: r = −0.06, p = 0.49, z = 3.89 p = 9.8 × 10−5).
FIGURE 3.
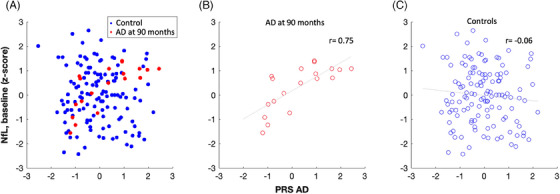
Correlation of NfL at baseline with PRS‐CS in the AD at 90 months and the control group. The association of NfL at baseline (z‐scores) and PRS‐CS (z‐scores) is depicted for all participants (A), participants with AD diagnosis at 90 months (B), and the control group (C). There is a significant difference in Spearman's correlation coefficient (r) between the AD at 90 months and the control group (AD r = 0.75, controls r = −0.06, p < 0.001). Correlations are visualized by least‐squares regression line. AD, Alzheimer's disease; NfL, neurofilament light chain; PRS‐CS, polygenic risk score continuous shrinkage.
In a logistic regression model PRS and baseline NfL together showed a significant effect (p = 0.006) in predicting AD diagnosis (AD/control group) with baseline BMI as an additional covariate. The significance of the combined predictive effect of PRS‐CS and baseline NfL was again slightly more pronounced when using NfL z‐scores (p = 0.0035). BMI and sex as covariates had no significant effect in this model (p = 0.23 and p = 0.69). Results using C+T PRS are presented in the supporting information.
All results remained significant after correction for multiple testing by controlling false discovery rate with the Benjamini–Hochberg procedure.
4. DISCUSSION
The search for indicators for the appraisal of individual AD risk susceptibility is a focus topic in the field as a more precise stratification of non‐demented participants for early therapeutic intervention trials is crucial with regard to the outcome.
In this prospective longitudinal study in a non‐demented old‐aged study population 19 out of 144 participants developed AD over the study duration of 7.5 years. This is in accordance with the literature on incidence and prevalence of sporadic AD in old‐aged individuals. 34 , 35 Here, we found an association of baseline plasma NfL as a sign of neuroaxonal damage with genetic risk for AD as indicated by PRS on the development of AD. In this cohort, this association was observable 60 months prior to median clinical disease onset. In the present study, neither PRS nor NfL alone at study inclusion as single biomarkers distinguished significantly between participants that did or did not develop AD during the 90 month study course. The observed correlation of baseline NfL with PRS in the group that developed AD over 90 months indicates that this group might already have been at a preclinical stage at baseline. This is supported by the significant difference in MMSE levels at baseline on a group level, while all participants were cognitively healthy at study entry. Our data indicate that the combination of PRS, particularly PRS‐CS, with plasma NfL could improve the assessment of individual AD risk susceptibility in non‐demented old‐aged persons several years prior to clinical disease manifestation.
Although power of our study with an n of 19 is limited, our findings are supported by a cross‐sectional study by Skoog et al., who showed an association of a non‐APOE AD‐PRS with NfL in CSF in a sample of 246 cognitively unimpaired 70‐year‐old individuals. 36 Two more recent studies also support an association of an APOE AD‐PRS with NfL in plasma in a mixed sample of old‐aged persons with cognitively normal status or AD 37 or with NfL in CSF in a mixed sample of cognitively unimpaired, MCI, and AD. 38 Also, a newly described deep‐learning–based PRS was associated with plasma NfL in old aged healthy control persons in a cross‐sectional analysis of the Chinese WGS 2 cohort. According to the authors these participants had “not yet developed AD” implying that these participants might be believed to be at a preclinical stage although no further information about this group is available. 39 Moreover, validity of our data is supported by the fact that the VITA sample itself is a very representative sample of 75‐year‐old “community‐dwelling” persons in central Europe. 14 The group of individuals that developed AD during the 90 month study course had a higher rate of APOE ε4 carriers. This is consistent with the literature and further supports validity of our data. 40 In our sample, the rate of APOE ε4 carriers in the control and the AD group was higher than in the literature, which might be attributable to a potential preselection bias due to possible motivators for study participation such as positive family history or subjective cognitive impairment.
