

Abstract
Isoxazolidine, isoxazole, and isoquinolinone rings are present in the structure of several natural products and/or pharmaceutically interesting compounds. In this work, facile and efficient pathways have been developed for the preparation of fused frameworks bearing those heterocycles. The successful approaches for both isoxazolidine/isoquinolinone and isoxazole/isoquinolinone hybrid syntheses relied initially on 1,3-dipolar cycloadditions of nitrones and nitrile oxides to indenone and 2-propargylbenzamide, respectively. The construction of the isoquinolinone lactam system followed by performing a selective Schmidt reaction for isoxazolidine derivatives (two steps overall), whereas the isoxazole lactams were reached via an Ullmann-type cyclisation (three steps overall). Key observations were made regarding the stereo- and regioselectivities of the reactions employed, and small libraries of the targeted hybrids were prepared, demonstrating the general applicability of these strategies.
Keywords: isoxazole; isoxazolidine; isoquinolinone; indanone; nitrone; nitrile oxide; 1,3-dipolar cycloaddition; Schmidt reaction; Ullmann reaction
1. Introduction
The 1,3-dipolar cycloaddition reaction (Huisgen cycloaddition) is established as a powerful tool for constructing five-membered heterocycles [1]. Among the various heterocyclic rings that are easily accessible via this strategy, isoxazolidines and isoxazoles, the cycloadducts obtained when nitrones or nitrile oxides are employed as 1,3-dipoles in reactions with alkenes and alkynes, respectively, are of great importance [2,3]. These scaffolds are present in the structure of several natural products, and in many instances, they were proved to be crucial for the observed biological activity [4] (Figure 1). A plethora of drugs and other compounds of interest to the pharmaceutical industry also contain those five-membered heterocycles [5,6], thus increasing the synthetic value of the 1,3-dipolar cycloaddition approach not only for the ease of preparing heterocyclic compounds but also for inserting high structural complexity in a straightforward manner.
Figure 1.

Representative examples of natural products and bioactive derivatives bearing the isoxazolidine or isoxazole moiety [7,8,9,10].
Another structural feature commonly appearing in biological active compounds, including either natural products or synthetic drugs, is the isoquinolinone or tetrahydroisoquinolinone moiety [11] (Figure 2). Such ring systems are often found in several pharmacophores, and positive results of Structure–Activity Relationship (SAR) studies support the design of new compounds bearing the aforementioned scaffold [12]. A convenient approach for the construction of the benzolactamic backbone present in those ring systems is the Schmidt reaction [13,14]. Apparently, the harsh acidic/thermal conditions required for the Schmidt protocol could be considered a drawback for the range of desired potential substrates. However, proper modifications may lead to a larger substrate pool being tolerated.
Figure 2.

Isoquinolinone or tetrahydroisoquinolinone scaffold in some natural products [15,16,17,18].
The facile preparation of libraries of novel hybrid compounds combining the two interesting structural features (isoxazolidine or isoxazole heterocycle and (tetra)isoquinolinone ring system) seems to be an appealing and challenging goal. A versatile synthetic methodology could provide potent lead compounds against various biological targets (enzyme inhibition, antifungal activity, etc.). Therefore, development of a concise synthetic sequence that simultaneously maintains a high level of versatility was the task we decided to address. Moreover, observations of the chemical behaviour and reactivity of both the heterocyclic intermediates and the targeted compounds could extract valuable information for the development of future synthetic routes towards other related compounds, so as to avoid unforeseen complications and the formation of unwanted side products in newly designed syntheses.
According to our original design, the employment of two key reactions was envisioned for the construction of the main core of the targeted compounds (Figure 3). The introduction of the amide functional group was planned via a Schmidt reaction to a ketone isoxazolidine or isoxazoline cycloadduct. The latter can be prepared utilising an 1,3-dipolar cycloaddition reaction between indenone, serving as the dipolarophile partner, and a nitrone or nitrile oxide 1,3-dipole. In the case of isoxazoline/isoquinolinones, a final oxidation step is required to obtain the target isoxazole derivatives. Both 1,3-dipoles and the dipolarophile can be accessed through known synthetic methodologies from commercially available reagents (Scheme 1).
Figure 3.

Design of targeted compounds.
Scheme 1.

Retrosynthetic plan for the targeted hybrids.
2. Results and Discussion
2.1. Synthesis of the Starting Materials
Due to its instability, indenone (3) was prepared from 3-bromoindanone (2) upon treatment with Et3N, each time prior to its use [19]. Bromide 2 was prepared from 1-indanone (1) at a large scale (up to 10 g) following a known procedure [20] and was kept in the freezer for months without any sign of decomposition (Scheme 2).
Scheme 2.

Preparation of dipolarophile 3.
For the first batch of 1,3-dipoles, we chose to proceed with N-benzyl nitrones of various commercially available aldehydes, consisting mostly of benzaldehydes. A modified procedure of a typical one reported in the literature [21] was followed, and, in total, 14 nitrones [22,23,24,25,26,27] were synthesised (Scheme 3A). In a similar manner, a variety of aldehydes were transformed to the corresponding oximes (Scheme 3B), following a known general protocol [28]. These oximes [29,30,31,32,33,34,35,36,37] served as the precursors of the required nitrile oxides.
Scheme 3.

(A). Preparation of nitrones 4. (B). Preparation of nitrile oxide precursors 5.
2.2. Synthesis of Isoxazolidine/Isoquinolinone Hybrids
2.2.1. Optimisation Studies for the 1,3-dipolar Cycloaddition Reaction between Indenone and Nitrones
In principle, four cycloadducts are to be expected as plausible products from the 1,3-dipolar cycloaddition reaction between a nitrone and a cyclic alkene. The E/Z-nitrone isomerisation in combination with the competing effects of secondary interactions, such as pi stacking and steric hindrance from bulky groups, can play a crucial role, thus rendering it rather hard to predict the stereoselective outcome (Scheme 4). Additionally, the issue of regioselectivity (regioisomers not shown) may complicate the situation even more. However, we presumed that in our case only one regio-orientation would be favoured due to the α,β-unsaturated system present in indenone.
Scheme 4.

The two possible stereoisomeric adducts for the reaction of indenone 3 and a nitrone.
To identify the preference towards the two competing stereoisomeric cycloadducts, several conditions were examined for the model reaction between N-benzyl nitrone of benzaldehyde (4a) and the in situ prepared indenone (3) (Table 1). Initially, simply refluxing the two partners in toluene afforded only two cycloadducts, 6a and 7a, with a good total yield (70%) (entry 1), whereas at lower temperatures the reaction seemed to be sluggish, regardless of the solvent used (only representative entries 1–3 are given in Table 1). The two adducts were successfully separated with column chromatography. 2D NMR spectroscopy (Figure 4) revealed that both cycloadducts emerged from the same regio orientation of the nitrone and were stereoisomers in regard to the relative geometry of the nitrone (E/Z isomerisation). Next, we settled with conducting a thorough screening in an attempt to determine conditions that may lead to the selective preparation of each of the two cycloadducts and further improve the yield. Metal triflates are widely used in such reactions with nitrones to control stereo- and regioselectivity because a lot of those compounds are commercially available [38,39,40]. Therefore, besides modifying the solvent, temperature, and reaction time, we also checked the influence of a reasonable number of metal triflates in substoichiometrical amounts as additives in order to investigate whether one of the cycloadducts is favoured over the other. Although we have not managed to establish a completely selective protocol, it is worth mentioning that Zn(OTf)2 (entries 7 and 8) led to the best ratio (almost 2:1 in favour of endo-adduct 6a) and an overall yield of 80%. The best results in terms of the overall yield of the reaction (90%) were obtained using AgOTf as the additive, but without a significant effect on selectivity regarding the formation of the two cycloadducts (6a/7a 1.25:1, entry 12). The silver salt counteranion proved to be of little importance because similar results for the stereoselectivity and the yield were observed with all silver salts employed (entries 12, 18, and 19). In an attempt to reverse the regioselectivity by taking advantage of their high affinity towards oxygen, titanium, and tin, Lewis acids were used (entries 16 and 17), but with no success. In general, heating the reaction mixture at 80 °C gave optimum results both in terms of productivity and time of completion.
Table 1.
Optimisation studies for the 1,3-dipolar cycloaddition reaction of 3 with nitrone 4a.

| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | 3 (equiv.) |
4a 1 (equiv.) |
Solvent 2 | Temp. (°C) |
Additive (0.25 equiv.) |
Time (h) |
6a 5 | 7a 5 | Yield 5 (comb.) |
| 1 | 1 | 1 | PhMe | 110 | - | 24 | 40% | 30% | 70% |
| 2 | 1 | 1 | DCM | 25 | - | 72 | 18% | 7% | 25% |
| 3 | 1 | 1 | none | 80 | - | 2 | 32.5% | 15.5% | 48% |
| 4 | 1 | 1 | DCM | 25 | Cu(OTf)2 | 24 | 10% | <1% | 10% |
| 5 | 1 | 1 | PhMe | 40 | Sc(OTf)3 | 72 | 43% | 20% | 63% |
| 6 | 1 | 1 | PhMe | 40 | Mg(OTf)2 | 72 | 39.5% | 25.5% | 65% |
| 7 | 1.2 | 1 | PhMe | 60 | Zn(OTf)2 | 24 | 50% | 28% | 78.5% |
| 8 | 1.2 | 1 | PhMe | 60 | Zn(OTf)2 | 48 | 54.5% | 25.5% | 80% |
| 9 | 1.2 | 1 | PhMe | 60 | In(OTf)3 | 48 | 35% | 20.5% | 55.5% |
| 10 | 1.2 | 1 | PhMe | 60 | AgOTf | 72 | 45.5% | 36% | 81.5% |
| 11 3 | 1.2 | 1 | PhMe | 80 | Zn(OTf)2 | 24 | 50% | 34% | 84% |
| 12 | 1.2 | 1 | PhMe | 80 | AgOTf | 24 | 50% | 40% | 90% |
| 13 | 1.2 | 1 | PhMe | 80 | Cu(OTf)2 | 24 | 31% | 19% | 50% |
| 14 4 | 1.2 | 1 | PhMe | 80 | Zn(OTf)2 | 3 | 34.5% | 18.5% | 53% |
| 15 4 | 1.2 | 1 | PhMe | 100 | AgOTf | 4 | 46% | 34% | 80% |
| 16 3 | 1 | 1 | PhMe | 25 | SnCl4 | 24 | - | - | - |
| 17 3 | 1 | 1 | PhMe | 25 | TiCl4 | 24 | - | - | - |
| 18 | 1.2 | 1 | PhMe | 80 | Ag2CO3 | 24 | 45% | 35.5% | 80.5% |
| 19 | 1.2 | 1 | PhMe | 80 | Ag2O | 24 | 45% | 35% | 80% |
1 1 mmole of 4a used; 2 7 mL/mmole of 4a; 3 1 equiv. of the additive was used; 4 the reaction was carried out under μW irradiation; 5 yield after purification through flash column chromatography based on the amount of nitrone used.
Figure 4.

Determining the structure of cycloadducts 6a and 7a using 2D NMR spectra.
2.2.2. Synthesis of Diverse Isoxazolidines
We then applied our optimised conditions (entry 12, Table 1) by using as dipoles the N-benzyl nitrones 4 shown in Scheme 3A (Table 2). For almost every nitrone examined, the endo cycloadduct 6 was the predominant one, in accordance with the optimisation studies. However, in two cases (entries 13 and 14), the exo cycloadduct was the main product of the reaction. It should be noted that for the o-substituted phenyl nitrones 4g and 4h (entries 7 and 8), the formation of the corresponding regioisomers 8 was also verified. 2D NMR studies were again used to prove that the latter originate from an exo-T.S. pathway (in a way similar to the analysis shown in Figure 4).
Table 2.
Synthesis of isoxazolidine cycloadducts using various nitrones.

| |||||
|---|---|---|---|---|---|
| Entry | R = | endo Cycloadduct | exo Cycloadduct | Regio Cycloadduct |
Yield 1 (comb.) |
| 1 |

|
6a (50%) | 7a (40%) | - | 90% |
| 2 |

|
6b (40%) | 7b (37%) | - | 77% |
| 3 |

|
6c (47%) | 7c (24%) | - | 71% |
| 4 |

|
6d (45%) | 7d (26%) | - | 71% |
| 5 |

|
6e (43%) | 7e (26%) | - | 69% |
| 6 |
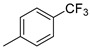
|
6f (54%) | 7f (24%) | - | 78% |
| 7 |

|
6g (31%) | 7g (24%) | 8g (16%) | 71% |
| 8 |

|
6h (28%) | 7h (20%) | 8h (23%) | 71% |
| 9 |

|
6i (42%) | 7i (34%) | - | 76% |
| 10 |

|
6j (55%) | 7j (27%) | - | 82% |
| 11 |

|
6k (32%) | 7k (20%) | - | 52% |
| 12 |

|
6l (40%) | - | - | 40% |
| 13 |

|
6m (18%) | 7m (37%) | - | 55% |
| 14 |

|
6n (21%) | 7n (55%) | - | 76% |
1 yield after purification through flash column chromatography based on the amount of nitrone used.
2.2.3. Optimisation of the Schmidt Reaction of Isoxazolidine Adducts
The Schmidt reaction represents a convenient method for the conversion of cyclic ketones to lactams. The nitrogen migration depends strongly on the acidic medium that the reaction is carried in (H2SO4, PPA, HCl) [13], while the respective substrate holds a decisive role as well (electronic effects of existing substituents may alter the outcome) [41,42,43].
To investigate the optimal conditions for the transformation of our ketone cycloadducts (6 and 7) to lactams (tetrahydroisoquinolinones), a number of experiments were conducted using endo cycloadduct 6a as the model substrate (Table 3). Interestingly enough, many of the more commonly used acids (HCl, H2SO4) failed to give any reaction, or the yield was meagre. Trifluoroacetic acid (TFA) and the extremely powerful triflic acid (TfOH) also failed to afford any product, and the starting material was recovered intact in both cases. The first encouraging result was obtained using methanesulfonic acid (MsOH), as shown in entry 9. Ultimately, proper modifications led to a protocol that furnished the desired tetrahydroisoquinolinone 9a, with a very good yield (entry 11). 2D NMR studies (Figure 5) confirmed the nitrogen migration to the sp3 (alkyl) carbon and retention of the relative stereochemistry of protons of the starting adduct 6a in hybrid 9a. Additionally, the formation of the tetrazole by-product 10a was observed. In order to test whether amide 9a could be formed exclusively, the Schmidt reaction was also carried out at a lower temperature (0 °C) in DCM or CHCl3, but the starting isoxazolidine 6a remained intact (experiments not shown in Table 3).
Table 3.
Investigation of the Schmidt reaction conditions on 6a.

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Acid | Reagent | Solvent 4 | Temp. (°C) |
Time (h) |
9a 5 | 10a 5 | Yield 5 (comb.) |
| 1 1 | aq. HCl (37%) | NaN3 | PhMe | 25 | 24 | - | - | rsm |
| 2 1 | aq. HCl (37%) | NaN3 | PhMe | 50 | 24 | - | - | rsm |
| 3 2 | aq. HCl (37%) | NaN3 | - | 80 | 24 | - | - | rsm |
| 4 2 | TFA | NaN3 | - | 25 | 24 | - | - | rsm |
| 5 2 | TFA | NaN3 | - | 72 | 24 | - | - | rsm |
| 6 1 | TfOH | NaN3 | PhMe | 25 | 24 | - | - | rsm |
| 7 2 | TfOH | NaN3 | - | 25 | 24 | - | - | rsm |
| 8 1 | H2SO4 | NaN3 | PhMe | 80 | 24 | 15% | - | 15% |
| 9 1 | MsOH | NaN3 | PhMe | 25 | 24 | <10% | - | ~10% + rsm |
| 10 3 | MsOH | HN3 | PhMe | 25 | 48 | 76% | 13% | 89% |
| 11 3 | MsOH | HN3 | DCM | 25 | 48 | 78% | 14% | 92% |
1 0.1 mmoles of 6a, 3 equiv. NaN3, 0.1 equiv. of the acid; 2 0.1 mmoles of 6a, 3 equiv. NaN3, acid is used as the solvent; 3 0.1 mmoles of 6a, 2.5M HN3 in solvent, ratio of solvent/MsOH 2:1; 4 20 mL/mmole of 6a; 5 yield after purification through flash column chromatography; rsm = recovered starting material.
Figure 5.

Determining the structure of lactam 9a using 2D NMR spectra.
It is known [44,45] that this type of tetrazole by-product may be derived via an alternative pathway of the Schmidt reaction, where the initially formed nitrilium ion is quenched by hydrazoic acid when the latter is present at high concentrations. To exclude the possibility of tetrazole ring formation via a post-amidation reaction, we left pure lactam 9a to react under our Schmidt protocol conditions, but the latter remained intact. Applying other methods that transform amides to tetrazoles at higher temperatures [46,47] led to the consumption of starting lactam and the formation of parent isoquinolinone 11 and the original nitrone 4a via an unexpected retro 1,3-dipolar cycloaddition reaction (Scheme 5). As it was further concluded, the same retro cycloaddition occurs when lactam 9a is simply heated above 80 °C in an appropriate solvent.
Scheme 5.

