

Abstract
INTRODUCTION
We investigated the association of inflammatory mechanisms with markers of Alzheimer's disease (AD) pathology and rates of cognitive decline in the AD spectrum.
METHODS
We studied 296 cases from the Deutsches Zentrum für Neurodegenerative Erkrankungen Longitudinal Cognitive Impairment and Dementia Study (DELCODE) cohort, and an extension cohort of 276 cases of the Alzheimer's Disease Neuroimaging Initiative study. Using Bayesian confirmatory factor analysis, we constructed latent factors for synaptic integrity, microglia, cerebrovascular endothelial function, cytokine/chemokine, and complement components of the inflammatory response using a set of inflammatory markers in cerebrospinal fluid.
RESULTS
We found strong evidence for an association of synaptic integrity, microglia response, and cerebrovascular endothelial function with a latent factor of AD pathology and with rates of cognitive decline. We found evidence against an association of complement and cytokine/chemokine factors with AD pathology and rates of cognitive decline.
DISCUSSION
Latent factors provided access to directly unobservable components of the neuroinflammatory response and their association with AD pathology and cognitive decline.
Keywords: amyloid, chemokine factors, complement, endothelial function, microglia, structural equation models, tau
1. INTRODUCTION
Inflammation is a central component of Alzheimer's disease (AD) pathophysiology, downstream of amyloid beta (Aβ) and tau pathology. 1 , 2 , 3 At the same time, inflammatory mechanisms influence Aβ and tau pathology in the brain. 4 , 5 Several studies associated single inflammatory cerebrospinal fluid (CSF) markers with Aβ pathology, tau pathology, or both in the AD spectrum. 6 , 7 Results become heterogeneous when one concentrates on single inflammatory markers. For the well‐established microglia marker soluble triggering receptor expressed on myeloid cells 2 (sTREM2) some studies found higher CSF sTREM2 levels associated with less Aβ pathology in the sporadic AD spectrum. 8 , 9 In contrast, several studies showed associations of sTREM2 levels mainly with phosphorylated tau (p‐tau) pathology. They found decreased or unchanged levels of CSF sTREM2 in Aβ‐positive but p‐tau negative cases, and increased levels in p‐tau positive cases, irrespective of Aβ status. 7 , 9 , 10 , 11
These variations may reflect different inflammatory mechanisms in different disease stages. However, a more parsimonious interpretation should take into account that CSF markers are only surrogate measures for an underlying construct, that is, inflammation, which cannot be directly observed in clinical studies. Mathematically, one can express this epistemological reservation in the framework of structural equation models (SEM). 12 We can construct the inflammatory response in the brain as a latent factor that eludes direct observation but can be assessed by observable proxy markers in the CSF. Confirmatory factor analysis is a readily available tool to form such latent factors.
Here, we constructed latent factors for a priori defined inflammatory domains, including synaptic integrity, microglia, complement factors, adhesion, and cytokines/chemokines. We had two goals: First, to determine latent factors of neuroinflammation based on an a priori assignment of single inflammatory markers to certain inflammatory domains. Second, to characterize these neuroinflammation factors in relation to AD pathology markers from CSF and longitudinal rates of cognitive decline. We tested latent factors in two independent cohorts that included individuals from the AD spectrum and cognitively healthy controls, the Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE) Longitudinal Cognitive Impairment and Dementia Study (DELCODE) 13 and the Alzheimer's Disease Neuroimaging Initiative (ADNI) 14 cohorts, respectively. Because of the small overlap of markers measured in both cohorts, we could not replicate the results across cohorts as originally planned, but used the ADNI data to extend the neuroinflammation domains from DELCODE. This provided us with access to the additional domain of endothelial adhesion function that may be related to blood–brain barrier dysfunction. We used a Bayesian analysis framework for statistical inference that allowed us to directly quantify both evidence for and against an effect. 15 This is different from the P value, which tells us the probability that a similar or even more extreme effect will occur in future experiments if the null hypothesis were true. 16 , 17 Additionally, the 95% credible interval of the posterior distribution of the parameter estimates is directly interpretable as the range in which the parameter lies with 95% probability given the data. This is different from the interpretation of the frequentist 95% confidence interval: the parameter estimate will lie in this interval in 95% of future repeated experiments. 15 , 17 , 18
2. MATERIAL AND METHODS
2.1. Data sources
Part of the data came from the DELCODE cohort, conducted by the DZNE. Another part of the data came from the ADNI cohorts, accessed via the ADNI database (http://adni.loni.usc.edu/). For a more detailed description of both cohorts see the supporting information.
2.2. Consent statement
For both studies, DELCODE and ADNI, written informed consent was obtained from all participants and/or authorized representatives. The study protocols for both studies had been approved by the local institutional review boards and ethical committees of the centers participating in the respective study. DELCODE and ADNI are being conducted in accord with the Declaration of Helsinki of 1975 and its later amendments.
2.3. Participants
For DELCODE, the baseline sample included n = 1079 participants. After excluding cases without an available CSF sample, we were left with n = 309 cases. After removing all cases without complete neuroinflammatory measures in the CSF, our baseline sample compromised n = 296 cases. We included the following diagnoses: older healthy controls, first‐degree relatives, subjective cognitive decline (SCD), mild cognitive impairment (MCI), and AD dementia.
From the ADNI cohort, we retrieved n = 279 cases who had complete measures on a previously reported inflammatory marker panel in CSF. 19 We included the following diagnoses: older healthy controls, MCI, and AD dementia.
Detailed diagnostic criteria for both cohorts are reported in the supporting information.
