

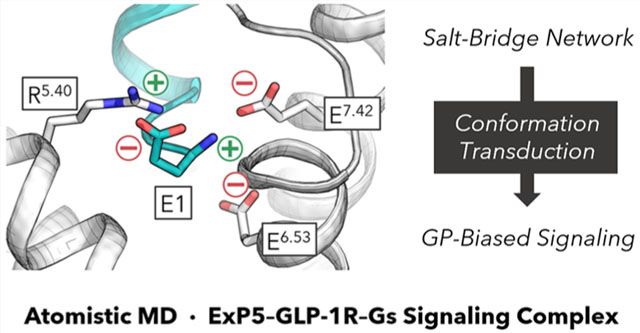
Abstract
The glucagon-like peptide-1 receptor (GLP-1R) is a key regulator of blood glucose and a prime target for the treatment of type II diabetes and obesity with multiple public drugs. Here we present a comprehensive computational analysis of the interactions of the activated GLP-1R–Gs signaling complex with a G protein biased agonist, Exendin P5 (ExP5), which possesses a unique N-terminal sequence responsible for the signal bias. Using a refined allatom model of the ExP5–GLP-1R–Gs complex in molecular dynamics (MD) simulations, we propose a novel mechanism of conformation transduction in which the unique interaction network of ExP5 N-terminus propagates the binding signal across an array of conserved residues at the transmembrane domain to enhance Gs protein coupling at the cytoplasmic end of the receptor. Our simulations reveal previously unobserved interactions important for activation by ExP5 toward GDP-GTP signaling, providing new insights into the mechanism of class B G protein-coupled receptor (GPCR) signaling. These findings offer a framework for the structure-based design of more effective therapeutics.
Graphical Abstract
INTRODUCTION
G protein-coupled receptors (GPCRs) constitute the largest family of transmembrane proteins, being responsible for transmitting extracellular signals to the cell interior to trigger various physiological processes. Class B GPCRs, a subfamily significant in hormonal homeostasis, are characterized by having long, extracellular N-terminal domains (NTDs) in addition to the seven transmembrane domains (TMD) that are structurally distinct from all GPCRs. GPCRs couple to a heterotrimeric G protein to increase or decrease cyclic adenosine monophosphate (cAMP) production by activating or inactivating a signaling pathway upon ligand binding. GPCRs can also lead to β-arrestin recruitment, which often leads to undesired side effects for therapeutics targeting GPCRs. Thus, there is growing interest in developing biased therapeutics that induce the receptor to selectively induce G protein activation over β-arrestin recruitment. One such target is the class B glucagon-like peptide-1 (GLP-1) receptor (GLP-1R).1
The GLP-1R plays an important role in inducing the secretion of insulin, making it a prime target for developing therapeutics to treat type II diabetes and obesity.1–3 Indeed, nearly 10 public drugs target GLP-1R, with a number of others under clinical trials.2 While named after its endogenous ligand, GLP-1, GLP-1R undergoes significant biased agonism with various peptidic and non-peptidic ligands,1,4–8 one of which is Exendin P5 (ExP5).9
The molecular mechanism of biased signaling is not well understood for class B GPCRs.4,10–12 Although a cryogenic electron microscopy (cryo-EM) model was reported for the activated ExP5–GLP-1R–Gs complex (PDB code: 6B3J),13 >350 residues were missing or partially resolved, and it contained an nanobody Nb35 added to improve stability.13 In addition, whereas a number of other structures have been reported for the active-state GLP-1R–Gs signaling complex,14–21 the peptide ligands all possess distinct N-terminal sequences resembling that of the endogenous GLP-1. Because the unique N-terminus of ExP5 is crucial to the biased agonism,9 a refined all-atom model for the ExP5-bound signaling complex is necessary to gain reliable insights.
We aim to establish additional understanding of the conformational dynamics responsible for the biased nature of agonist ExP5 bound to the GLP-1R–Gs protein complex in order to provide critical information underlying signal bias.10–12,22,23 We hereby report an in-depth analysis of the ligand–receptor interactions and the resulting structural reorganizations associated with the G protein biased behavior. We expand on existing models13,16,21 by providing a molecular mechanism of GLP-1R conformational transduction based on atomistic simulations of the signaling complex. We discuss how the unique N-terminal sequence of ExP5 increases the interactions that aid in rearranging the TMD. Our investigation uncovered a number of critical interactions that are closely related to biased agonism but were not resolved in the cryo-EM structure.13 This detailed understanding of the molecular interactions between ExP5 and GLP-1R offers important new insights into the biased signaling of class B GPCRs that should help guide the development of more effective treatments for type II diabetes and obesity.
RESULTS AND DISCUSSION
Molecular Dynamics (MD) Simulations Maintain Structural Integrity.
First we predicted the structures for the residues missing or not completely resolved in the 3.3 Å resolution cryo-EM structure of the ExP5 agonist-bound GLP-1R–Gs signaling complex (PDB ID: 6B3J).13 These include 29 residues at the N-terminus of GLP-1R, 8 located nearby the NTD (S129, K130, R131, G132, E133, R134, S135, and S136), and 6 in intracellular loop 3 (ICL3; N338, L339, M340, C341, K342, and T343). The side chains were not resolved for another 4 residues located in ECL3: D372, Q373, H374, and R376. Moreover, a total of 199 residues were not resolved in the Gαs subunit: residues 1–10, 48–204, 250–263, 296–307, and 365–370 inclusive. This covered the entire α-helical domain. In addition, residues 1–5 and 63–71 of the Gγ2 subunit were also missing, which included the C-terminus, where a lipid anchor should be attached. We used the protocols described in the Methods section to predict the structures of these residues by minimization, annealing simulations, and side-chain repacking. A graphical representation of the computationally refined regions is provided in Figure S1.
Using the reconstructed ExP5–GLP-1R–Gs complex, we performed all-atom MD simulations for a total of 1.5 μs (see the Methods section for details). Briefly, we immersed the ExP5–GLP-1R–Gs complex into a bilayer membrane of palmitoyloleoylphosphatidylcholine (POPC), and then we solvated the system in an ~120 × 120 × 200 Å3 water box, including 150 mM NaCl to achieve physiological salt conditions. In addition, we added two lipid anchors to the Gs protein, which included palmitoylation at C3 of Gαs and geranylgeranylation at C68 of Gγ2, with residues 69–71 of Gγ2 removed accordingly.24 Given the notable complexity of the signaling complex, our analysis prioritizes persistent interactions observed in the simulations as they are presumed to possess greater statistical and functional relevance.
This complex structure remains consistent throughout our MD simulation (Figures 1A and S2–S4). The total root-mean-square deviation (RMSD) of the whole protein backbone is 2.6 ± 0.5 Å vs cryo-EM, which is below the 3.3 Å resolution (Figure 1B). The average RMSDs of the receptor TMD (1.0 ± 0.1 Å), the ligand-binding pocket (1.4 ± 0.2 Å), and the Gs protein (1.8 ± 0.4 Å) are all quite low. Also, the two lipid anchors were able to penetrate into the bilayer membrane, aiding in the orientation and stabilization of the Gs protein. Together, these analyses demonstrate that our simulations preserve the integrity of the original cryo-EM template. Important differences from the cryo-EM structure are discussed below.
Figure 1.
