

Abstract
Pulmonary hypertension (PH) due to interstitial lung disease (ILD), a commonly encountered complication of fibrotic ILDs, is associated with significant morbidity and mortality. Until recently, the studies of pulmonary vasodilator therapy in PH‐ILD have been largely disappointing, with some even demonstrating the potential for harm. This paper is part of a series of Consensus Statements from the Pulmonary Vascular Research Institute's Innovative Drug Development Initiative for Group 3 Pulmonary Hypertension, with prior publications covering pathogenesis, prevalence, clinical features, phenotyping, clinical trials, and impact of PH‐ILD. It offers a comprehensive review of and a holistic approach to treatment of PH‐ILD, including the management of underlying interstitial lung diseases, importance of treating the comorbidities, emphasis on importance of exercise and palliation of dyspnea, and review of the most up‐to‐date guidelines for referral for potential lung transplant work up. It also summarizes the prior, ongoing, and possibly future studies in treatment of the vascular derangement of this morbid condition.
Keywords: idiopathic pulmonary fibrosis, management, parenchymal lung disease, pulmonary vascular disease
Abbreviations
- 6MWD
6‐min walk distance
- 6MWT
6‐min walk test
- BNP
brain natriuretic peptide
- CO
cardiac output
- DLCO
diffusion capacity for carbon monoxide
- ERA
endothelin receptor antagonist
- FC
functional class
- FVC
forced vital capacity
- IIP
idiopathic interstitial pneumonia
- ILD
interstitial lung disease
- iNO
inhaled nitric oxide
- IPF
Idiopathic pulmonary fibrosis
- mPAP
mean pulmonary artery pressure
- PAH
pulmonary arterial hypertension
- PH
pulmonary hypertension
- PVR
pulmonary vascular resistance
- QOL
quality of life
- RCT
randomized controlled trial
- RHC
right heart catheterization
- V/Q
ventilation/perfusion
INTRODUCTION
Pulmonary hypertension (PH) due to interstitial lung disease (PH‐ILD) is a commonly encountered clinical problem with an estimated prevalence of 10%–80% in patients with idiopathic pulmonary fibrosis (IPF) and is also common in other fibrotic ILDs. 1 , 2 Multiple studies have established that the development of PH is associated with substantial morbidity and reduced survival. 1 , 2 , 3 Until recently, the studies of pulmonary vasodilator therapy in PH‐ILD have been largely disappointing, with some even demonstrating the potential for harm. 3 , 4 , 5 , 6 This paper is part of a series of Consensus Statements from the Pulmonary Vascular Research Institute's Innovative Drug Development Initiative (IDDI) for Group 3 Pulmonary Hypertension, with prior publications covering pathogenesis, prevalence, clinical features, phenotyping, clinical trials, and impact of PH‐ILD. The IDDI is a transformational task force within the PVRI that facilitates the development of novel treatments for patients with PH and right ventricular failure. This is done by providing a worldwide platform that encourages an open discussion between academia, regulatory bodies, patient representatives, and pharmaceutical and device companies, thus driving innovative clinical and regulatory therapeutic development strategies. The present manuscript is a focused review providing an overview and update pertaining to the management of PH in the setting of ILD.
PATIENT PHENOTYPING
A principal diagnostic challenge in patients with a potential diagnosis of PH is to determine, based on a compendium of information including the history, physical signs, pulmonary function tests, imaging, and additional laboratory data, the most appropriate World Symposium Pulmonary Hypertension (WSPH) pulmonary vascular phenotype for an individual patient. This has obvious therapeutic implications since most (but not all) of the available agents are approved for Group 1 and/or Group 4 PH as primarily vascular driven diseases. An example of this frequent dilemma is in connective tissue disease (CTD)‐associated PH (particularly scleroderma) where patients with PH could fit into any of the first four groups. For example, Group 1 pulmonary arterial hypertension (PAH) if the lung parenchymal fibrosis is absent or very mild (as is often the case in limited scleroderma), Group 2 disease (with left ventricular dysfunction given the relatively older age of these patients), Group 3 (with more extensive parenchymal fibrosis, particularly in patients with diffuse scleroderma), and rarely in Group 4 (chronic clots) or Group 5 (e.g., renal failure) PH. Therefore, a careful integration and interpretation of all clinical and laboratory data is mandatory to determine what is the “best fit” group for an individual patient.
It is increasingly evident that patients may fit into more than one WSPH Group given the presence of associated comorbidities, as noted in several large registries and more recently by the PVDOMICS study group. 7 Distinguishing Group 1 from Group 3 PH is particularly challenging when lung function is preserved, and chest imaging shows mild parenchyma process (Figure 1), with many cases falling within a “grey zone.” 5 As part of this spectrum, a recently coined entity is iPAH with a “lung phenotype.” 8 These patients are typically elderly males with a heavy smoking history and may have occult parenchymal disease such as emphysema without airflow limitation or early stage ILD. In all cases, careful review of the HRCT and integration of all the clinical and laboratory data can help distinguish between Groups 1 and 3 PH (Figure 1).
Figure 1.
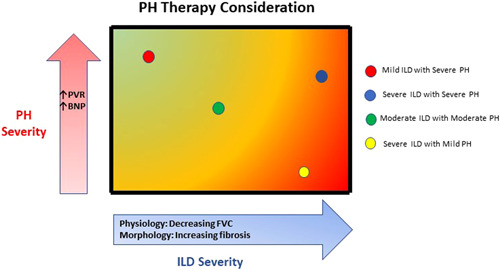
Pulmonary hypertension (PH) in interstitial lung disease (ILD) treatment according to disease phenotype. Patients with ILD and PH might present different phenotypes according to the severity of both ILD and PH. These phenotypes sit across a spectrum for both conditions; the phenotype should be taken into account when therapeutic strategies are considered for PH‐ILD patients.
A frequent lung function threshold employed in various clinical trials and suggested by the WSPH for Group 1 is a forced vital capacity (FVC) > 70% or an FVC between 60% and 70%, provided there are either no or limited parenchymal abnormalities on HRCT. 1 , 9 , 10 However, this recommendation is complicated by the fact that lung volume abnormalities correlate poorly with the extent of fibrosis, and that subtle fibrotic changes on HRCT can be associated with significant physiologic derangement. 11 , 12 A parameter that plays a significant role in the effort at unifying impact of the vascular disease among disparate diseases is the single breath diffusing capacity for carbon monoxide (DLCO). A low DLCO has been shown to be predictive of high mortality, both in iPAH and PH‐ILD. 13 , 14 , 15 A relative disconnect between the severity of PH and the extent of disease on lung function tests or fibrotic findings on HRCT might suggest a primary pulmonary vascular disease process and possibly Group 1 PAH phenotype of the disease. 1
TREATMENT OF UNDERLYING ILD
It makes intuitive sense that treating the underlying primary disease should have an impact on the trajectory of associated PH although this concept has not been studied in great depth. Immunosuppressive medications were a mainstay therapy for many forms of fibrotic ILDs, including IPF, until the PANTHER‐IPF study demonstrated that this approach was associated with worse outcomes including increased hospitalizations and mortality. 16 Thus, the use of immunosuppressive medications is not currently recommended in IPF and other fibrotic ILDs. 16 , 17 , 18 Immunosuppressive therapy is still recommended for patients with other forms of ILD when an inflammatory component is thought to be playing a role, such as some connective tissue‐related ILD, cellular or mixed nonspecific interstitial pneumonia and, in some instances, chronic hypersensitivity pneumonitis.
Previously, there were no approved therapies to halt or delay the progression of fibrotic lung diseases. However, in 2014 the US Food and Drug Administration approved two drugs, pirfenidone and nintedanib, for the treatment of IPF, ushering in a new era for the treatment of IPF. Subsequently, the 2015 updated ATS/ERS/JRS/ALAT treatment guidelines on the management of IPF included a conditional recommendation for the use of both pirfenidone and nintedanib. 17 Both agents are now variably available in many countries throughout the world. Subsequently, nintedanib was approved for both non‐IPF progressive fibrotic lung diseases and scleroderma‐related ILD. 18 , 19 , 20 The 2022 ATS Statement on IPF and Progressive Pulmonary Fibrosis (PFF) recommended antifibrotic therapy with nintedanib as a first‐line treatment for PFF, thus further cementing the role of antifibrotics in the management of fibrotic ILDs. 18
The direct effect of either of the antifibrotics on progression of PH has not been studied but warrants further investigation. It is a reasonable hypothesis that by halting disease progression and limiting parenchymal destruction, these medications may have an indirect effect on the development or progression of PH. Conversely, a somewhat fortuitous finding from two clinical trials of pulmonary vasodilatory drugs is that these agents may have a salutary effect on the progression of fibrosis. In the INSTAGE trial, patients treated with sildenafil and nintedanib had significantly less risk in meeting the composite endpoint of a 5% reduction in FVC or death. 21 In the INCREASE trial, the use of inhaled treprostinil was associated with a significant placebo‐corrected improvement in FVC over 16 weeks in the post‐hoc analysis of the data. 22 , 23 In addition, open label long term data showed sustained improvements in FVC over 52 weeks. 24 These results are hypothesis generating and FVC improvement associated with inhaled treprostinil is being prospectively evaluated in patients with IPF, agnostic to the presence or absence of PH, in the TETON studies. 25
Pirfenidone is a synthetic compound that probably has multiple different mechanisms of action that are still not fully understood; however, the drug appears to have antifibrotic properties via regulation of profibrotic growth factors such as transforming growth factor (TGF)‐β and tumor necrosis factor (TNF)‐α. 26 The conditional recommendation for the use of pirfenidone was based on three large randomized, double‐blinded, placebo‐controlled trials: the two CAPACITY studies and the ASCEND study. 27 , 28 Both CAPACITY trials were almost identical phase III, randomized, double‐blind, placebo‐controlled with change in the percentage predicted FVC from baseline to Week 72 as the primary endpoint. 27 Although the primary endpoint was met in only one of the studies, the two studies together did show a significant difference favoring pirfenidone which was sufficient for the European Medicines Agency to approve pirfenidone in 2011. However, the combined data set was not met with approval in the United States, and a third study was undertaken. The ASCEND study, the 3rd phase III trial, was a 52‐week study and was a positive trial based on the same primary endpoint. This subsequently enabled the approval of pirfenidone in the United States in 2014. 28
Neither the CAPACITY nor ASCEND trials were individually powered to show a difference in mortality, but combined data from these trials showed a beneficial effect in long‐term (up to 120 weeks) all‐cause, IPF‐related, and on‐treatment mortality. 29 Additionally, a post‐hoc analysis showed that patients who manifest progression of disease, as defined by either a 10% reduction in FVC, a 15% decrease in 6‐min walk distance (6MWD) or hospitalization, continue to derive significant benefit from ongoing therapy with pirfenidone. 30 A meta‐analysis also demonstrated that pirfenidone decreased disease progression and mortality in IPF. 31
Nintedanib is a triple tyrosine kinase inhibitor targeting fibroblast growth factor receptor in addition to platelet‐derived growth factor receptor and vascular endothelial growth factor (VEGF) receptor. 32 The Phase 2 TOMORROW trial found that nintedanib favorably impacted the decline in pulmonary function and time to acute exacerbation in patients with IPF. 33 The two replicate Phase III INPULSIS studies both demonstrated a positive effect of nintedanib on the primary endpoint of a reduction in the annual rate of decline in FVC. 34 Additionally, in INPULSIS‐2, a significant increase in the time to the first acute exacerbation was observed in the nintedanib arm. Neither the TOMORROW, nor the INPULSIS trials were powered to show a difference in mortality, but a meta‐analysis of these three trials demonstrated hazard ratios in favor of nintedanib for both all‐cause and on‐treatment mortality. 35 A subsequent subgroup analysis of pooled data from the two INPULSIS trials support the beneficial effect of nintedanib in IPF patients with well‐preserved lung function. 36 Also, the INPULSIS‐ON open‐label extension trial showed that IPF patients with severe physiologic impairment (FVC < 50% predicted) derived the same beneficial effects on FVC decline as those patients with less severe disease. 37
Patients with combined pulmonary fibrosis emphysema syndrome (CPFE) with significant emphysema (greater than the volume of fibrosis on HRCT) and/or significant airflow obstruction were excluded from the studies of both pirfenidone and nintedanib. 27 , 28 , 35 A subgroup analysis of the IPF INPULSIS trials with nintedanib found that the presence of mild‐to‐moderate emphysema did not have any impact on the magnitude of the antifibrotic treatment effect. 38 According to the most recent Official ATS/ERS/JRS/ALAT Research Statement published in 2022, 39 antifibrotic medications may have benefit in patients with CPFE with IPF and in other forms of pulmonary fibrosis. In addition, inhaled bronchodilators may have benefit in select patients with significant airflow limitation. 39
COMORBIDITIES COMMONLY ASSOCIATED WITH PH IN THE SETTING OF ILD
There are a number of comorbidities that can cause or contribute to the development of PH in ILD. Once PH has been diagnosed, it is incumbent to identify any comorbidity, potentially causative or contributory, since therapies directed at these may impact symptomatology and result in improved quality of life (QOL). Entities to consider include emphysema, sleep disordered breathing and/or obstructive sleep apnea (OSA), heart failure with preserved ejection fraction (HFpEF), coronary artery disease (CAD), cardiac arrhythmias, and acute or chronic thromboembolic disease. 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 Diuretics should be used aggressively to treat any fluid overload due to right‐sided heart failure. Finally, patients should be referred for pulmonary rehabilitation to address any deconditioning (Figure 2).
