

Abstract
Objectives: Jumonji C domain-containing (JMJD) 2B (JMJD2B) is a transcriptional cofactor and histone demethylase that is involved in prostate cancer formation. However, how its function is regulated by posttranslational modification has remained elusive. Hence, we examined if JMJD2B would be regulated by lysine methylation. Methods: Through in vitro methylation assays and Western blotting with methyl-lysine specific antibodies, we analyzed lysine methylation within JMJD2B. Identified methylated lysine residues were mutated to arginine residues and the respective impact on JMJD2B transcriptional activity measured with a reporter gene assay in human LNCaP prostate cancer cells. Results: We discovered that JMJD2B is methylated on up to six different lysine residues. Further, we identified the suppressor of variegation 3-9/enhancer of zeste/trithorax (SET) domain-containing protein 7/9 (SET7/9) as the methyltransferase being responsible for this posttranslational modification. Mutating the methylation sites in JMJD2B to arginine residues led to diminished coactivation of the Ju-nana (JUN) transcription factor, which is a known oncogenic protein in prostate tumors. In contrast, methylation of JMJD2B had no impact on its ability to coactivate another transcription factor associated with prostate cancer, the DNA-binding protein E26 transformation-specific (ETS) variant 1 (ETV1). Consistent with a potential joint action of JMJD2B, SET7/9 and JUN in prostate cancer, the expression of JMJD2B in human prostate tumors was positively correlated with both SET7/9 and JUN levels. Conclusions: The identified SET7/9-mediated methylation of JMJD2B appears to impact its cooperation with selected interacting transcription factors in prostate cancer cells. Given the implicated roles of JMJD2B beyond prostate tumorigenesis, SET7/9-mediated methylation of JMJD2B possibly also influences the development of other cancers, while its impairment might have relevance for obesity or a global developmental delay that can be elicited by reduced JMJD2B activity.
Keywords: Gene transcription, lysine demethylase 4B (KDM4B), posttranslational modification, prostate cancer, SET domain-containing 7 (SETD7)
Introduction
Jumonji C domain-containing (JMJD) 2B (JMJD2B), also known as lysine demethylase 4B (KDM4B), is an enzyme that targets lysine 9 and lysine 36 on histone H3 as well as lysine 26 on histone H1.4. It can demethylate H3K9me2, H3K36me3, H3K36me2 and H1.4K26me3, but most efficiently removes one methyl group from H3K9me3 [1-4]. Given that H3K9me3 is an eminent epigenetic mark for transcriptionally repressed genes [5], JMJD2B’s enzymatic activity is predicted to activate gene promoters.
JMJD2B performs pleiotropic functions and is a potential oncoprotein in various cancers [6-10]. This particularly pertains to prostate cancer, where JMJD2B is overexpressed and its downregulation compromised cell proliferation and sensitized to anti-androgen therapy. Mechanistically, this included the binding of JMJD2B to the androgen receptor, the myelocytomatosis (MYC) oncoprotein or the E26 transformation-specific (ETS) variant 1 (ETV1), thereby stimulating the ability of these DNA-binding proteins to upregulate gene transcription [11-19].
The transcription factor ETV1 can become overexpressed in prostate cancer especially when chromosomal translocations fuse its gene to androgen-inducible regulatory sequences [20-24], yet ETV1 overexpression may also be brought about by fusion-independent mechanisms [25]. Upon ETV1 overexpression in mouse prostates, development of prostatic intraepithelial neoplasia ensued [26-29], indicating that ETV1 overexpression is causally related to prostate cancer initiation. Moreover, ETV1 and JMJD2A, a close homolog of JMJD2B, synergized in promoting prostate tumorigenesis in a genetically engineered mouse model, and ETV1 cooperated with either JMJD2A or JMJD2B in stimulating gene transcription, including of the Yes-associated 1 (YAP1) proto-oncogene [14].
In this report, we explored how JMJD2B might be regulated in its activity through posttranslational modification. We discovered that JMJD2B can be methylated by the suppressor of variegation 3-9/enhancer of zeste/trithorax (SET) domain-containing protein 7/9 (SET7/9; also called SET domain-containing 7 (SETD7)), an enzyme that is reportedly overexpressed in human prostate tumors and capable of stimulating prostate cancer cell proliferation [30]. Our examinations suggest that methylation of JMJD2B by SET7/9 can affect its coactivation potential, providing a conceivable mechanism by which this posttranslational modification may promote prostate tumorigenesis.
Materials and methods
Cloning of JMJD2B expression vectors
Human JMJD2B cDNA was obtained from Origene (Rockville, MD, USA) and corresponded to the transcript variant X1 (GenBank accession number XM_005259521.4; and the cDNA deposited under GenBank BC144292.1), whose translation product contains 34 additional amino acids (position 373-406) compared to the translation product of the NCBI JMJD2B reference sequence NM_015015.3. To express N-terminally Flag-tagged JMJD2B in human cells, its open reading frame was cloned into pEV3S-Flag [31]. To express portions of JMJD2B as fusions with GST in bacteria, fragments of the JMJD2B open reading frame were cloned into a derivative of pGEX-2T [32]. All recombinant plasmids were validated by Sanger DNA sequencing [33]. This was done at the Oklahoma Medical Research Foundation DNA Sequencing Core (Oklahoma City, OK, USA) with sequencing primers specific to pEV3S-Flag (5’-GGGGGATCTTGGTGGCGTG-3’) or pGEX-2T (5’-CCAAAATCGGATCTGG-3’).
Immunoprecipitation and Western blotting
Human embryonic kidney 293T cells (CRL-3216; American Type Culture Collection, Manassas, VA, USA) were transiently transfected by the calcium phosphate coprecipitation method [34]. Cells were lysed 36 h after transfection as described [35] and immunoprecipitations performed with indicated antibodies [36]. Then, proteins were subjected to SDS polyacrylamide gel electrophoresis [37] and separated proteins blotted onto polyvinylidene difluoride membrane [38]. After incubation with primary and horseradish peroxidase-coupled secondary antibodies [39,40], detection of proteins was done with enhanced chemiluminescence and exposure to film [41]. The following antibodies were used at a 1:1000 dilution: rabbit polyclonal anti-p53-K372me (ab16033; Abcam, Cambridge, United Kingdom), rabbit polyclonal anti-JMJD2B (TA306832; Origene, Rockville, MD, USA), rabbit polyclonal anti-SET7/9 (07-314; Upstate, Boston, MA, USA), mouse monoclonal anti-Flag (F3165; Sigma, St. Louis, MO, USA), and rabbit polyclonal anti-Actin (A2066; Sigma, St. Louis, MO, USA).
Production of glutathione S-transferase (GST) fusion proteins
GST-JMJD2B and GST-SET7/9 expression plasmids were transformed into Escherichia coli BL21-CodonPlus bacteria (Stratagene, San Diego, CA, USA) [42]. These bacteria were grown in Luria broth media at 37°C to an optical density of 0.2-0.6 (measured at 595 nm) and then induced with 0.05-0.1 mM isopropyl β-D-thiogalactopyranoside and further grown for 8 h at 18°C. Purification of GST fusion proteins was then done utilizing glutathione agarose as described before [43,44]. These affinity purified proteins were frozen in liquid nitrogen and stored until usage at -80°C.