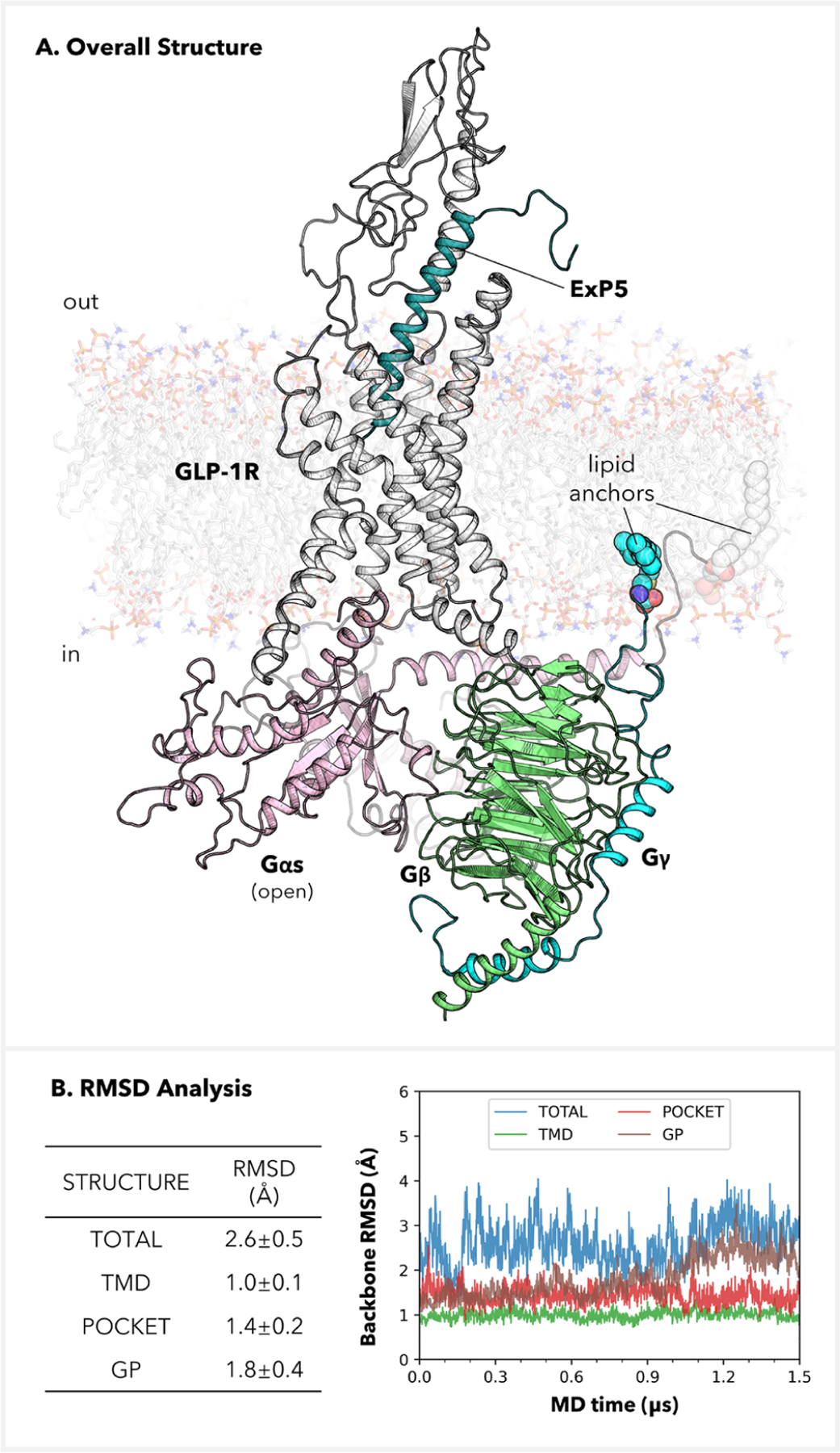
Completed atomistic level structure for the GLP-1 receptor in complex with the heterotrimeric Gs protein and the ExP5 biased agonist. (A) Side-view depiction. (B) RMSDs for the whole complex, the transmembrane domain, the ligand-binding pocket (all residues in 8.0 Å proximity with respect to ExP5), and the Gs protein. The RMSDs were computed after alignment of the selected components and were referenced against the cryo-EM structure (PDB code: 6B3J).13 All RMSDs are for non-hydrogen backbone atoms.
ExP5 Binding to GLP-1R.
The ExP5 peptide agonist is well-known for its bias toward G protein signaling of GLP-1R.9 This bias of ExP5 has been characterized to arise primarily from enhanced efficacy in cAMP signaling rather than a loss of β-arrestin coupling.9,13 Furthermore, a bioluminescence resonance energy transfer (BRET) assay has demonstrated that ExP5 induces a faster conformational change in Gαs compared to GLP-1 at equi-occupant concentrations.13 The primary objective of this study is to investigate how ExP5 modulates the conformations of the GLP-1R–Gs signaling complex at the active state to cause the observed behaviors.
We first analyzed the binding interactions of ExP5. Our MD simulations show that ExP5 maintains an α-helical fold from L2ExP5 to K28ExP5 (Figures 2A and S5). The bottom part of ExP5 binds to the extracellular pocket in the receptor TMD, while the top portion forms multiple anchors with TM1–2–3, ECL2, and NTD.13,25
Figure 2.
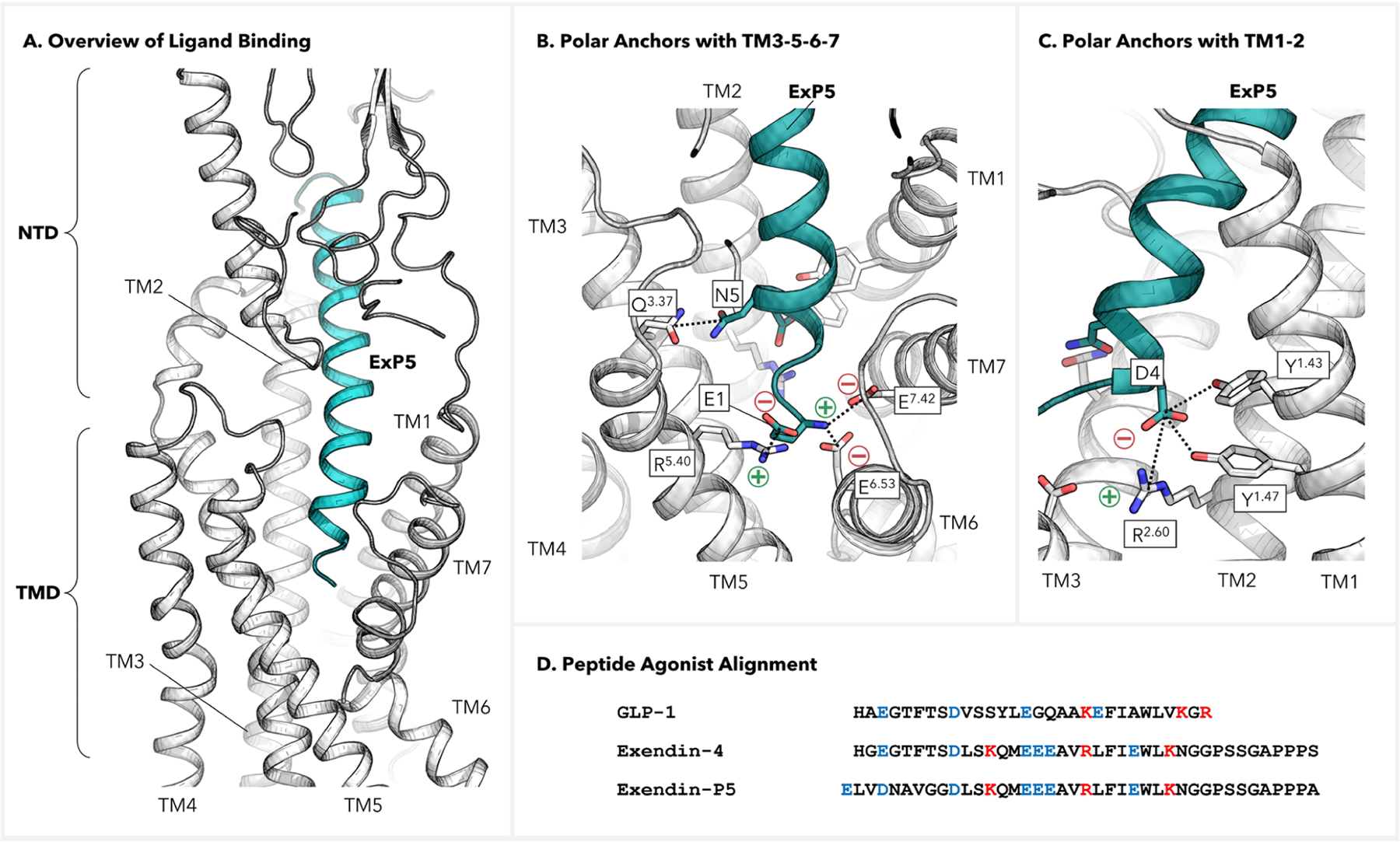
Interactions between the GLP-1 receptor and ExP5 peptide. (A) Side view of the overall interactions with the binding pocket. (B, C) Polar interactions of ExP5 with the binding pocket at the extracellular side of TMD. (D) Alignment of peptide agonists for GLP-1R. Residues with +/− charges are colored in red/blue, respectively.