Figure 2.
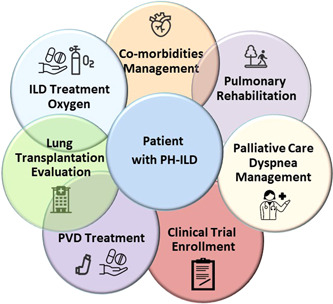
Holistic management of PH in ILD. When treating patients with ILD associated with PH it is recommended to take an holistic approach: in addition to considering pulmonary vasodilator drugs, several interventions such as lung transplant referral, optimal oxygen and antifibrotic therapy, the correct treatment for comorbidities, dyspnea and palliative care management, pulmonary rehabilitation and finally enrollment in available clinical trials may help improve the outcomes of these patients. ILD, interstitial lung disease; PDE5‐i, phosphodiesterase‐5 inhibitors; PH, pulmonary hypertension; PVD, pulmonary vascular disease.
Concomitant emphysema has been described in ~28% of IPF patients. A syndrome of ILD and concomitant emphysema is coined CPFE and is often associated with severe PH, which in turn portends a poor prognosis. 41 , 42
Hypoxemia should be actively sought and treated. Relying on information from the resting pulse oximetry is not sufficient, as it might not reflect the extent of nocturnal desaturation, or level of desaturation endured during patients' activities of daily living. 43 , 44 , 45 It is also important to be aware that pulse oximetry may underestimate the degree of desaturation and supplemental oxygen needs in patients with dark skin. 46 The 6‐min walk test (6MWT) and nocturnal oximetry are important to detect exertional and nocturnal desaturation, respectively. Patients with IPF have a higher prevalence of sleep disordered breathing, and further evaluation with overnight polysomnography should be considered in patients with nocturnal desaturation. 43 , 44 , 45 , 47
The role of anticoagulation is unclear as there are no studies evaluating this therapy in PH‐ILD. However, patients with idiopathic interstitial pneumonias (IIPs), including IPF, are at increased risk of pulmonary emboli possibly due to decreased mobility and/or a thrombotic predisposition which has been described in IPF. 40 , 48 An earlier study of anticoagulation in IPF without concomitant PH showed a potential mortality benefit of anticoagulation in IPF. 49 The follow‐up NIH IPF Network ACE‐IPF trial, however, failed to show any benefit for warfarin in the treatment of patients with progressive IPF. Moreover, treatment with warfarin was associated with an increased risk of mortality in IPF patients who lacked other indications for anticoagulation. 50 Nonetheless, if there are other reasons for anticoagulation, such as atrial fibrillation, this should not be withheld. In addition, an analysis of the Pulmonary Fibrosis Registry database did show that the use of direct oral anticoagulants is not associated with increased mortality. 51
The prevalence of CAD is high (60%) in the IIPs, specifically in IPF. 52 The risk of CAD appears to be independent of smoking, is frequently unrecognized and is associated with worse survival. 53 HFpEF has been described in between 15% and 28% of patients with IPF. 54 In addition, approximately 20% of patients with PH have a postcapillary phenotype due to underlying diastolic heart failure. 55 , 56 Therefore, a thorough evaluation for both coronary disease and HFpEF should be pursued. With regard to the latter, right heart catheterization (RHC) with fluid challenge can help determine the contribution of diastolic dysfunction to mechanism of PH and exercise limitation. 57
In addition to medical treatment of the underlying fibrotic lung disease, comorbidities, and associated PH, referral to palliative care should be considered throughout the course of disease. The morbidity associated with ILD alone is considerable and is made worse by the development of PH. Due to misconceptions that palliative care equates to end‐of‐life care, referral to palliative care is infrequent in patients with ILD as it is in those with PAH. 58 , 59 , 60 , 61 , 62 Palliative care should be considered as an integral part of a holistic approach to help patients (and their caregivers) live as well as possible with their conditions and therefore can be considered at any time in the disease course (Figure 2).
OVERVIEW OF PREVIOUSLY PUBLISHED TRIALS
To date, most clinical trials targeting ILD with pulmonary vasodilators have focused on ILD without PH (Table 1). Although subgroup analyses of patients with PH enrolled in ILD studies have revealed a few promising signals for efficacy, most studies have been negative with a few showing harm. 1 , 3 To date, in the United States, inhaled treprostinil is the only therapy approved by a regulatory body with an indication for PH‐ILD (Tyvaso U.S. package insert). 22
Table 1.
Clinical trials targeting ILD with pulmonary vasodilators.
| Lung disease | Investigator | Year | Study design | Pts | Therapy | Results | Comments |
|---|---|---|---|---|---|---|---|
| ILD/IIP/IPF trials | |||||||
| IPF | Collard et al. 63 | 2007 | Open label | 14 | Sildenafil (N = 14) |
57% improved 6MWT by ≥20% |
Median follow‐up of 91 days |
| IPF | Zisman et al. 64 | 2010 | RCT | 180 | Sildenafil (n = 89) |
Failed to improve 6MWT by ≥20% |
Improved oxygen saturation and QOL |
| IPF | Jackson et al. 65 | 2010 | RCT | 29 | Sildenafil (n = 14) | No difference in 6MWT or Borg score | |
| IPF | Gunther et al. 66 | 2007 | Open label | 12 | Bosentan (n = 12) | No worsening of gas exchange | |
| IPF | King et al. 67 | 2008 | RCT | 158 | Bosentan (n = 74) | Failed to improve 6MWT | Trend to delayed death or disease progression |
| IPF | King et al. 68 | 2011 | RCT | 610 | Bosentan (n = 407) | No effect on time to IPF worsening or death | |
| IPF | Raghu et al. 69 | 2013 | RCT | 492 | Ambrisentan (n = 330) | Terminated early for lack of efficacy in time to clinical worsening | In 32 patients with PH—no change in time to disease progression |
| IPF | Raghu et al. 70 | 2013 | RCT | 178 | Macitentan (n = 178) | Failed to alter primary endpoint of change in FVC | |
| IPF | Kolb et al. 71 | 2018 | RCT | 274 |
Nintedanib + Sildenafil (n = 137) |
Primary endpoint of change in SGRQ was not met | Enriched for PH (DLCO ≤ 35%) |
|
Fibrotic ILD |
Nathan et al. 72 | 2020 | RCT | 45 | iNO (n = 23) | Improvement in moderate to vigorous activity and overall activity | Enriched for PH (supplemental O2) |
| PH associated with ILD/IIP/IPF trials | |||||||
| PH‐ILD | Faria‐Urbina et al. 73 | 2018 | Retrospective | 22 |
Treprostinil (inhaled) (n = 22) |
Improvement in FC Improvement in 6MWD No change in resting O2 requirements |
|
| PH‐PF | Ghofrani et al. 74 | 2002 | Open label | 16 |
Sildenafil (n = 8) Epoprostenol (n = 8) |
Sildenafil improved V/Q matching and oxygenation | Epoprostenol worsened V/Q matching |
| PH‐IPF | Krowka et al. 75 | 2007 | RCT | 51 |
Iloprost (inhaled) (n = 26) |
No difference in 6MWT, NYHA FC, dyspnea score | |
| PH‐ILD | Chapman et al. 76 | 2009 | Retrospective | 5 | Sildenafil (n = 5) | Improved 6MWT | Decreased mPAP 2–12 months after start of treatment |
| PH‐ILD | Corte et al. 77 | 2010 | Retrospective | 15 | Sildenafil (n = 15) | Improved 6MWT and lower BNP | |
| PH‐ILD | Hoeper et al. 78 | 2013 | Open label | 22 | Riociguat (n = 22) | Improved CO and PVR but not mPAP | Arterial saturation decreased but mixed‐venous saturation increased |
| PH‐ILD | Zimmerman et al. 79 | 2014 | Open‐label, observational | 10 |
Sildenafil (n = 5) Tadalafil (n = 5) |
Increased CO and decreased PVR | No change in 6MWD or BNP |
| PH‐ILD | Corte et al. 80 | 2014 | RCT | 60 | Bosentan (n = 25) | No effect on hemodynamics, symptoms, or functional capacity | |
| PH‐ILD | Saggar et al. 81 | 2014 | Open label | 15 |
Treprostinil (parenteral) (n = 15) |
Improved hemodynamics without hypoxemia | All had mPAP ≥ 35 mmHg |
| PH‐ILD (CPFE & ILD) | Brewis et al. 82 | 2015 | Retrospective | 118 |
PDE‐5i (n = 31) ERAs (n = 11) Prostanoids (n = 1) |
Unchanged 6MWT, decreased BNP in ILD patients | |
| PH‐IIP | Nathan et al. 83 | 2019 | RCT | 147 | Riociguat (n = 73) | Terminated early for unfavorable risk/benefit profile | |
| PH‐ILD | Waxman et al. 22 | 2021 | RCT | 326 |
Treprostinil (inhaled) (n = 163) |
Improved 6MWD, NT‐proBNP, clinical worsening, and FVC | |
| PH‐ILD | Dawes et al. 84 | 2022 | Retrospective | 60 |
PDE‐5i (n = 50) ERAs (n = 10) |
Patients treated with sildenafil had longer survival | No effect on V/Q mismatching |
Abbreviations: 6MWT, 6‐min walk test; BNP, brain natriuretic peptide; CO, cardiac output; DLCO, diffusion capacity for carbon monoxide; ERA, endothelin receptor antagonist; FC, functional class; FVC, forced vital capacity; IIP, idiopathic interstitial pneumonia; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis; mPAP, mean pulmonary artery pressure; PDE‐5i, phosphodiesterase 5 inhibitor; PF, pulmonary fibrosis; PH, pulmonary hypertension; PVR, pulmonary vascular resistance; RCT, randomized controlled trial; RHC, right heart catheterization; SGRQ, St. George's Respiratory Quotient; QOL, quality of life; V/Q, Ventilation/Perfusion; WHO, World Health Organization.
