

Abstract
The gut microbiomes of human populations worldwide have many core microbial species in common. However, within a species, some strains can show remarkable population specificity. The question is whether such specificity arises from a shared evolutionary history (codiversification) between humans and their microbes. To test for codiversification of host and microbiota, we analyzed paired gut metagenomes and human genomes for 1225 individuals in Europe, Asia, and Africa, including mothers and their children. Between and within countries, a parallel evolutionary history was evident for humans and their gut microbes. Moreover, species displaying the strongest codiversification independently evolved traits characteristic of host dependency, including reduced genomes and oxygen and temperature sensitivity. These findings all point to the importance of understanding the potential role of population-specific microbial strains in microbiome-mediated disease phenotypes.
Across populations, humans share many of the same bacterial and archaeal species in their gut microbiomes (1–3). Within these cosmopolitan species, different strains can dominate in different populations (4–7). Strain variation can arise in several ways, from the uptake of new strains to their in situ evolution (8). When strains and their hosts evolve in parallel, they codiversify, and as a result, their phylogenies are congruent. Codiversification provides opportunities to develop intimate host-microbial relationships across multiple generations (9).
Previous work showed that a small subset of gut bacterial lineages speciated with hominid ancestors (10), but whether such patterns of codiversification extended within host species, and specifically within humans, remained to be demonstrated. There are reasons not to expect to see codiversification with humans: Our diets have changed with time, our populations have expanded across the world, and modern lifestyles may have blurred any signals (11). The identification of species that codiversified with humans has important implications for understanding how humans evolved with their microbiomes and how strains within species may interact with specific host populations (12).
Several human gut microbes are thought to have followed patterns of human migration out of Africa. A notable example is the stomach-dwelling bacterium Helicobacter pylori, the causative agent of gastritis and stomach cancer. Cultured isolates of H. pylori show spatial patterns of strain diversity consistent with human migration patterns (13). A few prevalent gut microbial species, including Prevotella copri, are also thought to have tracked human migration, given how patterns of metagenome-derived strain variation mapped onto continents (4–6). Strain distributions that map onto human migration patterns are suggestive of codiversification, as geographic origins tend to reflect human genetic origins, especially at the continental level (14, 15). But on finer geographic and population scales, such as within countries, the assumption that geography can stand in for genotype weakens (15). A host phylogeny is required to directly test for codiversification by comparison to microbial phylogenies. Such comparative phylogenetic analyses would also allow microbial taxa to be ranked by the degree of cophylogeny they display.
Given the paucity in the public domain of matched human genotype and gut metagenome datasets required for testing for codiversification, especially for undersampled regions (16), we generated new paired datasets from individuals that we sampled in Gabon, Vietnam, and Germany. We also leveraged existing datasets for subjects from Cameroon, the Republic of Korea (South Korea), and the UK by generating fecal metagenomes and/or host genotype data (17–20) (Fig. 1A and table S1). In addition, we collected fecal metagenomes from children whose mothers were study participants in Gabon, Vietnam, and Germany (Fig. 1A and table S2). Altogether, our combined dataset of 839 adults and 386 children allowed us to assess codiversification between humans and gut microbial species shared across and within populations.