2.4. Neuropsychological assessment
We aimed to cover similar cognitive domains across both cohorts. For the DELCODE and the ADNI cohort we used the Mini‐Mental State Examination (MMSE) score 20 as a measure of global cognition. For DELCODE we chose the Preclinical Alzheimer Cognitive Composite score plus verbal fluency (PACC5 score) 21 as a composite measure of memory and executive function, the Free and Cued Selective Reminding Test (FCSRT) 22 immediate free recall as measure of memory, and the ratio of the Trail Making Test B to A (TMTB/A) as measure of executive function. 23
For the ADNI cohort, we used the ADNI composite scores for memory 24 and executive function. 25
2.5. CSF sampling
For the DELCODE study details on standard operating procedures (SOP) for sampling and measurements of CSF can be found in Jessen et al. 13
RESEARCH IN CONTEXT
Systematic Review: We reviewed the literature using PubMed. Studies suggest an association of cerebrospinal fluid (CSF) inflammatory markers with CSF markers of Alzheimer's disease (AD) pathology and measures of brain integrity, but results were heterogeneous.
Interpretation: Heterogeneity of findings for individual inflammatory markers may be due to the fact that CSF markers are only surrogate measures for an underlying construct, that is, inflammation, which cannot be directly observed.
Future Directions: We have constructed latent factors for a priori defined domains of inflammation to determine the association of these factors with AD pathology and rates of cognitive change. This approach can integrate other domains of inflammation as well as other markers of inflammatory domains, such as molecular imaging modalities. This approach is also useful to determine the reproducibility of results across different cohorts that are harmonized at the level of inflammatory domains but not at the level of individual markers.
For the ADNI cohorts, details on SOPs for sampling and measurements of CSF can be found in the ADNI procedure manual (http://adni.loni.usc.edu/methods/).
For both DELCODE and ADNI, CSF samples were aliquoted after centrifugation and stored at −80°C before measurements were conducted.
2.6. CSF AD biomarker assessment
DELCODE features a centralized data management that provides access to the biomarker data. We used data for Aβ42, Aβ40, p‐tau 181, and total tau that were determined with the following commercially available kits: V‐PLEX Aβ Peptide Panel 1 (6E10) Kit (K15200E) and V‐PLEX Human Total Tau Kit (K151LAE; Mesoscale Diagnostics LLC), and Innotest Phospho‐Tau(181P) (81581; Fujirebio Germany GmbH). The measurements were conducted in accordance with the vendor specifications.
The Biomarker Core of ADNI at the Translational Research Laboratory, Department of Pathology & Laboratory Medicine at the University of Pennsylvania Medical School is in charge of processing CSF samples. We used data for Aβ42, p‐tau 181, and total tau measured by the Luminex multiplex immunoassay platform. A detailed description of the ADNI CSF Biomarker Core procedures can be found in the data primer, “An Overview of the first 8 ADNI CSF Batch Analyses” (http://adni.loni.usc.edu/methods/documents).
2.7. CSF inflammatory marker panel assessment
For the DELCODE cohort, a panel of markers had been defined that had been detected in previous studies with acceptable reliability 7 , 26 , 27 in CSF and serum, and is related to inflammation mechanisms. This panel includes the following markers: Ferritin, apolipoprotein E (apoE), sTrem2, serum‐soluble AXL, sTyro3, interleukin 6 (IL‐6), Interleukin 18 (IL‐18), monocyte chemoattractant protein 1, interferon gamma inducible protein 10, macrophage migration inhibitory factor, YKL‐40, C‐reactive protein, complement C1q, C3, C3b, C4, factor B, and factor H. In addition, it includes neurogranin and FABP‐3 as non‐tau neurodegeneration markers. 7 , 26 , 27
Detailed measurement procedures can be found in Brosseron et al. 7 , 26 , 27 Commercially available enzyme‐linked immunosorbent assays (ELISA) were used to measure all markers of the panel in CSF and serum. The DELCODE biomarker group used internal control samples and measured marker levels in duplicate. Measures were accepted only if the coefficient of variance (CV) was < 20%. We combined the CSF inflammatory markers into four categories: synaptic integrity markers, microglia markers, complement markers, and cytokine/chemokine markers, based on a literature review reported in Table S1 in supporting information.
In the ADNI cohort, we identified a marker panel in the ADNI database that had been reported before. 19 The Biomarker Core of ADNI measured CSF levels of the following inflammatory markers using multiplex immunoassays: interleukin 12 subunit p40 (IL‐12‐P40), interleukin 10 (IL‐10), IL‐6, tumor necrosis factor alpha (TNFα), tumor necrosis factor receptor 1 (TNFR1), transforming growth factor beta 1 (TGFβ1), vascular cell adhesion molecule 1 (VCAM‐1), and intercellular adhesion molecule 1 (ICAM‐1). We used one cluster of markers to create an adhesion factor (including TNFα, TNFR1, TGFβ1, ICAM‐1, and VCAM‐1), and another cluster of markers to create a cytokine/chemokine factor (IL‐6, IL‐10, IL‐12‐P40).
2.8. Statistical analysis
We compared demographic characteristics between diagnostic groups using Bayesian analysis of variance and contingency tables as required. For these calculations, we used Jeffreys's Amazing Statistics Program (JASP Version 0.11), available at jasp‐stats.org. We report the Bayes factor (BF10) quantifying evidence against the null hypotheses.
We used Bayesian confirmatory factor analysis as implemented in the library “blavaan” in R version 4.2.1 (2022‐06‐23), accessed through R Studio, to construct latent factors for the classes of neuroinflammatory response. Model fit was assessed using posterior predictive P values. Factor scores for each individual were derived for the latent factors using least square regression with the “predict” function of R library “lavaan.”
We compared the levels of latent factors across diagnostic groups, controlling for age and sex using Bayesian analysis of covariance (ANCOVA) in JASP. Furthermore, we analyzed the association of the latent factors with a latent factor of AD pathology constructed from CSF markers of Aβ42, Aβ40, total tau, and p‐tau, controlling for diagnosis, age, sex, and APOE ε4 genotype (none vs. at least one allele).