Attempting tetrazole formation from lactam 9a.
2.2.4. Synthesis of the Targeted Isoxazolidine/Tetrahydroisoquinolinone Hybrids
Having gathered critical insight, we proceeded to prepare a number of isozaxolidine hybrids. The optimised protocol for the Schmidt reaction was applied to both endo and exo cycloadducts, and the results are summarised in Table 4 and Table 5, respectively. Regardless of the stereoisomer used, this protocol furnished (in most cases, uneventfully) the desired tetrahydroisoquinolinones (9 and 12), although some rather distinct differences between the two can be underlined. Endo cycloadducts 6 gave the lactams in moderate to very good yields, whereas the exo ones (7) furnished the analogous lactams in excellent yields in almost every case. The reaction time was also notably shorter for exo cycloadducts compared to the endo ones. Furthermore, the tetrazole by-product of exo analogs was formed in almost every instance. Presumably, the difference in the conformation of the five-membered heterocyclic ring holds the answer to this overall quite different behaviour, but a complete explanation cannot be deduced from the existing data.
Table 4.
Synthesis of tetrahydroisoquinolinones 9.

| ||||
|---|---|---|---|---|
| Entry | R = | Lactam 1 | Tetrazole 1 | Combined Yield 1 |
| 1 |

|
9a (78%) | 10a (14%) | 89% |
| 2 |
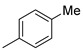
|
9b (59%) | 10b (19%) | 78% |
| 3 |

|
9c (62%) | - | 62% |
| 4 |

|
9d (54%) | - | 54% |
| 5 |

|
9e (81%) | - | 81% |
| 6 |

|
9f (70%) | - | 70% |
| 7 |

|
9g (65%) | 10g (12%) | 77% |
| 8 |

|
9h (65%) | 10h (17%) | 82% |
| 9 |

|
9i (51%) | - | 51% |
| 10 |

|
9j (53%) | - | 53% |
| 11 |
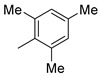
|
9k (90%) | - | 90% |
| 12 |

|
9l (72%) | 10l (13%) | 85% |
| 13 |

|
9m (65%) | - | 65% |
| 14 |

|
9n (69%) | 10n (11%) | 80% |
1 yield after purification through flash column chromatography.
Table 5.
Synthesis of tetrahydroisoquinolinones 12.

| ||||
|---|---|---|---|---|
| Entry | R = | Lactam 1 | Tetrazole 1 | Combined Yield 1 |
| 1 |

|
12a (82%) | 13a (14%) | 96% |
| 2 |

|
12b (72%) | 13b (14%) | 86% |
| 3 |

|
12c (83%) | 13c (14%) | 97% |
| 4 |

|
12d (75%) | - | 75% |
| 5 |

|
12e (84%) | 13e (14%) | 98% |
| 6 |

|
12f (85%) | 13f (14%) | 99% |
| 7 |

|
12g (82%) | 13g (14%) | 96% |
| 8 |

|
12h (87%) | 13h (12%) | 99% |
| 9 |

|
12i (84%) | 13i (8%) | 92% |
| 10 |

|
12j (64%) | - | 64% |
| 11 |

|
- | - | decomposed |
| 13 |

|
12m (81%) | 13m (12%) | 93% |
| 14 |

|
12n (90%) | 13n (9%) | 99% |
1 yield after purification through flash column chromatography.
2.3. Synthesis of Isoxazole/Isoquinolinone Hybrids
2.3.1. Model 1,3-dipolar Cycloaddition Reaction between Indenone and Benzonitrile Oxide and the Following Schmidt Reaction
The 1,3-dipolar cycloaddition reaction between indenone 3 and nitrile oxides was planned as a one-pot procedure. Treating oximes 6 with NCS in DMF furnished the corresponding hydroxamoyl chlorides 14 [48], which were then used without further purification. In the model reaction, one equivalent of Et3N was added to the solution of the in situ prepared indenone 3 before the dropwise addition of phenylhydroxamoyl chloride (Scheme 6). This reaction smoothly furnished the two regioisomeric adducts (15 and 16), which were isolated and characterised. Once again, the major regioisomer 15 was the one favoured due to the α,β-unsaturated system present in the dipolarophile.
Scheme 6.

1,3-dipolar cycloaddition between indenone 3 and benzonitrile oxide 14a.
Isoxazoline cycloadduct 15 was then submitted to the previously optimised Schmidt reaction protocol (i.e., entry 11, Table 3). To our surprise, this reaction did not furnish the desired lactam, but instead nitrile 17 was isolated as the main product. Traces of the corresponding isoxazole/benzamide 18a were also identified in the reaction mixture (Scheme 7). Alternatively, heating of 15 in the presence of sodium azide (NaN3) and concentrated sulfuric acid gave benzamide 18a as the major product. Obviously, the latter derived upon hydrolysis of the initially formed nitrile 17.
Scheme 7.

Schmidt reaction on ketone isoxazoline 15.
Both the Schmidt reaction and the Beckmann rearrangement share the same mechanistic pathway once the nitrogen migration occurs. Nitriles are common side products generated from the Beckmann rearrangement, and when those are the major products, the transformation is named “Beckmann fragmentation” instead [49]. We assume that in our case, this fragmentation is greatly favoured because the intermediate nitrilium ion 20 readily collapses to the significantly more stable aromatic isoxazole 17 (Scheme 8).
Scheme 8.

Nitrile/isoxazole formation as a result of a Beckmann fragmentation.
2.3.2. Revision of the Synthetic Plan
The unforeseen outcome of the Schmidt reaction on isoxazoline adduct 15 made us reconsider our synthetic approach towards the targeted isoxazole/isoquinolinone hybrids. Interestingly, derivative 17a attracted our attention because it incorporates the isoxazole/benzamide framework, which is ultimately present in our targeted hybrid compounds. Thus, a revised short, synthetic route involving isoxazole/benzamide derivatives was designed. According to this plan, cyclisation of the appropriate 4-iodo isoxazole intermediates via a copper-catalysed Ullmann-type reaction could afford the desired isoxazole/isoquinolinone hybrids. The required iodo derivatives could be prepared via an iodination reaction of the corresponding isoxazole/benzamides, which simply represent the adducts of 1,3-dipolar cycloadditions between 2-propargylbenzamide, now serving as the new dipolarophile, and various in situ prepared nitrile oxides (Scheme 9).
Scheme 9.

Revised retrosynthetic plan towards isoxazole/isoquinolinone hybrids.
2.3.3. Isoxazole/Benzamides via an 1,3-dipolar Cycloaddition Reaction
A Sonogashira cross coupling reaction to commercially available 2-iodobenzamide and subsequent cleavage of the TMS group, following a typical procedure, afforded 2-propargylbenzamide 21 [50]. A series of in situ generated nitrile oxides (see Scheme 6) reacted with propargyl dipolarophile 21 to give isoxazole/benzamides 18 as single products and in very good yields (Table 6).
Table 6.
Synthesis of isoxazole/benzamides 18.

| |||||
|---|---|---|---|---|---|
| Entry | R = | Yield 1 | Entry | R = | Yield 1 |
| 1 |

|
18a (84%) | 9 |

|
18i (83%) |
| 2 |

|
18b (89%) | 10 2 |

|
18j (90%) |
| 3 |

|
18c (87%) | 11 3 |

|
18k (84%) |
| 4 |
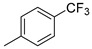
|
18d (86%) | 12 2 |

|
18l (93%) |
| 5 |

|
18e (88%) | 13 |

|
18m (80%) |
| 6 |

|
18f (91%) | 14 |

|
18n (79%) |
| 7 |

|
18g (92%) | 15 |

|
18o (92%) |
| 8 |

|
18h (89%) | |||
1 yield after purification through flash column chromatography; 2 PhMe was used instead of DCM and the reaction mixture was refluxed; 3 the reaction mixture was heated to 40 °C.
2.3.4. Iodination of the Isoxazole Ring
The isoxazole heterocycle is prone to undergoing various transformations, including halogenation. The protocol for the iodination of the 4-position of the isoxazole ring of our derivatives 18 involved treatment with NIS (N-iodosuccinimide) in trifluoroacetic acid under microwave irradiation [51]. This afforded the 4-iodoisoxazoles 22 in excellent yields for most of the substrates (Table 7). Exceptions were the analogs bearing the mesityl, furanyl, and thiophene groups (18j, 18m, and 18n, respectively), which failed to exclusively give the desired products. Instead, complicated mixtures of mono- and polyiodinated products were obtained, regardless of the equivalents of NIS used. It is also worth mentioning that two equivalents of NIS were used for isoxazole 18c, due to its facile concomitant iodination on the electronically rich p-methoxyphenyl ring.
Table 7.
Iodination of isoxazole derivatives 18.

| |||||
|---|---|---|---|---|---|
| Entry | R = | Yield 1 | Entry | R = | Yield 1 |
| 1 |

|
22a (97%) | 9 3 |

|
22i (99%) |
| 2 |

|
22b (99%) | 10 |

|
- |
| 3 2 |

|
22c (88%) | 11 |
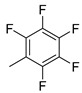
|
22k (99%) |
| 4 |

|
22d (99%) | 12 |

|
22l (98%) |
| 5 |

|
22e (97%) | 13 |

|
- |
| 6 |

|
22f (97%) | 14 |
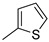
|
- |
| 7 |

|
22g (99%) | 15 |

|
22o (98%) |
| 8 |

|
22h (99%) | |||
1 yield after purification through flash column chromatography; 2 2 equiv. of NIS were used; 3 yield after purification through trituration with Et2O/n-hexane.
2.3.5. Optimisation Studies for the Ullmann Reaction
The copper-catalysed C–N bond formation (Ullmann reaction) remains an appealing alternative for the Buchwald–Hartwig reaction, because the latter depends on more expensive and less stable palladium catalysts. A thorough screening to determine the best conditions (copper catalyst, base, solvent, and temperature) was conducted on iodo-isoxazole 22a (Table 8). Copper(I) thiophene-2-carboxylate (CuTC), being a useful reagent in such reactions, gave inarguably superior results in comparison to the more commonly used copper(I) iodide (CuI) [52]. Typical inorganic bases and solvents were tested, and the best results were obtained upon using K2CO3 and DMF while maintaining the temperature at 80 °C (entry 10).
Table 8.
Ullmann cyclisation optimisation studies.