Fig. 1. The human phylogeny and selected bacterial phylogenies.
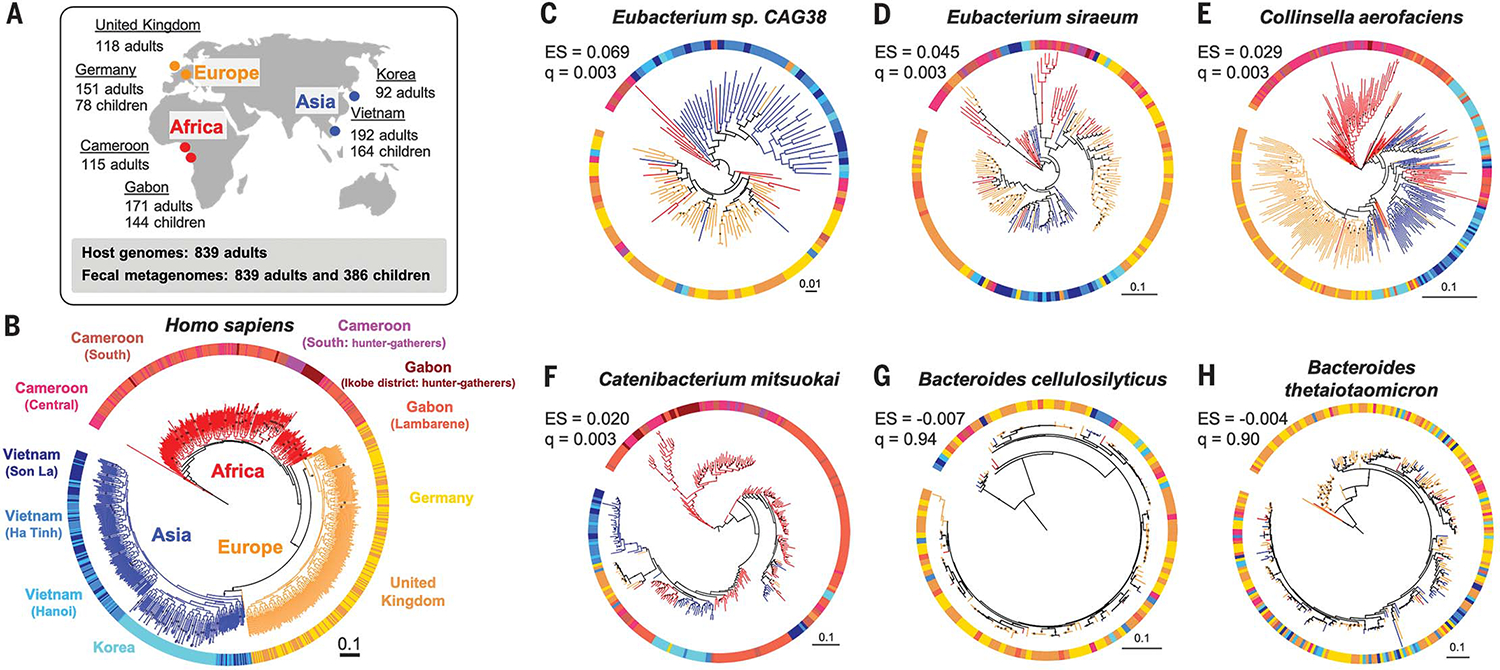
(A) Sampling locations and sizes. (B) A maximum likelihood phylogeny of human subjects based on 20,506 single-nucleotide polymorphisms. Tree branch colors indicate continental origins. Outer strip colors indicate finer geographic locations, and labels refer to sampling locations. (C to H) Maximum likelihood phylogenies for six bacterial species based on species-specific marker genes. PACo effect size (ES) and q values (q) are shown. Bootstrap values >50% are plotted on branches, and all phylogenies are rooted at the midpoint. Colors of branches and outer strips correspond to sampling locations shown in (B). The scale bars show substitutions per site for all phylogenies
Using 20,506 single-nucleotide polymorphisms, we created a maximum likelihood phylogeny to represent the genetic relatedness of the human subjects. As expected, humans clustered into three robust major groups matching their geographic origins (21), where individuals from Asia and Europe formed sister clades nested within individuals from Africa (Fig. 1B). We selected bacterial and archaeal species present in the guts of ≥100 adults with ≥10 individuals per major human group and ≥1 individual per country (see methods in the supplementary materials and table S3). We then created phylogenies for the resulting 59 species using two methods: (i) species-specific marker genes with StrainPhlan3 (7) and (ii) metagenome-assembled genomes (MAGs) using PhyloPhlAn (22). MAG-based trees were obtained for 33 of 59 taxa in adults.
Among the 59 taxa assessed for codiversification, 36 taxa have phylogenies that are more similar to the human host phylogeny than to a permuted host phylogeny [q < 0.05, PACo positive effect size (ES); see methods]. Eubacterium species showed the largest ES (q < 0.003; Fig. 1, C and D, and table S4). Similar results were obtained using two other methods, Parafit (23) and Phytools (24) (tables S4 and S5). Seven species that showed significant codiversification across all three tests included Collinsella aerofaciens (Fig. 1E), Catenibacterium mitsuokai (Fig. 1F), Eubacterium rectale, and P. copri (table S4). In contrast, Bacteroides, Alistipes, and Parabacteroides species generally showed the least evidence of cophylogeny (Fig. 1, G and H, and table S4). Results were robust to sample size (tables S6 and S7 and fig. S1) and to bootstrap support for 36 to 50% of taxa (tables S4 and S8). Overall, species within the Firmicutes phylum showed more evidence of cophylogeny (Wilcoxon rank sum test, P = 7.6 × 10−6) than others, and Bacteroidetes species showed least (Wilcoxon rank sum test, P = 1.5 × 10−6).
We also searched for a codiversification signal within each country. For a subset of taxa, we observed codiversification in multiple countries independently (table S9). Within-country tests included fewer individuals, so the codiversification signal tended to be weaker. Overall, 20 of 59 taxa showed positive ES with uncorrected P < 0.05 in at least one country, but only one taxon remained significant after false discovery rate (FDR) correction: P. copri within Gabon (q = 0.042; table S9). Notably, three species (P. copri, Coprococcus eutactus, and E. rectale) had uncorrected P < 0.05 in three countries independently (table S9). These within-country results indicate that codiversification is robust to hosts living in a shared environment and suggest that codiversification is not driven by continental-scale processes alone.