We identified persistent polar anchors at the N-terminus of ExP5 where it reaches deeply into the receptor extracellular cavity to establish favorable contacts with all transmembrane helices except for TM6. Particularly, the N-terminal E1ExP5, which is unique to the G protein biased agonist ExP5 compared to nonbiased agonist GLP-1, carries both + and − charges to form multiple salt bridges (SBs) involving R5.40, E6.53, and E7.42, which collectively create a strong electrostatic network (Figure 2B, bottom). This network is established early in the production trajectory (~40 ns) and remains stable thereafter for the entire 1.5 μs simulation (Figures S5 and S6). In addition, we observed stable H-bonds (HBs) between N5ExP5 and Q3.37 (Figure 2B, left) as well as a Y1.43–D4ExP5–R2.60–Y1.47 HB network (Figures 2C, S5, and S6). The abundance of these favorable anchors enhances the binding of ExP5 to promote subsequent TMD rearrangements.
Notably, the ionic locks of the −NH3+ group of E1ExP5 with E6.53 and E7.42 represent new interactions not resolved in the cryo-EM structure (Figures S7 and S8).13 Indeed, it is acknowledged that a cryo-EM resolution of 3.3 Å can be inadequate for accurately determining atomistic details of chemical interactions despite their critical roles in rationalizing signaling behaviors.26–28 Notably, the SBs formed by E1ExP5 with TM6–7 implicate a direct correlation to the movements of the extracellular portions of TM6–7 and ECL3,13,16,21 which exhibit the most significant differences between the GLP-1 and ExP5-bound GLP-1R structures. Moreover, the E1ExP5–R5.40 SB explains the almost complete abolishment of ExP5-mediated cAMP accumulation upon R5.40A mutation,13,21 and the E1ExP5–E7.42 SB explains why the E7.42A mutation leads to substantial decreases in binding affinity of ExP5 and decreased cAMP signaling efficiency.21 Also, R5.40A, E6.53A, and E7.42A all led to appreciable decrease in ExP5 binding potencies.21 Taken together, these findings suggest that the E1ExP5 SB network acts as a hub for the ExP5-mediated promotion of Gs signaling.
In fact, the structural alignment reveals that E1ExP5 is a one-residue extension at the N-terminus compared to GLP-1 and other typical GLP-1R peptide agonists (Figure 2D).21 Hence, no such E1-based SB network is observed at the N-terminal head of GLP-1 complexed with GLP-1R.15,16 In fact, as showcased in Figure S8, we have confirmed that a number of peptide ligands with reported cryo-EM structures—including GLP-1, Tirzepatide, Semaglutide, Taspoglutide, Ex4, and Oxyntomodulin15,16,18–21—all adopt the GLP-1-like binding mode, i.e., without the N-terminal SB network observed for ExP5. This highlights the unique binding of the ExP5 G protein biased agonist.
In addition, it is worth noting that single-point mutations R2.60A, Y1.43A, and Y1.47A all result in an appreciable decrease in cAMP response,13,21 underscoring the importance of the Y1.43–D4ExP5–R2.60–Y1.47 HB network for Gs protein signaling. However, this particular interaction pattern is commonly observed with GLP-1R peptide agonists15,16,18–21 and may be less directly linked to the signal bias exhibited by ExP5.
A Lipid-Aided Model for ECL3-Out Conformation.
It has been characterized that ExP5 induces an ECL3-out conformation of GLP-1R in the active complex.13,16,21 Indeed, superimposition of our simulated ExP5–GLP-1R–Gs complex structure onto a cryo-EM model for the GLP-1–GLP-1R–Gs complex (PDB code: 6X18)16 confirmed the outward positioning of ECL3 in the presence of ExP5 (Figure 3A). In fact, while the TM6 helix terminates at around I6.55 when bound to GLP-1,16 the presence of ExP5 and POPC elongates the TM6 α-helical folding until V6.59 (see right-hand side in Figure 3A). Meanwhile, the ECL3-TM7 transition occurs at G7.32 in the presence of GLP-1, whereas ExP5 will defer the first residue of the TM7 helix until R7.35. These features of ExP5 binding explains the enrichment of the ECL3-out state.
Figure 3.
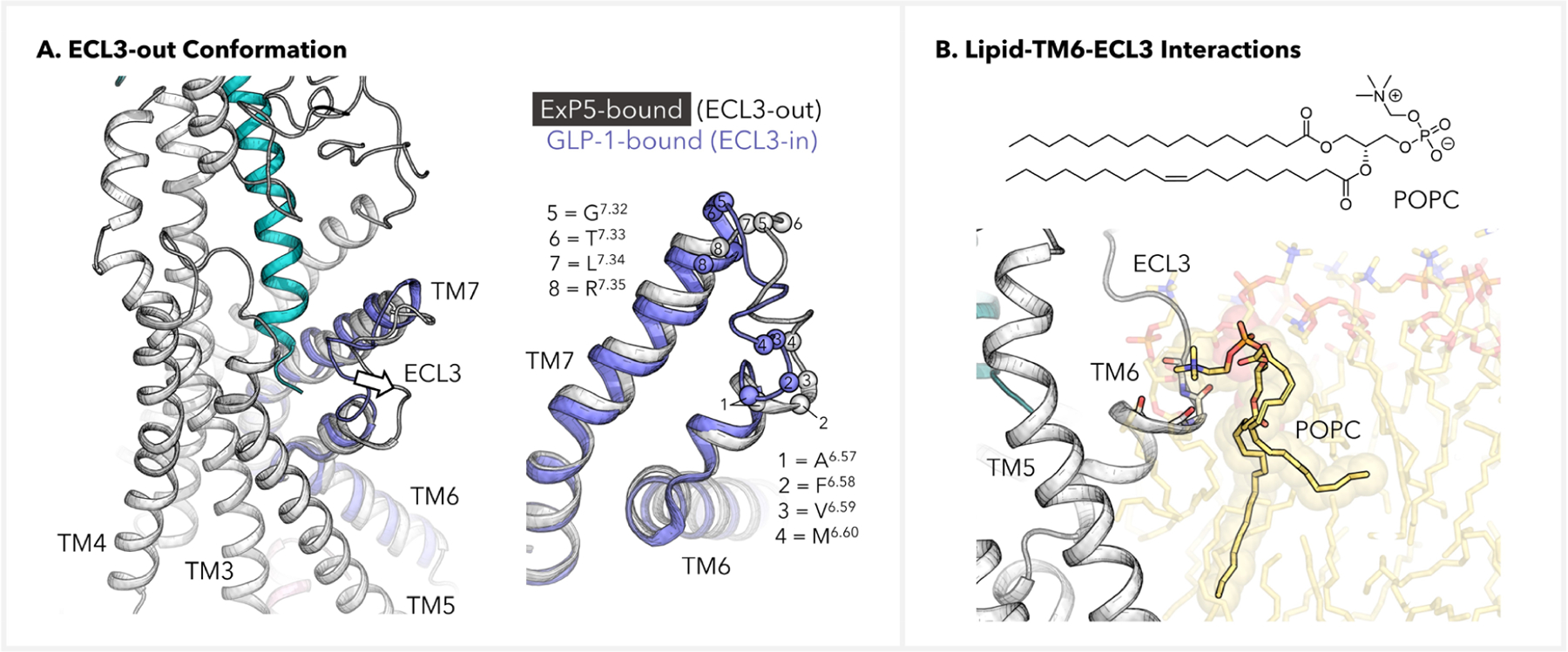
Lipid-aided induction of the ECL3-out GLP-1R conformation. (A) Depiction of the ECL3-out conformation in the ExP5-bound GLP-1R–Gs complex. Structure of the GLP-1-bound TM6-ECL3-TM7 (blue) was extracted from PDB code 6X18 and was based on alignment of the TMDs. (B) Backbone–POPC interactions at the TM6-ECL3 transition site, where the POPC–NMe3+ group is multianchored to A6.57, F6.58, V6.59, and M6.60. The backbone atoms of these residues are shown as sticks, and the side chains are omitted for clarity.