Subsequently, we estimated generalized mixed effects models in a Bayesian framework with time nested within individuals with random intercept and slope terms, and longitudinal cognitive scores as outcomes. The models contained the main effect of the inflammation factor and the interaction of the inflammation factor with time as well as the main effects of p‐tau, Aβ42, and diagnosis, and their interaction effects with time, and the main effects of age and sex. We compared fit of non‐Gaussian versus Gaussian models for the dependent variables using posterior predictive checks. We determined the posterior estimates of the inflammation factor by time interaction and its 90% and 95% credible intervals as primary outcomes of this analysis. These analyses were conducted using library “brms” in R version 4.2.1 (2022‐06‐23), accessed through R Studio.
Finally, we determined if neuroinflammation factors mediated the effects of AD pathology on rates of cognitive change, using library “blavaan” in R version 4.2.1 (2022‐06‐23). We report the median and 95% credible intervals of the posterior distribution of the indirect (mediating) effect.
Adherence to Bayesian Analysis Reporting Guidelines 28 is illustrated in Table S2 in supporting information.
3. RESULTS
3.1. Demographics
As shown in Table 1, participants from the DELCODE cohort were highly likely to differ in age and sex, but not in years of education across diagnostic groups. In the ADNI cohort, diagnostic groups were highly likely to differ in age, but not in years of education and sex. In the DELCOE cohort sample, 272 of the 296 baseline cases had at least one clinical follow up with a median follow‐up time of 4.0 years (range 0.9 to 6.3 years). From the 279 baseline cases of the ADNI sample, 213 had at least one clinical follow‐up examination with a median follow‐up time of 3.2 years (range 0.6 to 16.4 years). Number of cases per time points are shown in Figure S1 in supporting information.
TABLE 1.
Demographics.
| DELCODE (N = 296) | N(f/m) [%] a |
Age (years) b mean (95% CI) |
MMSE c mean (95% CI) |
Education (years) d mean (95% CI) |
|---|---|---|---|---|
| Controls | 39/31 [56/44] | 68.8 (67.6–69.9) | 29.4 (29.2–29.7) | 14.5 (13.9–15.2) |
| Relatives | 16/6 [73/27] | 64.9 (63.2–66.6) | 29.0 (28.5–29.5) | 13.5 (12.4–14.7) |
| SCD | 43/55 [44/56] | 71.1 (70.0–72.2) | 29.2 (29.0–29.4) | 15.1 (14.4–15.7) |
| MCI | 22/47 [32/68] | 72.3 (71.1–73.6) | 27.9 (27.5–28.4) | 14.0 (13.3–14.8) |
| AD | 23/14 [62/38] | 73.9 (71.8–76.0) | 24.0 (22.9–25.2) | 13.8 (12.9–14.8) |
| ADNI (N = 279) | N(f/m) [%] e |
Age (years) f mean (95% CI) |
MMSE g mean (95% CI) |
Education (years) h mean (95% CI) |
|---|---|---|---|---|
| Controls | 37/46 [45/55] | 78.5 (77.2–79.8) | 29 (28.8–29.2) | 15.4 (14.5–16.3) |
| MCI | 45/79 [36/64] | 75.9 (74.5–77.3) | 26.9 (26.6–27.2) | 14.1 (13.1–15.2) |
| AD | 31/41 [43/57] | 74.4 (72.5–76.4) | 23.6 (23.2–24) | 13.8 (12.4–15.1) |
Abbreviations: AD, Alzheimer's disease; ADNI, Alzheimer's Disease Neuroimaging Initiative; CI, credibility intervals; DELCODE, Deutsches Zentrum für Neurodegenerative Erkrankungen Longitudinal Cognitive Impairment and Dementia Study; MCI, mild cognitive impairment; MMSE Mini‐Mental State Examination; SCD, subjective cognitive decline.
Bayes factor in favor of a group effect (BF10) = 37.2; i.e., a group effect is 37.2 times more likely than the absence of such effect.
Bayes factor in favor of a group effect (BF10) = 2.2 * 107.
Bayes factor in favor of a group effect (BF10) = 2.6 * 1038.
Bayes factor in favor of the absence of a group effect (BF10) = 0.48.
Bayes factor in favor of the absence of a group effect (BF10) = 0.26.
Bayes factor in favor of a group effect (BF10) = 8.6.
Bayes factor in favor of a group effect (BF10) = 2.6*1056.
Bayes factor in favor of the absence of a group effect (BF10) = 0.08.
3.2. Inflammation latent factors and diagnoses
Fit indices of the SEM models for deriving the latent factors were high to very high (Table S3 in supporting information). The loading diagrams are plotted in Figure S2A in supporting information and the correlation matrices for the latent factors in Figure S3 in supporting information. In ANCOVA models, we compared the levels of latent factors across diagnoses, controlling for age and sex. We found strong to extremely strong evidence (BF10 between 49 and 1.8*106) for a difference of the synaptic integrity factor between AD patients and all other diagnostic groups (controls, MCI, SCD), but there was evidence against pairwise differences between the other diagnostic groups (BF10 < 2.2). For the microglia, complement, and cytokine/chemokine factors, we found evidence against diagnostic group differences (Figure 1).
FIGURE 1.
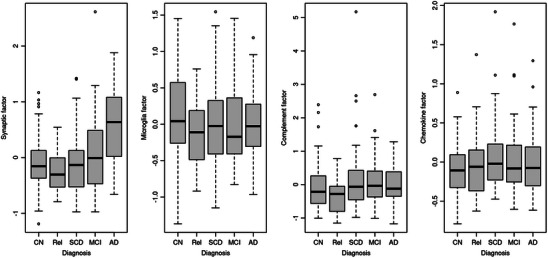
Levels of inflammatory latent factors across groups in the DELCODE data. Boxplots of factor scores of the inflammation factors in the healthy controls (CN), the cognitively normal relatives of people with dementia (Rel), the individuals with subjective cognitive decline (SCD), and the mild cognitive impairment (MCI) and Alzheimer's disease (AD) dementia cases
3.3. Inflammation latent factors and AD pathology
For DELCODE, we constructed a latent factor of AD pathology based on CSF Aβ42, Aβ40, tau, and p‐tau181 levels. P‐tau, tau, and Aβ40 contributed positively and Aβ42 contributed negatively to the latent factor of AD pathology (Figure 2). We found extreme evidence for a positive association of the AD pathology factor with the synaptic (BF10 = 2.6*1048) and the microglia factor (BF10 = 3.7*1034). For the complement and cytokine/chemokine factors, we found inconclusive evidence (BF10 = 0.536 and 0.562, respectively). Parameter estimates are shown in Table 2.