We sampled the gut metagenomes of children (average age = 7.4 months) of the genotyped participants in Gabon, Vietnam, and Germany. Using the mothers’ genotypes allowed us an unprecedented opportunity to test for codiversification of the child gut microbiome. Among the 20 most prevalent child taxa tested (table S10), nine showed evidence of codiversification (q < 0.05) (table S11). All four Bifidobacterium species tested showed significant PACo ES (q < 0.05) (Fig. 2, A to D, and table S11). According to MAG-based phylogenies, Bifidobacterium longum showed the strongest evidence of codiversification in children (table S12). The signals of codiversification for several taxa also extended within countries in children in Gabon and Germany, but not in Vietnam (table S11). These results show that microbes common to the gut in early childhood have also codiversified with humans.
Fig. 2. Bacterial phylogenies derived from children’s microbiomes and strain sharing with their mothers.
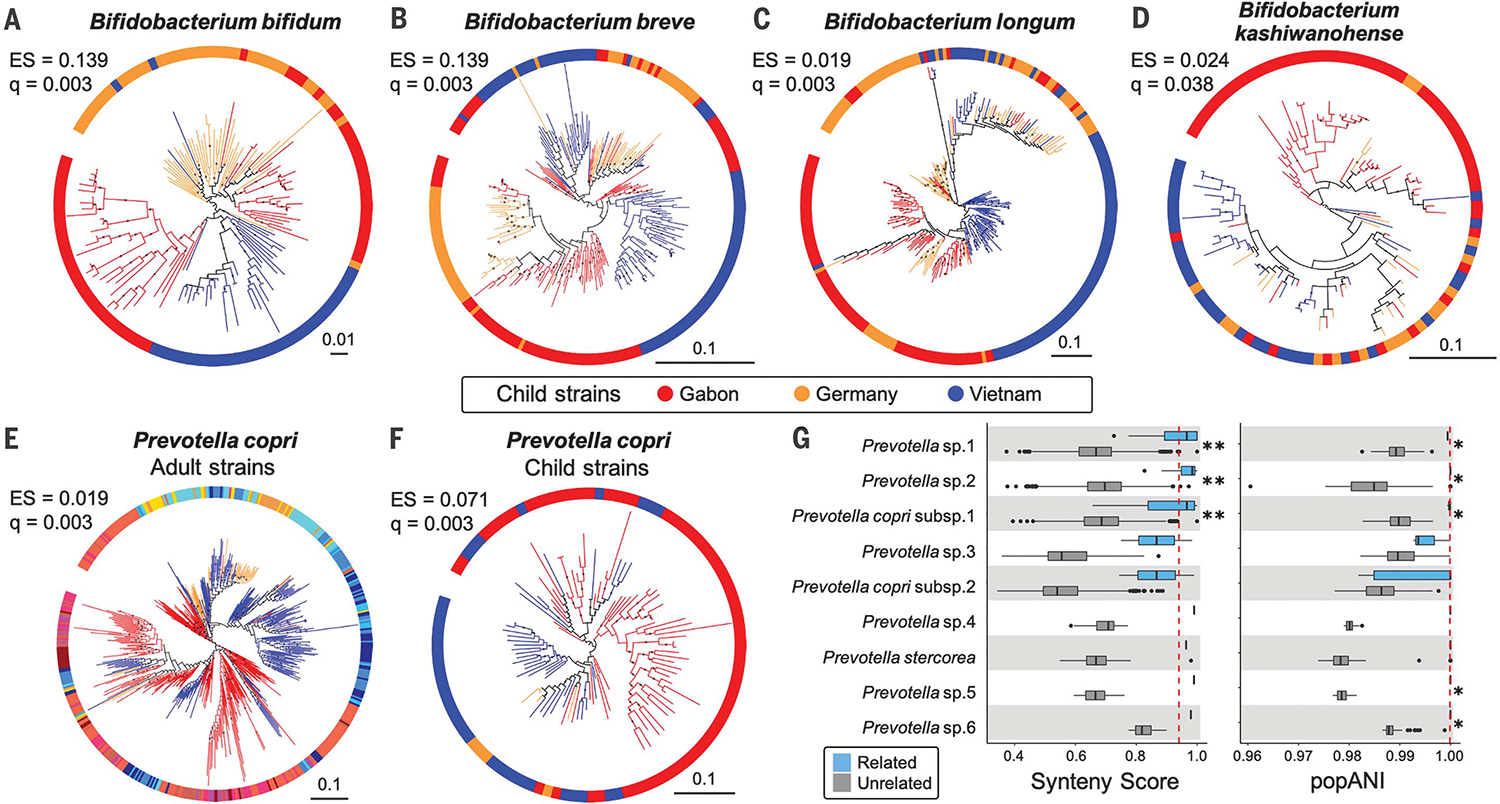
(A to D) Four species of Bifidobacterium show evidence of cophylogeny based on mothers’ genotypes. (E) Phylogeny of P. copri strains in adults and (F) in children. The colors in (E) correspond to those in Fig. 1B. Bootstrap values ≥50% are shown as black dots on branches, and the phylogenies are rooted at the midpoint. The scales show substitutions per site. (G) Prevotella strain sharing between mothers and their own children (“related,” blue boxplots) compared with sharing between women and unrelated children (“unrelated,” gray boxplots). (Left) Strain comparisons using SynTracker (39). (Right) Strain comparisons using inStrain (40). Dashed red lines indicate the thresholds for strain sharing events [0.96 for synteny; 0.99999 for popANI (population-level average nucleotide identity)]. *P < 0.05 and **P < 5 × 10−5 using Wilcoxon-Mann-Whitney test. Codiversification test results for all 20 common child taxa are reported in table S11.