Intriguingly, our simulations revealed lipid–TM6-ECL3 interactions associated with ECL3 conformation regulation (Figure 3B). While the conformations of the long hydrophobic tails of POPC appear flexible and dynamic, we found that the −NMe3+ ammonium head of a POPC lipid molecule can be multianchored to the TM6-ECL3 intersection by interacting with the backbone oxygen atoms of A6.57, F6.58, V6.59, and M6.60, giving rise to a lipid cap on the extracellular end of TM6. To optimize electrostatics, these backbone oxygen atoms all point toward the −NMe3+ group, which entails strong constraints on the folding state of this region. The proximity of this POPC to the ExP5 N-terminus led us to speculate that ExP5 might have utilized the N-terminal SB network to create a favorable local electrostatic environment that helped recruit and anchor the POPC.29 We note that the POPC–NMe3+ on–off events are rather rare in our trajectory (~5 switches over 1.5 μs, Figure S9), which implicates the stability of the multianchoring effect. These results offer a lipid-assisted allosteric model for understanding the ECL3-out conformation, a differential conformation change widely noted in the literature.13,16,21 Meanwhile, we envision that a more realistic representation of the cell membrane than the currently used POPC bilayer model would contribute to a more comprehensive understanding of the interactions observed here and their dynamics.
ExP5 Stabilization of GLP-1R Active Conformation.
To further probe how ExP5 stabilizes the active state of GLP-1R, we conducted a conformational analysis of the refined ExP5–GLP-1R–Gs complex using an apo GLP-1R structure as a reference. To obtain the relevant apo structure, we removed ExP5 and Gs protein and performed a separate 1.0 μs MD simulation for GLP-1R. By superimposing the resultant apo structure with the complex over TMD, we identified rearrangements of transmembrane helices that propagate to the cytoplasmic interface (Figure 4). Specifically, we found that ExP5 causes TM6 to move downward toward the intracellular side (Figure 4A, bottom left), whereas TM7 moves upward toward the extracellular side (Figure 4A, bottom right). Moreover, ExP5 engages TM6-ECL3-TM7 such that both helices are twisted at the extracellular side and undergo inward displacements (Figure 4A, bottom green arrows, and Figure 4B, top). Additionally, we observed an extension of the TM5 helical fold at the intracellular end, which results in a shortened ICL3 (Figure 4A, top; Figure 4B, bottom).
Figure 4.
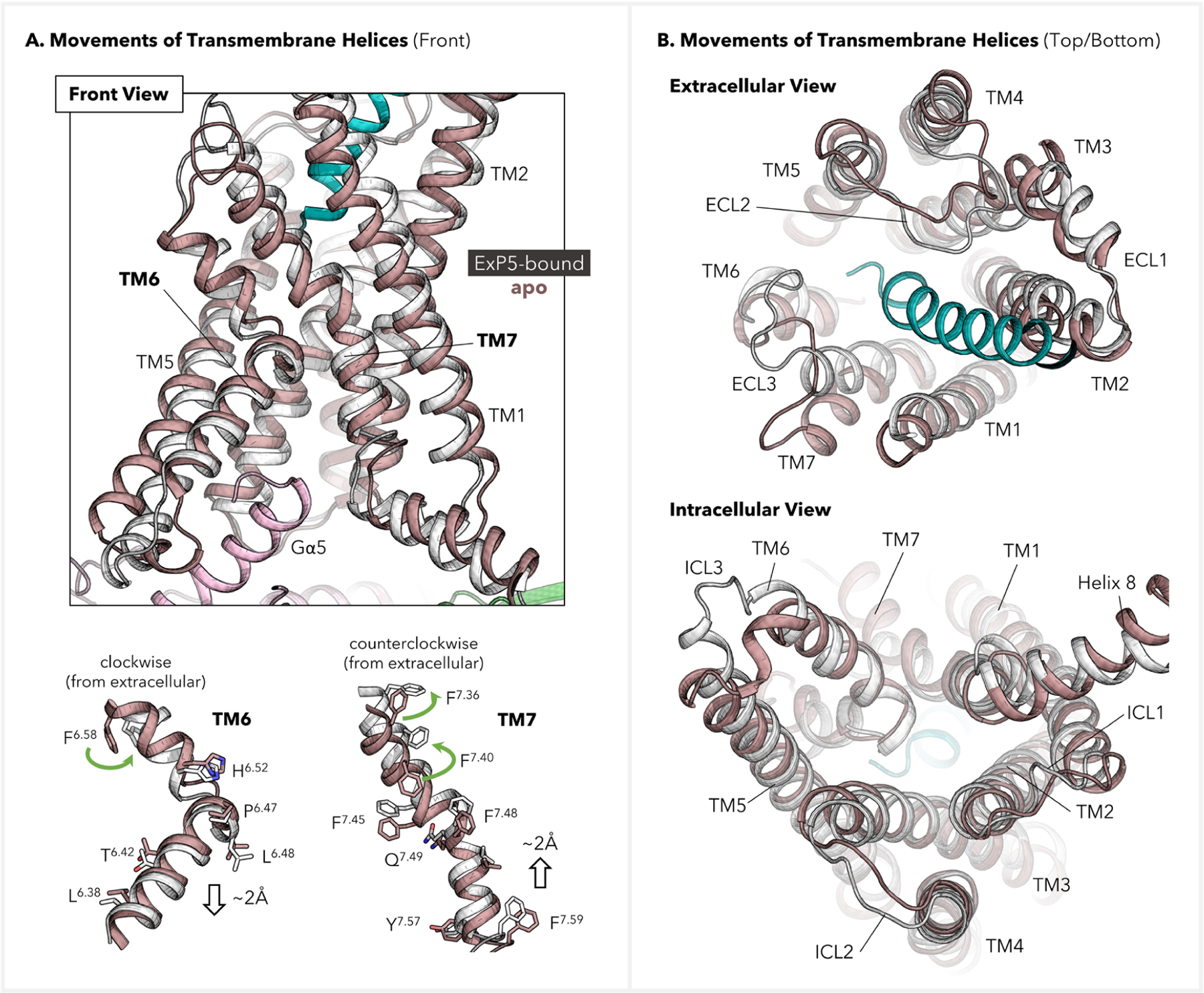
Displacements of transmembrane helices of GLP-1R before and after activation. The refined ExP5–GLP-1R–Gs signaling complex (color: ExP5 in green, GLP-1R in white, and Gs protein in pink) was used as the active-state structure, while the inactive structure was extracted from the MD simulation for an apo GLP-1R (color: brown). (A) Front view and residue movements in TM6–7. Selected residues are shown on sticks to indicate their movements upon activation. (B) Extra- and intracellular views for TMD displacements.
We postulated that the observed movements of the TMD might form a foundation for outside-in signal transduction. Indeed, our further analysis of the superimposed structures indicates that the E1ExP5 SB network plays a crucial role in promoting the aforementioned rearrangements by engaging and reorienting the extracellular parts of TM5–7. As presented in Figure 5A, the E1ExP5–E6.53 SB involves a rotamer switch of E6.53 that favors the downward translation of TM6. Notably, TM6 has formed a zigzag-shaped interface with neighboring TM5 utilizing a number of residues lying beneath the conserved P6.47xxG6.50 motif. This includes an HB between conserved residues L6.49 and F5.54 at the P6.47xxG6.50 motif, located in the middle of the two helices. More importantly, a large interface of hydrophobic residues results in a zipper-type TM5–TM6 coupling for their intracellular halves, which is based on bulky residues F5.54, I5.58, V5.61, L6.38, T6.42, and L6.45. Among them, (i) T6.42 and L6.45 are conserved across class B1; (ii) F5.54 and L6.38 are shared by 11 out of 15 class B1 GPCRs, while the two residues are highly synchronously replaced by L5.54 and A6.38, respectively, in the remaining 4 members; (iii) I5.58 and V5.61, despite higher sequence variations, occupy two positions with a hydrophobic sequence consensus, the variations limited to isoleucine (I), leucine (L), and valine (V). These observations led us to hypothesize that the TM5–TM6 collective motions can be relevant for a broader scope of class B GPCRs. Combined, these factors suggest that the E1ExP5 network utilizes an array of conserved residues to trigger the above-mentioned TMD rearrangements and therefore enriches the active conformation of GLP-1R, offering a basis for the signal bias.4,10–12,22,30–32 How this influences Gs protein coupling is explored in the next section.