FIGURE 2.
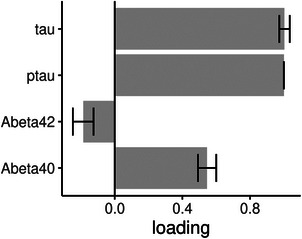
Latent factor Alzheimer's disease (AD) pathology and cerebrospinal fluid (CSF) pathology markers in the DELCODE data. Association of the CSF pathology markers with the latent factor AD pathology (bars are standard deviations of the estimates) in the DELCODE data
TABLE 2.
Inflammatory and AD pathology factors in the DELCODE data.
| (A) Association of synaptic and AD pathology factors | |||||
|---|---|---|---|---|---|
| 95% credible interval | |||||
| Variable | Level | Mean | SD | Lower | Upper |
| AD pathology | 0.492 | 0.026 | 0.439 | 0.544 | |
| Sex | f | −0.046 | 0.020 | −0.087 | −0.007 |
| m | 0.046 | 0.020 | 0.005 | 0.086 | |
| Age | 0.027 | 0.022 | −0.019 | 0.070 | |
| Diagnosis | AD | −0.048 | 0.049 | −0.150 | 0.048 |
| CN | 0.075 | 0.038 | −4.618×10−4 | 0.151 | |
| MCI | −0.035 | 0.037 | −0.109 | 0.037 | |
| Rel | 0.005 | 0.053 | −0.102 | 0.111 | |
| SCD | 0.003 | 0.034 | −0.066 | 0.069 | |
| APOE ε4 | 0 | −0.109 | 0.023 | −0.154 | −0.065 |
| 1 | 0.109 | 0.023 | 0.063 | 0.153 | |
| (B) Association of microglia and AD pathology factors | |||||
|---|---|---|---|---|---|
| AD pathology | 0.492 | 0.033 | 0.425 | 0.559 | |
| Sex | f | −0.006 | 0.025 | −0.056 | 0.044 |
| m | 0.006 | 0.025 | −0.045 | 0.055 | |
| Age | 0.062 | 0.027 | 0.005 | 0.115 | |
| Diagnosis | AD | −0.453 | 0.072 | −0.598 | −0.309 |
| CN | 0.262 | 0.050 | 0.159 | 0.361 | |
| MCI | −0.131 | 0.050 | −0.229 | −0.033 | |
| Rel | 0.196 | 0.078 | 0.041 | 0.353 | |
| SCD | 0.126 | 0.045 | 0.034 | 0.216 | |
| APOE ε4 | 0 | 0.082 | 0.028 | 0.027 | 0.136 |
| 1 | −0.082 | 0.028 | −0.136 | −0.027 | |
Abbreviations: AD, Alzheimer's disease; APOE, apolipoprotein E; CN, cognitively normal; f, female; m, male; MCI, mild cognitive impairment; SCD, subjective cognitive decline; SD, standard deviation.
AD—dummy variable for AD patient (1/0 – yes/no).
CN—dummy variable for normal control (1/0 – yes/no).
MCI—dummy variable for MCI (1/0 – yes/no).
Rel—dummy variable for relative of person with dementia (1/0 – yes/no).
SCD—dummy variable for subjective cognitive decline (1/0 – yes/no).
APOE ε4—APOE ε4 alleles; 0 = none, 1 = at least 1.
3.4. Longitudinal analysis
Posterior predictive checks and testing the normality of the distribution of the residuals suggested that the dependent variables PACC5 score, free recall from the FCSRT, and MMSE score were fit very well by a Gaussian distribution, the TMTB/A ratio by a skewed normal distribution. Rates of cognitive change by diagnosis are shown in Figure S4A in supporting information; correlations between rates of change in Table S4A in supporting information.
We found an association of the synaptic factor with rates of change in FCSRT free recall, PACC5 score, MMSE, and TMTB/A, with higher synaptic factor being associated with more pronounced cognitive decline. For PACC5 and MMSE, the 95% credible interval excluded zero, for FCSRT free recall and TMTB/A, it included zero (Figures 3A and 4).
FIGURE 3.