Methylation assays
In a buffer consisting of 50 mM Tris-HCl pH 8.5, 5 mM MgCl2, 4 mM dithiothreitol, 1 µM S-[methyl-3H] adenosyl-L-methionine (3H-SAM), approximately 2 µg of the GST-JMJD2B fusion proteins were incubated with GST-SET7/9 for 2 h at 30°C. Then, an equal volume of 2× Laemmli sample buffer was added and the samples incubated for 10 min at 95°C [45]. Denatured proteins were then separated via SDS polyacrylamide gel electrophoresis [46], which was followed by blotting onto polyvinylidene difluoride membrane and Ponceau S protein staining [47]. The dried polyvinylidene difluoride membranes were treated with EN3HANCE (Perkin Elmer, Waltham, MA, USA) four times and then exposed to film without intensifying screen at -80°C [48].
Luciferase assays
Human LNCaP prostate cancer cells (CRL-1740; American Type Culture Collection, Manassas, VA, USA) were seeded in poly-L-lysine coated 12-wells and grown in a humidified atmosphere with 5% CO2 to a confluency of approximately 25% [49,50]. The growth media consisted of Dulbecco’s modified Eagle medium (MT10013CV; Corning, Somerville, MA, USA) supplemented with 10% fetal bovine serum [51]. Cells were transfected employing 4 µg polyethylenimine [52] and the following amounts of DNA: 500 ng TORU luciferase reporter gene construct, 500 ng pBluescript KS+, indicated amounts of empty vector pEV3S or ETV1 or JUN expression vector, and indicated amounts of pEV3S or Flag-tagged JMJD2B expression vectors [53]. After 8-10 h, cells were washed once with 1 ml phosphate-buffered saline [54] and incubated in growth media for another ~40 h [55]. Then, cells were washed once again with 1 ml phosphate-buffered saline and lysed in 250 µl of 25 mM Tris-HCl pH 7.8, 10% glycerol, 2 mM EDTA, 1% Triton X-100, 2 mM dithiothreitol [56]. After removal of debris [57], luciferase activities were determined with the help of a luminometer (LB 9507; Berthold, Bad Wildbad, Germany) as described [58,59].
Statistical analysis
Prism 6 for Mac OS X (GraphPad Software, Boston, MA, USA) was used to determine statistical significance of experimental data. Averages (n=4) with standard deviations are displayed in case of luciferase activities, and the statistical significance was assessed through one-way analysis of variance (ANOVA) with post hoc Dunnett’s multiple comparison test. No data were excluded as outliers from the analysis. For the analysis of coexpression, the Spearman correlation coefficient was calculated. Statistical significance was assumed if P<0.05.
Results
SET7/9 methylates JMJD2B
In our quest to identify potential sites of posttranslational modification in JMJD2B, we noted that some of its lysine residues conform to a reported SET7/9 consensus methylation site, (K/R)(S/T/A)K [60]. One of the known targets of SET7/9 is the tumor suppressor p53, whose methylation on lysine 372 can be detected with respective p53-K372me antibodies [61]. Hence, we speculated that such an antibody may also recognize a methylated lysine residue(s) in JMJD2B. Indeed, when JMJD2B was coexpressed with SET7/9, we observed that a p53-K372me antibody recognized immunoprecipitated JMJD2B (Figure 1A). No methylation was observed in the absence of coexpressed SET7/9 or in the presence of the catalytically inactive H297A mutant of SET7/9 (Figure 1A). Further, we found that endogenous JMJD2B was also methylated in 293T cells (Figure 1B).
Figure 1.
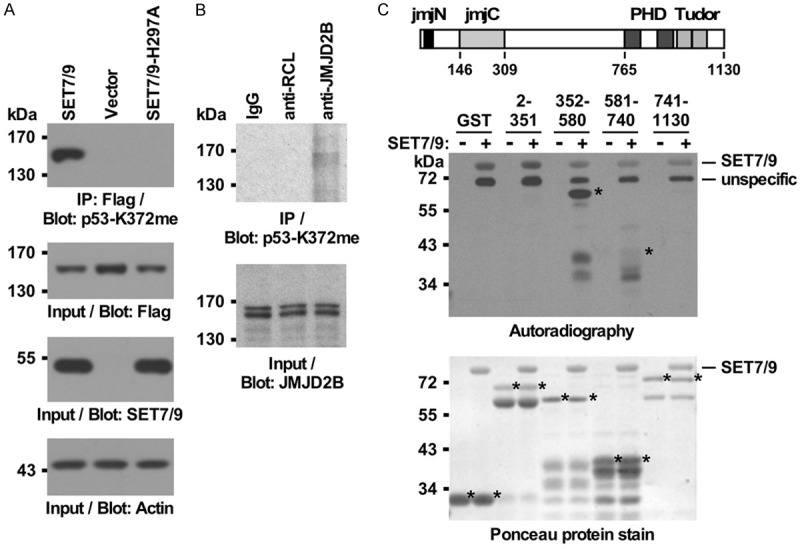
SET7/9 methylates JMJD2B. A. Immunoprecipitation (IP) of Flag-tagged JMJD2B with anti-Flag antibodies from 293T cells upon coexpression with wild-type SET7/9 or its catalytically inactive H297A mutant. Methylation was assessed with p53-K372me antibodies (top), while the bottom three panels show Western blots with the input levels of Flag-JMJD2B, SET7/9 or actin. B. Immunoprecipitation of endogenous JMJD2B from 293T cells; IgG and anti-RCL antibodies served as negative controls. C. In vitro methylation of JMJD2B by GST-SET7/9 utilizing radioactive 3H-SAM. Either GST or fusions of GST to indicated JMJD2B amino acids (asterisks mark the respective full-length proteins, but smaller polypeptides that were likely due to proteolytic degradation are also visible in the Ponceau stains) were used as substrates. The top shows a sketch of JMJD2B with its four known domains: catalytic jmjC, accessory jmjN, double PHD and double Tudor domains.
To identify which region(s) in JMJD2B could be methylated by SET7/9, we divided JMJD2B into four fragments and purified respective GST fusion proteins. These fragments were employed in an in vitro methylation experiment, where radioactively labeled 3H-SAM served as a methyl donor and purified GST-SET7/9 as the methyltransferase. We observed strong methylation of JMJD2B amino acids 352-580, but none with GST alone (Figure 1C). Also, JMJD2B amino acids 2-351 and 741-1130 did not serve as SET7/9 substrates, but there was a marginal methylation observable with JMJD2B amino acids 581-740. From these data, we conclude that substantial SET7/9-mediated methylation only occurs within JMJD2B amino acids 352-580. Notably, these amino acids are not part of any of the known functional domains of JMJD2B (see Figure 1C, top).
Identification of methylation sites within JMJD2B
To identify methylation sites within JMJD2B amino acids 352-580, we subdivided these amino acids into two parts (amino acids 352-430 and 431-580) and observed that both of these parts were substrates for SET7/9 (Figure 2A). Therefore, we constructed even smaller fragments representing JMJD2B amino acids 352-387, 388-430, 431-488 and 486-580 and noted that all of them could be methylated by SET7/9 in vitro (Figure 2B), indicating that there are at least four different methylation sites in JMJD2B.
Figure 2.

Identification of methylation sites in JMJD2B. A. Indicated JMJD2B amino acids fused to GST were employed as substrates in an in vitro methylation assay with purified SET7/9 and the radioactive methyl donor, 3H-SAM. B. Likewise with smaller JMJD2B fragments. C. Wild-type and indicated point mutants of GST-JMJD2B fusions were subjected to in vitro methylation assays. Below are listed all lysine residues (plus the two preceding amino acids) within the respective JMJD2B fragments. Only K368 and K485 match the reported SET7/9 consensus sequence, (K/R)(S/T/A)K.