A number of randomized, placebo‐controlled studies have evaluated endothelin receptor antagonists (ERAs) in patients with ILD. There is a rationale for the use of these agents over and above their role as vasodilators. Specifically, preclinical information indicated that endothelin‐1 (ET‐1) is both profibrotic and a mitogen. 85 ERAs were an obvious choice as treatments of PH‐ILD, to tackle both components. 86 In the BUILD‐1 study of IPF, bosentan did not significantly improve 6MWD over placebo. 67 In the BPHIT study of patients with fibrotic IIPs and associated PH, treatment with bosentan was not associated with improved hemodynamics, functional capacity, or PH symptoms. 87 The BUILD‐3 study failed to show an effect of bosentan on time to clinical worsening or death in patients with IPF. 68
In the ARTEMIS‐IPF study, treatment with ambrisentan was associated with an increased risk for disease progression and respiratory hospitalization resulting in the study being terminated early. 69 A subgroup analysis of patients with PH confirmed by RHC who enrolled in ARTEMIS‐IPF revealed similar results to the overall study population. 69 Based on the former study, the 2022 ESC/ERS Guidelines recommend against the use of ambrisentan in patients with IPF‐PH. 88 In the MUSIC trial, macitentan did not demonstrate any improvements in pulmonary function tests or time to disease worsening or death. 70 Combined with the bosentan data, it appears that the ERA class of medications is not useful and may be harmful in patients with PH‐ILD.
Clinical trial data for sildenafil is somewhat mixed. 63 , 64 , 65 , 71 , 74 , 76 , 77 , 89 The STEP‐IPF trial evaluated sildenafil in 180 patients with advanced IPF (defined by a DLCO ≤ 35% predicted) and did not meet its primary endpoint of a ≥20% improvement in the 6MWD. 64 There were small but statistically significant improvements in arterial oxygenation and DLCO in the sildenafil treatment arm with no notable safety signals. 64 Subjects with right ventricular dysfunction treated with sildenafil experienced less decrement in 6MWD and greater improvement in the quality‐of‐life assessments. 89 The same approach for trial enrichment (DLCO ≤ 35% predicted) was adopted in the INSTAGE trial, which evaluated sildenafil in combination with the antifibrotic nintedanib in patients with IPF. 71 This study failed to meet its primary endpoint of change in QOL as evaluated by the St. George's Respiratory Questionnaire at 12 weeks. One of the secondary endpoints, the prespecified composite endpoint of death and FVC decline of ≥5%, favored combination therapy with sildenafil and nintedanib. 71 A few studies of sildenafil in ILD (including IPF) and fibrotic ILD with associated PH have shown clinical improvements in 6MWD and brain natriuretic peptide (BNP), but these data are limited by their lack of placebo arms and small sample sizes. 63 , 65 , 74 , 76 , 77 According to the 2022 ESC/ERS Guidelines, phosphodiesterase 5 inhibitors may be considered in patients with PH associated with ILD on an individual basis, especially in those with severe vascular disease. 88
Riociguat was evaluated in the RISE‐IIP phase 3 study which enrolled patients with IIP and PH confirmed by RHC. 83 Treatment with riociguat did not improve 6MWD, the primary endpoint, and the study was terminated early due to increased serious adverse events and mortality in patients receiving riociguat, both in the randomized portion of the study as well as the open‐label extension. Therefore, the 2022 ESC/ERS Guidelines recommend against the use of riociguat in patients with IIP‐PH. 88 A subsequent post‐hoc analysis of chest CT scans from patients with available studies suggested that the group with advanced CPFE, and the presence of more emphysema than fibrosis, potentially drove the negative signal of the study. 90
Inhaled nitric oxide (iNO) has been investigated as a treatment to augment the endogenous nitric oxide pathway which may be altered in PH‐ILD. Phase 2b/3 trial of pulsed iNO in ILD patients on supplemental oxygen at risk of PH demonstrated that treatment with iNO was associated with improvements in moderate to vigorous physical activity (MVPA) measured by actigraphy when compared with placebo. 72 Furthermore, during the open label extension patients maintained their activity levels including placebo subjects who transitioned from a decline to maintenance in moderate to vigorous activity. 72 At the time of the submission of this manuscript, according to the company press release, Phase 3 REBUILD trial of iNO45 in patients with fibrotic ILD on supplemental oxygen did not meet its primary endpoint of the change in MVPA. 91
Prostanoids have also been evaluated in PH‐ILD patients. Small studies using parenteral prostanoids have been shown to improve functional class (FC), 6MWD, echocardiographic parameters, and hemodynamics. 81 , 92 One theoretical drawback of systemic vasodilator therapy in ILD is decreased oxygenation due to worsening ventilation/perfusion (V/Q) mismatch, as was observed in one small study of IV epoprostenol and oral sildenafil. 77 Inhaled therapies may circumvent this concern by causing vasodilation in well‐ventilated areas of the lung, thus preserving V/Q matching. This hypothesis was first investigated by Olschewski and colleagues comparing the effects of intravenous prostacyclin, systemic calcium channel blockers versus iNO and aerosolized prostacyclin/iloprost in a randomized order in 8 patients with severe PH secondary to lung fibrosis in 1999. 93 Inhalation of prostacyclin/iloprost or NO caused preferential pulmonary vasodilatation with a decrease in mean pulmonary arterial and pulmonary vascular resistance (PVR) without deleterious effects on oxygenation, systemic blood pressure or pulmonary right‐to‐left shunt flow, as measured by multiple inert gas analysis. In contrast, both intravenous prostacyclin and systemic calcium antagonists were not pulmonary vasculature selective, resulting in a significant drop in arterial pressure. In addition, prostacyclin infusion caused a marked increase in shunt flow. One patient received long‐term treatment with inhaled iloprost showing a tendency toward improvement of hemodynamics and 6MWD during 12 months of treatment. This observation was further supported by a retrospective review of 22 patients with severe PH (mean pulmonary artery pressure [mPAP] ≥35 mmHg or mPAP ≥ 25 mmHg with PVR > 4 WU) associated with parenchymal lung disease (36% COPD, 41% ILD and 23% CPFE) treated with inhaled treprostinil which showed significant improvements in FC and 6MWD without adverse effects on peripheral oxygen saturation. 73
Despite these early promising reports of inhaled prostanoids in severe PH‐ILD, it took another two decades to demonstrate efficacy for this condition. The INCREASE trial, a 16‐week randomized controlled trial (RCT) of inhaled treprostinil in patients with PH‐ILD, met its primary endpoint of change in 6MWD at Week 16. 22 The study enrolled patients with various etiologies of ILD, including IPF (44.8%), chronic hypersensitivity pneumonitis (5.8%), CPFE (25.2%), and CTDs (22.1%). 22 The study's secondary endpoints including change in NT‐proBNP and time to clinical worsening were also met. Inhaled treprostinil was found to be safe and well tolerated in this patient population and demonstrated the known side effects of the drug, including throat irritation, cough, headache, and diarrhea. The positive outcome of INCREASE trial resulted in FDA approval of inhaled treprostinil for PH‐ILD in the United States in early 2021. The ESC/ERS 2022 Guidelines state that inhaled treprostinil may be considered in patients with PH‐ILD. 88 In the forward‐looking section, the Guidelines asked for long‐term data for inhaled treprostinil, which since has been published. 24 , 88 Treatment of PH in ILD is not addressed in the most recent ATS/ERS/JLA/ALAT 2022 IPF Guidelines. 18 Thus, there are currently no guideline recommendations addressing the use of inhaled treprostinil in the United States, which remains the only country where this therapy is available.
Several post‐hoc analyses of the INCREASE trial have suggested that treatment with inhaled treprostinil has additional clinical benefits. One analysis demonstrated that patients across all the ILD etiologies who received inhaled treprostinil were less likely to experience multiple disease progression events compared with placebo. 94 The therapeutics benefits of inhaled treprostinil may be dose‐related, as patients who reached a dose of over nine breaths per session four times per day had both a lower rate of clinical worsening, and a higher rate of clinical improvement. 95 More recently, long‐term data from the INCREASE open‐label extension demonstrated stabilization of exercise capacity and a durable improvement in the FVC. 24 It should be noted that proven efficacy and safety of inhaled treprostinil in both PAH or PH‐ILD should not be extrapolated to other PH indications or other causes of Group 3 PH, and separate trials are needed.