Several studies have supported validity of peripheral NfL measures for inferring neuronal damage in the nervous system. 2 , 41 , 42 Associations between elevated peripheral NfL and pathology of many neuro‐cognitive diseases have been reported. 43 A recent study reported a negative correlation of plasma NfL measured several years prior to death with post mortem NfL staining in brain tissue of AD patients, indicating a predictive value of peripheral NfL for neuronal loss. 44 However, as NfL and neuronal loss are not specific for a certain etiology, peripheral NfL values might not have sufficient predictive value for the development of AD in non‐demented elderly persons as a single biomarker. Also, considering the well‐established general increase of plasma NfL levels with age, a distinct attribution to a specific disease‐pathology—particularly those diseases that are linked to increased age—appears challenging, unless reliable reference data adjusting for age are available. 32 Therefore, validity of the results in the present study was confirmed with age‐ and BMI‐adjusted NfL z‐scores.
Consistent with earlier reports, in our study on “community‐dwelling” persons of the age of 75 years, we were able to reproduce a significant increase of plasma NfL levels over the 90 month study duration, supporting validity of our data. Interestingly, this increase in plasma NfL levels was observable both in persons that developed AD, and those that did not develop AD. While to our knowledge this is the first study to find this uniform increase of plasma NfL in a prospective longitudinal cohort of old‐aged persons, some participants might have developed AD after termination of this study, and as such may have escaped diagnosis due to the limitation of the present study on a duration of 90 months.
Our findings might thus be consistent with a notion of increased NfL levels being linked to increased age, which might result from general alterations of the central nervous system during aging that remain below a threshold at which resulting clinical symptoms could be expected. 45 , 46 , 47 Earlier work of ours supports the idea that genetic risk for sporadic AD, as conferred by APOE ε4, might be reflected by cerebrovascular dysfunction 48 and possibly also changed iron metabolism, 49 which might be related to neuroaxonal damage.
The cumulative weight of genetic variations distributed over the entire human genome, as integrated in individual PRS, may be one of the most conclusive results of GWAS. 50 , 51 Recent studies have demonstrated the validity of PRS to assess an individual's genetic risk for age‐related sporadic AD. 11 , 19 , 52 , 53 Yet, PRS in their current application are not recommended to be applied as single markers to assess AD risk. In our study, the lack of significant difference between PRS in the AD developing and control group and the lack of association of AD diagnosis with NfL levels in the linear mixed regression model might also have been impacted by the limited power (n = 19 AD cases). While a C+T PRS with a single p‐value of 1 only showed a trend (p = 0.054) in predicting AD diagnosis combined with baseline NfL, we found a highly significant predictive effect for AD diagnosis for PRS‐CS combined with baseline NfL. The more recently described PRS‐CS has previously been shown to outperform other PRS formulations. 54 , 55 The use of continuous shrinkage priors and the blockwise adjustment of LD into the posterior inference might contribute to its better predictive property compared to the other methods. 28 Also, the sample size might have impacted the predictive value of the C+T PRS in the present study.
A particular advantage might result from the combination of PRS with NfL measures, as future advancements in genetic profiling might lead the way to potential molecular subclassification of study participants based on genetic information. 12 Considering that NfL might serve as a marker of therapeutic response, 10 the combination with PRS might allow for precision medicine interventions in neuro‐cognitive diseases such as AD.