Figure 5.
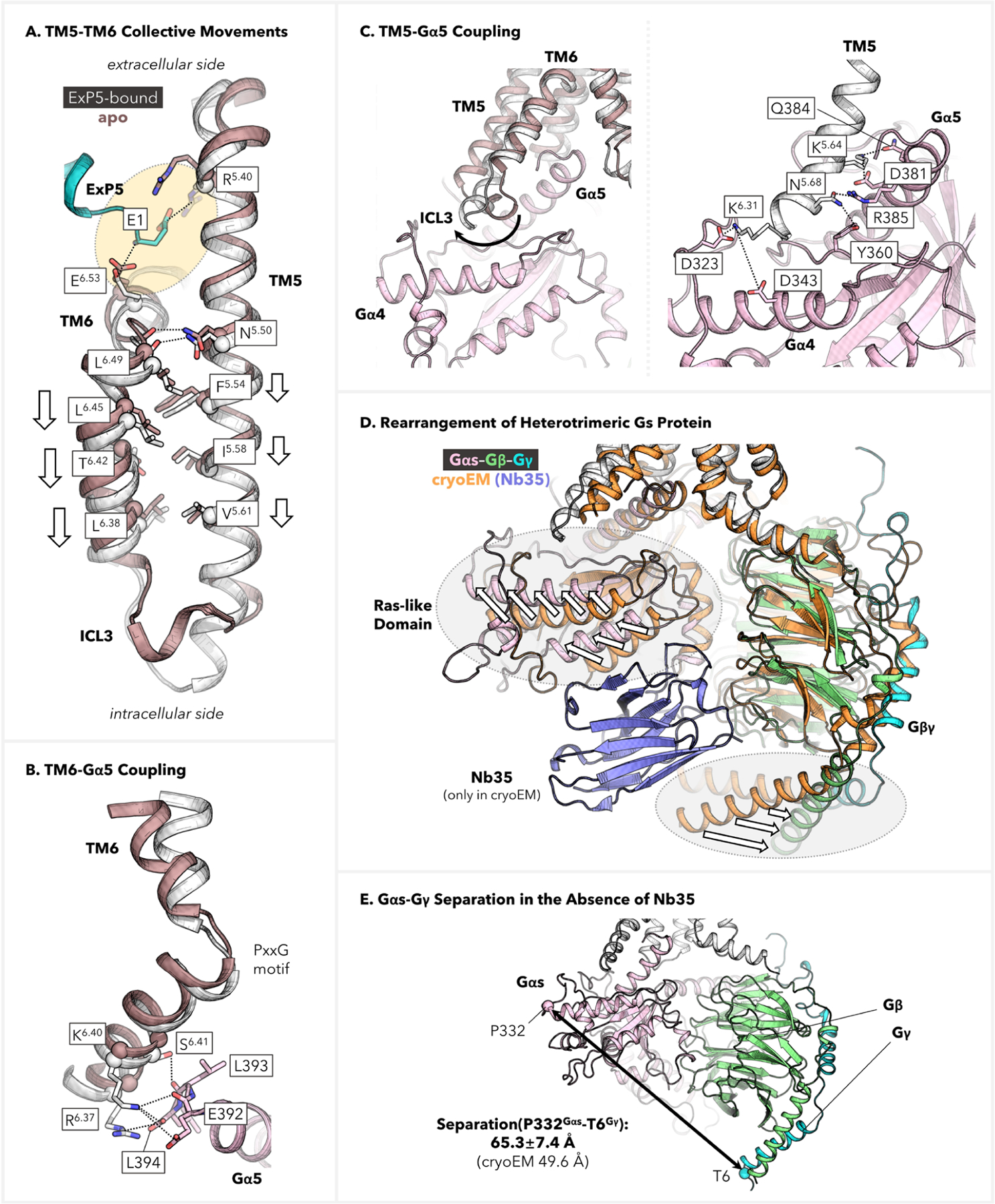
Residue-level mapping of E1ExP5-triggered conformational transduction. (A) TM5–TM6 collective displacements toward the cytoplasmic end as compared with the apo GLP-1R structure. (B) Depiction of TM6–Gα5 coupling, which involves multiple SBs and H-bond anchors at the C-terminus of the Gα5 helix. (C) Depiction of TM5–Gαs coupling, including (left) comparison with the apo structure showcasing the critical movement of ICL3 and (right) detailed description of the anchors involved in the Gs protein coupling. (D, E) Structure rearrangement of the heterotrimeric Gs protein in the absence of Nb35. The cryo-EM structure of the ExP5–GLP-1R–Gs complex (PDB code: 6B3J),13 which was stabilized by Nb35 during determination, is used here for comparison.
GLP-1R–Gs Protein Interactions.
Most importantly, the ExP5-stabilized collective movements propagate from the extracellular side of the TMD to the intracellular side, giving rise to enhanced coupling with the Gαs subunit, which forms a crucial basis for Gs protein signaling. As shown in Figures 5B,C and S10, our simulations point to the TM5-ICL3-TM6–Gαs interface as a pivotal site for stabilizing the activated Ras-like domain, offering abundant coupling interactions. Our simulations show that TM6 is stabilized at a position advantageous for forming multiple ionic locks and H-bond anchors between conserved residues R6.37, K6.40, and S6.41 and the C-terminus of the deeply inserted Gα5 helix (Figure 5B). In addition, we discovered a closer and more robust coupling of the Gαs Ras-like domain that was not captured in the cryo-EM model.13 Remarkably, we identified an extension of the TM5 helix at the intracellular end that could stretch to K6.31 (Figure 5C, left). As a result, GLP-1R can utilize the TM5 end and ICL3 to interact efficiently with Gαs. This includes persistent ionic interactions from conserved residue K5.64 to D381Gαs as well as a flexible SB that alternates between K6.31–D323Gαs and K6.31–D343Gαs. Moreover, TM5 forms a significant number of polar anchors to further couple with Gα5, which is also described in Figure 5C (right). Besides stabilizing the fully activated conformation, these interactions likely establish an increased number of metastable states in the course of G protein activation during ExP5-mediated signaling.
We emphasize here the significance of the differential Gs protein coupling that we observe in our simulations as compared to the cryo-EM structure.13 As presented in Figure 5D, the cryo-EM determination employed nanobody Nb35 to engage the heterotrimeric G protein and stabilize the signaling complex,13 whereas our MD simulations did not incorporate Nb35. As a result, we observed a remarkable increase by ~16 Å in the P332Gαs Cα–T6Gγ Cα distance, which provides a measure of the separation between the Gαs Ras-like domain and the Gβγ subunits (Figure 5E). This ~16 Å further separation reflects the dynamic movement of the Ras-like domain, which facilitates its enhanced interactions with TM5-ICL3-TM6. Hence, the placement of the artificial Nb35 beneath Gαs probably increased the strain energy required for Gαs to engage the ICL3 and TM5–6 residing above it. Indeed, similar observations were made upon superimposing our ExP5-bound structure onto a high-resolution GLP-1-bound cryo-EM structure that also contains the nanobody (Figure S11).16 Because Nb35 has been widely used in related determinations,13–21 these results highlight the importance of exercising caution when interpreting such modified cryo-EM structures to gain insight into G protein biased agonism.
CONCLUSION
The class B GLP-1R is a major therapeutic target for treating type 2 diabetes mellitus and obesity with multiple clinically approved drugs.1–3 The molecular mechanism of GLP-1R activation has been interpreted in terms of critical TMD reorganizations in the active state,10,13,15 but the basis was not well understood. Our simulations now establish a new explanation for how ExP5, a Gs protein biased agonist,9 might exploit its unique N-terminal E1ExP5 SB network to transmit its binding signal across the TMD to enhance Gs protein coupling. The network induces TM5–TM6 collective movement toward the intracellular end, allowing extensive TM5–ICL3–TM6 interactions with the Gαs subunit that was only partially captured by the experimental determination. We also identified a possible lipid-aided allosteric effect, which might prove useful in understanding membrane composition effects and help to design new GLP-1R allosteric modulators. Notably, our simulations have identified the majority of the most influential single-point alanine mutagenesis in the systematic assays by Liang et al.13 and Deganutti et al.,21 which closely align with the proposed R5.40–E1ExP5–E6.53–E7.42 and Y1.43–D4ExP5–R2.60–Y1.47 interaction hubs. Our study provides a unifying view of many aforementioned observations with a number of implications.