Posterior distribution of predictors of cognitive decline in the DELCODE data. Plots of the posterior distribution of the parameter estimates for the cognitive outcomes (FCSRT‐FR, PACC5, MMSE, and TMTB/A, respectively), including effects of time and time by predictor interactions. The circle indicates the mean value, the thick segments the 90% credible intervals, and the thinner outer lines the 95% credible intervals of the posterior distribution. Of note, the time by diagnosis interactions (time:CN, controls; time:Rel, first‐degree relatives; time:SCD, subjective cognitive decline; time:MCI, mild cognitive impairment) are plotted against the reference of the AD group. So positive values for time:CN indicate that rates of cognitive decline were less pronounced in the controls than the AD group. A, Synaptic factor. B, Microglia factor. C, Cytokine/chemokine factor. D, Complement factor. AD, Alzheimer's disease; CN, cognitively normal; FCSRT‐FR, Free and Cued Selective Reminding Test Free Recall; MMSE, Mini‐Mental State Examination; PACC5, Preclinical Alzheimer Cognitive Composite score plus verbal fluency; TMTB/A, Trail Making Test Parts B to A
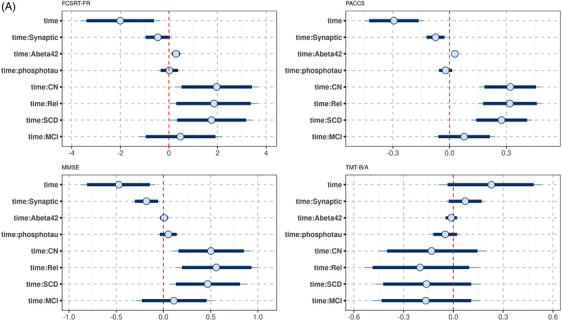
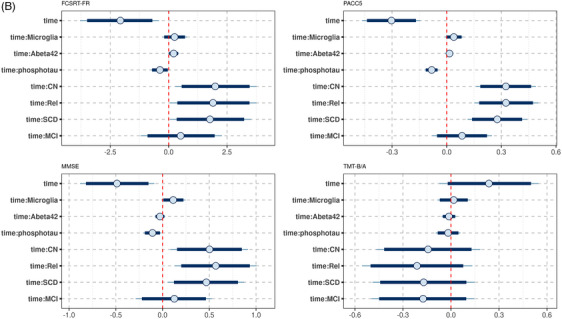
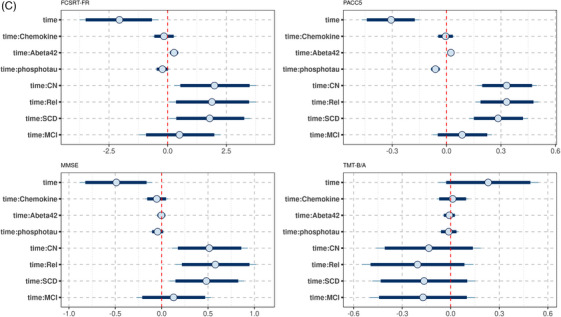
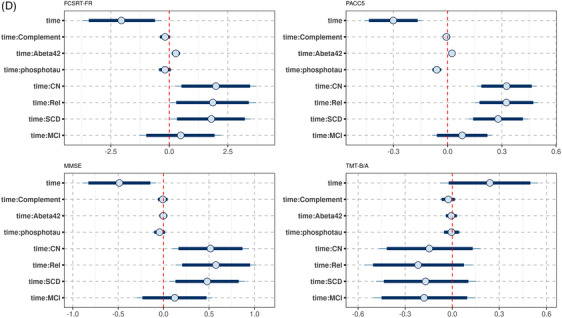
FIGURE 4.
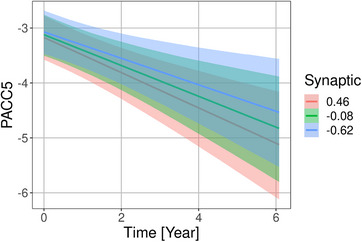
Associations of synaptic factor with rates of Preclinical Alzheimer Cognitive Composite score plus verbal fluency (PACC5) change in the DELCODE cohort. Marginal interaction effect of time with synaptic factor for PACC5 change as dependent variable in linear mixed effect models predicting cognitive scores by diagnosis, amyloid beta 42 levels, phosphorylated tau levels, synaptic factor, and their interaction with time with a random intercept term, nested within individuals. The depicted values for the synaptic factor were chosen as the mean, and the mean plus and minus one standard deviation. This is used to illustrate the effect of the continuous synaptic variable on the rates of change
Lower levels of the microglia factor were associated with more pronounced cognitive decline in FCSRT free recall, PACC5, and MMSE. For MMSE and PACC5 scores the 90% credible intervals of the posterior distribution excluded zero (Figures 3B and 5).
FIGURE 5.
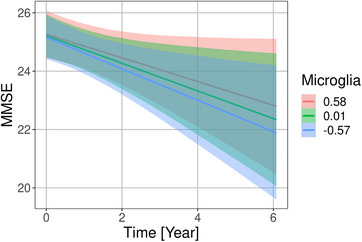
Associations of microglia factor with rates of Mini‐Mental State Examination (MMSE) score change in the DELCODE cohort. Marginal interaction effect of time with microglia factor for MMSE score as dependent variable in a generalized mixed effect model predicting MMSE score by diagnosis, amyloid beta 42 levels, phosphorylated tau levels, microglia factor, and their interaction with time with a random intercept term, nested within individuals. The depicted values for the microglia factor were chosen as the mean, and the mean plus and minus one standard deviation. This is used to illustrate the effect of the continuous microglia variable on the rates of change
For cytokine/chemokine and complement factors, the 90% credibility intervals excluded zero for the associations with all cognitive scores (Figure 3C and D).
3.5. ADNI sample
The factor loading diagrams are plotted in Figure 2. We found evidence against an association of diagnosis with the adhesion and the cytokine/chemokine factors (Figure S5 in supporting information). P‐tau and tau contributed positively and Aβ42 contributed negatively to a latent factor of AD pathology constructed from the ADNI data (Figure S6 in supporting information).We found very strong evidence for a positive association of the AD pathology factor with the adhesion factor (BF10 = 202.4), and moderate evidence against an association with the cytokine/chemokine factor (BF10 = 0.25). Parameter estimates are shown in Table S5 in supporting information.
In the longitudinal data, the dependent variables ADNI memory and executive function as well as MMSE scores were fit very well by a Gaussian distribution. Rates of cognitive change by diagnosis are shown in Figure S4B; correlations between rates of change in Table S4B.
Lower levels of the adhesion factor were associated with more pronounced cognitive decline in ADNI memory and executive function and MMSE scores. For ADNI memory and executive function scores the 90% credible interval excluded zero; for MMSE score, it included zero (Figures S7A and S8 in supporting information).
For the cytokine/chemokine factor, the posterior distribution suggested no association with change in ADNI memory and executive function and MMSE scores (Figure S7B).
3.6. Mediation analysis
In the DELCODE data, we found a consistent mediation effect of the microglia factor for the effect of AD pathology on rates of cognitive change for FCSRT free recall, PACC5, MMSE, and TMTB/A (Table S6 in supporting information). One example is shown in Figure S9 in supporting information. For all other factors and cognitive scores in the DELOCDE and ADNI samples, there was no mediation effect of the neuroinflammation factors on the effect of AD pathology on rates of cognitive change (Table S6).