| |||||
|---|---|---|---|---|---|
| Entry 1 | Copper Source (equiv.) |
Base (2 equiv.) |
Solvent 2 | Temp. (°C) | Yield 5 |
| 1 | CuI (0.15) | tBuONa | tBuOH | 100 | 16% |
| 2 | CuI (0.15) | tBuONa | 1,4-dioxane | 100 | 20% |
| 3 3 | CuI (0.15) | Cs2CO3 | DMF | 100 | 15% |
| 4 | CuI (1) | Cs2CO3 | DMF | 100 | 56% |
| 5 | CuTC (0.15) | K3PO4 | DMF | 100 | 50% |
| 6 | CuTC (0.25) | K3PO4 | DMF | 80 | 61% |
| 7 | CuTC (0.15) | tBuONa | DMF | 100 | 55% |
| 8 | CuTC (0.15) | Cs2CO3 | DMF | 100 | 56% |
| 9 | CuTC (0.25) | Cs2CO3 | DMF | 80 | 73% |
| 10 | CuTC (0.25) | K2CO3 | DMF | 80 | 78% |
| 11 | CuTC (0.25) | K2CO3 | DMF | 120 | - |
| 12 | CuTC (0.25) | K2CO3 | DMSO | 80 | 31% |
| 13 4 | CuTC (0.25) | K2CO3 | DMF | 80 | 48% |
1 0.1 mmoles of iodo-benzamide 22a was used; 2 10 mL/mmole of 22a; 3 1 equiv. of DMEDA was used as additive; 4 1 equiv. of base was used; 5 yield after purification through trituration with Et2O/n-hexane.
2.3.6. Building a Library of Isoxazole/Isoquinolinone Hybrids via the Ullmann-Type Cyclisation
The optimum conditions for Ullmann cyclisation were, thereafter, applied to the rest of the iodo-isoxazoles (Table 9). Analogs bearing a p-substituted aryl group afforded the desired isoquinolinones in very good yields (67–93%). Isoquinolinones where the aryl group is ortho-substituted were isolated in moderate yields (58–61%), whereas analogs with polysubstituted aryl groups furnished the targeted products in generally low yields (29–46%). Nitro analog 22i failed to give the Ullmann cyclisation because it gradually decomposed during the reaction. Moreover, isoquinolinone derivative 23c was subjected to a reductive deiodination employing 10% Pd/C to furnish isoquinolinone 24.
Table 9.
Synthesis of isoquinolinones 23.

| |||||
|---|---|---|---|---|---|
| Entry | R = | Yield 1 | Entry | R = | Yield 1 |
| 1 |