There is little overlap in species composition between adult and child microbiomes; nevertheless, of the overlapping 12 species detected, P. copri (Fig. 2, E and F) and Blautia wexlerae showed evidence of codiversification in both adults (q < 0.01; table S4) and children (q < 0.01; table S11). In addition, we observed that mothers and their children share the same strains of P. copri (Fig. 2G and fig. S2). For mother-child pairs, strain sharing is often interpreted as vertical transmission, but acquisition of strains from a shared environment cannot be excluded (8). Indeed, our data also support strain sharing between community members: Within sampling locations in Gabon and Vietnam, we observed instances of the same strains in the microbiomes of mothers and unrelated children (fig. S2 and tables S13 and S14). Strain sharing is known among families and socially engaging individuals in the human species (25) and other social animal species (26, 27). Although vertical transmission from parents to offspring over long time periods can result in patterns of codiversification, strain transmission between related individuals in the same communities may also contribute to these patterns (28).
Modern humans emerged in Africa before colonizing the rest of the world (29). Microbial species that migrated with their human hosts may also show signatures of out-of-Africa patterns, and, indeed, some bacterial species exhibit such patterns (4–6). To test for an African origin for the species tested here for cophylogeny, we quantified the number and direction of strain transfer events by applying stochastic character mapping. Consistent with out-of-Africa migration events, when the 10 most highly and 10 least highly ranked taxa (by PACo ES) were compared, the top 10 had significantly greater proportions of transfer events from Africa to the rest of the regions compared with the bottom 10 (Wilcoxon rank sum test, P = 0.029) (fig. S3). Because this analysis does not require host genotype data, we added data from 1219 public fecal metagenomes derived from other human populations and from wild primates (tables S15 and S16). Trends were the same with the expanded dataset (Wilcoxon rank sum test, P = 0.089) (Fig. 3, A to D, and fig. S4). Additionally, the top taxa also showed significantly more transfer events from Asia to other regions (Fig. 3, A, E, and F) and fewer transfer events from America to other regions (Fig. 3A). Our results underscore the fact that each species has its own story. Caveats include inaccurate assumptions of host genetic origin based on sampling locations, or a complex history of strain transmission events among different human populations. As expected from codiversification patterns observed in some bacterial families with hominids (10), for the top taxa, primate strains tend to be basal in relation to all human strains (Fig. 3, D and F). In contrast, taxa with least evidence of cophylogeny had primate strains nested within human strains (fig. S5).
Fig. 3. Strain transfer events and microbial phylogenies including data from public metagenomes.