4. DISCUSSION
We have determined latent factors of neuroinflammation based on an a priori assignment of individual inflammatory markers and characterized them in relation to AD pathology and longitudinal cognitive decline. We found high to very high fit of the factor models. This suggests that the derived models are consistent with the a priori assumptions about the assignments of the individual markers, but of course does not prove their correctness.
We found very strong to extreme evidence for an association of the synaptic integrity, microglia, and adhesion factors with AD pathology. In contrast, evidence was against an association of complement and cytokine/chemokine factors with AD pathology. In addition, the posterior distributions of the parameter estimates suggested that the synaptic, microglia, and adhesion factors, but not the complement and cytokine/chemokine factors, were associated with rates of cognitive change.
The association of the microglia factor with AD pathology parallels our previous observation that early innate immune responses of microglia seemed to be associated with tau pathology markers. 7 Of note, the microglia factor was mainly associated with AD pathology but not with clinical diagnosis, that is, stage of disease. This is also reflected in the association of the microglia factor with rates of cognitive decline but not with baseline cognitive performance. However, the association was in an a priori unexpected direction, that is, higher levels of microglia factor were associated with less cognitive decline. Studies suggest a negative impact of aberrant microglia activation on cognitive performance. 29 At the same time, a neuropathological study demonstrated higher microglial activation and TREM2 expression in brains of non‐demented people than demented people with AD neuropathology 30 suggesting that microglia activation may contribute to synaptic integrity and resilience. In a mediation model, microglia factor mediated the effect of AD pathology on rates of change. The mediation effect had the same direction as the bilateral associations, that is, higher AD pathology was associated with higher microglia factor, which in turn was associated with less cognitive decline. This would suggest that in the presence of higher pathology, microglia response serves as a protective factor. However, such a model would require independent replication as it implies an a priori unexpected direction of the effects.
Associations of the synaptic factor with disease stage, AD pathology, and rates of cognitive decline are consistent with previous single‐marker studies. 31 , 32 With an entirely different approach, fluorodeoxyglucose positron emission tomography (PET) as marker of neuronal metabolism and synaptic integrity showed strong association with AD pathology and rates of cognitive decline as well. 33 The absence of a mediation effect would suggest that the contributions of AD pathology and synaptic integrity to rates of cognitive decline are independent from each other. Because we lack previous evidence, however, the finding of absence of a mediation effect needs independent replication.
Adhesion as marker of cerebrovascular integrity has not yet widely been studied. One previous study reported an association of plasma markers of adhesion with AD pathology and cross‐sectional measures of cognition in MCI and AD cases. 34 Our study extends these findings toward a strong association of the adhesion factor in CSF with AD pathology. In addition, the adhesion factor had a strong contribution to rates of memory and executive function change on top of p‐tau and Aβ42 effects. These data encourage the further study of cerebrovascular endothelial dysfunction as potential driver of AD‐related cognitive decline and may also highlight a role of extravasation of peripheral immune cells and systemic inflammation that has been suggested in experimental preclinical AD models. 35 , 36 Indeed, a range of previous studies reported blood–brain barrier dysfunction in AD, reviewed in Kurz et al., 37 which may contribute to AD pathology through two related mechanisms: impaired clearance of Aβ and other toxic proteins and reduced protection against peripheral and central inflammatory response.
Interestingly, we found evidence for no association of the cytokine/chemokine and complement factors with AD pathology, and associations with rates of cognitive decline were not detectable in the DELCODE and the ADNI data. Chemokine levels were increased in AD and were related to the level of Aβ42 pathology in a previous study. 38 However, in these previous results, just one among several selected chemokine markers showed such an association with the majority of markers showing no effect. These heterogeneous findings may be related to the short half‐life of chemokines so that CSF levels may not always reflect tissue concentration. This underscores that the latent factor approach may lead to a more robust assessment with less propensity to false positive findings.
For a detailed discussion of the Bayesian framework of the analysis, we refer to the supporting information (pages 2 to 4).
A limitation and at the same time a strength of our study is the use of a priori assignment of markers to specific latent factors, such as synaptic integrity or microglial response. This was based on a review of the literature for each CSF marker. At the same time, a single marker typically is not only related to one single mechanism but contributes to several mechanisms of inflammation. Therefore, this a priori separation was to some extent arbitrary. At the same time the fit of the confirmatory factor models (including multiple factor models) was very high, supporting the a priori assignment of markers. In an alternative approach one could have used an unsupervised approach for creating composites of markers, for example with principal component analysis. If this approach is heuristically blind, that is, no specific domain of inflammation is being assigned to the resulting factors, such as in Clark et al., 19 one cannot draw conclusions on the differential involvement of various domains of the inflammatory response. Alternatively, one may assign specific inflammatory mechanisms to the resulting factors post hoc based on the loadings of the original variables on the new composites. However, this approach is at least as arbitrary as the a priori clustering and less likely to be reproducible across different cohorts because of the post hoc assignment. The data‐driven approach has the advantage that it can uncover unexpected clusters. However, these are difficult to interpret, and their validity depends heavily on replication in an independent cohort. Both cohorts, DELCODE and ADNI, include selected cases from memory clinic and research center contexts so that our results cannot be generalized to the general population of older people.