|
23a (78%) | 7 |

|
23g (58%) |
| 2 |

|
23b (67%) | 8 |

|
23h (61%) |
| 3 |

|
23c (93%) | 9 |

|
23k (29%) |
| 4 |

|
23d (81%) | 10 |

|
23l (46%) |
| 5 |

|
23e (89%) | 11 |

|
23o (80%) |
| 6 |

|
23f (78%) | |||
1 yield after purification through recrystallisation (dissolved in 10% TFA in DCM and crystallised upon adding a few drops of MeOH).
3. Experimental Section
3.1. General Information
All anhydrous reactions were performed under an argon atmosphere using oven-dried (120 °C) or flame-dried glassware (under vacuum) with dry solvents under anhydrous conditions. THF, 1,4-dioxane, and toluene were distilled over sodium/benzophenone under an argon atmosphere into a dry Schlenk Kjeldahl storage flask containing activated molecular sieves (4 Å), and they were allowed to stand for at least for 24 h. DCM was distilled over calcium hydride (CaH2) before use. Carbon tetrachloride was distilled over phosphorus pentoxide before use. All reactions requiring high temperatures were conducted using silicon oil baths as the heating medium. Flash column chromatography was performed by employing silica gel 60 (40–63 μm, Merck). Reactions were monitored through TLC using 0.25 mm silica gel 60 F254 plates purchased from Merck. TLC plates were visualised through exposure to ultraviolet light (UV) and/or exposure to an acidic solution of p-anisaldehyde or a solution of ninhydrin stain, followed by heating with a heat gun (400 °C). All commercially available reagents and solvents were purchased from Fluorochem, Sigma-Aldrich & Merck, Fischer Scientific, and TCI Chemicals and used as such. Molecular sieves (3 Å and 4 Å) were dried under a high vacuum by being heated with a propane torch in a round-bottom flask for 1–2 min, and the procedure was repeated 2–3 times. Celite®® 545 was purchased from Fluorochem. 1H, 13C, 19F, and 2D NMR spectra were recorded with an Agilent-500/54 spectrometer. Unless otherwise stated, all NMR spectra were recorded at 25 °C. Proton chemical shifts are reported in parts per million (δ scale) and are calibrated relative to a residual nondeuterated solvent as an internal reference (CDCl3: δ 7.26, DMSO-d6: δ 2.5 ppm). Carbon chemical shifts are reported in parts per million (δ scale) and are referenced from the central peak of the carbon resonance of the solvent (CDCl3: 77.00, DMSO-d6: 40.00 ppm). Infrared (IR) data were recorded in a scan range from 400 to 4000 cm−1 on a Thermo Scientific Nicolet 6700 FT-IR spectrometer equipped with a diamond attenuated total reflection (ATR) stage. HRMS data were acquired using an Agilent 6540 HRMS-QTOF model equipped with a Dual AJS ESI-MS system or with a Q-TOF (Time of Flight Mass Spectrometry) Maxis Impact (Bruker Daltonics, Bremen, Germany) with ESI source and U-HPLC Thermo Dionex UltiMate 3000 RSLC (ThermoFisher Scientific, Dreieich, Germany) pump and autosampler. Melting points were determined on a A.KRÜSS Optronic Melting Point Meters KSP1N model apparatus. Reactions under microwave irradiation were carried out using a Biotage Initiator+ microwave synthesiser.
3.2. Synthesis of 3-Bromo-2,3-dihydro-1H-inden-1-one (2)
Compound 2 was prepared following a modified procedure of that reported in the literature [20]. 1-Indanone (10.04 g, 76.0 mmoles, 1.0 equiv.) was dissolved in anhydrous CCl4 (150 mL), and then N-bromosuccinimide (13.52 g, 76.0 mmoles, 1.0 equiv.) and AIBN (0.125 g, 0.76 mmoles, 0.01 equiv.) were sequentially added at room temperature. The resulting suspension was refluxed for 4 h in an oil bath protected from light, allowed to cool to room temperature, and then filtered through a short pad of Celite. After evaporation of the solvent under reduced pressure, the crude mixture was purified through flash column chromatography (silica gel, n-hexane/Et2O 35:1 v/v) to give 2 (11.23 g, 70% yield) as an orange oil, which solidified in the freezer. Rf = 0.43 (n-hexane/EtOAc 4:1 v/v). All spectroscopic data were in accordance with those reported in the literature [53].
3.3. General Procedure for the Preparation of Nitrones 4
To a round bottom flask containing N-benzylhydroxylamine hydrochloride (2.0 equiv.), anhydrous DCM (40 mL/g) was added under an argon atmosphere. To the resulting suspension of MgSO4 (2.0 equiv.), the corresponding aldehyde (1.1 equiv.) and NaHCO3 (1.1 equiv.) were added in that order, and the mixture was stirred for 24 h at room temperature. The reaction mixture was then filtered through a short pad of Celite, and the solvent was evaporated under reduced pressure. The crude mixture was purified through flash column chromatography (silica gel, n-hexane/EtOAc 5:1 v/v to 1:1 v/v) to give the desired nitrones 4.
3.4. General Procedure for the Preparation of Oximes 5
Oximes 5 were prepared following a procedure reported previously in the literature [28].
3.5. General Procedure for the Preparation of Isoxazolidine Cycloadducts 6 and 7
3-Bromo-1-indanone (2, 0.726 g, 3.44 mmoles, 1.2 equiv.) was dried azeotropically through evaporation with PhMe (3x10 mL) and dissolved in anhydrous PhMe (20 mL) under an argon atmosphere. The mixture was placed in an ice bath, and anhydrous Et3N (0.48 mL, 3.44 mmoles, 1.2 equiv.) was added dropwise. After stirring for 15 min at 0 °C, AgOTf (0.185 g, 0.72 mmoles, 0.25 equiv.) and a solution of the corresponding nitrone (2.87 mmoles, 1 equiv.) in anhydrous PhMe (3 mL) were added to the reaction mixture, which was then placed in an oil bath at 80 °C. After stirring for 24 h at that temperature, the reaction mixture was cooled to room temperature, diluted with a small amount of DCM, and filtered through a short pad of Celite. The solvent was evaporated under reduced pressure, and the crude mixture was purified through flash column chromatography (silica gel, n-hexane/EtOAc 20:1 v/v to 12:1 v/v) to give the corresponding cycloadducts 6 and 7 (and, in some cases, regioisomers 8).
3.6. General Procedure for the Preparation of Hybrids 9 and 12
A solution of HN3 (2.5 M) in DCM was prepared by dissolving NaN3 (0.650 g, 10 mmoles) in H2O (1 mL), adding the organic solvent (4 mL), cooling the mixture to −10 °C, and slowly adding MsOH (1 mL) over a period of 20 min under vigorous stirring. The mixture was stirred for another 10 min, and the organic layer was then separated. The starting ketone (6 or 7, 0.15 mmoles) was dissolved to this solution (3 mL), and the mixture was stirred for 30 min at room temperature. MsOH (1.5 mL) was added dropwise over a period of 3 h. The resulting mixture was stirred for 24–72 h at room temperature, diluted with DCM (5 mL), placed in an ice bath, and slowly quenched through the addition of a saturated aqueous Na2CO3 solution until effervescence ceased. The organic layer was separated, and the aqueous layer was extracted with DCM (3 × 10 mL). The organic layers were combined, washed with brine (40 mL), and dried over anhydrous Na2SO4. After evaporation of the solvent under reduced pressure, the crude mixture was purified through flash column chromatography (silica gel, n-hexane/EtOAc 5:1 to 1:1 v/v) to give the desired isoxazolidine/tetrahydroisoquinolinone hybrids 9 and 12 (and tetrazole by-products 10 and 13, respectively).
3.7. Synthesis of Isoxazoline Cycloadducts 15 and 16
3-Bromo-1-indanone (2, 1.00 g, 4.74 mmoles, 1 equiv.) was dissolved in DCM (50 mL), and the mixture was placed in an ice bath. Et3N (0.79 mL, 5.69 mmoles, 1.2 equiv.) was added dropwise, and the yellow solution was stirred for 15 min at 0 °C. A DCM solution (100 mL) of phenylhydroxamoyl chloride 14a (0.811 g, 5.21 mmoles, 1.1 equiv.) was then added via a pressure-equalising dropping funnel over a period of 1 h, while the temperature was maintained at 0 °C. After the addition, the reaction mixture was left stirring for another 1 h, and the reaction temperature was gradually allowed to reach 20 °C. The reaction mixture was quenched through the addition of aqueous semi-saturated aqueous NaCl solution (150 mL), the organic layer was separated, and the aqueous layer was extracted with DCM (3 × 150 mL). The organic layers were combined, washed with brine (500 mL), and dried over anhydrous Na2SO4. After evaporation of the solvent under reduced pressure, the crude mixture was purified through flash column chromatography (silica gel, n-hexane/EtOAc 12:1 to 10:1 v/v) to give cycloadducts 15 (1.04 g, 88% yield) and 16 (0.071 g, 6% yield).