A meta‐analysis published before the INCREASE trial investigated the effect of PAH therapy on 6MWT in two RCT and four single arm studies in patients with PH‐ILD. 96 In the single arm studies, there was a significant improvement in 6MWD (46.2 m; 95% CI, 27.9–64.4), whereas in the RCTs the improvement did not reach statistical significance (21.6 m; 95% CI, −17.8 to 61.0).
INVESTIGATIONAL THERAPEUTIC AND DEVICE‐BASED THERAPIES
Whereas an inhaled compound from the prostacyclin pathway have shown efficacy and safety in PH‐ILD recently, several inhaled compounds that work on the cGMP/sGC are being further investigated in various forms of PH. Two independent soluble guanylate cyclase (sGC) modulators delivered via dry powder inhalers designed to selectively target the lung vasculature are being studied clinically in Group 1 PAH and CTEPH 97 , 98 and may also have a potential for the treatment of PH due to chronic lung disease. However, at this time it is unknown if they will be studied in PH‐ILD in the future. Other inhalation antiproliferative products are currently being investigated in Phase 2 and 3 trials for the treatment of Group 1 PAH, including tyrosine kinase inhibitors imatinib and seralutinib. 99 , 100 , 101 It is unknown if these compounds will be studied in patients with PH due to ILD, although such studies are encouraged.
Bardoxolone methyl, an oral once‐daily antioxidant inflammation modulator that acts via Nrf2 pathway, was investigated in a Phase 2 LARIAT study for safety and efficacy in patients with PH. As per a company press release, eight patients with IPF‐PH were included, and bardoxolone was found to improve exercise capacity with a 6MWD change of 38 m in this small cohort. 102 It is unknown if further studies of bardoxolone in PH‐ILD will be pursued.
Hyaluronan (HA) is a large glycosaminoglycan composed of repeating units of glucuronic acid and N‐acetyl‐glucosamine. Expression of HA was shown to promote an invasive fibroblast phenotype that increases lung fibrosis. In addition, HA overexpression also results in vascular remodeling and the increased presence of inflammatory cells in the lung. 103 Furthermore, the pathological deposition of HA in the alveolar regions and surrounding small arterioles correlates with the presence of PH. 104 Hymecromone (H01), also known as a 4‐methylumbelliferone, is a coumarin derivative that inhibits the synthesis of HA. It is currently being studied in a proof‐of‐concept Phase 2 safety and efficacy study in patients with PH, including a subset of patients with ILD. 105
Several medical devices are under investigation for the treatment of PH. In general, they are intended for use in Group 1 PAH, but their mechanisms of action may also be beneficial in Group 3 physiology. In one example, early clinical work suggests that a device‐based enhancement of compliance in the pulmonary vasculature may improve cardiac output and unload the right ventricle without worsening V/Q mismatch. 106 In the INCREASE trial, inhaled treprostinil/Tyvaso was delivered via an iNEB device. 22 The FDA recently approved a new dry powder formulation of treprostinil/Tyvaso delivered via a hand‐held inhalation device for both Group 1 and PH‐ILD patients. 107 LIQ861 is another dry powder formulation of treprostinil palmitil tested in patients with Group 1 PAH in the Phase 3 open label INSPIRE trial. 108 It was recently granted a tentative approval by the FDA but remains commercially unavailable. A small safety study of this inhaled formulation of treprostinil in patients with PH‐ILD is currently recruiting. 109
LUNG TRANSPLANTATION
Lung transplantation (LTx) is the only treatment for fibrotic ILD that improves QOL and survival. However, the median survival after LTx for patients with IPF is only ~4.5 years, which is shorter than the average survival after LTx in general. 110 , 111 , 112 , 113 Additionally, only a small proportion of patients qualify for this intervention because of the high risks associated with advanced age, significant comorbidities, and the complexity of the surgery, coupled with the judicious use of a scarce resource. The current recommendation is to refer patients with fibrotic ILD for LTx work up early in the course of disease, ideally at diagnosis, due to the unpredictable nature of disease progression, the risk of acute exacerbations, and the associated high mortality. 114 Since the presence of even mild PH significantly increases the risk of death in patients with fibrotic ILD, the development of PH is one of the listing criteria for LTx. 111 , 112 , 113 , 114 Specific referral and listing guidelines are summarized in Table 2. 114
Table 2.
Recommendations for referral and work up for lung transplantation in patients with fibrotic lung diseases.
| Timing of referral |
|
| Timing of listing |
|
Note: 6MWT, 6‐min walking test; DLCO, diffusion of carbon monoxide; FVC, forced vital capacity; PH, pulmonary hypertension; UIP, usual interstitial pneumonia.
Source: Adopted from Leard et al. Consensus document for the selection of lung transplant candidates: an update from the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2021; 40(11):1349–1379. 114
Overall, posttransplant survival rates continue to improve for all disease categories, including fibrosing ILDs. 110 Although there have been conflicting single center reports on the impact of pretransplant PH on posttransplant outcomes in ILD patients, a large UNOS registry analysis showed that there is no deleterious effect on posttransplant survival in patients who receive BLTx. 115 , 116
The choice of the type of procedure (bilateral [BLTx] vs. single [SLTx] lung transplant) in patients with ILD needs further study. BLTx clearly provides a long‐term survival advantage in IPF, even after adjusting for other variables. 117 , 118 , 119 In a recent retrospective UNOS analysis of 9191 patients with IPF, 10‐year survival rates were 55% for BLTx and 32% for SLTx (p < 0.001). 119 When stratified by age, BLTx recipients had improved survival at all age cutoffs, except age ≥70 years. In addition, BLTx recipients had improved survival across all lung allocation scores (<45, ≥45, ≥60, ≥75) and all pulmonary artery pressure categories (<25, ≥25, ≥30, ≥40 mmHg). 119 However, any posttransplant survival advantage of BLTx versus SLTx needs to be weighed against a longer wait time for a suitable pair of donor lungs (BLTx) versus one good lung (SLTx), and hence a higher risk of pretransplant mortality. 120 , 121 Furthermore, for patients with very severe PH‐ILD (arbitrarily defined as mPAP ≥ 45 mmHg in this study) unrestricted listing for either a BLTx or SLTx versus only BLTx was associated with a 17% improved rate of the composite primary outcome, defined as the number of days between listing and death, removal from the list for clinical deterioration, or need for retransplantation. 119
PH TREATMENT APPROACH
While contemplating treatment of PH‐ILD, it is imperative to do due diligence to phenotype a patient to the best of one's ability. 6 While it is beyond the scope of this paper to delve into the distinction between patients who are more of the Group 1 versus Group 3 phenotype, there are certainly some patients, especially those with underlying CTD‐ILD, where phenotyping is difficult (Figure 1). Although this distinction is somewhat arbitrary, every effort should be made in characterizing the patient with PFTs, chest CT imaging, ECHO, and RHC to identify patients with a pulmonary vascular phenotype. HFpEF should be considered in patients with significant risk factors as treatment with pulmonary vasodilators may precipitate pulmonary edema. For patients who are better categorized as Group 1 PAH, the “toolbox” of treatment options is much broader with treatment based on risk stratification reflecting the severity of the underlying pulmonary vascular disease. 88
Concomitant with the treatment of pulmonary vasculopathy, providers should ensure that the underlying fibrotic lung disease is optimally managed, although no evidence exists that antifibrotic therapy will improve PH specifically. 6 Exertional desaturation needs to be reevaluated on a serial basis and supplemental oxygen prescribed when appropriate. Screening for sleep‐disordered breathing and nocturnal hypoxemia should be considered and treated if present. If there is evidence of volume overload, then diuretics should be used to optimize fluid balance. Pulmonary rehabilitation, early lung transplant evaluation, and timely referral to palliative care are all important considerations in the holistic care of these patients (Figure 2).
The recent ESC/ERS guidelines state that the treatment approach to the patients with lung disease and PH needs to be individualized and recommend a referral to a PH center. 88 In addition, the use of PAH medications, other than inhaled treprostinil, is not recommended in patients with nonsevere precapillary PH. While those patients with more severe hemodynamic derangements clearly have worse outcomes than those with mild to moderate PH, the latter group still has a poor prognosis. 111 , 112 The INCREASE study enrolled patients with mPAP ≥ 25 mmHg with a PVR ≥ 3 wood units. A prespecified subgroup analysis of the subgroup of patients with PVR < 4 wood units did not show a benefit in 6MWD with a major limitation being the small sample size of patients with milder disease (n = 52). 22 In addition, this analysis was not subjected to a multivariate analysis, and other benefits aside from the 6MWD were not evaluated. The ESC/ERS guidelines also state that inhaled treprostinil may be considered in patients with PH‐ILD, irrespective of severity. 88
In addition, sildenafil appears to be safe, well tolerated, and has some efficacy data for treatment of these patients and may be considered where inhaled treprostinil is not available. 63 , 65 , 71 , 74 , 76 , 77 , 79 , 82 , 84 , 89 It is unknown if a combination of sildenafil and inhaled treprostinil provides incremental efficacy. However, given safety profile of both drugs in this population, consideration can be given to dual therapy in patients with severe PH on an individual basis with careful and frequent follow‐up after an open discussion with the patient. Based on the published data and ESC/ERS guidelines, riociguat and ERAs are not recommended. 88 Enrollment in clinical trials should be offered whenever possible. For those patients who are potential transplant candidates, early referral to a transplant center for further evaluation and work‐up is strongly encouraged. Finally, for patients who are not transplant candidates, involvement of palliative care to help manage dyspnea should be strongly considered.
SUMMARY
Treatment of Group 3 PH, in general, has relied on the principle of optimizing the treatment of the underlying lung disease and contributory factors, such as heart failure, sleep disordered breathing, thromboembolism, and deconditioning. Given the quality of prior reports, as well as the negative or equivocal results from clinical trials, until recently systemic pulmonary vasodilators have not been recommended in the treatment of PH associated with lung disease. The INCREASE study of inhaled treprostinil has renewed interest in the use of inhaled pulmonary vasodilators in patients with PH due to underlying ILD, with hope for more studies to come in the next several years.
AUTHOR CONTRIBUTIONS
Oksana A. Shlobin, Paul M. Hassoun, Stephen J. Wort, John A. Scandurra, and Eric Shen were involved in the conception and design of the manuscript, conducted the searches and data extraction as well as wrote the first draft of the manuscript. Oksana A. Shlobin, Lucilla Piccari, and Steven D. Nathan contributed to creation of the figures. All authors contributed to edits of the manuscript, analyzed and interpreted the data, revised the manuscript critically for important intellectual content, approved the final manuscript, and agreed to be accountable for its overall content.