Within JMJD2B amino acids 352-387, there are four lysine residues (see Figure 2C), but only the one at position 368 matches the SET7/9 consensus methylation site, (K/R)(S/T/A)K [60]. Hence, we mutated K368 to arginine and this R368 mutation abolished SET7/9-mediated methylation of JMJD2B amino acids 352-387 (Figure 2C), identifying K368 as the one and only methylation site within JMJD2B amino acids 352-387. Then, we individually mutated all four lysine residues within JMJD2B amino acids 388-408 and found that only mutation of K397 and K401 resulted into reduced methylation; further, joint mutation of these two lysine residues (R397/401 mutant) completely suppressed SET7/9-mediated methylation (Figure 2C), identifying K397 and K401 as further methylation sites in JMJD2B. Next, we focused on amino acids 404-430 that harbor five lysine residues. Yet, mutating K415 alone already abolished SET7/9-mediated methylation (Figure 2C), indicating that K415 is the only relevant methylation site within amino acids 404-430. Similarly, K485 that matches the (K/R)(S/T/A)K SET7/9 consensus site is the only substrate for SET7/9 in JMJD2B amino acids 431-488 (Figure 2C), indicating that K485 is a fifth methylation site in JMJD2B. Lastly, we analyzed JMJD2B amino acids 486-580. Mutation of K490 to arginine completely abolished SET7/9-mediated methylation, while mutation of K489 and K491 led to somewhat reduced methylation (Figure 2C). We hypothesize that mutation of the latter two lysine residues alters the affinity of K490 for SET7/9 and thereby indirectly reduced methylation of JMJD2B amino acids 486-580; accordingly, K489 and K491 are not considered SET7/9 methylation sites, while K490 is. Altogether, we have identified six lysine residues (K368, K397, K401, K415, K485 and K490) that can be methylated by SET7/9 in vitro.
We then introduced respective K→R mutations into full-length JMJD2B, expressed these mutants in 293T cells, and purified them by virtue of a N-terminal Flag-tag through immunoprecipitation. These proteins were incubated in vitro with SET7/9 and methylation assessed. As shown in Figure 3A, mutation of any single lysine residue had no obvious effect except for the R485 mutation. But joint mutation of K485 and K490 greatly reduced in vitro methylation, while the R368/397/401/415 mutant was just slightly affected in its ability to become methylated. Joint mutation of all six lysine residues completely abrogated SET7/9-mediated methylation (Figure 3A). Altogether, these data implicate that K485 and K490 are major methylation sites, while K368, K397, K401 and K415 are minor ones. Please note that the fact that the R368/397/401/415/485/490 mutant was no longer methylated strongly suggests that the marginal amount of methylation observed with JMJD2B amino acids 581-740 (see Figure 1C) was likely an artefact in this truncation due to non-natural exposure of lysine residue(s) towards SET7/9.
Figure 3.

Confirmation of methylation sites in full-length JMJD2B. A. Flag-tagged JMJD2B or indicated mutants thereof (marked by # in Ponceau stain) were immunoprecipitated from 293T cells and then methylated by GST-SET7/9 (marked by * in Ponceau stain) in vitro. B. Likewise, indicated Flag-JMJD2B proteins were coexpressed with Flag-SET7/9 in 293T cells and methylation assessed after anti-Flag immunoprecipitation with p53-K372me antibodies (top). Bottom two panels show input levels for JMJD2B and SET7/9.
We also expressed the K→R mutants of JMJD2B together with SET7/9 in 293T cells and probed for in vivo methylation with the p53-K372me antibody (Figure 3B). Mutation of K485, either alone or in combination with other K→R mutations, led to JMJD2B no longer being recognized by this antibody, indicating that this antibody exclusively recognized JMJD2B when methylated on K485. This substantiated that K485 is an in vivo methylation site for SET7/9. Unfortunately, we were unable to find commercially available anti-methyl-lysine antibodies that would recognize methylation on K368, K397, K401, K415 or K490 in vivo.
JMJD2B methylation modulates its transactivation potential
To test how methylation of JMJD2B would affect its transactivation potential, we utilized a reporter gene construct, in which the firefly luciferase gene is driven by the 12-O-Tetradecanoylphorbol-13-acetate Oncogene Responsive Unit (TORU) derived from the polyomavirus enhancer that can be bound and cooperatively induced by ETS and activator protein-1 (AP1) proteins [62]. Specifically, the ETS transcription factor ETV1 has previously been shown to activate the TORU-luciferase reporter gene [63-65]. And accordingly, ETV1 stimulated the TORU reporter gene by ~50-fold in human LNCaP prostate cancer cells (Figure 4). In contrast, JMJD2B had only a moderate 3-fold effect, probably due to recruitment of JMJD2B to the TORU reporter by endogenous ETS or AP1 proteins. However, when ETV1 and JMJD2B were coexpressed, transcriptional synergy was observed (>200-fold activation). No synergy was observable with a catalytically impaired JMJD2B protein, the H189A/E191Q mutant (Figure 4), indicating that catalytic activity of JMJD2B is needed for its ability to coactivate ETV1, similarly as described before [14]. We also tested the ability of the transcription factor JUN, which is one component of AP1 [66,67] and that is also capable of binding to JMJD2B [68], to cooperate with JMJD2B. Likewise, we found that wild-type JMJD2B, but not its H189A/E191Q catalytic mutant, synergized with JUN in activating the TORU reporter (Figure 4). Hence, we have established that the TORU luciferase reporter assay is a suitable means to test the cooperation of JMJD2B with either ETV1 or JUN.
Figure 4.

Synergy between JMJD2B and either ETV1 or JUN in human LNCaP prostate cancer cells. 6 ng JMJD2B (wild-type or H189A/E191Q catalytic mutant) expression vector was cotransfected with 15 ng of ETV1 or JUN expression vector. Shown is the activation of the cotransfected TORU luciferase reporter (mean with standard deviation; n=4). ****, P<0.0001; n.s., not significant (one-way ANOVA with Dunnett’s multiple comparisons).
Then, we determined how mutation of its methylation sites would modulate the JMJD2B coactivation potential in LNCaP cells. In the absence of coexpressed ETV1 or JUN, none of the tested methylation site mutants was significantly different from wild-type JMJD2B (Figure 5). Likewise, we did not observe any change in synergy with ETV1 upon mutation of JMJD2B methylation sites (Figure 5). In contrast, the synergy between JMJD2B and JUN was affected by methylation of JMJD2B. The most severe effects, a downregulation by ~60%, were observed with the R485/490 double mutant and the 6xR mutant, in which all mapped JMJD2B methylation sites were mutated (Figure 5); in contrast, joint mutation of the four minor methylation sites (K368, K397, K401 and K415) did not significantly reduce the ability of JMJD2B to coactivate JUN (Figure 5). Further, single mutation of K485 or K490 also did not significantly affect the coactivation potential of JMJD2B (not shown). Altogether, these data indicate that SET7/9-mediated methylation of JMJD2B has selective effects: in case of coactivation of ETV1, it appears irrelevant, but in case of modulating JUN, methylation of JMJD2B is required for efficient coactivation.
Figure 5.

Activity of JMJD2B methylation site mutants. Wild-type or methylation site mutants of JMJD2B (4 ng expression vector) were cotransfected with either ETV1 or JUN (each 10 ng expression vector) into LNCaP cells and resultant TORU luciferase activity measured. Shown are means with standard deviation (n=4). The statistical significance between respective wild-type and methylation site mutants was determined with one-way ANOVA (Dunnett’s multiple comparisons). **, P<0.01; n.s., not significant.