3-Phenyl-3a,8b-dihydro-4H-indeno [2,1-d]isoxazol-4-one (15): white solid; m.p. 153–154 °C; Rf = 0.31 (n–hexane/EtOAc 4:1 v/v); 1H NMR (500 MHz, CDCl3): δ = 8.03–7.96 (m, 2H), 7.82 (d, J = 7.7 Hz, 1H), 7.79–7.73 (m, 2H), 7.55 (t, J = 7.4 Hz, 1H), 7.46–7.41 (m, 3H), 6.31 (d, J = 8.3 Hz, 1H), 4.75 (d, J = 8.3 Hz, 1H) ppm; 13C NMR (125 MHz, CDCl3): δ = 197.2, 152.6, 150.7, 136.3, 134.5, 130.6, 130.4, 128.6, 128.0, 127.9, 126.9, 124.2, 83.0, 60.6 ppm; FT-IR (neat): ν = 3057, 2941, 1723, 1604, 1590, 1334, 1269, 884, 761, 696 cm–1; HRMS (ESI), m/z: [M + Na]+ calcd for C16H11NNaO2+ 272.0682; 272.0679.
3-Phenyl-3a,8a-dihydro-8H-indeno [1,2-d]isoxazol-8-one (16): white solid; m.p. 172–173 °C; Rf = 0.18 (n–hexane/EtOAc 4:1 v/v); 1H NMR (500 MHz, CDCl3): δ = 7.85 (d, J = 7.7 Hz, 1H), 7.83–7.79 (m, 2H), 7.53 (td, J = 7.5, 1.3 Hz, 1H), 7.50–7.47 (m, 3H), 7.43 (t, J = 7.9 Hz, 2H), 5.42 (d, J = 8.6 Hz, 1H), 5.33 (d, J = 8.6 Hz, 1H) ppm; 13C NMR (125 MHz, CDCl3): δ = 198.7, 156.9, 150.3, 136.1, 134.4, 130.6, 129.3, 128.1, 128.0, 127.6, 126.2, 125.7, 84.7, 53.1 ppm; FT-IR (neat): ν = 3058, 2962, 1716, 1592, 1444, 1347, 1251, 881, 759, 694 cm–1. HRMS (ESI), m/z: [M + Na]+ calcd for C16H11NNaO2+ 272.0682; 272.0686.
3.8. Synthesis of Nitrile 17 and Benzamide 18a from 15
Procedure A: The solution of HN3 (2.5 M) in DCM was prepared as described in Section 3.6. Isoxazoline adduct 15 (0.424 g, 1.7 mmoles, 1 equiv.) was dissolved in DCM (6 mL, 2.5 M HN3 solution) and left stirring for 30 min at room temperature. MsOH (3 mL) was added dropwise over a period of 3 h. The resulting mixture was stirred for 24 h at room temperature, diluted with DCM (15 mL), placed in an ice bath, and slowly quenched through the addition of saturated aqueous Na2CO3 solution until effervescence ceased. The organic layer was separated, and the aqueous layer was extracted with DCM (3 × 20 mL). The organic layers were combined, washed with brine (80 mL), and dried over anhydrous Na2SO4. After evaporation of the solvent under reduced pressure, the crude mixture was purified through flash column chromatography (silica gel, n-hexane/EtOAc 10:1 to 1:1 v/v) to give nitrile 17 (0.247 g, 68% yield) and benzamide 18a (4 mg, 1%).
Procedure B: Cycloadduct 15 (0.112 g, 0.45 mmoles, 1 equiv.) was dissolved in PhMe (5 mL), and the clear solution was placed in an ice bath. NaN3 (0.088 g, 1.35 mmoles, 3 equiv.) and H2SO4 (98%, 3 µL, 0.045 mmoles, 0.1 equiv.) were sequentially added at 0 °C, and the reaction mixture was then placed in an oil bath at 80 °C. After stirring for 2 h, the mixture was allowed to cool to toom temperature, placed in an ice bath, diluted with a small amount of EtOAc, and slowly quenched through the addition of saturated aqueous Na2CO3 solution until effervescence ceased. The organic layer was separated, and the aqueous layer was extracted with EtOAc (3 × 10 mL). The organic layers were combined, washed with brine (30 mL), and dried over anhydrous Na2SO4. After evaporation of the solvent under reduced pressure, the crude mixture was purified through flash column chromatography (silica gel, n-hexane/EtOAc 10:1 to 1:1 v/v) to give nitrile 17 (0.013 g, 12% yield) and benzamide 18a (0.081 g, 61%).
2-(3-phenylisoxazol-5-yl)benzonitrile (17): white solid; Rf = 0.53 (n-hexane/EtOAc 3:1 v/v). All spectroscopic data are in accordance with those reported in the literature [54].
2-(3-phenylisoxazol-5-yl)benzamide (18a): white solid; m.p. 159–160 °C; Rf = 0.11 (n-hexane/EtOAc 1:1 v/v); 1H NMR (500 MHz, DMSO-d6): δ = 7.99 (br s, 1H), 7.89 (dd, J = 7.8, 1.8 Hz, 2H), 7.83–7.79 (m, 1H), 7.63–7.50 (m, 7H), 7.25 (s, 1H) ppm; 13C NMR (125 MHz, DMSO-d6): δ = 170.6, 169.8, 162.6, 137.5, 130.8, 130.7, 130.0, 129.7, 129.0, 128.9, 128.3, 127.0, 124.7, 101.1 ppm. FT-IR (neat): ν = 3332, 3158, 2804, 1663, 1623, 1464, 1401, 1137, 950, 758, 690 cm–1. HRMS (ESI), m/z: [M + Na]+ calcd for C16H12N2NaO2+ 287.0791; found 287.0795.
3.9. Synthesis of 2-Propargylbenzamide (21)
Compound 21 was prepared following a procedure reported previously in the literature [51].
3.10. General Procedure for the Preparation of Hydroxamoyl Chlorides 14
Hydroxamoyl chlorides 14 were prepared following a procedure reported previously in the literature [49].
3.11. General Procedure for the Preparation of Isoxazole/Benzamides 18
2-propargyl benzamide (21, (0.164 g, 1.13 mmoles) was dissolved in DCM (6 mL) and placed in an ice bath. Et3N (0.19 mL, 1.36 mmoles, 1.2 equiv.) was added, and the reaction mixture was stirred for 10 min at 0 °C. A DCM solution (4 mL) of hydroxamoyl chloride (14, 1.36 mmoles, 1.2 equiv.) was then added dropwise over a period of 15 min, and the mixture was left stirring at 0 °C for 1 h. The reaction mixture was gradually allowed to reach room temperature and stirred for another 4 h before it was quenched through the addition of semi-saturated aqueous NaCl solution (20 mL). The organic layer was separated, and the aqueous layer was extracted with DCM (3 × 15 mL). The organic layers were combined, washed with brine (60 mL), and dried over anhydrous Na2SO4. After evaporation of the solvent under reduced pressure, the crude mixture was purified through flash column chromatography (silica gel, n-hexane/EtOAc 5:1 to 1:2 v/v) to afford the corresponding isoxazole/benzamides 18.
3.12. General Procedure for the Preparation of 4-Iodo Isoxazoles 22
To a proper heavy-wall microwave reaction vial containing an isoxazole/benzamide 18 (0.58 mmoles), TFA (2 mL) and N-iodosuccinimide (0.137 g, 0.61 mmoles, 1.05 equiv.) were added at room temperature. The vial was then sealed and stirred under microwave irradiation at 80 °C for 10 min. The light purple solution was diluted with DCM (10 mL), placed in an ice bath, and neutralised with a saturated, aqueous NaHCO3 solution. The organic layer was separated, and the aqueous layer was extracted with DCM (2 × 15 mL). The organic layers were combined, washed with brine (40 mL), and dried over anhydrous Na2SO4. After evaporation of the solvent under reduced pressure, the crude mixture was purified through flash column chromatography (silica gel, n-hexane/EtOAc 5:1 to 1:2 v/v) to give the corresponding 4-iodo isoxazole/benzamides 22.
3.13. General Procedure for the Preparation of Isoxazole/Isoquinolinones 23
DMF (3 mL) was added to a heavy-wall sealed tube containing a 4-iodo isoxazole/benzamide 22 (0.29 mmoles) at room temperature. K2CO3 (0.08 g, 0.58 mmoles, 2 equiv.) and CuTC (0.014 g, 0.072 mmoles, 0.25 equiv.) were sequentially added, and the light green suspension was flushed with argon for 2–3 min. The tube was tightly sealed, and the mixture was stirred at 80 °C for 24 h. The dark greenish/brown suspension was diluted with EtOAc (5 mL) and quenched with the addition of an aqueous NH4OH-NH4Cl solution (10 mL). The organic layer was separated, and the aqueous layer was extracted with EtOAc (3 × 10 mL). The organic layers were combined, washed with brine (40 mL), and dried over anhydrous Na2SO4. After evaporation of the solvent under reduced pressure, the resulting solid residue was purified through recrystallisation (dissolved in 10% TFA in DCM and crystallised upon adding a few drops of MeOH) to afford the corresponding isoxazole/isoquinolinones 23.
3.14. Synthesis of Isoquinolinone 24 via Reductive Deiodination
DMF (1 mL) was added to a Schlenk tube containing isoquinolinone 23c (21 mg, 0.051 mmoles) at room temperature. Then, 10% Pd/C (3 mg) and a couple of drops of Et3N were sequentially added, and the reaction vessel was purged with H2 and placed under an atmosphere of H2. After stirring for 1 h at room temperature, the resultant slurry was diluted with EtOAc (10 mL), filtered through a short a pad of Celite, and semi-saturated, aqueous NaCl solution (15 mL) was added. The organic layer was separated, and the aqueous layer was extracted with EtOAc (3 × 10 mL). The organic layers were combined, washed with brine (40 mL), and dried over anhydrous Na2SO4. After evaporation of the solvent under reduced pressure, the resulting solid residue was purified through recrystallisation (dissolved in 10% TFA in DCM and crystallised upon adding a few drops of MeOH) to give pure 24 (15 mg, 98% yield).
3-(4-methoxyphenyl)isoxazolo [4,5-c]isoquinolin-5(4H)-one (24): white solid; m.p. > 250 °C (decomposed); Rf = 0.4 (n-hexane/EtOAc 1:1 v/v); 1H NMR (500 MHz, 4% TFA-d in CDCl3): δ = 8.54 (d, J = 8.1 Hz, 1H), 8.19 (d, J = 7.9 Hz, 1H), 7.99 (t, J = 7.6 Hz, 1H), 7.83–7.77 (m, 3H), 7.11 (d, J = 8.3 Hz, 2H), 3.91 (s, 3H) ppm; 13C NMR (125 MHz, 4% TFA-d in CDCl3): δ = 164.4, 161.9, 152.2, 151.4, 135.0, 130.3, 129.4, 129.3, 125.6, 123.9, 121.6, 117.8, 117.3, 115.1, 55.5 ppm; FT-IR (neat): ν = 3089, 2981, 1663, 1600, 1486, 1346, 1259, 1182, 826, 769 cm–1. HRMS (ESI), m/z: [M + Na]+ calcd for C17H12N2NaO3+ 315.0740; found 315.0746.