CONFLICT OF INTEREST STATEMENT
Dr. Oksana A. Shlobin has consulted for United Therapeutics, Bayer, Altavant, Aerovate, Jenssen&Jenssen and Merck, and is on the speaker bureau for UT, Bayer, and J&J. Mr. Eric Shen is an employee of United Therapeutics Corporation and owns stocks/shares in the company. Dr. Lucilla Piccari has received research funding from and served as a speaker for Janssen and Ferrer, advised Janssen, Ferrer, and United Therapeutics as well as received support for attending congresses from Janssen, MSD, and Ferrer, all of which not related to this manuscript. Dr. John A. Scandurra is an employee of Aria CV Inc. and owns stocks/shares in the company. Dr. Paul M. Hassoun serves on a scientific advisory board for Merck, an activity unrelated to the current work. Dr. Stephen J. Wort received honoraria from Janssen, MSD, Bayer, and Acceleron for advisory boards, received honoraria from Janssen for educational activity, received unrestricted research grants from Janssen and Bayer, and travel grants, conference registration and accommodation from Actelion and GSK. Dr. Sylvia M. Nikkho is an employee of Bayer AG. Dr. Steven D. Nathan a consultant for United Therapeutics, Bellerophon, Third Pole, Roche, Boehringer‐Ingelheim, Merck, and Daewoong.
ETHICS STATEMENT
Not applicable.
ACKNOWLEDGMENTS
The authors would like to all the members of the Innovative Drug Development Initiative Group 3 Pulmonary Hypertension workstream for their dedication and commitment to the work of the group: Steven H. Abman, Peter Fernandes, Howard M. Lazarus, Horst Olschewski, Mitchell Psotka, Manuel J. Richter, Norman Stockbridge, Carmine Dario Vizza, Brian Allwood, Katerina Antoniou, Jonathan H. Chung and, Patrizio Vitulo. Finally, thank you to the IDDI Leads: Paul Corris, Raymond Benza, Mark Toshner and to the Pulmonary Vascular Research Institute for supporting innovative research in pulmonary vascular disease. The authors have no funding information to disclose.
Shlobin OA, Shen E, Wort SJ, Piccari L, Scandurra JA, Hassoun PM, Nikkho SM, Nathan SD. Pulmonary hypertension in the setting of interstitial lung disease: approach to management and treatment. A consensus statement from the Pulmonary Vascular Research Institute's Innovative Drug Development Initiative—Group 3 Pulmonary Hypertension. Pulm Circ. 2024;14:e12310. 10.1002/pul2.12310
REFERENCES
- 1. Nathan SD, Barbera JA, Gaine SP, Harari S, Martinez FJ, Olschewski H, Olsson KM, Peacock AJ, Pepke‐Zaba J, Provencher S, Weissmann N, Seeger W. Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J. 2019;53(1):1801914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nikkho SM, Richter MJ, Shen E, Abman SH, Antoniou K, Chung J, Fernandes P, Hassoun P, Lazarus HM, Olschewski H, Piccari L, Psotka M, Saggar R, Shlobin OA, Stockbridge N, Vitulo P, Vizza CD, Wort SJ, Nathan SD. Clinical significance of pulmonary hypertension in interstitial lung disease: a consensus statement from the Pulmonary Vascular Research Institute's innovative drug development initiative—Group 3 pulmonary hypertension. Pulm Circ. 2022;12(3):e12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. King CS, Shlobin OA. The trouble with group 3 pulmonary hypertension in interstitial lung disease: dilemmas in diagnosis and the conundrum of treatment. Chest. 2020;158(4):1651–1664. [DOI] [PubMed] [Google Scholar]
- 4. Jurgen Behr, Nathan SD. Pulmonary hypertension in interstitial lung diseases: screening, diagnosis and treatment. Curr Opin Med. 2021;27:396–404. [DOI] [PubMed] [Google Scholar]
- 5. Piccari L, Allwood B, Antoniou K, Chung JH, Hassoun PM, Nikkho SM, Saggar R, Shlobin OA, Vitulo P, Nathan SD, Wort SJ. Pathogenesis, clinical features, and phenotypes of pulmonary hypertension associated with interstitial lung disease: a consensus statement from the Pulmonary Vascular Research Institute's Innovative Drug Development Initiative—Group 3 Pulmonary Hypertension. Pulm Circ. 2023;13(2):e12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nathan SD. Progress in the treatment of pulmonary hypertension associated with interstitial lung disease. Am J Respir Crit Care Med. 2023;208:238–246. 10.1164/rccm.202212-2342CI [DOI] [PubMed] [Google Scholar]
- 7. Hemnes AR, Beck GJ, Newman JH, Abidov A, Aldred MA, Barnard J, Berman Rosenzweig E, Borlaug BA, Chung WK, Comhair SAA, Erzurum SC, Frantz RP, Gray MP, Grunig G, Hassoun PM, Hill NS, Horn EM, Hu B, Lempel JK, Maron BA, Mathai SC, Olman MA, Rischard FP, Systrom DM, Tang WHW, Waxman AB, Xiao L, Yuan JXJ, Leopold JA. PVDOMICS: a multi‐center study to improve understanding of pulmonary vascular disease through phenomics. Circ Res. 2017;121(10):1136–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hoeper MM, Dwivedi K, Pausch C, Lewis RA, Olsson KM, Huscher D, Pittrow D, Grünig E, Staehler G, Vizza CD, Gall H, Distler O, Opitz C, Gibbs JSR, Delcroix M, Park DH, Ghofrani HA, Ewert R, Kaemmerer H, Kabitz HJ, Skowasch D, Behr J, Milger K, Lange TJ, Wilkens H, Seyfarth HJ, Held M, Dumitrescu D, Tsangaris I, Vonk‐Noordegraaf A, Ulrich S, Klose H, Claussen M, Eisenmann S, Schmidt KH, Swift AJ, Thompson AAR, Elliot CA, Rosenkranz S, Condliffe R, Kiely DG, Halank M. Phenotyping of idiopathic pulmonary arterial hypertension: a registry analysis. Lancet Respir Med. 2022;10:937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Frost A, Badesch D, Gibbs JSR, Gopalan D, Khanna D, Manes A, Oudiz R, Satoh T, Torres F, Torbicki A. Diagnosis of pulmonary hypertension. Eur Respir J. 2019;53(1):1801904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fulmer JD, Roberts WC, von Gal ER, Crystal RG. Morphologic‐physiologic correlates of the severity of fibrosis and degree of cellularity in idiopathic pulmonary fibrosis. J Clin Invest. 1979;63:665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Doyle TJ, Hunninghake GM, Rosas IO. Subclinical interstitial lung disease: why you should care. Am J Respir Crit Care Med. 2012;185:1147–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Trip P, Nossent EJ, de Man FS, van den Berk IAH, Boonstra A, Groepenhoff H, Leter EM, Westerhof N, Grünberg K, Bogaard HJ, Vonk‐Noordegraaf A. Severely reduced diffusion capacity in idiopathic pulmonary arterial hypertension: patient characteristics and treatment responses. Eur Respir J. 2013;42:1575–1585. [DOI] [PubMed] [Google Scholar]
- 14. Lewis RA, Thompson AAR, Billings CG, Charalampopoulos A, Elliot CA, Hamilton N, Hill C, Hurdman J, Rajaram S, Sabroe I, Swift AJ, Kiely DG, Condliffe R. Mild parenchymal lung disease and/or low diffusion capacity impacts survival and treatment response in patients diagnosed with idiopathic pulmonary arterial hypertension. Eur Respir J. 2020;55:2000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rose L, Prins KW, Archer SL, Pritzker M, Weir EK, Misialek JR, Thenappan T. Survival in pulmonary hypertension due to chronic lung disease: influence of low diffusion capacity of the lungs for carbon monoxide. J Heart Lung Transplant. 2019;38:145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Raghu G, Anstrom KJ, King TE, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N‐acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Raghu G, Rochwerg B, Zhang Y, Garcia CAC, Azuma A, Behr J, Brozek JL, Collard HR, Cunningham W, Homma S, Johkoh T, Martinez FJ, Myers J, Protzko SL, Richeldi L, Rind D, Selman M, Theodore A, Wells AU, Hoogsteden H, Schünemann HJ. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192:e3–e19. [DOI] [PubMed] [Google Scholar]
- 18. Raghu G, Remy‐Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, Kreuter M, Lynch DA, Maher TM, Martinez FJ, Molina‐Molina M, Myers JL, Nicholson AG, Ryerson CJ, Strek ME, Troy LK, Wijsenbeek M, Mammen MJ, Hossain T, Bissell BD, Herman DD, Hon SM, Kheir F, Khor YH, Macrea M, Antoniou KM, Bouros D, Buendia‐Roldan I, Caro F, Crestani B, Ho L, Morisset J, Olson AL, Podolanczuk A, Poletti V, Selman M, Ewing T, Jones S, Knight SL, Ghazipura M, Wilson KC. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205(9):e18–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, Richeldi L, Kolb M, Tetzlaff K, Stowasser S, Coeck C, Clerisme‐Beaty E, Rosenstock B, Quaresma M, Haeufel T, Goeldner RG, Schlenker‐Herceg R, Brown KK. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381:1718–1727. [DOI] [PubMed] [Google Scholar]
- 20. Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, Raghu G, Sauter W, Girard M, Alves M, Clerisme‐Beaty E, Stowasser S, Tetzlaff K, Kuwana M, Maher TM. Nintedanib for systemic sclerosis–associated interstitial lung disease. N Engl J Med. 2019;380:2518–2528. [DOI] [PubMed] [Google Scholar]
- 21. Kolb M, Raghu G, Wells AU, Behr J, Richeldi L, Schinzel B, Quaresma M, Stowasser S, Martinez FJ. Nintedanib plus sildenafil in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2018;379:1722–1731. [DOI] [PubMed] [Google Scholar]
- 22. Waxman A, Restrepo‐Jaramillo R, Thenappan T, Ravichandran A, Engel P, Bajwa A, Allen R, Feldman J, Argula R, Smith P, Rollins K, Deng C, Peterson L, Bell H, Tapson V, Nathan SD. Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med. 2021;384(4):325–334. [DOI] [PubMed] [Google Scholar]
- 23. Nathan SD, Waxman A, Rajagopal S, Case A, Johri S, DuBrock H, De La Zerda DJ, Sahay S, King C, Melendres‐Groves L, Smith P, Shen E, Edwards LD, Nelsen A, Tapson VF. Inhaled treprostinil and forced vital capacity in patients with interstitial lung disease and associated pulmonary hypertension: a post‐hoc analysis of the INCREASE study. Lancet Respir Med. 2021;9(11):1266–1274. [DOI] [PubMed] [Google Scholar]
- 24. Waxman AB, Restrepo Jaramillo R, Thenappan T, Engel PP, Bajwa A, Ravichandran AK, Feldman JP, Hajari Case A, Tapson VF, Smith P, Deng C, Shen E, Nathan SD. Long‐term effects of inhaled treprostinil in patients with pulmonary hypertension due to interstitial lung disease: the increase study open‐label extension. In B96, Fisherman's Wharf: Omics and translational approaches in pulmonary vascular disease. American Thoracic Society; 2022. p. A5671. [Google Scholar]
- 25. https://clinicaltrials.gov/ct2/show/NCT04708782. Accessed October 2, 2023.