Discussion
In this report, we have identified a new substrate for the SET7/9 methyltransferase, namely the JMJD2B epigenetic regulator. This further supports the notion that SET7/9 is primarily a non-histone methyltransferase [69-71] rather than - as originally purported - a histone H3 lysine 4 methyltransferase [72,73]. Two major methylation sites, K485 and K490, were identified in JMJD2B, whose joint mutation reduced the ability of JMJD2B to coactivate JUN, an established oncoprotein [74,75]. However, mutation of JMJD2B methylation sites had no impact on coactivating the ETV1 transcription factor, indicating that SET7/9-mediated methylation of JMJD2B will only affect a limited cadre of JMJD2B-regulated genes. Given that JMJD2 proteins can not only demethylate lysine residues on histones, but also on non-histone proteins [6,7,10], it is conceivable that JMJD2B demethylates repressive methyl marks on histones within nucleosomes as well as on to-be-identified coactivators binding to JUN in order to stimulate JUN-dependent gene transcription (Figure 6A). Interestingly, none of the six JMJD2B methylation sites are conserved in its closest homologs, JMJD2A and JMJD2C (Figure 6B), suggesting that this could be one means to differentiate the often overlapping functions of these three JMJD2 proteins [6,7,10].
Figure 6.

A. Model of how SET7/9 methylates and thereby stimulates the coactivator potential of JMJD2B. Please note that JMJD2B may in part coactivate JUN-dependent transcription through demethylation (indicated by ---| in the figure) of methylated histones within nucleosomes and/or of methylated coactivators binding to JUN. B. No conservation of JMJD2B methylation sites in JMJD2A or JMJD2C. Amino acid sequences for JMJD2A (amino acids 331-619), JMJD2B (amino acids 332-670) and JMJD2C (amino acids 333-596) were derived from the NCBI entries NP_055478.2, XP_005259578.2 and NP_055876.2, respectively. Please note that the utilized JMJD2B protein was the X1 isoform, which has a 34 amino acids insert (amino acids 373-406) compared to the frequently used JMJD2B isoform 1 (NP_055830.1), and that isoform 1 also encompasses four of the identified six SET7/9 methylation sites, including the two major ones corresponding to K485 and K490. C and D. Correlation between JMJD2B mRNA levels and either SET7/9 or JUN mRNA levels in human prostate adenocarcinomas. Data were derived from The Cancer Genome Atlas (TCGA) through the GEPIA (Gene Expression Profiling Interactive Analysis) webtool. Shown are log2 values of transcripts per million; R = Spearman correlation coefficient.
Similar to JMJD2B and SET7/9, JUN is overexpressed in prostate tumors and can promote disease development [76-78]. Hence, these three proteins together may cooperate in the neoplastic transformation of prostate cells. Consistently, these three proteins are likely co-overexpressed in prostate adenocarcinomas, since JMJD2B mRNA levels were positively correlated with both SET7/9 (Figure 6C) and JUN mRNA levels (Figure 6D), the latter possibly reflecting that JUN is a known JMJD2B target gene [79]. And JMJD2B’s cooperation with SET7/9 and/or JUN may extend to other cancers such as acute myeloid leukemia, clear cell renal carcinoma, breast, gastric, colorectal or liver cancer, where JMJD2B has also been found to be overexpressed [8,9,80,81] and accordingly could be inactivated with small molecule inhibitors targeting its catalytic center [82-84] in order to ameliorate disease severity and outcome. Our data suggest that suppression of SET7/9-mediated methylation of JMJD2B represents another strategy to curtail the oncogenic effects of JMJD2B in prostate and other cancers.
Aside from its role in cancer, JMJD2B performs many functions in normal cells and other diseases. Although two different Jmjd2b whole-body knockout mouse models displayed no obvious defects during embryogenesis [85,86], pathologic changes were observable upon Jmjd2b knockout in adult mice. For instance, tissue-specific Jmjd2b knockout in mammary glands led to their delayed development [85], while knockout in adipocytes or in the whole body caused metabolic abnormalities and obesity [87,88] and knockout in neurons led to defective spine maturation, deficits in working memory and hyperactivity in a novel environment [89]. Moreover, mono- and bi-allelic JMJD2B pathogenic mutations were found in patients afflicted with global developmental delay or autism, and heterozygous Jmjd2b+/- mice showed a reduction in brain volume which may cause the aforementioned phenotypes [90-92]. These latter data indicate that haploinsufficiency of JMJD2B is already enough to elicit serious defects, and accordingly the observed ~60% reduction in coactivation of JUN with the JMJD2B-R485/490 mutant strongly suggests that SET7/9-mediated methylation will profoundly affect JMJD2B-dependent biological processes.
Another role of JMJD2B is in stem cell biology. In part in conjunction with other histone demethylases, JMJD2B was required for embryonic stem cell renewal or reprogramming into pluripotency, but on the other hand also promoted the osteogenic differentiation of mesenchymal stem cells [93-95]. As SET7/9 appears to promote pluripotency in human embryonic stem cells [96], one may speculate whether this also involves SET7/9-mediated methylation of JMJD2B. Lastly, while JMJD2B is a true histone demethylase that acts in the cell nucleus, it has also been found in the cytoplasm where its enzymatic activity regulated the unfolded protein response [97]. This suggests that JMJD2B is not only demethylating histones, but potentially also other (nuclear and cytoplasmic) proteins. Given that some JMJD proteins have been shown to hydroxylate various proteins [98-100], one can as well imagine that SET7/9-mediated methylation of JMJD2B not only regulates its demethylation but also hydroxylation activity, thereby providing another mechanism how JMJD2B may pleiotropically affect the proteome and in so doing could modulate cell homeostasis and pathological changes.
Conclusion
The identification of SET7/9-mediated lysine methylation of the JMJD2B protein and how it can selectively stimulate the cooperation with DNA-binding factors such a JUN has provided new insights how posttranslational modification may stimulate JMJD2B transcriptional activity and thereby prostate cancer development. However, some limitations of our study that should be addressed in the future are: (i) the absence of animal models testing how JMJD2B methylation on particularly K485 and K490 would modulate its physiological functions in development, homeostasis and malignancy; (ii) the unanswered question whether methylation of JMJD2B affects its ability to impose epigenetic changes; or (iii) if SET7/9-mediated methylation of JMJD2B leads to enhanced JUN-dependent gene transcription and thereby stimulation of tumor cells not only in prostate carcinomas, but also in the many other cancers where SET7/9, JMJD2B and JUN exert cancer-critical functions [9,67,101,102].
Acknowledgements
This work was partially funded by a grant from the National Institutes of Health/National Cancer Institute (R03 CA223615) to RJ. Also, RJ has been supported in part by the Oklahoma Tobacco Settlement Endowment Trust through an award made to the University of Oklahoma/Stephenson Cancer Center.
Disclosure of conflict of interest
None.