As outlined above, a clear limitation of the current study is the limited number of individuals that developed AD during the 90 month observation period (n = 19), while a majority of the study sample did not develop AD during the study course. Although in line with findings by Skoog et al., 36 our findings therefore need to be interpreted with caution, and relevance for developing AD needs to be confirmed in future and larger longitudinal cohorts. Another limitation is the fact that the sample was only stratified by development of AD and not by other neurodegenerative diseases. Thus, it cannot be ruled out that the observed increase of NfL in all participants (both with and without AD at 90 months), might also have been impacted by undetected brain pathology due to diseases other than AD. Yet, excluding participants with concurrent vascular pathology and high baseline NfL z‐scores did not change significance levels of our results, which might support the notion that plasma NfL is rather associated with neurodegenerative disorders than vascular pathology. Moreover, we might have missed individuals that developed AD after the 90 month study duration.
In this study, for the calculation of NfL z‐scores only BMI and age as important influences were considered, as creatinine levels, which also might impact NfL levels, were unavailable. Yet, both BMI and creatinine have been reported to influence NfL serum levels but not to have clinically relevant confounding effects in predicting CSF NfL levels or clinical conversion to dementia. 56
Also, the median clinical manifestation of 60 months is only an approximation as the presence of the clinical disease was only assessed at the follow‐up visits at 30, 60, and 90 months. The clinical onset in real life might have between these visits.
Taken together, our study supports a combined assessment of plasma NfL and PRS for increased precision to identify cognitively healthy elderly persons at risk for sporadic AD. Additional studies are needed to assess benefit for future disease‐modifying therapeutic interventions at very early stages of AD. Such early therapeutic interventions could particularly benefit affected individuals without significant cognitive impairment, as studied in the cohort described here.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All human subjects provided informed consent.
Supporting information
Supporting Information
{kind=link}
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
We wish to thank all participants and their families in joining the VITA study. We thank Synapsis Alzheimer Research Switzerland for the support of this project. We also thank the Ludwig Boltzmann Institute of Aging Research Vienna, Austria for the support of the VITA study. Moreover, we wish to acknowledge the GWAS data availability by the EADB study (https://www.neurodegenerationresearch.eu/survey/eadb‐a‐european‐dna‐bank‐for‐deciphering‐the‐missing‐heritability‐of‐alzheimer%C2%92s‐disease/), supported by the JPND research. This work was supported by Swiss National Research Foundation (SNF) Grants #190510 and #192480.
Kagerer SM, Awasthi S, Ripke S, et al. Polygenic risk for Alzheimer's disease is associated with neuroaxonal damage before onset of clinical symptoms. Alzheimer's Dement. 2024;16:e12504. 10.1002/dad2.12504
REFERENCES
- 1. Moscoso A, Grothe MJ, Ashton NJ, et al. Longitudinal associations of blood phosphorylated Tau181 and neurofilament light chain with neurodegeneration in Alzheimer disease. JAMA Neurol. 2021;78(4):396‐406. doi: 10.1001/jamaneurol.2020.4986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. 2018;14(10):577‐589. doi: 10.1038/s41582-018-0058-z [DOI] [PubMed] [Google Scholar]
- 3. Kuhle J, Barro C, Andreasson U, et al. Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: eLISA, electrochemiluminescence immunoassay and Simoa. Clin Chem Lab Med. 2016;54(10):1655‐1661. doi: 10.1515/cclm-2015-1195 [DOI] [PubMed] [Google Scholar]