- 26. Conte E, Gili E, Fagone E, Fruciano M, Iemmolo M, Vancheri C. Effect of pirfenidone on proliferation, TGF‐β‐induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur J Pharm Sci. 2014;58:13–19. [DOI] [PubMed] [Google Scholar]
- 27. Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, King TE, Lancaster L, Sahn SA, Szwarcberg J, Valeyre D, du Bois RM. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–1769. [DOI] [PubMed] [Google Scholar]
- 28. King TE, Bradford WZ, Castro‐Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, Lederer DJ, Nathan SD, Pereira CA, Sahn SA, Sussman R, Swigris JJ, Noble PW. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–2092. [DOI] [PubMed] [Google Scholar]
- 29. Nathan SD, Albera C, Bradford WZ, Costabel U, Glaspole I, Glassberg MK, Kardatzke DR, Daigl M, Kirchgaessler KU, Lancaster LH, Lederer DJ, Pereira CA, Swigris JJ, Valeyre D, Noble PW. Effect of pirfenidone on mortality: pooled analyses and meta‐analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med. 2017;5(1):33–41. [DOI] [PubMed] [Google Scholar]
- 30. Nathan SD, Albera C, Bradford WZ, Costabel U, du Bois RM, Fagan EA, Fishman RS, Glaspole I, Glassberg MK, Glasscock KF, King TE, Lancaster L, Lederer DJ, Lin Z, Pereira CA, Swigris JJ, Valeyre D, Noble PW, Wells AU. Effect of continued treatment with pirfenidone following clinically meaningful declines in forced vital capacity: analysis of data from three phase 3 trials in patients with idiopathic pulmonary fibrosis. Thorax. 2016;71(5):429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aravena C, Labarca G, Venegas C, Arenas A, Rada G. Pirfenidone for idiopathic pulmonary fibrosis: a systematic review and meta‐analysis. PLoS One. 2015;10(8):e0136160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch‐Grunt U, Garin‐Chesa P, Bader G, Zoephel A, Quant J, Heckel A, Rettig WJ. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68(12):4774–4782. [DOI] [PubMed] [Google Scholar]
- 33. Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, Brown KK, Flaherty KR, Noble PW, Raghu G, Brun M, Gupta A, Juhel N, Klüglich M, du Bois RM. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365(12):1079–1087. [DOI] [PubMed] [Google Scholar]
- 34. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, Kim DS, Kolb M, Nicholson AG, Noble PW, Selman M, Taniguchi H, Brun M, Le Maulf F, Girard M, Stowasser S, Schlenker‐Herceg R, Disse B, Collard HR. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. [DOI] [PubMed] [Google Scholar]
- 35. Richeldi L, Cottin V, du Bois RM, Selman M, Kimura T, Bailes Z, Schlenker‐Herceg R, Stowasser S, Brown KK. Nintedanib in patients with idiopathic pulmonary fibrosis: combined evidence from the TOMORROW and INPULSIS® trials. Respir Med. 2016;113:74–79. [DOI] [PubMed] [Google Scholar]
- 36. Kolb M, Richeldi L, Behr J, Maher TM, Tang W, Stowasser S, Hallmann C, du Bois RM. Nintedanib in patients with idiopathic pulmonary fibrosis and preserved lung volume. Thorax. 2017;72(4):340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wuyts WA, Kolb M, Stowasser S, Stansen W, Huggins JT, Raghu G. First data on efficacy and safety of nintedanib in patients with idiopathic pulmonary fibrosis and forced vital capacity of ≤50% of predicted value. Lung. 2016;194(5):739–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cottin V, Azuma A, Raghu G, Stansen W, Stowasser S, Schlenker‐Herceg R, Kolb M. Therapeutic effects of nintedanib are not influenced by emphysema in the INPULSIS trials. Eur Respir J. 2019;53:1801655. [DOI] [PubMed] [Google Scholar]
- 39. Cottin V, Selman M, Inoue Y, Wong AW, Corte TJ, Flaherty KR, Han MK, Jacob J, Johannson KA, Kitaichi M, Lee JS, Agusti A, Antoniou KM, Bianchi P, Caro F, Florenzano M, Galvin L, Iwasawa T, Martinez FJ, Morgan RL, Myers JL, Nicholson AG, Occhipinti M, Poletti V, Salisbury ML, Sin DD, Sverzellati N, Tonia T, Valenzuela C, Ryerson CJ, Wells AU. Syndrome of combined pulmonary fibrosis and emphysema: an official ATS/ERS/JRS/ALAT research statement. Am J Respir Crit Care Med. 2022;206(4):e7–e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Panos RJ, Mortenson RL, Niccoli SA, King TE. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med. 1990;88(4):396–404. [DOI] [PubMed] [Google Scholar]
- 41. Mejía M, Carrillo G, Rojas‐Serrano J, Estrada A, Suárez T, Alonso D, Barrientos E, Gaxiola M, Navarro C, Selman M. Idiopathic pulmonary fibrosis and emphysema. Chest. 2009;136(1):10–15. [DOI] [PubMed] [Google Scholar]
- 42. Cottin V, Selman M, Inoue Y, Wong AW, Corte TJ, Flaherty KR, Han MK, Jacob J, Johannson KA, Kitaichi M, Lee JS, Agusti A, Antoniou KM, Bianchi P, Caro F, Florenzano M, Galvin L, Iwasawa T, Martinez FJ, Morgan RL, Myers JL, Nicholson AG, Occhipinti M, Poletti V, Salisbury ML, Sin DD, Sverzellati N, Tonia T, Valenzuela C, Ryerson CJ, Wells AU. Syndrome of combined pulmonary fibrosis and emphysema: an official ATS/ERS/JRS/ALAT research statement. Am J Respir Crit Care Med. 2022;206(4):e7–e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bye PT, Issa F, Berthon‐Jones M, Sullivan CE. Studies of oxygenation during sleep in patients with interstitial lung disease. Am Rev Respir Dis. 1984;129:27–32. [DOI] [PubMed] [Google Scholar]
- 44. Perez‐Padilla R, West P, Lertzman M, Kryger MH. Breathing during sleep in patients with interstitial lung disease. Am Rev Respir Dis. 1985;132:224–229. [DOI] [PubMed] [Google Scholar]
- 45. Shitrit D, Rusanov V, Peled N, Amital A, Fuks L, Kramer MR. The 15‐step oximetry test: a reliable tool to identify candidates for lung transplantation among patients with idiopathic pulmonary fibrosis. J Heart Lung Transplant. 2009;28:328–333. [DOI] [PubMed] [Google Scholar]
- 46. Sjoding MW, Dickson RP, Iwashyna TJ, Gay SE, Valley TS. Racial bias in pulse oximetry measurement. N Engl J Med. 2020;383:2477–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schiza S, Mermigkis C, Margaritopoulos GA, Daniil Z, Harari S, Poletti V, Renzoni EA, Torre O, Visca D, Bouloukaki I, Sourvinos G, Antoniou KM. Idiopathic pulmonary fibrosis and sleep disorders: no longer strangers in the night. Eur Respir Rev. 2015;24:327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nathan SD, Barnett SD, Urban BA, Nowalk C, Moran BR, Burton N. Pulmonary embolism in idiopathic pulmonary fibrosis transplant recipients. Chest. 2003;123:1758–1763. [DOI] [PubMed] [Google Scholar]
- 49. Kubo H, Nakayama K, Yanai M, Suzuki T, Yamaya M, Watanabe M, Sasaki H. Anticoagulant therapy for idiopathic pulmonary fibrosis. Chest. 2005;128:1475–1482. [DOI] [PubMed] [Google Scholar]
- 50. Noth I, Anstrom KJ, Calvert SB, de Andrade J, Flaherty KR, Glazer C, Kaner RJ, Olman MA. A placebo‐controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186(1):88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. King CS, Freiheit E, Brown AW, Shlobin OA, Aryal S, Ahmad K, Khangoora V, Flaherty KR, Venuto D, Nathan SD. Association between anticoagulation and survival in interstitial lung disease. Chest. 2021;159(4):1507–1516. [DOI] [PubMed] [Google Scholar]
- 52. Kizer JR, Zisman DA, Blumenthal NP, Kotloff RM, Kimmel SE, Strieter RM, Arcasoy SM, Ferrari VA, Hansen‐Flaschen J. Association between pulmonary fibrosis and coronary artery disease. Arch Intern Med. 2004;164:551–556. [DOI] [PubMed] [Google Scholar]
- 53. Nathan SD, Basavaraj A, Reichner C, Shlobin OA, Ahmad S, Kiernan J, Burton N, Barnett SD. Prevalence and impact of coronary artery disease in idiopathic pulmonary fibrosis. Respir Med. 2010;104:1035–1041. [DOI] [PubMed] [Google Scholar]
- 54. Papadopoulis CE, Pitsiou G, Karamitsos TD, Karvounis HI, Kontakiotis T, Giannakoulas G, Efthimiadis GK, Argyropoulou P, Parharidis GE, Bouros D. Left ventricular diastolic dysfunction in idiopathic pulmonary fibrosis: a tissue Doppler echocardiographic [corrected] study. Eur Respir J. 2008;31:701–706. [DOI] [PubMed] [Google Scholar]
- 55. Nathan SD, Barnett SD, King CS, Provencher S, Barbera JA, Pastre J, Shlobin OA, Seeger W. Impact of the new definition for pulmonary hypertension in patients with lung disease: an analysis of the United Network for Organ Sharing database. Pulm Circ. 2021;11(2):2045894021999960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Teramachi R, Taniguchi H, Kondoh Y, Kimura T, Kataoka K, Yokoyama T, Furukawa T, Yagi M, Sakamoto K, Hashimoto N, Hasegawa Y. Impact of post‐capillary pulmonary hypertension on mortality in interstitial lung disease. Respir Investig. 2021;59(3):342–349. [DOI] [PubMed] [Google Scholar]
- 57. Ewert R, Heine A, Müller‐Heinrich A, Bollmann T, Obst A, Desole S, Knaak C, Stubbe B, Opitz CF, Habedank D. Exercise and fluid challenge during right heart catheterisation for evaluation of dyspnoea. Pulm Circ. 2020;10(3):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liang Z, Hoffman LA, Nouraie M, Kass DJ, Donahoe MP, Gibson KF, Saul MI, Lindell KO. Referral to palliative care infrequent in patients with idiopathic pulmonary fibrosis admitted to an intensive care unit. J Palliat Med. 2017;20(2):134–140. [DOI] [PubMed] [Google Scholar]
- 59. Kreuter M, Bendstrup E, Russell AM, Bajwah S, Lindell K, Adir Y, Brown CE, Calligaro G, Cassidy N, Corte TJ, Geissler K, Hassan AA, Johannson KA, Kairalla R, Kolb M, Kondoh Y, Quadrelli S, Swigris J, Udwadia Z, Wells A, Wijsenbeek M. Palliative care in interstitial lung disease: living well. Lancet Respir Med. 2017;5(12):968–980. [DOI] [PubMed] [Google Scholar]