References
- 1.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 2.Fodor BD, Kubicek S, Yonezawa M, O’Sullivan RJ, Sengupta R, Perez-Burgos L, Opravil S, Mechtler K, Schotta G, Jenuwein T. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006;20:1557–1562. doi: 10.1101/gad.388206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hillringhaus L, Yue WW, Rose NR, Ng SS, Gileadi C, Loenarz C, Bello SH, Bray JE, Schofield CJ, Oppermann U. Structural and evolutionary basis for the dual substrate selectivity of human KDM4 histone demethylase family. J Biol Chem. 2011;286:41616–41625. doi: 10.1074/jbc.M111.283689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trojer P, Zhang J, Yonezawa M, Schmidt A, Zheng H, Jenuwein T, Reinberg D. Dynamic histone H1 isotype 4 methylation and demethylation by histone lysine methyltransferase G9a/KMT1C and the Jumonji domain-containing JMJD2/KDM4 proteins. J Biol Chem. 2009;284:8395–8405. doi: 10.1074/jbc.M807818200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao S, Allis CD, Wang GG. The language of chromatin modification in human cancers. Nat Rev Cancer. 2021;21:413–430. doi: 10.1038/s41568-021-00357-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. 2013;73:2936–2942. doi: 10.1158/0008-5472.CAN-12-4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Labbe RM, Holowatyj A, Yang ZQ. Histone lysine demethylase (KDM) subfamily 4: structures, functions and therapeutic potential. Am J Transl Res. 2013;6:1–15. [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson C, Krieg AJ. KDM4B: a nail for every hammer? Genes (Basel) 2019;10:134. doi: 10.3390/genes10020134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z, Cai H, Zhao E, Cui H. The diverse roles of histone demethylase KDM4B in normal and cancer development and progression. Front Cell Dev Biol. 2022;9:790129. doi: 10.3389/fcell.2021.790129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang J, Hu Y, Zhang B, Liang X, Li X. The JMJD family histone demethylases in crosstalk between inflammation and cancer. Front Immunol. 2022;13:881396. doi: 10.3389/fimmu.2022.881396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coffey K, Rogerson L, Ryan-Munden C, Alkharaif D, Stockley J, Heer R, Sahadevan K, O’Neill D, Jones D, Darby S, Staller P, Mantilla A, Gaughan L, Robson CN. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res. 2013;41:4433–4446. doi: 10.1093/nar/gkt106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chu CH, Wang LY, Hsu KC, Chen CC, Cheng HH, Wang SM, Wu CM, Chen TJ, Li LT, Liu R, Hung CL, Yang JM, Kung HJ, Wang WC. KDM4B as a target for prostate cancer: structural analysis and selective inhibition by a novel inhibitor. J Med Chem. 2014;57:5975–5985. doi: 10.1021/jm500249n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duan L, Rai G, Roggero C, Zhang QJ, Wei Q, Ma SH, Zhou Y, Santoyo J, Martinez ED, Xiao G, Raj GV, Jadhav A, Simeonov A, Maloney DJ, Rizo J, Hsieh JT, Liu ZP. KDM4/JMJD2 histone demethylase inhibitors block prostate tumor growth by suppressing the expression of AR and BMYB-regulated genes. Chem Biol. 2015;22:1185–1196. doi: 10.1016/j.chembiol.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim TD, Jin F, Shin S, Oh S, Lightfoot SA, Grande JP, Johnson AJ, van Deursen JM, Wren JD, Janknecht R. Histone demethylase JMJD2A drives prostate tumorigenesis through transcription factor ETV1. J Clin Invest. 2016;126:706–720. doi: 10.1172/JCI78132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duan L, Chen Z, Lu J, Liang Y, Wang M, Roggero CM, Zhang QJ, Gao J, Fang Y, Cao J, Lu J, Zhao H, Dang A, Pong RC, Hernandez E, Chang CM, Hoang DT, Ahn JM, Xiao G, Wang RT, Yu KJ, Kapur P, Rizo J, Hsieh JT, Luo J, Liu ZP. Histone lysine demethylase KDM4B regulates the alternative splicing of the androgen receptor in response to androgen deprivation. Nucleic Acids Res. 2019;47:11623–11636. doi: 10.1093/nar/gkz1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sha J, Han Q, Chi C, Zhu Y, Pan J, Dong B, Huang Y, Xia W, Xue W. Upregulated KDM4B promotes prostate cancer cell proliferation by activating autophagy. J Cell Physiol. 2020;235:2129–2138. doi: 10.1002/jcp.29117. [DOI] [PubMed] [Google Scholar]
- 17.Tang DE, Dai Y, He JX, Lin LW, Leng QX, Geng XY, Fu DX, Jiang HW, Xu SH. Targeting the KDM4B-AR-c-Myc axis promotes sensitivity to androgen receptor-targeted therapy in advanced prostate cancer. J Pathol. 2020;252:101–113. doi: 10.1002/path.5495. [DOI] [PubMed] [Google Scholar]
- 18.Wu MJ, Chen CJ, Lin TY, Liu YY, Tseng LL, Cheng ML, Chuu CP, Tsai HK, Kuo WL, Kung HJ, Wang WC. Targeting KDM4B that coactivates c-Myc-regulated metabolism to suppress tumor growth in castration-resistant prostate cancer. Theranostics. 2021;11:7779–7796. doi: 10.7150/thno.58729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duan L, Chen YA, Liang Y, Chen Z, Lu J, Fang Y, Cao J, Lu J, Zhao H, Pong RC, Hernandez E, Kapur P, Tran TAT, Smith T, Martinez ED, Ahn JM, Hsieh JT, Luo JH, Liu ZP. Therapeutic targeting of histone lysine demethylase KDM4B blocks the growth of castration-resistant prostate cancer. Biomed Pharmacother. 2023;158:114077. doi: 10.1016/j.biopha.2022.114077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, Pienta KJ, Rubin MA, Chinnaiyan AM. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 21.Oh S, Shin S, Janknecht R. ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochim Biophys Acta. 2012;1826:1–12. doi: 10.1016/j.bbcan.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nicholas TR, Strittmatter BG, Hollenhorst PC. Oncogenic ETS factors in prostate cancer. Adv Exp Med Biol. 2019;1210:409–436. doi: 10.1007/978-3-030-32656-2_18. [DOI] [PubMed] [Google Scholar]
- 23.Qi T, Qu Q, Li G, Wang J, Zhu H, Yang Z, Sun Y, Lu Q, Qu J. Function and regulation of the PEA3 subfamily of ETS transcription factors in cancer. Am J Cancer Res. 2020;10:3083–3105. [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Huang Z, Sun M, Huang W, Xia L. ETS transcription factors: multifaceted players from cancer progression to tumor immunity. Biochim Biophys Acta Rev Cancer. 2023;1878:188872. doi: 10.1016/j.bbcan.2023.188872. [DOI] [PubMed] [Google Scholar]