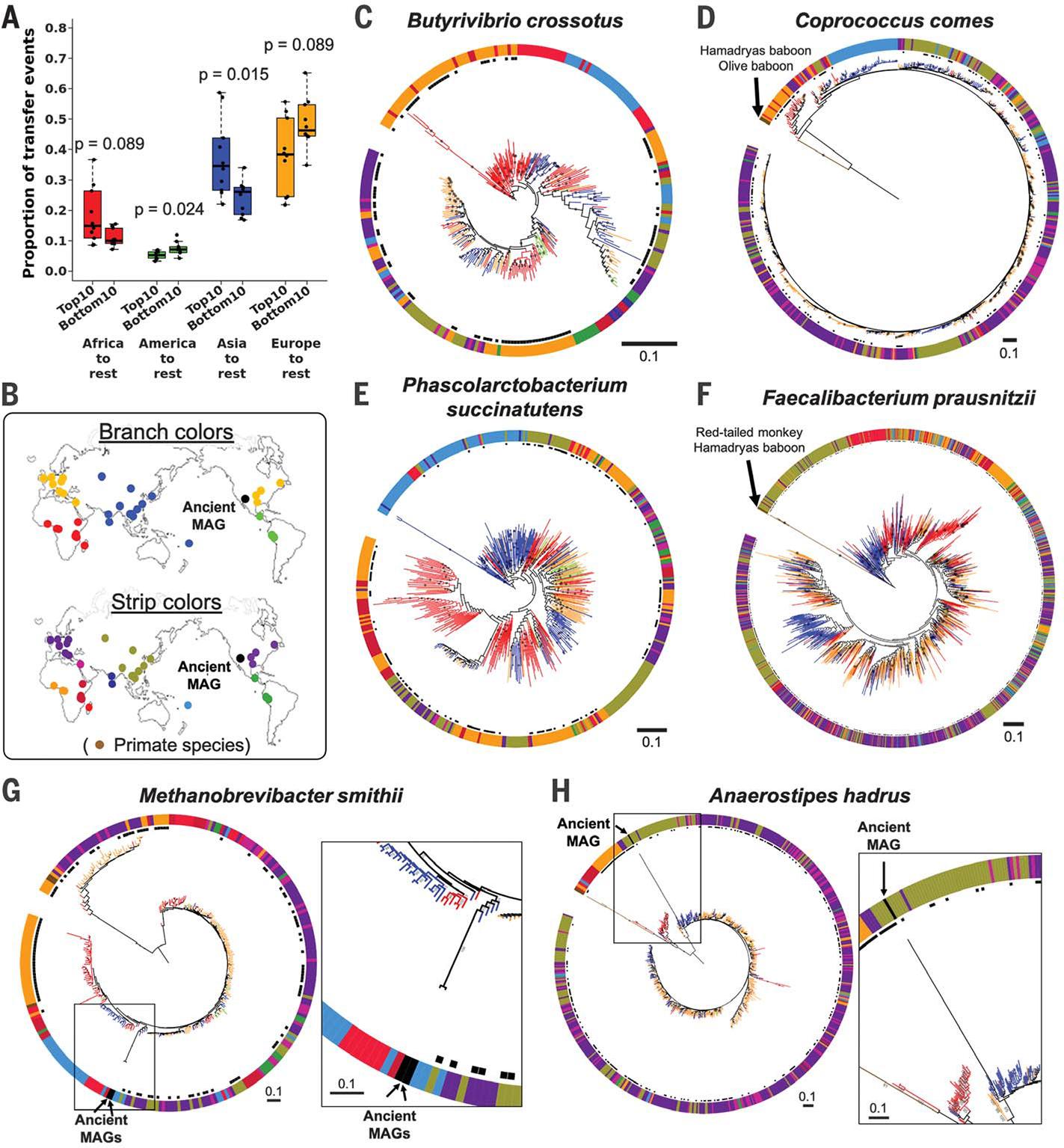
(A) Results from stochastic character mapping on microbial phylogenies including six countries from this study and public metagenomes. The boxplots compare the occurrence of transfer events between sampling regions between the top 10 and bottom 10 taxa identified by PACo ES. P values are based on the Wilcoxon rank sum test. (B) Sampling locations and color keys correspond to the panels that follow. The colors of the branches and outer color strip indicate the estimated host genetic structure based on sampling locations (21). Black dots next to the color strip indicate samples from the original six countries. Example phylogenies: (C) Butyrivibrio crossotus, where African strains are basal; (D) Coprococcus comes, where primate strains are basal, followed by African strains; (E) Phascolarctobacterium succinatutens, where Asian strains are basal; (F) Faecalibacterium prausnitzii, where strains from primates are basal, followed by strains from Asia. (G and H) Examples of microbial phylogenies with ancient MAGs recovered from paleofeces of Native Americans. Bootstrap values ≥50% are shown on branches. All trees were rooted at the midpoint. The scale bars show substitutions per site
Our inference of strain transfer events is based on present-day strains, yet ancient DNA analysis can provide a snapshot directly from the past. We added high-quality ancient MAGs recovered from 1000- to 2000-year-old paleofeces of Native North American tribes (30) to the phylogenies of five gut microbial species (Fig. 3, G and H, and fig. S6). Consistent with the known migration history of the Americas (31), the ancient MAGs were most closely related to strains from modern East Asians, with high bootstrap values for species with significant codiversification (Methanobrevibacter smithii and Anaerostipes hadrus; Fig. 3, G and H); this was not the case for taxa that did not show significant codiversification (fig. S6).
We hypothesized that species that codiversified with their hosts are better adapted to the host environment than those that did not. Therefore, we predicted codiversified species (high PACo ES) to be enriched in features characteristic of host adaptation, including genome reduction, enrichment in AT content and essential functions, and depletion of nonessential functions (32). To test for these traits genomically, we collected publicly available genome sequences for the 59 species (table S17). As expected, the degree of codiversification was inversely correlated to genome size (Fig. 4A) and positively correlated with genomic AT content (fig. S7A). The relationship with genome size, but not with AT content, remained significant after correcting for phylogenetic relatedness (see methods), indicating that genome size reduction arose independently in codiversified taxa.