In summary, confirmatory factor analysis revealed strong associations of synaptic integrity, microglia response, and cerebrovascular endothelial dysfunction with AD pathology. Even when AD pathology and stage of disease were accounted for, these three inflammation‐related mechanisms contributed at least moderately to rates of cognitive decline on top of the effects of Aβ and p‐tau. The direction of effect for microglia, however, requires independent confirmation. Cytokine/chemokine and complement factors were robustly unrelated to AD pathology and rates of cognitive decline, suggesting a less important role of these particular innate components of the inflammatory response to AD pathology and course of disease. Future extensions of this approach can easily incorporate other inflammatory markers, such as translocator protein (TSPO) PET 39 imaging findings as microglia marker, or other domains of inflammatory response. Despite having > 250 cases in both cohorts, the number of cases did not allow for a meaningful substratification, for example, according to APOE genotype or amyloid positivity versus negativity, because a reliable estimate of latent factors requires a high enough number of cases. In the future, larger samples per cohort and better harmonized assessments of inflammation markers across cohorts may become available, allowing a more differentiated analysis of inflammatory domains. In addition, the associations of the adhesion factor, if replicated in another cohort, may point to an important link between inflammation and protein clearance from the brain. This indicates a promising research line that will deserve more in‐depth study once in vivo measures of brain clearance, less invasive than current methods, 40 become available.
CONFLICT OF INTEREST STATEMENT
S.J.T. participated in scientific advisory boards of Roche Pharma AG, Biogen, Grifols, and MSD, and received lecture fees from Roche and MSD. M.T.H. serves as a scientific board member of IFM Therapeutics, Novo Nordisk, and Alector and has received lecture honoraria from NovoNordisk. The other authors state no competing interests. Author disclosures are available in the supporting information.
Supporting information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The DELCODE study (Study‐ID: BN012) was supported and conducted by the Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE). The data samples were provided by the DELCODE study group. Details and participating sites can be found at www.dzne.de/en/research/studies/clinical‐studies/delcode. The DELCODE study was supported with respect to the MR imaging by Max Delbrück Center for Molecular Medicine in the Helmholtz Association (MDC), Berlin; Center for Cognitive Neuroscience Berlin (CCNB) at Freie Universität Berlin; Bernstein Center for Computational Neuroscience (BCCN), Berlin; Core Facility MR‐Research in Neurosciences, University Medical Center Goettingen; Institute for Clinical Radiology, Ludwig Maximilian University, Munich; Institute of Diagnostic and Interventional Radiology, Pediatric Radiology and Neuroradiology, Rostock University Medical Center; and Magnetic Resonance research center, University Hospital Tuebingen.
Part of the data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI; National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen Idec Inc.; Bristol‐Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
The work was supported by a grant to S.J.T. within the CureDem funding of the Federal Ministry of Research (BMBF), grant number 01KX2130, and additionally by a grant from the German Research Council (DFG), grant number 390873048 (Cluster of Excellence EXC 2151 “Immunosensation”) to M.T.H.
Open access funding enabled and organized by Projekt DEAL.
Teipel SJ, Dyrba M, Kleineidam L, et al. Association of latent factors of neuroinflammation with Alzheimer's disease pathology and longitudinal cognitive decline. Alzheimer's Dement. 2024;16:e12510. 10.1002/dad2.12510
DATA AVAILABILITY STATEMENT
Anonymized data used in this article will be made available by request from any qualified investigator through the DELCODE Steering Board for purposes of replicating procedures and results.
REFERENCES
- 1. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement (N Y). 2018;4:575‐590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Heneka MT, O'Banion MK. Inflammatory processes in Alzheimer's disease. J Neuroimmunol. 2007;184:69‐91. [DOI] [PubMed] [Google Scholar]
- 3. Calsolaro V, Edison P. Neuroinflammation in Alzheimer's disease: current evidence and future directions. Alzheimers Dement. 2016;12:719‐732. [DOI] [PubMed] [Google Scholar]
- 4. Cai Z, Hussain MD, Yan LJ. Microglia, neuroinflammation, and beta‐amyloid protein in Alzheimer's disease. Int J Neurosci. 2014;124:307‐321. [DOI] [PubMed] [Google Scholar]
- 5. Heneka MT, O'Banion MK, Terwel D, Kummer MP. Neuroinflammatory processes in Alzheimer's disease. J Neural Transm (Vienna). 2010;117:919‐947. [DOI] [PubMed] [Google Scholar]
- 6. Schuitemaker A, Dik MG, Veerhuis R, et al. Inflammatory markers in AD and MCI patients with different biomarker profiles. Neurobiol Aging. 2009;30:1885‐1889. [DOI] [PubMed] [Google Scholar]
- 7. Brosseron F, Maass A, Kleineidam L, et al. Soluble TAM receptors sAXL and sTyro3 predict structural and functional protection in Alzheimer's disease. Neuron. 2022;110:1009‐1022.e4. [DOI] [PubMed] [Google Scholar]
- 8. Ewers M, Biechele G, Suarez‐Calvet M, et al. Higher CSF sTREM2 and microglia activation are associated with slower rates of beta‐amyloid accumulation. EMBO Mol Med. 2020;12:e12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ma LZ, Tan L, Bi YL, et al. Dynamic changes of CSF sTREM2 in preclinical Alzheimer's disease: the CABLE study. Mol Neurodegener. 2020;15:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Knapskog AB, Henjum K, Idland AV, et al. Cerebrospinal fluid sTREM2 in Alzheimer's disease: comparisons between clinical presentation and AT classification. Sci Rep. 2020;10:15886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suarez‐Calvet M, Morenas‐Rodriguez E, Kleinberger G, et al. Early increase of CSF sTREM2 in Alzheimer's disease is associated with tau related‐neurodegeneration but not with amyloid‐beta pathology. Mol Neurodegener. 2019;14:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bollen KA. Structural Equations with Latent Variables. John Wiley & Sons, Inc.; 1989. [Google Scholar]