Our findings highlight that agonist–TM5–6–7 interactions are a hot spot for exerting conformational control on GLP-1R toward biased signaling. While studies on class A GPCRs have also revealed differential conformational dynamics in TM5, TM6, and/or TM7 associated with signal bias,22,30–33 class B GPCRs exhibit distinct activation rearrangements,4,10,23,34,35 for which only limited molecular insights have been established relative to biased agonism. Our identification of a new interaction hub at E1ExP5 may provide a framework for achieving more deliberate control of ligand bias for class B GPCRs. It also offers a novel viewpoint for connecting the behaviors of class B GPCRs to those observed in class A, whose structures are vastly different.
Moreover, GLP-1R belongs to the (class B1) glucagon receptor (GCGR) family. Previously, this family has attracted intense interest in developing bi- and triagonists, which have proved to give superior therapeutic efficacy.2 In the E1ExP5 SB network, R5.40 and E6.53 are shared by all four GPCRs in the GCGR family, and E7.42 is also present in the gastric inhibitory polypeptide receptor (GIPR) within the same family. Hence, the new model for ExP5–GLP-1R interactions may have implications for the development of more effective multitarget agonists for these closely related GPCRs.
We expect that our findings will aid the design of new small-molecule biased agonists that more readily allow oral formulation and reduce nausea as well as other gastrointestinal problems seen in current GLP-1R drugs, which are mostly peptides. Comparative studies of small-molecule vs peptide agonists might also shed more light on the nature of GLP-1R signaling and ligand bias.16
As discussed above, our findings are based upon out new structure, which displays a series of critical differences from the cryo-EM model,13 including the E1ExP5 SB network and the mode of Gs protein coupling. This emphasizes the importance of computational structure refinement in correcting the artifacts in the cryo-EM structures.
It is important to acknowledge that our studies are currently limited to the GLP-1R–Gs signaling complex. However, we anticipate that gaining access to the GLP-1R–arrestin complex structure, in both the presence and absence of agonists, could provide valuable insights into the underlying mechanisms of biased signaling. Additionally, despite the extensive duration of our MD simulations, it is crucial to recognize the limitations arising from the restricted number of replicas due to the computational demands associated with simulating the extended signaling system under investigation. Lastly, we acknowledge that our representation of the cell membrane remains simplified. While such simplifications have been commonly employed, it is crucial to recognize that GPCR activities are known to depend on the specific characteristics of the membrane. Therefore, it is highly desirable for future studies to conduct more detailed investigations into these limitations in order to assess the potential bias that may emerge as a result.
In summary, we provide new structural insights into ExP5 activation of GLP-1R, many of which were not resolved in cryo-EM or were influenced by the additional stabilizing components (the nanobody).13–21 This work advances existing models by offering a plausible atomistic pathway for conformational transduction. In addition to various mechanistic implications, this study has potential implications for structure-based therapeutic design for class B GPCRs to provide greater efficacy and specificity with reduced side effects.
METHODS
Modeling of ExP5–GLP-1R–Gs Complex.
Using the cryo-EM structure of the ExP5–GLP-1R–Gs complex (PDB code: 6B3J),13 we completed the following residues using the Maestro Software (ver. 12.4.075; Schrödinger):36 S127, K128, R129, G130, E131, R132, S133, S134, N338, L339, M340, C341, K342, and T343. The disulfide bridges—C46 to C71, C62 to C104, C85 to C126, and C226 to C296—were reconnected during simulations. We then annealed residues 127 to 137 and 338 to 344 by heating from 50 to 600 K and then cooled to 50 K over 10 ps, keeping all other residues fixed. We subsequently minimized the whole structure, keeping the backbone fixed to relax any steric clash that might have been introduced during our modeling. In the Gαs subunit, a grand total of ~200 residues were not resolved in the cryo-EM structure: residues 1 to 10, 48 to 204, 250 to 263, 296 to 307, and 365 to 370 inclusive. These residues, with the exception of residues 71–84, were spliced in by superimposing the Gαs from our previous modeling of a β2-adrenergic receptor–Gs protein complex (original PDB code: 6NI3).37,38 Then, Maestro software was used to remove overlapping structures and connect the peptide segments. Residues 71–84 were not included in either crystal structure, and so they were added using the same software. We then optimized the Gαs subunit with annealing simulations: residues 9–11, 45–51, 68–87, 200–205, 255–263, 290–310, 362–370, and 394–290 were permitted to move while heated from 50 to 600 K and then cooled to 50 K for 10 ps. The now completed ExP5–GLP-1R–Gs complex was then recombined and was subject to the SCREAM procedure39 to optimize side chain rotamer orientations. We note that because adding lipid anchors requires the full C-terminus of the Gγ subunit, which was not resolved in the cryo-EM, we used the structure model documented in the AlphaFold Protein Structure Database40,41 (accessed using UniProt code: P59768; model v4 was retrieved and used in our work). This completed complex structure was used for subsequent simulations.
MD Simulations.
All MD simulations were performed using the GROMACS software package.42 Using the CHARMM-GUI web server,43–45 we immersed the reconstructed complex into a bilayer membrane of 370 POPC molecules, added the two lipid anchors, solvated with 67102 water molecules, and neutralized with 203 Na+ and 182 Cl− (150 mM salt concentration), giving rise to a periodic box of ~120 × 120 × 200 Å3. The CHARMM36m46 force field was used with the TIP3P47 water force field and the CHARMM36 lipid force field.48 The system was relaxed by steepest-descent energy minimization, followed by a total of ~6.4 ns of pre-equilibration with gradually decreasing restraints. These included positional restraints for backbones that reduced step by step from ~9.6 to ~0.1 kcal mol−1 Å−2, side chains from ~4.8 to 0.0 kcal mol−1 Å−2, and lipids from ~2.4 to 0.0 kcal mol−1 Å−2. During pre-equilibration, we also applied ~1.2 kcal mol−1 Å−2 harmonic restraints to the following residue pairs, which formed salt bridges in the initial structure: K28ExP5–E128NTD, E18ExP5–R134NTD, D10ExP5–R3807.35, D4ExP5–R1902.60, E1ExP5–R3105.40, R1762.46–E4088.49, E2624.38–R38Gαs, R3486.37–L394Gαs, and K4158.56–D312Gβ. The system was then subjected to a further 30 ns pre-equilibration with only the salt bridge restraints. Lastly, production run was conducted for 1.5 μs. The LINCS algorithm49 was used to fix the bond distances involving H atoms. The temperature was maintained at 310 K using the Nosé−Hoover thermostat,50,51 and the pressure was controlled at 1 bar using a semi-isotropic Parrinello–Rahman barostat52 with a 5.0 ps damping constant and a 4.5 × 10−5 bar−1 compressibility.
MD simulations of the apo GLP-1R had the ExP5 agonist and the entire heterotrimeric Gs protein removed. The system included 255 POPC lipid molecules, 33420 water molecules, 92 Na+, and 90 Cl−, reaching 150 mM salt concentration, and the box size was ~100 × 100 × 145 Å3. Minimization and pre-equilibration (~6.4 ns) employed identical parameters for the gradually released restraints for different parts of the system. However, no salt-bridge restraint was applied, and the 30 ns additional pre-equilibration with only the salt-bridge restraints was skipped accordingly. Production was performed for 1.0 μs with identical simulation settings. Molecular visualization was conducted in the open-source PyMOL program.53
Supplementary Material
ACKNOWLEDGMENTS
B.L. was supported by a graduate research funding from Caltech. K.M. and A.V. were supported by the Caltech Summer Undergraduate Research Fellowship program. S.-K.K., M.Y.Y., and W.A.G. received support from NIH (R01HL155532 and R35HL150807).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c05996.
Supplementary computational results and simulation details (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.3c05996
The authors declare no competing financial interest.