Fig. 4. Genomic and functional features correlated with codiversification.
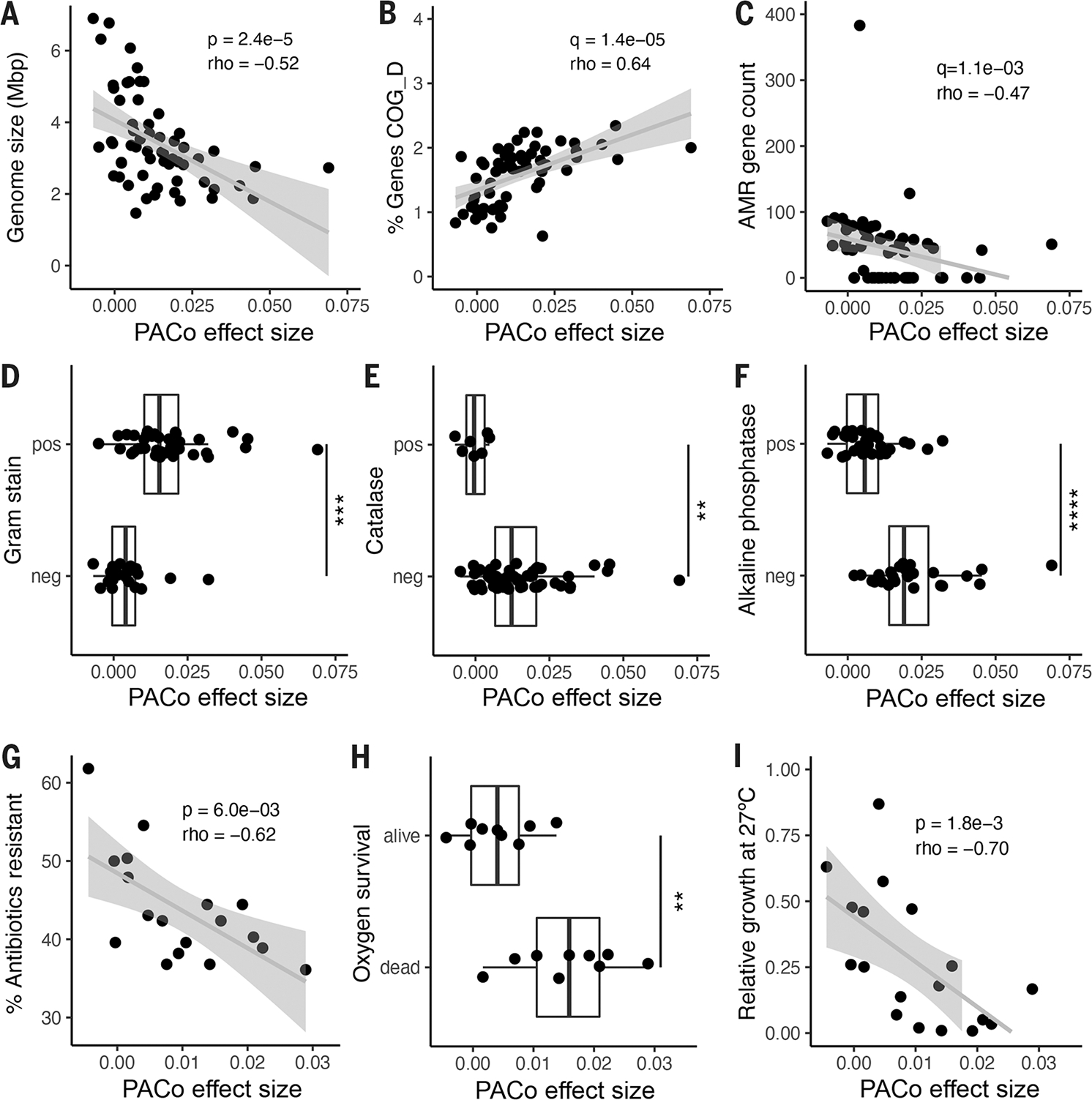
PACo effect size correlated with: (A) Median genome size per species. (B) Percentage of total genes in the genome annotated to COG D for cell cycle and replication. (C) Number of antimicrobial resistance (AMR) markers annotated per genome. (D) Predicted Gram stain per species. (E) Predicted catalase activity per species. (F) Predicted alkaline phosphatase activity. (G) Percentage of antibiotics to which the species was resistant in vitro in a panel of 144 common antimicrobials. (H) Survival in vitro after 48 hours of O2 exposure for a subset of culturable species. (I) Relative growth of each species in vitro at 27°C compared with 37°C. (A) to (F): n = 59 species; (G) to (I): n = 18 species. Statistical significance was determined by Spearman’s correlation [(A) to (C), (G), and (I)] or by Wilcoxon rank sum test [(D) to (F) and (H)]. Exploratory analyses were corrected using FDR across all gene categories and predicted traits [q value; (B) to (F)]. pos, positive for the given trait; neg, negative for the given trait. **P < 0.01, ***P < 0.001, ****P < 0.0001. Exact P values are reported in table S18.