- 60. Lindell K, Raghu G. Palliative care for patients with pulmonary fibrosis: symptom relief is essential. Eur Respir J. 2018;52(6):1802086. [DOI] [PubMed] [Google Scholar]
- 61. Rohlfing AB, Bischoff KE, Kolaitis NA, Kronmal RA, Kime NA, Gray MP, Bartolome S, Chakinala MM, Frantz RP, Ventetuolo CE, Mathai SC, De Marco T. Palliative care referrals in patients with pulmonary arterial hypertension: the Pulmonary Hypertension Association Registry. Respir Med. 2023;206:107066. [DOI] [PubMed] [Google Scholar]
- 62. Wislon M, Anguiano RH, Awdish RLA, Coons JC, Kimber A, Morrison M, Paulus S, Schmit A, Spexarth F, Swetz KM, Verlinden NJ, Whittenhall ME, Sketch MR, Broderick M, Brewer J. An expert panel Delphi consensus statement on the use of palliative care in the management of patients with pulmonary arterial hypertension. Pulm Circ. 2022;12(1):e12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Collard HR, Anstrom KJ, Schwarz MI, Zisman DA. Sildenafil improves walk distance in idiopathic pulmonary fibrosis. Chest. 2007;131(3):897–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zisman DA, Schwarz M, Anstrom KJ, Collard HR, Flaherty KR, Hunninghake GW. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med. 2010;363(7):620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jackson RM, Glassberg MK, Ramos CF, Bejarano PA, Butrous G, Gómez‐Marín O. Sildenafil therapy and exercise tolerance in idiopathic pulmonary fibrosis. Lung. 2010;188(2):115–123. [DOI] [PubMed] [Google Scholar]
- 66. Gunther A, Enke B, Markart P, Hammerl P, Morr H, Behr J, Stahler G, Seeger W, Grimminger F, Leconte I, Roux S, Ghofrani HA. Safety and tolerability of bosentan in idiopathic pulmonary fibrosis: an open label study. Eur Respir J. 2007;29(4):713–719. [DOI] [PubMed] [Google Scholar]
- 67. King, Jr. TE , Behr J, Brown KK, du Bois RM, Lancaster L, de Andrade JA, Stähler G, Leconte I, Roux S, Raghu G. BUILD‐1: a randomized placebo‐controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177(1):75–81. [DOI] [PubMed] [Google Scholar]
- 68. King TE Jr., Brown KK, Raghu G, Lynch DA, Martinez F, Valeyre D, Leconte I, Morganti A, Roux S, Behr J. BUILD‐3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(1):92–99. [DOI] [PubMed] [Google Scholar]
- 69. Raghu G. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med. 2013;158(9):641–649. [DOI] [PubMed] [Google Scholar]
- 70. Raghu G, Million‐Rousseau R, Morganti A, Perchenet L, Behr J. Macitentan for the treatment of idiopathic pulmonary fibrosis: the randomised controlled MUSIC trial. Eur Respir J. 2013;42(6):1622–1632. [DOI] [PubMed] [Google Scholar]
- 71. Kolb M, Raghu G, Wells AU, Behr J, Richeldi L, Schinzel B, Quaresma M, Stowasser S, Martinez FJ. Nintedanib plus sildenafil in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2018;379:1722–1731. [DOI] [PubMed] [Google Scholar]
- 72. Nathan SD, Flaherty KR, Glassberg MK, Raghu G, Swigris J, Alvarez R, Ettinger N, Loyd J, Fernandes P, Gillies H, Kim B, Shah P, Lancaster L. A randomized, double‐blind, placebo‐controlled study of pulsed, inhaled nitric oxide in subjects at risk of pulmonary hypertension associated with pulmonary fibrosis. Chest. 2020;158:637–645. [DOI] [PubMed] [Google Scholar]
- 73. Faria‐Urbina M, Oliveira RKF, Agarwal M, Waxman AB. Inhaled treprostinil in pulmonary hypertension associated with lung disease. Lung. 2018;196(2):139–146. [DOI] [PubMed] [Google Scholar]
- 74. Ghofrani HA, Wiedemann R, Rose F, Schermuly RT, Olschewski H, Weissmann N, Gunther A, Walmrath D, Seeger W, Grimminger F. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. The Lancet. 2002;360(9337):895–900. [DOI] [PubMed] [Google Scholar]
- 75. Krowka MJ, Ahmad S, de Andrade JA, Frost A, Glassberg MK, Lancaster LH, Lasky J, Mathier MA, Stocks J. A randomized, double‐blind, placebo‐controlled study to evaluate the safety and efficacy of iloprost inhalation in adults with abnormal pulmonary arterial pressure and exercise limitation associated with idiopathic pulmonary fibrosis. Chest. 2007;132:633A. [Google Scholar]
- 76. Chapman TH, Wilde M, Sheth A, Madden BP. Sildenafil therapy in secondary pulmonary hypertension: is there benefit in prolonged use? Vascul Pharmacol. 2009;51(2–3):90–95. [DOI] [PubMed] [Google Scholar]
- 77. Corte TJ, Gatzoulis MA, Parfitt L, Harries C, Wells AU, Wort SJ. The use of sildenafil to treat pulmonary hypertension associated with interstitial lung disease. Respirology. 2010;15(8):1226–1232. [DOI] [PubMed] [Google Scholar]
- 78. Hoeper MM, Halank M, Wilkens H, Günther A, Weimann G, Gebert I, Leuchte HH, Behr J. Riociguat for interstitial lung disease and pulmonary hypertension: a pilot trial. Eur Respir J. 2013;41(4):853–860. [DOI] [PubMed] [Google Scholar]
- 79. Zimmermann GS, von Wulffen W, Huppmann P, Meis T, Ihle F, Geiseler J, Leuchte HH, Tufman A, Behr J, Neurohr C. Haemodynamic changes in pulmonary hypertension in patients with interstitial lung disease treated with PDE‐5 inhibitors. Respirology. 2014;19(5):700–706. [DOI] [PubMed] [Google Scholar]
- 80. Corte TJ, Keir GJ, Dimopoulos K, Howard L, Corris PA, Parfitt L, Foley C, Yanez‐Lopez M, Babalis D, Marino P, Maher TM, Renzoni EA, Spencer L, Elliot CA, Birring SS, O'Reilly K, Gatzoulis MA, Wells AU, Wort SJ. Bosentan in pulmonary hypertension associated with fibrotic idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2014;190(2):208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Saggar R, Khanna D, Vaidya A, Derhovanessian A, Maranian P, Duffy E, Belperio JA, Weigt SS, Dua S, Shapiro SS, Goldin JG, Abtin F, Lynch JP, Ross DJ, Forfia PR, Saggar R. Changes in right heart haemodynamics and echocardiographic function in an advanced phenotype of pulmonary hypertension and right heart dysfunction associated with pulmonary fibrosis. Thorax. 2014;69:123–129. [DOI] [PubMed] [Google Scholar]
- 82. Brewis MJ, Church AC, Johnson MK, Peacock AJ. Severe pulmonary hypertension in lung disease: phenotypes and response to treatment. Eur Respir J. 2015;46(5):1378–1389. [DOI] [PubMed] [Google Scholar]
- 83. Nathan SD, Behr J, Collard HR, Cottin V, Hoeper MM, Martinez FJ, Corte TJ, Keogh AM, Leuchte H, Mogulkoc N, Ulrich S, Wuyts WA, Yao Z, Boateng F, Wells AU. Riociguat for idiopathic interstitial pneumonia‐associated pulmonary hypertension (RISE‐IIP): a randomised, placebo‐controlled phase 2b study. Lancet Respir Med. 2019;7(9):780–790. [DOI] [PubMed] [Google Scholar]
- 84. Dawes TJW, McCabe C, Dimopoulos K, Stewart I, Bax S, Harries C, Samaranayake CB, Kempny A, Molyneaux PL, Seitler S, Semple T, Li W, George PM, Kouranos V, Chua F, Renzoni EA, Kokosi M, Jenkins G, Wells AU, Wort SJ, Price LC. Phosphodiesterase 5 inhibitor treatment and survival in interstitial lung disease pulmonary hypertension: a Bayesian retrospective observational cohort study. Respirology. 2022;28:262–272. [DOI] [PubMed] [Google Scholar]
- 85. Pulito‐Cueto V, Genre F, López‐Mejías R, Mora‐Cuesta VM, Iturbe‐Fernández D, Portilla V, Sebastián Mora‐Gil M, Ocejo‐Vinyals JG, Gualillo O, Blanco R, Corrales A, Ferraz‐Amaro I, Castañeda S, Cifrián Martínez JM, Atienza‐Mateo B, Remuzgo‐Martínez S, González‐Gay MÁ. Endothelin‐1 as a biomarker of idiopathic pulmonary fibrosis and interstitial lung disease associated with autoimmune diseases. Int J Mol Sci. 2023;24(2):1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gunther A, Enke B, Markart P, Hammerl P, Morr H, Behr J, Stahler G, Seeger W, Grimminger F, Leconte I, Roux S, Ghofrani HA. Safety and tolerability of bosentan in idiopathic pulmonary fibrosis: an open label study. Eur Respir J. 2007;29(4):713–719. [DOI] [PubMed] [Google Scholar]
- 87. Corte TJ, Keir GJ, Dimopoulos K, Howard L, Corris PA, Parfitt L, Foley C, Yanez‐Lopez M, Babalis D, Marino P, Maher TM, Renzoni EA, Spencer L, Elliot CA, Birring SS, O'Reilly K, Gatzoulis MA, Wells AU, Wort SJ. Bosentan in pulmonary hypertension associated with fibrotic idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2014;190:208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Humbet M, Kovacs G, Hoeper MM, Badagliacca R, Berger RM, Brida M, Carlsen J, Coats AJ, Escribano‐Subias P, Ferrari P, Ferreira DS, Ghofrani HA, Giannakoulas G, Kiely DG, Mayer E, Meszaros G, Nagavci B, Olsson KM, Pepke‐Zaba J, Quint JK, Rådegran G, Simonneau G, Sitbon O, Tonia T, Toshner M, Vachiery JL, Noordegraaf AV, Delcroix M, Rosenkran S. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;61:2200879. [Google Scholar]
- 89. Han MK, Bach DS, Hagan PG, Yow E, Flaherty KR, Toews GB, Anstrom KJ, Martinez FJ. Sildenafil preserves exercise capacity in patients with idiopathic pulmonary fibrosis and right‐sided ventricular dysfunction. Chest. 2013;143(6):1699–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Nathan SD, Cottin V, Behr J, Hoeper MM, Martinez FJ, Corte TJ, Keogh AM, Leuchte H, Mogulkoc N, Ulrich S, Wuyts WA, Yao Z, Ley‐Zaporozhan J, Müller‐Lisse UG, Scholle FD, Brüggenwerth G, Busse D, Nikkho S, Wells AU. Impact of lung morphology on clinical outcomes with riociguat in patients with pulmonary hypertension and idiopathic interstitial pneumonia: a post hoc subgroup analysis of the RISE‐IIP study. J Heart Lung Transplant. 2021;40:494–503. [DOI] [PubMed] [Google Scholar]
- 91. Bellopheron Press Release . Bellerophon announces top‐line data from Phase 3 REBUILD clinical trial of INOpulse® for treatment of fibrotic interstitial lung disease May 6, 2023. https://investors.bellerophon.com/news-releases/news-release-details/Bellerophon-announces-top-line-data-phase-3-rebuild-clinical. Accessed July 6, 2023.