- 4. Hendricks R, Baker D, Brumm J, et al. Establishment of neurofilament light chain Simoa assay in cerebrospinal fluid and blood. Bioanalysis. 2019;11(15):1405‐1418. doi: 10.4155/bio-2019-0163 [DOI] [PubMed] [Google Scholar]
- 5. Forgrave LM, Ma M, Best JR, DeMarco ML. The diagnostic performance of neurofilament light chain in CSF and blood for Alzheimer's disease, frontotemporal dementia, and amyotrophic lateral sclerosis: a systematic review and meta‐analysis. Alzheimers Dement. 2019;11:730‐743. doi: 10.1016/j.dadm.2019.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Preische O, Schultz SA, Apel A, et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer's disease. Nat Med. 2019;25(2):277‐283. doi: 10.1038/s41591-018-0304-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mattsson N, Andreasson U, Zetterberg H, Blennow K. Alzheimer's disease neuroimaging i. association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2017;74(5):557‐566. doi: 10.1001/jamaneurol.2016.6117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gerards M, Schild AK, Meiberth D, et al. Alzheimer's disease plasma biomarkers distinguish clinical diagnostic groups in memory clinic patients. Dement Geriatr Cogn Disord. 2022:1‐11. doi: 10.1159/000524390 [DOI] [PubMed] [Google Scholar]
- 9. Cianflone A, Coppola L, Mirabelli P, Salvatore M. Predictive accuracy of blood‐derived biomarkers for amyloid‐beta brain deposition along with the Alzheimer's disease continuum: a systematic review. J Alzheimers Dis. 2021;84(1):393‐407. doi: 10.3233/JAD-210496 [DOI] [PubMed] [Google Scholar]
- 10. Mielke MM, Syrjanen JA, Blennow K, et al. Plasma and CSF neurofilament light: relation to longitudinal neuroimaging and cognitive measures. Neurology. 2019;93(3):e252‐e260. doi: 10.1212/WNL.0000000000007767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baker E, Escott‐Price V. Polygenic risk scores in Alzheimer's disease: current applications and future directions. Front Digit Health. 2020;2:14. doi: 10.3389/fdgth.2020.00014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med. 2016;18(5):421‐430. doi: 10.1038/gim.2015.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ibanez LF, FHG, Dube U, Mihindukulasuriya KA, Harari O>. Polygenic risk scores in neurodegenerative Diseases: a review. Curr Genet Med Rep. 2019;7:22‐29. doi: 10.1007/s40142-019-0158-0 [DOI] [Google Scholar]
- 14. Fischer P, Jungwirth S, Krampla W, et al. Vienna Transdanube Aging “VITA”: study design, recruitment strategies and level of participation. J Neural Transm Suppl. 2002(62):105‐116. doi: 10.1007/978-3-7091-6139-5_11 [DOI] [PubMed] [Google Scholar]
- 15. Grunblatt E, Zehetmayer S, Bartl J, et al. Genetic risk factors and markers for Alzheimer's disease and/or depression in the VITA study. J Psychiatr Res. 2009;43(3):298‐308. doi: 10.1016/j.jpsychires.2008.05.008 [DOI] [PubMed] [Google Scholar]
- 16. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA work group under the auspices of department of health and human services task force on Alzheimer's disease. Neurology. 1984;34(7):939‐944. doi: 10.1212/wnl.34.7.939 [DOI] [PubMed] [Google Scholar]
- 17. Scholz CJ, Weber H, Jungwirth S, et al. Explorative results from multistep screening for potential genetic risk loci of Alzheimer's disease in the longitudinal VITA study cohort. J Neural Transm. 2018;125(1):77‐87. doi: 10.1007/s00702-017-1796-6 [DOI] [PubMed] [Google Scholar]
- 18. Harp C, Thanei GA, Jia X, et al. Development of an age‐adjusted model for blood neurofilament light chain. Ann Clin Transl Neurol. 2022. doi: 10.1002/acn3.51524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. de Rojas I, Moreno‐Grau S, Tesi N, et al. Common variants in Alzheimer's disease and risk stratification by polygenic risk scores. Nature communications. 2021;12(1):3417. doi: 10.1038/s41467-021-22491-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alzheimer Europe . A European DNA bank for deciphering the missing heritability of Alzheimer's disease. https://www.alzheimer‐europe.org/research/projects/european‐dna‐bank‐deciphering‐missing‐heritability‐alzheimers‐disease