To further explore the genomic signatures of codiversification, we tested for differences in 67 genomic features, including 23 functional categories [clusters of orthologous groups (COGs)], in addition to pseudogenes, antibiotic resistance markers, plasmid markers, and 41 traits predicted from genomic content (see methods and table S18). Overall, ES correlated with 24 of 67 genomic features (FDR-adjusted P < 0.05), and five retained significance after correction for phylogenetic relatedness (table S18). A random forest model including these genomic characteristics accurately predicted ES (PACo q < 0.01), with a mean area under the curve of 0.83 ± 0.22 (SD) across a fivefold cross-validation.
PACo ES was significantly correlated with the proportion of the genome dedicated to essential functions such as replication, transcription, and translation (Fig. 4B, fig. S7B, and table S18). In contrast, greater ES was associated with fewer pseudogenes (fig. S7C) and antibiotic resistance markers Fig. 4C, and a smaller proportion of the genome dedicated for nonessential functions, such as secretion and cell wall biogenesis (fig. S7D and table S18). Gram-positive species were enriched for higher ES overall (Fig. 4D). A number of predicted traits related to environmental survival, including oxygen sensitivity (Fig. 4E), inorganic phosphate scavenging (Fig. 4F), and the use of diverse energy sources (fig. S7, E to I, and table S18), were reduced on average in species with high ES.
To directly test the functions predicted from genome-based observations, we assessed in vitro phenotypes of a representative set of 18 culturable species (see methods and table S19). Consistent with the reduction in antibiotic resistance markers, codiversified species exhibited significantly reduced antibiotic resistance in a previously published drug screen of 144 antimicrobial compounds (Fig. 4G) (33). Consistent with the predicted loss of catalase activity, species with higher ES were significantly more likely to die upon exposure to atmospheric oxygen (Fig. 4H). Increased temperature sensitivity has been associated with coevolved insect symbionts and is expected in gut microbes that enjoy a temperature-stable niche (34). Accordingly, we observed that ES was significantly correlated with poor relative growth at below-host temperature (27°C) (Fig. 4I). All in vitro phenotypic associations retained significance after correction for phylogenetic relatedness, even in cases where the corresponding genomic prediction did not (table S18).
Taken together, the features that associate with cophylogeny are highly reminiscent of those commonly seen in host-associated microbes (35–37) and coevolved insect symbionts (32). Patterns of host-microbial codiversification alone do not necessarily imply interactions or adaptations between hosts and microbes (9, 28). However, together with the observed functional attributes, such as smaller genomes and oxygen and temperature sensitivity, codiversified species likely evolved host dependency.
By expanding metagenome collections into poorly characterized populations, and pairing metagenome and host genomic data obtained from the same individuals, we have identified common members of the human gut microbiota that have independently codiversified with human populations. These codiversified species have repeatedly and independently acquired traits that suggest limited survival capabilities outside of the host (2, 28, 35). Loss of an environmental reservoir can facilitate dependence on resources produced by other gut microbes and/or the host and lead to reduced genome size (35–37). The selection pressure on efficient host-to-host transmission could result in strain sharing between related individuals, or those living in proximity, such as we observed in our populations. Many of the traits characteristic of codiversified species likely adapted to the niche of the animal gut (not necessarily human), and whether humans reciprocally adapted to these microbial species or strains remains to be investigated. The list of codiversified species provides a starting point to investigate host-microbial coevolution in humans (12).
The list of human health conditions linked to the microbiome ranges from mulnutrition to allergies and cardiovascular disease. The incidence of these diseases is population specific, and the diversity of microbiomes is also population specific. Several of the species that codiversified with humans, such as P. copri (4), E. rectale (5), and B. longum (38), are known to vary in their functional capacity according to population. An awareness of differences in gut microbial strains between populations has already led to the notion that probiotics for treating malnutrition should be locally sourced (38). The microbiome is a therapeutic target for personalized medicine, and our results underscore the importance of a population-specific approach to microbiome-based therapies.
Supplementary Material
ACKNOWLEDGMENTS
We thank T. H. Nguyen, E. Cosgrove, A. Clark, A. Kostic, M. Taylor, Native American tribe officers (M. Bremer, J. Aguilar, J. Charlie, Williams, B. Lewis, S. Anton, A. Garcia-Lewis, and B. Bernstein), Dauser and members of the Department of Microbiome Science, and four anonymous reviewers.