- 25.Gupta N, Song H, Wu W, Ponce RK, Lin YK, Kim JW, Small EJ, Feng FY, Huang FW, Okimoto RA. The CIC-ERF co-deletion underlies fusion-independent activation of ETS family member, ETV1, to drive prostate cancer progression. Elife. 2022;11:e77072. doi: 10.7554/eLife.77072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, Menon A, Jing X, Cao Q, Han B, Yu J, Wang L, Montie JE, Rubin MA, Pienta KJ, Roulston D, Shah RB, Varambally S, Mehra R, Chinnaiyan AM. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 27.Shin S, Kim TD, Jin F, van Deursen JM, Dehm SM, Tindall DJ, Grande JP, Munz JM, Vasmatzis G, Janknecht R. Induction of prostatic intraepithelial neoplasia and modulation of androgen receptor by ETS variant 1/ETS-related protein 81. Cancer Res. 2009;69:8102–8110. doi: 10.1158/0008-5472.CAN-09-0941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baena E, Shao Z, Linn DE, Glass K, Hamblen MJ, Fujiwara Y, Kim J, Nguyen M, Zhang X, Godinho FJ, Bronson RT, Mucci LA, Loda M, Yuan GC, Orkin SH, Li Z. ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev. 2013;27:683–698. doi: 10.1101/gad.211011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oh S, Shin S, Song H, Grande JP, Janknecht R. Relationship between ETS transcription factor ETV1 and TGF-beta-regulated SMAD proteins in prostate cancer. Sci Rep. 2019;9:8186. doi: 10.1038/s41598-019-44685-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaughan L, Stockley J, Wang N, McCracken SR, Treumann A, Armstrong K, Shaheen F, Watt K, McEwan IJ, Wang C, Pestell RG, Robson CN. Regulation of the androgen receptor by SET9-mediated methylation. Nucleic Acids Res. 2011;39:1266–1279. doi: 10.1093/nar/gkq861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mooney SM, Grande JP, Salisbury JL, Janknecht R. Sumoylation of p68 and p72 RNA helicases affects protein stability and transactivation potential. Biochemistry. 2010;49:1–10. doi: 10.1021/bi901263m. [DOI] [PubMed] [Google Scholar]
- 32.Goel A, Janknecht R. Concerted activation of ETS protein ER81 by p160 coactivators, the acetyltransferase p300 and the receptor tyrosine kinase HER2/Neu. J Biol Chem. 2004;279:14909–14916. doi: 10.1074/jbc.M400036200. [DOI] [PubMed] [Google Scholar]
- 33.Kim J, Shin S, Subramaniam M, Bruinsma E, Kim TD, Hawse JR, Spelsberg TC, Janknecht R. Histone demethylase JARID1B/KDM5B is a corepressor of TIEG1/KLF10. Biochem Biophys Res Commun. 2010;401:412–416. doi: 10.1016/j.bbrc.2010.09.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu J, Janknecht R. Regulation of the ETS transcription factor ER81 by the 90-kDa ribosomal S6 kinase 1 and protein kinase A. J Biol Chem. 2002;277:42669–42679. doi: 10.1074/jbc.M205501200. [DOI] [PubMed] [Google Scholar]
- 35.Kim TD, Fuchs JR, Schwartz E, Abdelhamid D, Etter J, Berry WL, Li C, Ihnat MA, Li PK, Janknecht R. Pro-growth role of the JMJD2C histone demethylase in HCT-116 colon cancer cells and identification of curcuminoids as JMJD2 inhibitors. Am J Transl Res. 2014;6:236–247. [PMC free article] [PubMed] [Google Scholar]
- 36.Oh S, Shin S, Lightfoot SA, Janknecht R. 14-3-3 proteins modulate the ETS transcription factor ETV1 in prostate cancer. Cancer Res. 2013;73:5110–5119. doi: 10.1158/0008-5472.CAN-13-0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oh S, Song H, Freeman WM, Shin S, Janknecht R. Cooperation between ETS transcription factor ETV1 and histone demethylase JMJD1A in colorectal cancer. Int J Oncol. 2020;57:1319–1332. doi: 10.3892/ijo.2020.5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papoutsopoulou S, Janknecht R. Phosphorylation of ETS transcription factor ER81 in a complex with its coactivators CREB-binding protein and p300. Mol Cell Biol. 2000;20:7300–7310. doi: 10.1128/mcb.20.19.7300-7310.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oh S, Janknecht R. Histone demethylase JMJD5 is essential for embryonic development. Biochem Biophys Res Commun. 2012;420:61–65. doi: 10.1016/j.bbrc.2012.02.115. [DOI] [PubMed] [Google Scholar]
- 40.Goueli BS, Janknecht R. Regulation of telomerase reverse transcriptase gene activity by upstream stimulatory factor. Oncogene. 2003;22:8042–8047. doi: 10.1038/sj.onc.1206847. [DOI] [PubMed] [Google Scholar]
- 41.Kim TD, Oh S, Lightfoot SA, Shin S, Wren JD, Janknecht R. Upregulation of PSMD10 caused by the JMJD2A histone demethylase. Int J Clin Exp Med. 2016;9:10123–10134. [PMC free article] [PubMed] [Google Scholar]
- 42.DiTacchio L, Bowles J, Shin S, Lim DS, Koopman P, Janknecht R. Transcription factors ER71/ETV2 and SOX9 participate in a positive feedback loop in fetal and adult mouse testis. J Biol Chem. 2012;287:23657–23666. doi: 10.1074/jbc.M111.320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knebel J, De Haro L, Janknecht R. Repression of transcription by TSGA/Jmjd1a, a novel interaction partner of the ETS protein ER71. J Cell Biochem. 2006;99:319–329. doi: 10.1002/jcb.20945. [DOI] [PubMed] [Google Scholar]
- 44.Mooney SM, Goel A, D’Assoro AB, Salisbury JL, Janknecht R. Pleiotropic effects of p300-mediated acetylation on p68 and p72 RNA helicase. J Biol Chem. 2010;285:30443–30452. doi: 10.1074/jbc.M110.143792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li X, Moon G, Shin S, Zhang B, Janknecht R. Cooperation between ETS variant 2 and Jumonji domain-containing 2 histone demethylases. Mol Med Rep. 2018;17:5518–5527. doi: 10.3892/mmr.2018.8507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sui Y, Li X, Oh S, Zhang B, Freeman WM, Shin S, Janknecht R. Opposite roles of the JMJD1A interaction partners MDFI and MDFIC in colorectal cancer. Sci Rep. 2020;10:8710. doi: 10.1038/s41598-020-65536-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oh S, Shin S, Janknecht R. Sumoylation of transcription factor ETV1 modulates its oncogenic potential in prostate cancer. Int J Clin Exp Pathol. 2021;14:795–810. [PMC free article] [PubMed] [Google Scholar]
- 48.Janknecht R. Regulation of the ER81 transcription factor and its coactivators by mitogen- and stress-activated protein kinase 1 (MSK1) Oncogene. 2003;22:746–755. doi: 10.1038/sj.onc.1206185. [DOI] [PubMed] [Google Scholar]
- 49.De Haro L, Janknecht R. Functional analysis of the transcription factor ER71 and its activation of the matrix metalloproteinase-1 promoter. Nucleic Acids Res. 2002;30:2972–2979. doi: 10.1093/nar/gkf390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sui Y, Jiang H, Kellogg CM, Oh S, Janknecht R. Promotion of colorectal cancer by transcription factor BHLHE40 involves upregulation of ADAM19 and KLF7. Front Oncol. 2023;13:1122238. doi: 10.3389/fonc.2023.1122238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li X, Oh S, Song H, Shin S, Zhang B, Freeman WM, Janknecht R. A potential common role of the Jumonji C domain-containing 1A histone demethylase and chromatin remodeler ATRX in promoting colon cancer. Oncol Lett. 2018;16:6652–6662. doi: 10.3892/ol.2018.9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim TD, Shin S, Janknecht R. ETS transcription factor ERG cooperates with histone demethylase KDM4A. Oncol Rep. 2016;35:3679–3688. doi: 10.3892/or.2016.4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bosc DG, Goueli BS, Janknecht R. HER2/Neu-mediated activation of the ETS transcription factor ER81 and its target gene MMP-1. Oncogene. 2001;20:6215–6224. doi: 10.1038/sj.onc.1204820. [DOI] [PubMed] [Google Scholar]