4. Conclusions
Herein, we presented our investigation of syntheses of novel isoxazolidine and isoxazole isoquinolinone hybrids. According to our original retrosynthesis, those fused heterocyclic compounds could be prepared via an 1,3-dipolar cycloaddition of indenone with nitrones and nitrile oxides and a subsequent Schmidt reaction. The cycloaddition with nitrones was found to be regioselective and, to some extent, depending on the reaction conditions, stereoselective, thus favouring the endo-adducts. Both endo- and exo-stereoisomers were uneventfully subjected to the Schmidt reaction to give the corresponding desired lactams (isoxazolidine/isoquinolinone hybrids). Although this scenario proved successful for isoxazolidine derivatives, a Beckmann fragmentation occurred when the Schmidt reaction protocol was applied on the indenone–benzonitrile oxide adduct. Thus, an alternative approach was adopted, which first involved an 1,3-dipolar cycloaddition reaction of an appropriate alkyne with nitrile oxides, and then an Ullmann type cyclisation of the corresponding iodinated isoxazoles, to furnish the desired isoxazole/isoquinolinone hybrids. Differentially substituted dipoles were used to obtain small libraries of each category of hybrids. The overall syntheses represent short and relatively straightforward pathways towards these new classes of compounds, which will be evaluated in the future for their biological activity.
Acknowledgments
This research work was supported by the Hellenic Foundation for Research and Innovation (HFRI) under the 3rd Call for HFRI PhD Fellowships (Fellowship Number: 5748). K.A.O. is grateful for this fellowship. The authors express their thanks to Maroula G. Kokotou, Laboratory of Chemistry, Department of Food Science and Human Nutrition, Agricultural University of Athens, for performing the HRMS analyses of several compounds.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29010091/s1. It contains data for compounds 4–10, 12, 13, 18b–18o, 22, and 23 and copies of 1H, 13C, 19F, and 2D NMR spectra of all new compounds.
Author Contributions
Conceptualisation, A.E.K.; methodology, K.A.O. and S.R.R.; formal analysis, K.A.O.; data curation, K.A.O.; writing—original draft preparation, K.A.O. and A.E.K.; writing—review and editing, A.E.K.; project administration, A.E.K. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data supporting the findings of this study are available within the paper and within its Supplementary Materials published online.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding Statement
This research received no external funding.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Breugst M., Reissig H.-U. The Huisgen Reaction: Milestones of the 1,3-Dipolar Cycloaddition. Angew. Chem. Int. Ed. 2020;59:12293–12307. doi: 10.1002/anie.202003115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berthet M., Cheviet T., Dujardin G., Parrot I., Martinez J. Isoxazolidine: A Privileged Scaffold for Organic and Medicinal Chemistry. Chem. Rev. 2016;116:15235–15283. doi: 10.1021/acs.chemrev.6b00543. [DOI] [PubMed] [Google Scholar]
- 3.Sysak A., Obmińska-Mrukowicz O. Isoxazole Ring as A Useful Scaffold in A Search for New Therapeutic Agents. Eur. J. Med. Chem. 2017;137:292–309. doi: 10.1016/j.ejmech.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 4.Wang X., Hu Q., Tang H., Pan X. Isoxazole/Isoxazoline Skeleton in the Structural Modification of Natural Products: A Review. Pharmaceuticals. 2023;16:228. doi: 10.3390/ph16020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiacchioa M.A., Giofrèb S.V., Romeob R., Romeob G., Chiacchio U. Isoxazolidines as Biologically Active Compounds. Curr. Org. Synth. 2016;13:726–749. doi: 10.2174/1570179412666150914195807. [DOI] [Google Scholar]
- 6.Perrone M.G., Vitale P., Panella A., Ferorelli S., Contino M., Lavecchia A., Scilimati A. Isoxazole-Based-Scaffold Inhibitors Targeting Cyclooxygenases (COXs) ChemMedChem. 2016;11:1172–1187. doi: 10.1002/cmdc.201500439. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi S., Kakinuma N., Uchida K., Hashimoto R., Yanagisawa T., Nakagawa A. Pyridovericin and Pyridomacrolidin: Novel Metabolites from Entomopathogenic Fungi, Beauveria Bassiana. J. Antibiot. 1998;51:596–598. doi: 10.7164/antibiotics.51.596. [DOI] [PubMed] [Google Scholar]
- 8.Nge C.-E., Sim K.-S., Lim S.-H., Thomas N.F., Low Y.-Y., Kam T.-S. A Hexacyclic, Iboga-Derived Monoterpenoid Indole with a Contracted Tetrahydroazepine C-Ring and Incorporation of an Isoxazolidine Moiety, a Seco-Corynanthean, an Aspidosperma- Aspidosperma Bisindole with Anticancer Properties, and the Absolute Configuration of the Pyridopyrimidine Indole Alkaloid, Vernavosine. J. Nat. Prod. 2016;79:2709–2717. doi: 10.1021/acs.jnatprod.6b00674. [DOI] [PubMed] [Google Scholar]
- 9.Ma L., Miao D., Lee J.J., Li T., Chen Y., Su G., Zhao Y. Synthesis and Biological Evaluation of Heterocyclic Ring-Fused Dammarane-Type Ginsenoside Derivatives as Potential Anti-Tumor Agents. Bioorg. Chem. 2021;116:105365. doi: 10.1016/j.bioorg.2021.105365. [DOI] [PubMed] [Google Scholar]
- 10.Arya J.S., Joseph M.M., Sherin D.R., Nair J.B., Manojkumar T.K., Maiti K.K. Exploring Mitochondria-Mediated Intrinsic Apoptosis by New Phytochemical Entities: An Explicit Observation of Cytochrome c Dynamics on Lung and Melanoma Cancer Cells. J. Med. Chem. 2019;62:8311–8329. doi: 10.1021/acs.jmedchem.9b01098. [DOI] [PubMed] [Google Scholar]
- 11.Rao L.B., Sreenivasulu C., Kishore D.R., Satyanarayana G. Trending Strategies for the Synthesis of Quinolinones and Isoquinolinones. Tetrahedron. 2022;127:133093. doi: 10.1016/j.tet.2022.133093. [DOI] [Google Scholar]
- 12.Humphries P.S., Benbow J.W., Bonin P.D., Boyer D., Doran S.D., Frisbie R.K., Piotrowski D.W., Balan G., Bechle B.M., Conn E.L., et al. Synthesis and SAR of 1,2,3,4-Tetrahydroisoquinolin-1-ones as Novel G-Protein-Coupled Receptor 40 (GPR40) Antagonists. Bioorg. Med. Chem. Lett. 2009;19:2400–2403. doi: 10.1016/j.bmcl.2009.03.082. [DOI] [PubMed] [Google Scholar]
- 13.Crosby I.T., Shin J.K., Capuano B. The Application of the Schmidt Reaction and Beckmann Rearrangement to the Synthesis of Bicyclic Lactams: Some Mechanistic Considerations. Aust. J. Chem. 2010;63:211–226. doi: 10.1071/CH09402. [DOI] [Google Scholar]
- 14.López L., Selent J., Ortega R., Masaguer C.F., Domínguez E., Areias F., Brea J., Loza M.I., Sanz F., Pastor M. Synthesis, 3D-QSAR, and Structural Modeling of Benzolactam Derivatives with Binding Affinity for the D2 and D3 Receptors. ChemMedChem. 2010;5:1300–1317. doi: 10.1002/cmdc.201000101. [DOI] [PubMed] [Google Scholar]
- 15.Pailee P., Prachyawarakorn V., Mahidol C., Ruchirawat S., Kittakoop P. Protoberberine Alkaloids and Cancer Chemopreventive Properties of Compounds from Alangium salviifolium. Eur. J. Org. Chem. 2011;2011:3809–3814. doi: 10.1002/ejoc.201100423. [DOI] [Google Scholar]
- 16.Arthur H.R., Hui W.H., Ng Y.L. An Examination of the Rutaceae of Hong Kong. Part III. The Alkaloid, Avicine, from Zanthoxylum Avicennae. J. Chem. Soc. 1959;803:4007–4009. doi: 10.1039/jr9590004007. [DOI] [Google Scholar]
- 17.Zhan G., Zhou J., Liu R., Liu T., Guo G., Wang J., Xiang M., Xue Y., Luo Z., Zhang Y., et al. Galanthamine, Plicamine, and Secoplicamine Alkaloids from Zephyranthes candida and Their Anti-acetylcholinesterase and Anti-inflammatory Activities. J. Nat. Prod. 2016;79:760–766. doi: 10.1021/acs.jnatprod.5b00681. [DOI] [PubMed] [Google Scholar]
- 18.Manpadi M., Kornienko A. Total Syntheses of Pancratistatin. A Review. Org. Prep. Proced. Int. 2008;40:107–161. doi: 10.1080/00304940809458083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hauser F.M., Zhou M., Sun Y. Facile Synthesis of Indenones from Indanones: A New Procedure. Synth. Commun. 2001;31:77–80. doi: 10.1081/SCC-100000182. [DOI] [Google Scholar]
- 20.Lu X., Schneider U. Aza-Morita-Baylis-Hillman Reactions Catalyzed by a Cyclopropenylidene. Chem. Commun. 2016;52:12980–12983. doi: 10.1039/C6CC06201F. [DOI] [PubMed] [Google Scholar]
- 21.Chandrasekhar B., Ahn S., Ryu J.-S. Synthesis of 4-Isoxazolines through Gold(I)-Catalyzed Cyclization of Propargylic N-Hydroxylamines. J. Org. Chem. 2016;81:6740–6749. doi: 10.1021/acs.joc.6b01499. [DOI] [PubMed] [Google Scholar]