Funding:
This work was supported by the Max Planck Society. T.D.S. was funded by the Wellcome Trust, Medical Research Council, European Union, Chronic Disease Research Foundation, Zoe Global Ltd., the National Institute for Health Research–funded BioResource, and the Clinical Research Facility and Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London. L.S. was supported by an Agence Nationale de la Recherche grant (MICROREGAL, ANR-15-CE02–0003). R.B. was supported by NIH grant R35-GM128716.
Footnotes
Competing interests: G.K. is the founder and a board member of KoBioLabs, Inc. T.D.S is a cofounder of ZOE Ltd., a personalized nutrition company.
Data and materials availability: All data used in this study are free to access. The raw sequence data and MAGs are available from the European Nucleotide Archive under the study accession numbers PRJEB40256, PRJEB9584, PRJEB32731, PRJEB27005, PRJEB30834, and PRJEB46788. All sample metadata used in this study are provided in the supplementary materials. The code used in data analysis is outlined on Zenodo (41), and phylogenies and alignments are available in Dryad (42).
REFERENCES AND NOTES
- 1.Gupta VK, Paul S, Dutta C, Front. Microbiol. 8, 1162 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Costea PI et al. Mol. Syst. Biol. 13, 960 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pasolli E et al. Cell 176, 649–662.e20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tett A et al. Cell Host Microbe 26, 666–679.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karcher N et al. Genome Biol. 21, 138 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merrill BD et al. bioRxiv 2022.03.30.486478 [Preprint] (2022). 10.1101/2022.03.30.48647. [DOI]
- 7.Truong DT, Tett A, Pasolli E, Huttenhower C, Segata N, Genome Res. 27, 626–638 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Enav H, Bäckhed F, Ley RE, Cell Host Microbe 30, 627–638 (2022). [DOI] [PubMed] [Google Scholar]
- 9.Moran NA, Sloan DB, PLOS Biol. 13, e1002311 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moeller AH et al. Science 353, 380–382 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishida AH, Ochman H, Nat. Commun. 12, 5632 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki TA, Ley RE, Science 370, eaaz6827 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Falush D et al. Proc. Natl. Acad. Sci. U.S.A. 98, 15056–15061 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Novembre J et al. Nature 456, 98–101 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosenberg NA et al. Science 298, 2381–2385 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Abdill RJ, Adamowicz EM, Blekhman R, PLOS Biol. 20, e3001536 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lokmer A et al. PLOS ONE 14, e0211139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Even G et al. Front. Cell. Infect. Microbiol. 11, 533528 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim MY et al. Gut 66, 1031–1038 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Xie H et al. Cell Syst. 3, 572–584.e3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duda P, Zrzavý Jan, Sci. Rep. 6, 29890 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asnicar F et al. Nat. Commun. 11, 2500 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Legendre P, Desdevises Y, Bazin E, Syst. Biol. 51, 217–234 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Revell LJ, Methods Ecol. Evol. 3, 217–223 (2012). [Google Scholar]
- 25.Brito IL et al. Nat. Microbiol. 4, 964–971 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tung J et al. eLife 4, e05224 (2015).25774601 [Google Scholar]
- 27.Moeller AHAH et al. Sci. Adv. 2, e1500997–e1500997 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Groussin M, Mazel F, Alm EJ, Cell Host Microbe 28, 12–22 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Nielsen R et al. Nature 541, 302–310 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wibowo MC et al. Nature 594, 234–239 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goebel T, Waters MR, O’Rourke DH, Science 319, 1497–1502 (2008). [DOI] [PubMed] [Google Scholar]
- 32.McCutcheon JP, Moran NA, Nat. Rev. Microbiol. 10, 13–26 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Maier L et al. Nature 555, 623–628 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huus KE, Ley RE, mSystems 6, e0070721 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Browne HP et al. Genome Biol. 22, 204 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nayfach S, Shi ZJ, Seshadri R, Pollard KS, Kyrpides NC, Nature 568, 505–510 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frese SA et al. PLOS Genet. 7, e1001314 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barratt MJ et al. Sci. Transl. Med. 14, eabk1107 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Enav H, Ley RE, bioRxiv 2021.10.06.463341 [Preprint] (2021). 10.1101/2021.10.06.463341. [DOI]
- 40.Olm MR et al. Nat. Biotechnol. 39, 727–736 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki TA, Fitzstevens L, Huus K, Youngblut N, leylabmpi/codiversification: Zenodo release, version 1.0.1, Zenodo; (2022); 10.5281/zenodo.6947454. [DOI] [Google Scholar]
- 42.Suzuki TA, Fitzstevens JL, Youngblut ND, Ley RE, Phylogenies related to “Codiversification of gut microbiota with humans,” Dryad; (2022); 10.5061/dryad.qrfj6q5k2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.