- 54.Kim TD, Shin S, Janknecht R. Repression of Smad3 activity by histone demethylase SMCX/JARID1C. Biochem Biophys Res Commun. 2008;366:563–567. doi: 10.1016/j.bbrc.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 55.Shin S, Bosc DG, Ingle JN, Spelsberg TC, Janknecht R. Rcl is a novel ETV1/ER81 target gene upregulated in breast tumors. J Cell Biochem. 2008;105:866–874. doi: 10.1002/jcb.21884. [DOI] [PubMed] [Google Scholar]
- 56.Kim TD, Oh S, Shin S, Janknecht R. Regulation of tumor suppressor p53 and HCT116 cell physiology by histone demethylase JMJD2D/KDM4D. PLoS One. 2012;7:e34618. doi: 10.1371/journal.pone.0034618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shin S, Oh S, An S, Janknecht R. ETS variant 1 regulates matrix metalloproteinase-7 transcription in LNCaP prostate cancer cells. Oncol Rep. 2013;29:306–314. doi: 10.3892/or.2012.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dowdy SC, Mariani A, Janknecht R. HER2/Neu- and TAK1-mediated up-regulation of the transforming growth factor beta inhibitor Smad7 via the ETS protein ER81. J Biol Chem. 2003;278:44377–44384. doi: 10.1074/jbc.M307202200. [DOI] [PubMed] [Google Scholar]
- 59.Rossow KL, Janknecht R. The Ewing’s sarcoma gene product functions as a transcriptional activator. Cancer Res. 2001;61:2690–2695. [PubMed] [Google Scholar]
- 60.Couture JF, Collazo E, Hauk G, Trievel RC. Structural basis for the methylation site specificity of SET7/9. Nat Struct Mol Biol. 2006;13:140–146. doi: 10.1038/nsmb1045. [DOI] [PubMed] [Google Scholar]
- 61.Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, McKinney K, Tempst P, Prives C, Gamblin SJ, Barlev NA, Reinberg D. Regulation of p53 activity through lysine methylation. Nature. 2004;432:353–360. doi: 10.1038/nature03117. [DOI] [PubMed] [Google Scholar]
- 62.Wasylyk B, Wasylyk C, Flores P, Begue A, Leprince D, Stehelin D. The c-ets proto-oncogenes encode transcription factors that cooperate with c-Fos and c-Jun for transcriptional activation. Nature. 1990;346:191–193. doi: 10.1038/346191a0. [DOI] [PubMed] [Google Scholar]
- 63.Monte D, Coutte L, Baert JL, Angeli I, Stehelin D, de Launoit Y. Molecular characterization of the ets-related human transcription factor ER81. Oncogene. 1995;11:771–779. [PubMed] [Google Scholar]
- 64.Goel A, Janknecht R. Acetylation-mediated transcriptional activation of the ETS protein ER81 by p300, P/CAF, and HER2/Neu. Mol Cell Biol. 2003;23:6243–6254. doi: 10.1128/MCB.23.17.6243-6254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goueli BS, Janknecht R. Upregulation of the catalytic telomerase subunit by the transcription factor ER81 and oncogenic HER2/Neu, Ras, or Raf. Mol Cell Biol. 2004;24:25–35. doi: 10.1128/MCB.24.1.25-35.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bejjani F, Evanno E, Zibara K, Piechaczyk M, Jariel-Encontre I. The AP-1 transcriptional complex: local switch or remote command? Biochim Biophys Acta Rev Cancer. 2019;1872:11–23. doi: 10.1016/j.bbcan.2019.04.003. [DOI] [PubMed] [Google Scholar]
- 67.Brennan A, Leech JT, Kad NM, Mason JM. Selective antagonism of cJun for cancer therapy. J Exp Clin Cancer Res. 2020;39:184. doi: 10.1186/s13046-020-01686-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu MC, Cheng HH, Yeh TS, Li YC, Chen TJ, Sit WY, Chuu CP, Kung HJ, Chien S, Wang WC. KDM4B is a coactivator of c-Jun and involved in gastric carcinogenesis. Cell Death Dis. 2019;10:68. doi: 10.1038/s41419-019-1305-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dhayalan A, Kudithipudi S, Rathert P, Jeltsch A. Specificity analysis-based identification of new methylation targets of the SET7/9 protein lysine methyltransferase. Chem Biol. 2011;18:111–120. doi: 10.1016/j.chembiol.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 70.Gu Y, Zhang X, Yu W, Dong W. Oncogene or tumor suppressor: the coordinative role of lysine methyltransferase SET7/9 in cancer development and the related mechanisms. J Cancer. 2022;13:623–640. doi: 10.7150/jca.57663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chiang C, Yang H, Zhu L, Chen C, Chen C, Zuo Y, Zheng D. The epigenetic regulation of nonhistone proteins by SETD7: new targets in cancer. Front Genet. 2022;13:918509. doi: 10.3389/fgene.2022.918509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang H, Cao R, Xia L, Erdjument-Bromage H, Borchers C, Tempst P, Zhang Y. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol Cell. 2001;8:1207–1217. doi: 10.1016/s1097-2765(01)00405-1. [DOI] [PubMed] [Google Scholar]
- 73.Nishioka K, Chuikov S, Sarma K, Erdjument-Bromage H, Allis CD, Tempst P, Reinberg D. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002;16:479–489. doi: 10.1101/gad.967202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 75.Song D, Lian Y, Zhang L. The potential of activator protein 1 (AP-1) in cancer targeted therapy. Front Immunol. 2023;14:1224892. doi: 10.3389/fimmu.2023.1224892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Edwards J, Krishna NS, Mukherjee R, Bartlett JM. The role of c-Jun and c-Fos expression in androgen-independent prostate cancer. J Pathol. 2004;204:153–158. doi: 10.1002/path.1605. [DOI] [PubMed] [Google Scholar]
- 77.Ouyang X, Jessen WJ, Al-Ahmadie H, Serio AM, Lin Y, Shih WJ, Reuter VE, Scardino PT, Shen MM, Aronow BJ, Vickers AJ, Gerald WL, Abate-Shen C. Activator protein-1 transcription factors are associated with progression and recurrence of prostate cancer. Cancer Res. 2008;68:2132–2144. doi: 10.1158/0008-5472.CAN-07-6055. [DOI] [PubMed] [Google Scholar]
- 78.Riedel M, Berthelsen MF, Cai H, Haldrup J, Borre M, Paludan SR, Hager H, Vendelbo MH, Wagner EF, Bakiri L, Thomsen MK. In vivo CRISPR inactivation of Fos promotes prostate cancer progression by altering the associated AP-1 subunit Jun. Oncogene. 2021;40:2437–2447. doi: 10.1038/s41388-021-01724-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Berry WL, Kim TD, Janknecht R. Stimulation of beta-catenin and colon cancer cell growth by the KDM4B histone demethylase. Int J Oncol. 2014;44:1341–1348. doi: 10.3892/ijo.2014.2279. [DOI] [PubMed] [Google Scholar]
- 80.Ueda T, Kanai A, Komuro A, Amano H, Ota K, Honda M, Kawazu M, Okada H. KDM4B promotes acute myeloid leukemia associated with AML1-ETO by regulating chromatin accessibility. FASEB Bioadv. 2021;3:1020–1033. doi: 10.1096/fba.2021-00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tang H, Guan Y, Yuan Z, Guo T, Tan X, Fan Y, Zhang E, Wang X. Histone demethylase KDM4B contributes to advanced clear cell renal carcinoma and association with copy number variations and cell cycle progression. Epigenetics. 2023;18:2192319. doi: 10.1080/15592294.2023.2192319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee DH, Kim GW, Jeon YH, Yoo J, Lee SW, Kwon SH. Advances in histone demethylase KDM4 as cancer therapeutic targets. FASEB J. 2020;34:3461–3484. doi: 10.1096/fj.201902584R. [DOI] [PubMed] [Google Scholar]