- 21. Lam M, Awasthi S, Watson HJ, et al. RICOPILI: rapid imputation for consortias pipeline. Bioinformatics. 2020;36(3):930‐933. doi: 10.1093/bioinformatics/btz633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Loh PR, Palamara PF, Price AL. Fast and accurate long‐range phasing in a UK Biobank cohort. Nature genetics. 2016;48(7):811‐816. doi: 10.1038/ng.3571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Das S, Forer L, Schonherr S, et al. Next‐generation genotype imputation service and methods. Nature genetics. 2016;48(10):1284‐1287. doi: 10.1038/ng.3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome‐wide association studies through pre‐phasing. Nature genetics. 2012;44(8):955‐959. doi: 10.1038/ng.2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. European genome‐phenome archive. Dataset: Haplotype Reference Consortium Release 1.1. https://ega‐archive.org/datasets/EGAD00001002729
- 26. Jansen IE, Savage JE, Watanabe K, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nature genetics. 2019;51(3):404‐413. doi: 10.1038/s41588-018-0311-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Complex Trait Genetics lab. GWAS Summary Statistics. https://ctg.cncr.nl/software/summary_statistics
- 28. Ge T, Chen CY, Ni Y, Feng YA, Smoller JW. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nature communications. 2019;10(1):1776. doi: 10.1038/s41467-019-09718-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Genomes Project C , Auton A, Brooks LD, et al, Genomes Project C . A global reference for human genetic variation. Nature. 2015;526(7571):68‐74. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fisher RA. Frequency distribution of the values of the correlation coefficient in samples from an indefinitely large population. Biometrika. 1915;10(4):522‐529. [Google Scholar]
- 31. Edwards AL. An Introduction to Linear Regression and Correlation. W. H. Freeman & Co.; 1984. [Google Scholar]
- 32. Benkert P, Meier S, Schaedelin S, et al. Serum neurofilament light chain for individual prognostication of disease activity in people with multiple sclerosis: a retrospective modelling and validation study. Lancet Neurol. 2022;21(3):246‐257. doi: 10.1016/S1474-4422(22)00009-6 [DOI] [PubMed] [Google Scholar]
- 33. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B. 1995;57(1):289‐300. [Google Scholar]
- 34. Evans DA, Funkenstein HH, Albert MS, et al. Prevalence of Alzheimer's disease in a community population of older persons. Higher than previously reported. JAMA. 1989;262(18):2551‐2556. [PubMed] [Google Scholar]
- 35. Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(8). doi: 10.1101/cshperspect.a006239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Skoog I, Kern S, Najar J, et al. A Non‐APOE polygenic risk score for alzheimer's disease is associated with cerebrospinal fluid neurofilament light in a representative sample of cognitively unimpaired 70‐year olds. J Gerontol A Biol Sci Med Sci. 2021;76(6):983‐990. doi: 10.1093/gerona/glab030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiao B, Xiao X, Yuan Z, et al. Associations of risk genes with onset age and plasma biomarkers of Alzheimer's disease: a large case‐control study in mainland China. Neuropsychopharmacology. 2022;47(5):1121‐1127. doi: 10.1038/s41386-021-01258-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kumar A, Janelidze S, Stomrud E, et al. beta‐Amyloid‐Dependent and ‐Independent genetic pathways regulating CSF tau biomarkers in Alzheimer disease. Neurology. 2022;99(5):e476‐e487. doi: 10.1212/WNL.0000000000200605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhou X, Chen Y, Ip FCF, et al. Deep learning‐based polygenic risk analysis for Alzheimer's disease prediction. Commun Med. 2023;3(1):49. doi: 10.1038/s43856-023-00269-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta‐analysis. APOE and Alzheimer disease meta analysis consortium. JAMA. 1997;278(16):1349‐1356. 22‐29. [PubMed] [Google Scholar]