- 22.He C.-T., Han X.-L., Zhang Y.-X., Du Z.-T., Si C.-M., Wei B.-G. Sc(OTf)3-Catalyzed [3 + 2]-Cycloaddition of Nitrones with Ynones. Org. Biomol. Chem. 2021;19:457–466. doi: 10.1039/D0OB02158J. [DOI] [PubMed] [Google Scholar]
- 23.Bortolini o., Mulani I., De Niro A., Maiuolo L., Nardi M., Russo B., Avnet S. Efficient Synthesis of Isoxazolidine-Substituted Bisphosphonates By 1,3-Dipolar Cycloaddition Reactions. Tetrahedron. 2011;67:5635–5641. doi: 10.1016/j.tet.2011.05.098. [DOI] [Google Scholar]
- 24.Poulsen P.H., Vergura S., Monleón A., Jørgensen D.K.B., Jørgensen K.A. Controlling Asymmetric Remote and Cascade 1,3-Dipolar Cycloaddition Reactions by Organocatalysis. J. Am. Chem. Soc. 2016;138:6412–6415. doi: 10.1021/jacs.6b03546. [DOI] [PubMed] [Google Scholar]
- 25.Delso I., Terejo T. 1H–15N HMBC as a Valuable Tool for the Identification and Characterization of Nitrones. Tetrahedron Lett. 2007;48:4101–4104. doi: 10.1016/j.tetlet.2007.04.006. [DOI] [Google Scholar]
- 26.Chakraborty B., Chettri E. Synthesis of Some Novel Class of Regioselective Spiro Isoxazolidine Derivatives via 1,3-Dipolar Cycloaddition Reaction of N-Benzyl-C-fluorosubstituted Phenyl Nitrones in Ionic Liquid. J. Heterocycl. Chem. 2018;55:1157–1165. doi: 10.1002/jhet.3148. [DOI] [Google Scholar]
- 27.Katahara S., Kobayashi S., Fujita K., Matsumoto T., Sato T., Chida N. An Iridium-Catalyzed Reductive Approach to Nitrones from N-Hydroxyamides. J. Am. Chem. Soc. 2016;138:5246–5249. doi: 10.1021/jacs.6b02324. [DOI] [PubMed] [Google Scholar]
- 28.Mukhopadhyay S., Batra S. Direct Transformation of Arylamines to Aryl Halides via Sodium Nitrite and N-Halosuccinimide. Chem. Eur. J. 2018;24:14622–14626. doi: 10.1002/chem.201803347. [DOI] [PubMed] [Google Scholar]
- 29.Schierle S., Neumann S., Heitel P., Willems S., Kaiser A., Pollinger J., Merk D. Design and Structural Optimization of Dual FXR/PPARδ Activators. J. Med. Chem. 2020;63:8369–8379. doi: 10.1021/acs.jmedchem.0c00618. [DOI] [PubMed] [Google Scholar]
- 30.Di Nunno L., Vitale P., Scilimati A., Simonea L., Capitelli F. Stereoselective Dimerization of 3-Arylisoxazoles to Cage-Shaped Bis-b-lactams syn 2,6-Diaryl-3,7-diazatricyclo[4.2.0.02,5]-octan-4,8-diones Induced by Hindered Lithium Amides. Tetrahedron. 2007;63:12388–12395. doi: 10.1016/j.tet.2007.09.040. [DOI] [Google Scholar]
- 31.Tambara K., Dan Pantos G. Conversion of Aldoximes into Nitriles and Amides Under Mild Conditions. Org. Biomol. Chem. 2013;11:2466–2472. doi: 10.1039/c3ob27362h. [DOI] [PubMed] [Google Scholar]
- 32.Steiger S.A., Li C., Backos D.S., Reigan P., Natale N.R. Dimeric Isoxazolyl-1,4-Dihydropyridines Have Enhanced Binding at the Multi-Drug Resistance Transporter. Bioorg. Med. Chem. 2017;25:3223–3234. doi: 10.1016/j.bmc.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu J., Lu M. Metal-Free: A Novel and Efficient Aerobic Oxidation of Primary Amines to Oximes Using N,N′,N″-Trihydroxyisocyanuric Acid and Acetaldoxime as Catalysts in Water. Synlett. 2014;25:1873–1878. doi: 10.1002/chin.201506084. [DOI] [Google Scholar]
- 34.McIntosh M.L., Naffziger M.R., Ashburn B.O., Zakharov L.N., Carter R.G. Highly Regioselective Nitrile Oxide Dipolar Cycloadditions with ortho-Nitrophenyl Alkynes. Org. Biomol. Chem. 2012;10:9204–9213. doi: 10.1039/c2ob26267c. [DOI] [PubMed] [Google Scholar]
- 35.Jawalekar A.M., Reubsaet E., Rutjes F.P.J.T., van Delft F.L. Synthesis of Isoxazoles by Hypervalent Iodine-Induced Cycloaddition of Nitrile Oxides to Alkynes. Chem. Commun. 2011;47:3198–3200. doi: 10.1039/c0cc04646a. [DOI] [PubMed] [Google Scholar]
- 36.Kanemasa S., Matsuda H., Kamimurac A., Kakinami T. Synthesis of Hydroximoyl Chlorides from Aldoximes and Benzyltrimethylammonium Tetrachloroiodate (BTMA ICl4) Tetrahedron. 2000;56:1057–1064. doi: 10.1016/S0040-4020(99)01047-9. [DOI] [Google Scholar]
- 37.Suzuki K., Watanabe T., Murahashi S.-I. Oxidation of Primary Amines to Oximes with Molecular Oxygen using 1,1-Diphenyl-2-picrylhydrazyl and WO3/Al2O3 as Catalysts. J. Org. Chem. 2013;78:2301–2310. doi: 10.1021/jo302262a. [DOI] [PubMed] [Google Scholar]
- 38.Palomo C., Oiarbide M., Arceo E., García J.M., López R., González A., Linden A. Lewis Acid Catalyzed Asymmetric Cycloadditions of Nitrones: A’-Hydroxy Enones as Efficient Reaction Partners. Angew. Chem. Int. Ed. 2005;44:6187–6190. doi: 10.1002/anie.200502308. [DOI] [PubMed] [Google Scholar]
- 39.Cao G., Zhou S., Teng D. Synthesis of Spiroisoxazolidinyl-Benzoisothiazolines by 1,3-Dipolar Cycloaddition of Benzoisothiazole-2,2-dioxide-3-ylidenes with Nitrones. Tetrahedron. 2017;73:2329–2333. doi: 10.1016/j.tet.2017.03.024. [DOI] [Google Scholar]
- 40.Śnieżek M., Stecko S., Panfil I., Furman B., Urbańczyk-Lipkowska Z., Chmielewski M. Thermal and Sc(OTf)3 Catalyzed 1,3-Dipolar Cycloaddition of Open-Chain Nitrones to α,β-Unsaturated Lactones: Combined Experimental and Computational Studies. Tetrahedron Asymmetry. 2013;24:89–103. doi: 10.1016/j.tetasy.2012.12.006. [DOI] [Google Scholar]
- 41.Minami S., Tomita M., Takamatsu H., Uyeo S. The Schmidt Reaction with Some Tetralone and Indanone Derivatives. Chem. Pharm. Bull. 1965;13:1084–1091. doi: 10.1248/cpb.13.1084. [DOI] [PubMed] [Google Scholar]
- 42.Evans D., Lockhart I.M. The Schmidt Reaction with Aromatic Ketones. J. Chem. Soc. 1965:4806–4812. doi: 10.1039/jr9650004806. [DOI] [Google Scholar]
- 43.Conley R.T. Schmidt Reactions in Polyphosphoric Acid. I. Rearrangement of Ketones. J. Org. Chem. 1958;23:1330–1333. doi: 10.1021/jo01103a023. [DOI] [Google Scholar]
- 44.Škorić D.Đ., Klisurić O.R., Jakimov D.S., Sakač M.N., Csanádi J.J. Synthesis of New Bile Acid-Fused Tetrazoles Using the Schmidt Reaction. Beilstein J. Org. Chem. 2021;17:2611–2620. doi: 10.3762/bjoc.17.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koldobskii G.I., Ostrovskii V.A. Application of the Schmidt Reaction for the Preparation of Tetrazoles. Chem. Heterocycl. Compd. 1975;11:626–635. doi: 10.1007/BF00959947. [DOI] [Google Scholar]
- 46.Ishihara K., Shioiri T., Matsugi M. An Expeditious Approach to Tetrazoles from Amides Utilizing Phosphorazidates. Org. Lett. 2020;22:6244–6247. doi: 10.1021/acs.orglett.0c01890. [DOI] [PubMed] [Google Scholar]
- 47.Wan Z.-K., Wacharasindhu S., Levins G.C., Lin M., Tabei K., Mansour T.S. The Scope and Mechanism of Phosphonium-Mediated SNAr Reactions in Heterocyclic Amides and Ureas. J. Org. Chem. 2007;72:10194–10210. doi: 10.1021/jo7020373. [DOI] [PubMed] [Google Scholar]
- 48.Stotani S., Gatta V., Medda F., Padmanaban M., Karawajczyk A., Tammela P., Giordanetto F., Tzalis D., Collina S. A Versatile Strategy for the Synthesis of 4,5-Dihydroxy-2,3-Pentanedione (DPD) and Related Compounds as Potential Modulators of Bacterial Quorum Sensing. Molecules. 2018;23:2545. doi: 10.3390/molecules23102545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.VerHaeghe D.G., Weber G.S., Pappalardo P.A. Beckmann Fragmentation Versus Beckmann Rearrangement in Dehydronorcamphor Derivatives. Tetrahedron Lett. 1989;30:4041–4044. doi: 10.1016/S0040-4039(00)99316-X. [DOI] [Google Scholar]
- 50.Brahmchari D., Verma A.K., Mehta S. Regio- and Stereoselective Synthesis of Isoindolin-1-ones through BuLi-Mediated Iodoaminocyclization of 2-(1-Alkynyl)benzamides. J. Org. Chem. 2018;83:3339–3347. doi: 10.1021/acs.joc.7b02903. [DOI] [PubMed] [Google Scholar]
- 51.Morita T., Fuse S., Nakamura H. Generation of an 4-Isoxazolyl Anion Species: Facile Access to Multifunctionalized Isoxazoles. Angew. Chem. Int. Ed. 2016;55:1–6. doi: 10.1002/anie.201608039. [DOI] [PubMed] [Google Scholar]
- 52.Quana Z.-J., Xiaa H.-D., Zhanga Z., Daa Y.-X. Ligand-free CuTC-catalyzed N-arylation of Amides, Anilines and 4-aminoantipyrine: Synthesis of N-arylacrylamides, 4- amido-N-phenylbenzamides and 4-amino (N-phenyl)antipyrenes. Appl. Organometal. Chem. 2014;28:81–85. doi: 10.1002/aoc.3080. [DOI] [Google Scholar]
- 53.Cantillo D., de Frutos O., Rincon J.A., Mateos C., Kappe C.O. A Scalable Procedure for Light-Induced Benzylic Brominations in Continuous Flow. J. Org. Chem. 2014;79:223–229. doi: 10.1021/jo402409k. [DOI] [PubMed] [Google Scholar]
- 54.Wang A., Lv P., Liu Y. 4,5-Dihydro-1,2,4-oxadiazole as a Single Nitrogen Transfer Reagent: Synthesis of Functionalized Isoxazoles Assisted by Sc(OTf)3 or Au(I)/ Sc(OTf)3 Synergistic Catalysis. Org. Lett. 2023;25:4377–4382. doi: 10.1021/acs.orglett.3c01566. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper and within its Supplementary Materials published online.