Contributor Information
Bo Li, Division of Chemistry and Chemical Engineering and Materials Process and Simulation Center, California Institute of Technology, Pasadena, California 91125, United States.
Krystyna Maruszko, Division of Chemistry and Chemical Engineering and Materials Process and Simulation Center, California Institute of Technology, Pasadena, California 91125, United States.
Soo-Kyung Kim, Division of Chemistry and Chemical Engineering and Materials Process and Simulation Center, California Institute of Technology, Pasadena, California 91125, United States.
Moon Young Yang, Division of Chemistry and Chemical Engineering and Materials Process and Simulation Center, California Institute of Technology, Pasadena, California 91125, United States.
Amy-Doan P. Vo, Division of Chemistry and Chemical Engineering and Materials Process and Simulation Center, California Institute of Technology, Pasadena, California 91125, United States;
William A. Goddard, III, Division of Chemistry and Chemical Engineering and Materials Process and Simulation Center, California Institute of Technology, Pasadena, California 91125, United States;.
REFERENCES
- (1).Graaf C. de; Donnelly D; Wootten D; Lau J; Sexton PM; Miller LJ; Ahn J-M; Liao J; Fletcher MM; Yang D; Brown AJH; Zhou C; Deng J; Wang M-W Glucagon-Like Peptide-1 and Its Class B G Protein-Coupled Receptors: A Long March to Therapeutic Successes. Pharmacol Rev. 2016, 68 (4), 954–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Yang D; Zhou Q; Labroska V; Qin S; Darbalaei S; Wu Y; Yuliantie E; Xie L; Tao H; Cheng J; Liu Q; Zhao S; Shui W; Jiang Y; Wang M-W G Protein-Coupled Receptors: Structure- and Function-Based Drug Discovery. Sig Transduct Target Ther 2021, 6 (1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hauser AS; Attwood MM; Rask-Andersen M; Schiöth HB; Gloriam DE Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discov 2017, 16 (12), 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wootten D; Miller LJ; Koole C; Christopoulos A; Sexton PM Allostery and Biased Agonism at Class B G Protein-Coupled Receptors. Chem. Rev 2017, 117 (1), 111–138. [DOI] [PubMed] [Google Scholar]
- (5).Hager MV; Clydesdale L; Gellman SH; Sexton PM; Wootten D Characterization of Signal Bias at the GLP-1 Receptor Induced by Backbone Modification of GLP-1. Biochem. Pharmacol 2017, 136, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Koole C; Wootten D; Simms J; Valant C; Sridhar R; Woodman OL; Miller LJ; Summers RJ; Christopoulos A; Sexton PM Allosteric Ligands of the Glucagon-Like Peptide 1 Receptor (GLP-1R) Differentially Modulate Endogenous and Exogenous Peptide Responses in a Pathway-Selective Manner: Implications for Drug Screening. Mol. Pharmacol 2010, 78 (3), 456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wootten D; Savage EE; Willard FS; Bueno AB; Sloop KW; Christopoulos A; Sexton PM Differential Activation and Modulation of the Glucagon-Like Peptide-1 Receptor by Small Molecule Ligands. Mol. Pharmacol 2013, 83 (4), 822–834. [DOI] [PubMed] [Google Scholar]
- (8).van der Velden WJC; Smit FX; Christiansen CB; Møller TC; Hjortø GM; Larsen O; Schiellerup SP; Bräuner-Osborne H; Holst JJ; Hartmann B; Frimurer TM; Rosenkilde MM GLP-1 Val8: A Biased GLP-1R Agonist with Altered Binding Kinetics and Impaired Release of Pancreatic Hormones in Rats. ACS Pharmacol. Transl. Sci 2021, 4 (1), 296–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhang H; Sturchler E; Zhu J; Nieto A; Cistrone PA; Xie J; He L; Yea K; Jones T; Turn R; Di Stefano PS; Griffin PR; Dawson PE; McDonald PH; Lerner RA Autocrine Selection of a GLP-1R G-Protein Biased Agonist with Potent Antidiabetic Effects. Nat. Commun 2015, 6 (1), 8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wootten D; Christopoulos A; Marti-Solano M; Babu MM; Sexton PM Mechanisms of Signalling and Biased Agonism in G Protein-Coupled Receptors. Nat. Rev. Mol. Cell Biol 2018, 19 (10), 638–653. [DOI] [PubMed] [Google Scholar]
- (11).Latorraca NR; Venkatakrishnan AJ; Dror RO GPCR Dynamics: Structures in Motion. Chem. Rev 2017, 117 (1), 139–155. [DOI] [PubMed] [Google Scholar]
- (12).Wingler LM; Lefkowitz RJ Conformational Basis of G Protein-Coupled Receptor Signaling Versatility. Trends in Cell Biology 2020, 30 (9), 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Liang Y-L; Khoshouei M; Glukhova A; Furness SGB; Zhao P; Clydesdale L; Koole C; Truong TT; Thal DM; Lei S; Radjainia M; Danev R; Baumeister W; Wang M-W; Miller LJ; Christopoulos A; Sexton PM; Wootten D Phase-Plate Cryo-EM Structure of a Biased Agonist-Bound Human GLP-1 Receptor-Gs Complex. Nature 2018, 555 (7694), 121–125. [DOI] [PubMed] [Google Scholar]
- (14).Johnson RM; Zhang X; Piper SJ; Nettleton TJ; Vandekolk TH; Langmead CJ; Danev R; Sexton PM; Wootten D Cryo-EM Structure of the Dual Incretin Receptor Agonist, Peptide-19, in Complex with the Glucagon-like Peptide-1 Receptor. Biochem. Biophys. Res. Commun 2021, 578, 84–90. [DOI] [PubMed] [Google Scholar]
- (15).Zhang Y; Sun B; Feng D; Hu H; Chu M; Qu Q; Tarrasch JT; Li S; Sun Kobilka T; Kobilka BK; Skiniotis G Cryo-EM Structure of the Activated GLP-1 Receptor in Complex with a G Protein. Nature 2017, 546 (7657), 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhang X; Belousoff MJ; Zhao P; Kooistra AJ; Truong TT; Ang SY; Underwood CR; Egebjerg T; Šenel P; Stewart GD; Liang Y-L; Glukhova A; Venugopal H; Christopoulos A; Furness SGB; Miller LJ; Reedtz-Runge S; Langmead CJ; Gloriam DE; Danev R; Sexton PM; Wootten D Differential GLP-1R Binding and Activation by Peptide and Non-Peptide Agonists. Mol. Cell 2020, 80 (3), 485–500 e7. [DOI] [PubMed] [Google Scholar]
- (17).Cary BP; Deganutti G; Zhao P; Truong TT; Piper SJ; Liu X; Belousoff MJ; Danev R; Sexton PM; Wootten D; Gellman SH Structural and Functional Diversity among Agonist-Bound States of the GLP-1 Receptor. Nat. Chem. Biol 2022, 18 (3), 256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sun B; Willard FS; Feng D; Alsina-Fernandez J; Chen Q; Vieth M; Ho JD; Showalter AD; Stutsman C; Ding L; Suter TM; Dunbar JD; Carpenter JW; Mohammed FA; Aihara E; Brown RA; Bueno AB; Emmerson PJ; Moyers JS; Kobilka TS; Coghlan MP; Kobilka BK; Sloop KW Structural Determinants of Dual Incretin Receptor Agonism by Tirzepatide. Proc. Natl. Acad. Sci. U. S. A 2022, 119 (13), No. e2116506119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhang X; Belousoff MJ; Liang Y-L; Danev R; Sexton PM; Wootten D Structure and Dynamics of Semaglutide- and Taspoglutide-Bound GLP-1R-Gs Complexes. Cell Reports 2021, 36 (2), 109374. [DOI] [PubMed] [Google Scholar]