- 13. Jessen F, Spottke A, Boecker H, et al. Design and first baseline data of the DZNE multicenter observational study on predementia Alzheimer's disease (DELCODE). Alzheimers Res Ther. 2018;10:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weiner MW, Aisen PS. The Alzheimer's disease neuroimaging initiative: progress report and future plans. Alzheimers Dement. 2010;6:202‐211.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wagenmakers EJ, Marsman M, Jamil T, et al. Bayesian inference for psychology. Part I: theoretical advantages and practical ramifications. Psychon B Rev. 2018;25:35‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Windish DM, Huot SJ, Green ML. Medicine residents' understanding of the biostatistics and results in the medical literature. JAMA. 2007;298:1010‐1022. [DOI] [PubMed] [Google Scholar]
- 17. Goodman S. A dirty dozen: twelve P‐value misconceptions. Semin Hematol. 2008;45:135‐140. [DOI] [PubMed] [Google Scholar]
- 18. Temp AGM, Lutz MW, Trepel D, et al. How Bayesian statistics may help answer some of the controversial questions in clinical research on Alzheimer's disease. Alzheimers Dement. 2021;17:917‐919. [DOI] [PubMed] [Google Scholar]
- 19. Clark AL, Weigand AJ, Thomas KR, et al. Elevated inflammatory markers and arterial stiffening exacerbate tau but not amyloid pathology in older adults with mild cognitive impairment. J Alzheimers Dis. 2021;80:1451‐1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Folstein MF, Folstein SE, McHugh PR. Mini‐mental‐state: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189‐198. [DOI] [PubMed] [Google Scholar]
- 21. Papp KV, Rentz DM, Orlovsky I, Sperling RA, Mormino EC. Optimizing the preclinical Alzheimer's cognitive composite with semantic processing: the PACC5. Alzheimers Dement (N Y). 2017;3:668‐677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lemos R, Cunha C, Maroco J, Afonso A, Simoes MR, Santana I. Free and Cued Selective Reminding Test is superior to the Wechsler Memory Scale in discriminating mild cognitive impairment from Alzheimer's disease. Geriatr Gerontol Int. 2015;15:961‐968. [DOI] [PubMed] [Google Scholar]
- 23. Arbuthnott K, Frank J. Trail making test, part B as a measure of executive control: validation using a set‐switching paradigm. J Clin Exp Neuropsychol. 2000;22:518‐528. [DOI] [PubMed] [Google Scholar]
- 24. Crane PK, Carle A, Gibbons LE, et al. Development and assessment of a composite score for memory in the Alzheimer's Disease Neuroimaging Initiative (ADNI). Brain Imaging Behav. 2012;6:502‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gibbons LE, Carle AC, Mackin RS, et al. A composite score for executive functioning, validated in Alzheimer's Disease Neuroimaging Initiative (ADNI) participants with baseline mild cognitive impairment. Brain Imaging Behav. 2012;6:517‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brosseron F, Traschutz A, Widmann CN, et al. Characterization and clinical use of inflammatory cerebrospinal fluid protein markers in Alzheimer's disease. Alzheimers Res Ther. 2018;10:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brosseron F, Kolbe CC, Santarelli F, et al. Multicenter Alzheimer's and Parkinson's disease immune biomarker verification study. Alzheimers Dement. 2020;16:292‐304. [DOI] [PubMed] [Google Scholar]
- 28. Kruschke JK. Bayesian Analysis Reporting Guidelines. Nat Hum Behav. 2021;5:1282‐1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Duggan MR, Parikh V. Microglia and modifiable life factors: potential contributions to cognitive resilience in aging. Behav Brain Res. 2021;405:113207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fracassi A, Marcatti M, Tumurbaatar B, Woltjer R, Moreno S, Taglialatela G. TREM2‐induced activation of microglia contributes to synaptic integrity in cognitively intact aged individuals with Alzheimer's neuropathology. Brain Pathol. 2023;33:e13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kvartsberg H, Duits FH, Ingelsson M, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer's disease. Alzheimers Dement. 2015;11:1180‐1190. [DOI] [PubMed] [Google Scholar]
- 32. Tarawneh R, D'Angelo G, Crimmins D, et al. Diagnostic and prognostic utility of the synaptic marker neurogranin in Alzheimer disease. JAMA Neurol. 2016;73:561‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kato T, Inui Y, Nakamura A, Ito K. Brain fluorodeoxyglucose (FDG) PET in dementia. Ageing Res Rev. 2016;30:73‐84. [DOI] [PubMed] [Google Scholar]
- 34. Drake JD, Chambers AB, Ott BR, Daiello LA. Alzheimer's Disease Neuroimaging I. Peripheral markers of vascular endothelial dysfunction show independent but additive relationships with brain‐based biomarkers in association with functional impairment in Alzheimer's disease. J Alzheimers Dis. 2021;80:1553‐1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zenaro E, Pietronigro E, Della Bianca V, et al. Neutrophils promote Alzheimer's disease‐like pathology and cognitive decline via LFA‐1 integrin. Nat Med. 2015;21:880‐886. [DOI] [PubMed] [Google Scholar]
- 36. Tejera D, Mercan D, Sanchez‐Caro JM, et al. Systemic inflammation impairs microglial Abeta clearance through NLRP3 inflammasome. EMBO J. 2019;38:e101064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kurz C, Walker L, Rauchmann BS, Perneczky R. Dysfunction of the blood‐brain barrier in Alzheimer's disease: evidence from human studies. Neuropathol Appl Neurobiol. 2022;48:e12782. [DOI] [PubMed] [Google Scholar]
- 38. Correa JD, Starling D, Teixeira AL, Caramelli P, Silva TA. Chemokines in CSF of Alzheimer's disease patients. Arq Neuropsiquiatr. 2011;69:455‐459. [DOI] [PubMed] [Google Scholar]
- 39. Gouilly D, Saint‐Aubert L, Ribeiro MJ, et al. Neuroinflammation PET imaging of the translocator protein (TSPO) in Alzheimer's disease: an update. The Eur J Neurosci. 2022;55(5):1322‐1343. [DOI] [PubMed] [Google Scholar]
- 40. Ringstad G, Valnes LM, Dale AM, et al. Brain‐wide glymphatic enhancement and clearance in humans assessed with MRI. JCI Insight. 2018;3:e121537. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
Anonymized data used in this article will be made available by request from any qualified investigator through the DELCODE Steering Board for purposes of replicating procedures and results.