- 41. Bergman J, Dring A, Zetterberg H, et al. Neurofilament light in CSF and serum is a sensitive marker for axonal white matter injury in MS. Neurol Neuroimmunol Neuroinflamm. 2016;3(5):e271. doi: 10.1212/NXI.0000000000000271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hoffman PN, Cleveland DW, Griffin JW, Landes PW, Cowan NJ, Price DL. Neurofilament gene expression: a major determinant of axonal caliber. Proc Natl Acad Sci U S A. 1987;84(10):3472‐3476. doi: 10.1073/pnas.84.10.3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alirezaei Z, Pourhanifeh MH, Borran S, Nejati M, Mirzaei H, Hamblin MR. Neurofilament Light chain as a biomarker, and correlation with magnetic resonance imaging in diagnosis of cns‐related disorders. Mol Neurobiol. 2020;57(1):469‐491. doi: 10.1007/s12035-019-01698-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ashton NJ, Leuzy A, Lim YM, et al. Increased plasma neurofilament light chain concentration correlates with severity of post‐mortem neurofibrillary tangle pathology and neurodegeneration. Acta Neuropathol Commun. 2019;7(1):5. doi: 10.1186/s40478-018-0649-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Long X, Liao W, Jiang C, Liang D, Qiu B, Zhang L. Healthy aging: an automatic analysis of global and regional morphological alterations of human brain. Acad Radiol. 2012;19(7):785‐793. doi: 10.1016/j.acra.2012.03.006 [DOI] [PubMed] [Google Scholar]
- 46. Kagerer SM, Schroeder C, van Bergen JMG, et al. Low Subicular Volume as an indicator of dementia‐risk susceptibility in old age. Frontiers in aging neuroscience. 2022;14:811146. doi: 10.3389/fnagi.2022.811146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kagerer SM, van Bergen JMG, Li X, et al. APOE4 moderates effects of cortical iron on synchronized default mode network activity in cognitively healthy old‐aged adults. Alzheimers Dement. 2020;12(1):e12002. doi: 10.1002/dad2.12002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hua J, Lee S, Blair NIS, et al. Increased cerebral blood volume in small arterial vessels is a correlate of amyloid‐beta‐related cognitive decline. Neurobiology of aging. 2019;76:181‐193. doi: 10.1016/j.neurobiolaging.2019.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. van Bergen JM, Li X, Hua J, et al. Colocalization of cerebral iron with amyloid beta in mild cognitive impairment. Sci Rep. 2016;6:35514. doi: 10.1038/srep35514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Khera AV, Chaffin M, Aragam KG, et al. Genome‐wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nature genetics. 2018;50(9):1219‐1224. doi: 10.1038/s41588-018-0183-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lewis CM, Vassos E. Polygenic risk scores: from research tools to clinical instruments. Genome Med. 2020;12(1):44. doi: 10.1186/s13073-020-00742-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tan CH, Hyman BT, Tan JJX, et al. Polygenic hazard scores in preclinical Alzheimer disease. Ann Neurol. 2017;82(3):484‐488. doi: 10.1002/ana.25029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Desikan RS, Fan CC, Wang Y, et al. Genetic assessment of age‐associated Alzheimer disease risk: development and validation of a polygenic hazard score. PLoS Med. 2017;14(3):e1002258. doi: 10.1371/journal.pmed.1002258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ni G, Zeng J, Revez JA, et al. A comparison of ten polygenic score methods for psychiatric disorders applied across multiple cohorts. Biological psychiatry. 2021;90(9):611‐620. doi: 10.1016/j.biopsych.2021.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pain O, Glanville KP, Hagenaars SP, et al. Evaluation of polygenic prediction methodology within a reference‐standardized framework. PLoS Genet. 2021;17(5):e1009021. doi: 10.1371/journal.pgen.1009021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pichet Binette A, Janelidze S, Cullen N, et al. Confounding factors of Alzheimer's disease plasma biomarkers and their impact on clinical performance. Alzheimers Dement. 2023;19(4):1403‐1414. doi: 10.1002/alz.12787 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information