- (20).Zhao F; Zhou Q; Cong Z; Hang K; Zou X; Zhang C; Chen Y; Dai A; Liang A; Ming Q; Wang M; Chen L-N; Xu P; Chang R; Feng W; Xia T; Zhang Y; Wu B; Yang D; Zhao L; Xu HE; Wang M-W Structural Insights into Multiplexed Pharmacological Actions of Tirzepatide and Peptide 20 at the GIP, GLP-1 or Glucagon Receptors. Nat. Commun 2022, 13 (1), 1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Deganutti G; Liang Y-L; Zhang X; Khoshouei M; Clydesdale L; Belousoff MJ; Venugopal H; Truong TT; Glukhova A; Keller AN; Gregory KJ; Leach K; Christopoulos A; Danev R; Reynolds CA; Zhao P; Sexton PM; Wootten D Dynamics of GLP-1R Peptide Agonist Engagement Are Correlated with Kinetics of G Protein Activation. Nat. Commun 2022, 13 (1), 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Suomivuori C-M; Latorraca NR; Wingler LM; Eismann S; King MC; Kleinhenz ALW; Skiba MA; Staus DP; Kruse AC; Lefkowitz RJ; Dror RO Molecular Mechanism of Biased Signaling in a Prototypical G Protein-Coupled Receptor. Science 2020, 367 (6480), 881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Glukhova A; Draper-Joyce CJ; Sunahara RK; Christopoulos A; Wootten D; Sexton PM Rules of Engagement: GPCRs and G Proteins. ACS Pharmacol. Transl. Sci 2018, 1 (2), 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wedegaertner PB; Wilson PT; Bourne HR Lipid Modifications of Trimeric G Proteins. J. Biol. Chem 1995, 270 (2), 503–506. [DOI] [PubMed] [Google Scholar]
- (25).Mann R; Nasr N; Hadden D; Sinfield J; Abidi F; AlSabah S; de Maturana RL; Treece-Birch J; Willshaw A; Donnelly D Peptide Binding at the GLP-1 Receptor. Biochem. Soc. Trans 2007, 35 (4), 713–716. [DOI] [PubMed] [Google Scholar]
- (26).Danev R; Belousoff M; Liang Y-L; Zhang X; Eisenstein F; Wootten D; Sexton PM Routine Sub-2.5 Å Cryo-EM Structure Determination of GPCRs. Nat. Commun 2021, 12 (1), 4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Mafi A; Kim S-K; Goddard WA The G Protein-First Activation Mechanism of Opioid Receptors by Gi Protein and Agonists. QRB Discovery 2021, 2, No. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Mafi A; Kim S-K; Goddard WA The Atomistic Level Structure for the Activated Human κ-Opioid Receptor Bound to the Full Gi Protein and the MP1104 Agonist. Proc. Natl. Acad. Sci. U. S. A 2020, 117 (11), 5836–5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Dror RO; Green HF; Valant C; Borhani DW; Valcourt JR; Pan AC; Arlow DH; Canals M; Lane JR; Rahmani R; Baell JB; Sexton PM; Christopoulos A; Shaw DE Structural Basis for Modulation of a G-Protein-Coupled Receptor by Allosteric Drugs. Nature 2013, 503 (7475), 295–299. [DOI] [PubMed] [Google Scholar]
- (30).Teng X; Chen S; Nie Y; Xiao P; Yu X; Shao Z; Zheng S Ligand Recognition and Biased Agonism of the D1 Dopamine Receptor. Nat. Commun 2022, 13 (1), 3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Qu Q; Huang W; Aydin D; Paggi JM; Seven AB; Wang H; Chakraborty S; Che T; DiBerto JF; Robertson MJ; Inoue A; Suomivuori C-M; Roth BL; Majumdar S; Dror RO; Kobilka BK; Skiniotis G Insights into Distinct Signaling Profiles of the ΜOR Activated by Diverse Agonists. Nat. Chem. Biol 2023, 19, 423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).McCorvy JD; Butler KV; Kelly B; Rechsteiner K; Karpiak J; Betz RM; Kormos BL; Shoichet BK; Dror RO; Jin J; Roth BL Structure-Inspired Design of β-Arrestin-Biased Ligands for Aminergic GPCRs. Nat. Chem. Biol 2018, 14 (2), 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wingler LM; Elgeti M; Hilger D; Latorraca NR; Lerch MT; Staus DP; Dror RO; Kobilka BK; Hubbell WL; Lefkowitz RJ Angiotensin Analogs with Divergent Bias Stabilize Distinct Receptor Conformations. Cell 2019, 176 (3), 468–478 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Bortolato A; Doré AS; Hollenstein K; Tehan BG; Mason JS; Marshall FH Structure of Class B GPCRs: New Horizons for Drug Discovery: Structure of Class B GPCRs Enable Drug Discovery. Br. J. Pharmacol 2014, 171 (13), 3132–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Cong Z; Liang Y-L; Zhou Q; Darbalaei S; Zhao F; Feng W; Zhao L; Xu HE; Yang D; Wang M-W Structural Perspective of Class B1 GPCR Signaling. Trends Pharmacol. Sci 2022, 43 (4), 321–334. [DOI] [PubMed] [Google Scholar]
- (36).Schrödinger Release: Maestro (ver. 12.4.075); Schrödinger, LLC: New York, 2023. [Google Scholar]
- (37).Nguyen AH; Thomsen ARB; Cahill TJ; Huang R; Huang L-Y; Marcink T; Clarke OB; Heissel S; Masoudi A; Ben-Hail D; Samaan F; Dandey VP; Tan YZ; Hong C; Mahoney JP; Triest S; Little J; Chen X; Sunahara R; Steyaert J; Molina H; Yu Z; Des Georges A; Lefkowitz RJ Structure of an Endosomal Signaling GPCR-G Protein-β-Arrestin Megacomplex. Nat. Struct. Mol. Biol 2019, 26 (12), 1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Mafi A; Kim S-K; Goddard WA The Dynamics of Agonist-Β2-Adrenergic Receptor Activation Induced by Binding of GDP-Bound Gs Protein. Nat. Chem 2023, 15, 1127–1137. [DOI] [PubMed] [Google Scholar]
- (39).Tak Kam VW; Goddard WA Flat-Bottom Strategy for Improved Accuracy in Protein Side-Chain Placements. J. Chem. Theory Comput 2008, 4 (12), 2160–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Žídek A; Potapenko A; Bridgland A; Meyer C; Kohl SAA; Ballard AJ; Cowie A; Romera-Paredes B; Nikolov S; Jain R; Adler J; Back T; Petersen S; Reiman D; Clancy E; Zielinski M; Steinegger M; Pacholska M; Berghammer T; Bodenstein S; Silver D; Vinyals O; Senior AW; Kavukcuoglu K; Kohli P; Hassabis D Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596 (7873), 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Dobson L; Szekeres LI; Gerdán C; Langó T; Zeke A; Tusnády GE TmAlphaFold Database: Membrane Localization and Evaluation of AlphaFold2 Predicted Alpha-Helical Transmembrane Protein Structures. Nucleic Acids Res. 2023, 51 (D1), D517–D522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Abraham MJ; Murtola T; Schulz R; Páll S; Smith JC; Hess B; Lindahl E GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar]
- (43).Jo S; Kim T; Iyer VG; Im W CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem 2008, 29 (11), 1859–1865. [DOI] [PubMed] [Google Scholar]
- (44).Lee J; Cheng X; Swails JM; Yeom MS; Eastman PK; Lemkul JA; Wei S; Buckner J; Jeong JC; Qi Y; Jo S; Pande VS; Case DA; Brooks CL; MacKerell AD; Klauda JB; Im W CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput 2016, 12 (1), 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Wu EL; Cheng X; Jo S; Rui H; Song KC; Dávila-Contreras EM; Qi Y; Lee J; Monje-Galvan V; Venable RM; Klauda JB; Im W CHARMM-GUI Membrane Builder toward Realistic Biological Membrane Simulations. J. Comput. Chem 2014, 35 (27), 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmüller H; MacKerell AD CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14 (1), 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79 (2), 926–935. [Google Scholar]
- (48).Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD; Pastor RW Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114 (23), 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Hess B; Bekker H; Berendsen HJC; Fraaije JGEM LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem 1997, 18 (12), 1463–1472. [Google Scholar]
- (50).Nosé S A Molecular Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys 1984, 52 (2), 255–268. [Google Scholar]
- (51).Hoover WG Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31 (3), 1695–1697. [DOI] [PubMed] [Google Scholar]
- (52).Parrinello M; Rahman A Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys 1981, 52 (12), 7182–7190. [Google Scholar]
- (53).Delano W The PyMOL Molecular Graphics System, ver. 2.6.0a0; Schrodinger, LLC.: 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.