- 92. Minai OA, Sahoo D, Chapman JT, Mehta AC. Vaso‐active therapy can improve 6‐min walk distance in patients with pulmonary hypertension and fibrotic interstitial lung disease. Respir Med. 2008;102(7):1015–1020. [DOI] [PubMed] [Google Scholar]
- 93. Olschewski H, Ardeschir Ghofrani H, Walmrath D, Schermuly R, Temmesfeld‐Wollbrück B, Grimminger F, Seeger W. Inhaled prostacyclin and iloprost in severe pulmonary hypertension secondary to lung fibrosis. Am J Respir Crit Care Med. 1999;160(2):600–607. [DOI] [PubMed] [Google Scholar]
- 94. Nathan SD, Tapson VF, Elwing J, Rischard F, Mehta J, Shapiro S, Shen E, Deng C, Smith P, Waxman A. Efficacy of inhaled treprostinil on multiple disease progression events in patients with pulmonary hypertension due to parenchymal lung disease in the increase trial. Am J Respir Crit Care Med. 2022;205(2):198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Nathan SD, Deng C, King CS, DuBrock HM, Elwing J, Rajagopal S, Rischard F, Sahay S, Broderick M, Shen E, Smith P, Tapson VF, Waxman AB. Inhaled treprostinil dosage in pulmonary hypertension associated with interstitial lung disease and its effects on clinical outcomes. Chest. 2023;163:398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Prins KW, Duval S, Markowitz J, Pritzker M, Thenappan T. Chronic use of PAH‐specific therapy in World Health Organization Group III Pulmonary Hypertension: a systematic review and meta‐analysis. Pulm Circ. 2017;7(1):145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bajwa EK, Cislak D, Palcza J, Feng H, Messina EJ, Reynders T, Denef JF, Corcea V, Lai E, Stoch SA. Effects of an inhaled soluble guanylate cyclase (sGC) stimulator MK‐5475 in pulmonary arterial hypertension (PAH). Respir Med. 2023;206:107065. [DOI] [PubMed] [Google Scholar]
- 98. Becker‐Pelster EM, Hahn MG, Delbeck M, Dietz L, Hüser J, Kopf J, Kraemer T, Marquardt T, Mondritzki T, Nagelschmitz J, Nikkho SM, Pires PV, Tinel H, Weimann G, Wunder F, Sandner P, Schuhmacher J, Stasch JP, Truebel HKF. Inhaled mosliciguat (BAY 1237592): targeting pulmonary vasculature via activating apo‐sGC. Respir Res. 2022;23(1):272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. https://www.clinicaltrials.gov/ct2/show/NCT05036135. Accessed July 2, 2023.
- 100. https://clinicaltrials.gov/ct2/show/NCT04903730. Accessed July 2, 2023.
- 101. https://www.clinicaltrials.gov/ct2/show/NCT04456998. Accessed July 2, 2023.
- 102. https://www.clinicaltrials.gov/ct2/show/NCT02036970. Accessed July 2, 2023.
- 103. Li Y, Jiang D, Liang J, Meltzer EB, Gray A, Miura R, Wogensen L, Yamaguchi Y, Noble PW. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J Exp Med. 2011;208(7):1459–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Collum SD, Molina JG, Hanmandlu A, Bi W, Pedroza M, Mertens TCJ, Wareing N, Wei W, Wilson C, Sun W, Rajadas J, Bollyky PL, Philip KM, Ren D, Thandavarayan RA, Bruckner BA, Xia Y, Blackburn MR, Karmouty‐Quintana H. Adenosine and hyaluronan promote lung fibrosis and pulmonary hypertension in combined pulmonary fibrosis and emphysema. Dis Models & Mech. 2019;12(50):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. https://clinicaltrials.gov/ct2/show/NCT05128929. Accessed July 2, 2023.
- 106. Gerges C, Vollmers K, Pritzker M, Gainor J, Scandurra J, Weir EK, Lang IM. Pulmonary artery endovascular device compensates for loss of vascular compliance in PAH. J Am Coll Cardiol. 2020;76(19):2282–2287. [DOI] [PubMed] [Google Scholar]
- 107. Spikes LA, Bajwa AA, Burger CD, Desai SV, Eggert MS, El‐Kersh KA, Fisher MR, Johri S, Joly JM, Mehta J, Palevsky HI, Ramani GV, Restrepo‐Jaramillo R, Sahay S, Shah TG, Deng C, Miceli M, Smith P, Shapiro SM. BREEZE: open‐label clinical study to evaluate the safety and tolerability of treprostinil inhalation powder as Tyvaso DPI™ in patients with pulmonary arterial hypertension. Pulm Circ. 2022;12(2):e12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. https://clinicaltrials.gov/ct2/show/NCT03399604. Accessed July 2, 2023.
- 109. https://clinicaltrials.gov/ct2/show/NCT05176951. Accessed July 2, 2023.
- 110. Chambers DC, Cherikh WS, Goldfarb SB, Hayes D Jr., Kucheryavaya AY, Toll AE, Khush KK, Levvey BJ, Meiser B, Rossano JW, Stehlik J, International Society for Heart and Lung T. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: thirty‐fifth adult lung and heart‐lung transplant report—2018; focus theme: multiorgan transplantation. J Heart Lung Transplant. 2018;37:1169–1183. [DOI] [PubMed] [Google Scholar]
- 111. Piccari L, Wort SJ, Meloni F, Rizzo M, Price LC, Martino L, Salvaterra E, Scelsi L, López Meseguer M, Blanco I, Callari A, Pérez González V, Tuzzolino F, McCabe C, Rodríguez Chiaradía DA, Vitulo P. The effect of borderline pulmonary hypertension on survival in chronic lung disease. Respiration. 2022;101(8):717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kimura M, Taniguchi H, Kondoh Y, Kimura T, Kataoka K, Nishiyama O, Aso H, Sakamoto K, Hasegawa Y. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Respiration. 2013;85:456–463. [DOI] [PubMed] [Google Scholar]
- 113. Hayes D Jr., Black SM, Tobias JD. Influence of pulmonary hypertension on patients with idiopathic pulmonary fibrosis awaiting lung transplantation. Ann Thorac Surg. 2016;101(1):246–252. [DOI] [PubMed] [Google Scholar]
- 114. Leard L, Holm A, Valapour A, Glanville AR, Attawar S, Aversa M, Campos SV, Christon LM, Cypel M, Dellgren G, Hartwig MG, Kapnadak SG, Kolaitis NA, Kotloff RM, Patterson CM, Shlobin OA, Smith PJ, Solé A, Solomon M, Weill D, Ramos KJ. Consensus document for the selection of lung transplant candidates: an update from the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2021;40(11):1349–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Whelan TPM, Dunitz JM, Kelly RF, Edwards LB, Herrington CS, Hertz MI, Dahlberg PS. Effect of preoperative pulmonary artery pressure on early survival after lung transplantation for idiopathic pulmonary fibrosis. J Heart Lung Transplant. 2005;24:1269–1274. [DOI] [PubMed] [Google Scholar]
- 116. Hayes D, Higgins RS, Black SM, Wehr AM, Lehman AM, Kirkby S, Whitson BA. Effect of pulmonary hypertension on survival in patients with idiopathic pulmonary fibrosis after lung transplantation: an analysis of the United Network of Organ Sharing Registry. J Heart Lung Transplant. 2015;34(3):430–437. [DOI] [PubMed] [Google Scholar]
- 117. Spratt JR, Tomic R, Brown RZ, Rudser K, Loor G, Hertz M, Shumway S, Kelly RF. Single versus bilateral lung transplantation for idiopathic pulmonary fibrosis in the Lung Allocation Score Era. J Surg Res. 2019;234:84–95. [DOI] [PubMed] [Google Scholar]
- 118. Schaffer JM, Singh SK, Reitz BA, Zamanian RT, Mallidi HR. Single‐vs double‐lung transplantation in patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis since the implementation of lung allocation based on medical need. JAMA. 2015;313:936–948. [DOI] [PubMed] [Google Scholar]
- 119. Villavicencio MA, Axtell AL, Osho A, Astor T, Roy N, Melnitchouk S, D'Alessandro D, Tolis G, Raz Y, Neuringer I, Sundt TM. Single versus double lung transplantation in pulmonary fibrosis: impact of age and pulmonary hypertension. Ann Thorac Surg. 2018;106(3):856–863. [DOI] [PubMed] [Google Scholar]
- 120. Nathan SD, Shlobin OA, Ahmad S, Burton NA, Barnett SD, Edwards E. Comparison of wait times and mortality for idiopathic pulmonary fibrosis patients listed for single or bilateral lung transplantation. J Heart Lung Transplant. 2010;29:1165–1171. [DOI] [PubMed] [Google Scholar]
- 121. Anderson MR, Tabah A, RoyChoudhury A, Lederer DJ. Procedure preference and intention‐to‐treat outcomes after listing for lung transplantation among U.S. adults. A cohort study. Ann Am Thorac Soc. 2019;16:231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]