- 83.Lee J, Kim JS, Cho HI, Jo SR, Jang YK. JIB-04, a pan-inhibitor of histone demethylases, targets histone-lysine-demethylase-dependent AKT pathway, leading to cell cycle arrest and inhibition of cancer stem-like cell properties in hepatocellular carcinoma cells. Int J Mol Sci. 2022;23:7657. doi: 10.3390/ijms23147657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Singh S, Abu-Zaid A, Jin H, Fang J, Wu Q, Wang T, Feng H, Quarni W, Shao Y, Maxham L, Abdolvahabi A, Yun MK, Vaithiyalingam S, Tan H, Bowling J, Honnell V, Young B, Guo Y, Bajpai R, Pruett-Miller SM, Grosveld GC, Hatley M, Xu B, Fan Y, Wu G, Chen EY, Chen T, Lewis PW, Rankovic Z, Li Y, Murphy AJ, Easton J, Peng J, Chen X, Wang R, White SW, Davidoff AM, Yang J. Targeting KDM4 for treating PAX3-FOXO1-driven alveolar rhabdomyosarcoma. Sci Transl Med. 2022;14:eabq2096. doi: 10.1126/scitranslmed.abq2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kawazu M, Saso K, Tong KI, McQuire T, Goto K, Son DO, Wakeham A, Miyagishi M, Mak TW, Okada H. Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS One. 2011;6:e17830. doi: 10.1371/journal.pone.0017830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pedersen MT, Kooistra SM, Radzisheuskaya A, Laugesen A, Johansen JV, Hayward DG, Nilsson J, Agger K, Helin K. Continual removal of H3K9 promoter methylation by Jmjd2 demethylases is vital for ESC self-renewal and early development. EMBO J. 2016;35:1550–1564. doi: 10.15252/embj.201593317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kang C, Saso K, Ota K, Kawazu M, Ueda T, Okada H. JMJD2B/KDM4B inactivation in adipose tissues accelerates obesity and systemic metabolic abnormalities. Genes Cells. 2018;23:767–777. doi: 10.1111/gtc.12627. [DOI] [PubMed] [Google Scholar]
- 88.Cheng Y, Yuan Q, Vergnes L, Rong X, Youn JY, Li J, Yu Y, Liu W, Cai H, Lin JD, Tontonoz P, Hong C, Reue K, Wang CY. KDM4B protects against obesity and metabolic dysfunction. Proc Natl Acad Sci U S A. 2018;115:E5566–E5575. doi: 10.1073/pnas.1721814115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fujiwara K, Fujita Y, Kasai A, Onaka Y, Hashimoto H, Okada H, Yamashita T. Deletion of JMJD2B in neurons leads to defective spine maturation, hyperactive behavior and memory deficits in mouse. Transl Psychiatry. 2016;6:e766. doi: 10.1038/tp.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Duncan AR, Vitobello A, Collins SC, Vancollie VE, Lelliott CJ, Rodan L, Shi J, Seman AR, Agolini E, Novelli A, Prontera P, Guillen Sacoto MJ, Santiago-Sim T, Trimouille A, Goizet C, Nizon M, Bruel AL, Philippe C, Grant PE, Wojcik MH, Stoler J, Genetti CA, van Dooren MF, Maas SM, Alders M, Faivre L, Sorlin A, Yoon G, Yalcin B, Agrawal PB. Heterozygous variants in KDM4B lead to global developmental delay and neuroanatomical defects. Am J Hum Genet. 2020;107:1170–1177. doi: 10.1016/j.ajhg.2020.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, Shih-Chen F, Aleksic B, Biscaldi M, Bolton PF, Brownfeld JM, Cai J, Campbell NG, Carracedo A, Chahrour MH, Chiocchetti AG, Coon H, Crawford EL, Curran SR, Dawson G, Duketis E, Fernandez BA, Gallagher L, Geller E, Guter SJ, Hill RS, Ionita-Laza J, Jimenz Gonzalez P, Kilpinen H, Klauck SM, Kolevzon A, Lee I, Lei I, Lei J, Lehtimaki T, Lin CF, Ma’ayan A, Marshall CR, McInnes AL, Neale B, Owen MJ, Ozaki N, Parellada M, Parr JR, Purcell S, Puura K, Rajagopalan D, Rehnstrom K, Reichenberg A, Sabo A, Sachse M, Sanders SJ, Schafer C, Schulte-Ruther M, Skuse D, Stevens C, Szatmari P, Tammimies K, Valladares O, Voran A, Li-San W, Weiss LA, Willsey AJ, Yu TW, Yuen RK DDD Study; Homozygosity Mapping Collaborative for Autism; UK10K Consortium. Cook EH, Freitag CM, Gill M, Hultman CM, Lehner T, Palotie A, Schellenberg GD, Sklar P, State MW, Sutcliffe JS, Walsh CA, Scherer SW, Zwick ME, Barett JC, Cutler DJ, Roeder K, Devlin B, Daly MJ, Buxbaum JD. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Takada S, Silva S, Zamorano I, Perez A, Iwabuchi C, Miyake N. Human phenotype caused by biallelic KDM4B frameshift variant. Clin Genet. 2024;105:72–76. doi: 10.1111/cge.14409. [DOI] [PubMed] [Google Scholar]
- 93.Ye L, Fan Z, Yu B, Chang J, Al Hezaimi K, Zhou X, Park NH, Wang CY. Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell Stem Cell. 2012;11:50–61. doi: 10.1016/j.stem.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Das PP, Shao Z, Beyaz S, Apostolou E, Pinello L, De Los Angeles A, O’Brien K, Atsma JM, Fujiwara Y, Nguyen M, Ljuboja D, Guo G, Woo A, Yuan GC, Onder T, Daley G, Hochedlinger K, Kim J, Orkin SH. Distinct and combinatorial functions of jmjd2b/kdm4b and jmjd2c/kdm4c in mouse embryonic stem cell identity. Mol Cell. 2014;53:32–48. doi: 10.1016/j.molcel.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wei J, Antony J, Meng F, MacLean P, Rhind R, Laible G, Oback B. KDM4B-mediated reduction of H3K9me3 and H3K36me3 levels improves somatic cell reprogramming into pluripotency. Sci Rep. 2017;7:7514. doi: 10.1038/s41598-017-06569-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim SK, Lee H, Han K, Kim SC, Choi Y, Park SW, Bak G, Lee Y, Choi JK, Kim TK, Han YM, Lee D. SET7/9 methylation of the pluripotency factor LIN28A is a nucleolar localization mechanism that blocks let-7 biogenesis in human ESCs. Cell Stem Cell. 2014;15:735–749. doi: 10.1016/j.stem.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang W, Oguz G, Lee PL, Bao Y, Wang P, Terp MG, Ditzel HJ, Yu Q. KDM4B-regulated unfolded protein response as a therapeutic vulnerability in PTEN-deficient breast cancer. J Exp Med. 2018;215:2833–2849. doi: 10.1084/jem.20180439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Markolovic S, Wilkins SE, Schofield CJ. Protein hydroxylation catalyzed by 2-oxoglutarate-dependent oxygenases. J Biol Chem. 2015;290:20712–20722. doi: 10.1074/jbc.R115.662627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Islam MS, Leissing TM, Chowdhury R, Hopkinson RJ, Schofield CJ. 2-Oxoglutarate-dependent oxygenases. Annu Rev Biochem. 2018;87:585–620. doi: 10.1146/annurev-biochem-061516-044724. [DOI] [PubMed] [Google Scholar]
- 100.Oh S, Shin S, Janknecht R. The small members of the JMJD protein family: enzymatic jewels or jinxes? Biochim Biophys Acta Rev Cancer. 2019;1871:406–418. doi: 10.1016/j.bbcan.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Monteiro FL, Williams C, Helguero LA. A systematic review to define the multi-faceted role of lysine methyltransferase SETD7 in cancer. Cancers (Basel) 2022;14:1414. doi: 10.3390/cancers14061414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang S, Wang X, Bai J, Duan B. The role of SET domain containing lysine methyltransferase 7 in tumorigenesis and development. Cell Cycle. 2023;22:269–275. doi: 10.1080/15384101.2022.2122257. [DOI] [PMC free article] [PubMed] [Google Scholar]