

Abstract
Rare genetic variations contribute to the heterogeneity of autism spectrum disorder (ASD) and the responses to various interventions for ASD probands. However, the associated molecular underpinnings remain unclear. Herein, we estimated the association between rare genetic variations in 410 vitamin A (VA)-related genes (VARGs) and ASD aetiology using publicly available de novo mutations (DNMs), rare inherited variants, and copy number variations (CNVs) from about 50,000 ASD probands and 20,000 normal controls (discovery and validation cohorts). Additionally, given the functional relevance of VA and oxytocin, we systematically compared the similarities and differences between VA and oxytocin with respect to ASD aetiology and evaluated their potential for clinical applications. Functional DNMs and pathogenic CNVs in VARGs contributed to ASD pathogenesis in the discovery and validation cohorts. Additionally, 324 potential VA-related biomarkers were identified, 243 of which were shared with previously identified oxytocin-related biomarkers, while 81 were unique VA biomarkers. Moreover, multivariable logistic regression analysis revealed that both VA- and oxytocin-related biomarkers were able to predict ASD aetiology for individuals carrying functional DNM in corresponding biomarkers with an average precision of 0.94. As well as, convergent and divergent functions were also identified between VA- and oxytocin-related biomarkers. The findings of this study provide a basis for future studies aimed at understanding the pathophysiological mechanisms underlying ASD while also defining a set of potential molecular biomarkers for adjuvant diagnosis and intervention in ASD.
Keywords: Autism spectrum disorder, Rare genetic variants, Vitamin A, Oxytocin, Biomarker, Combined model
Graphical Abstract
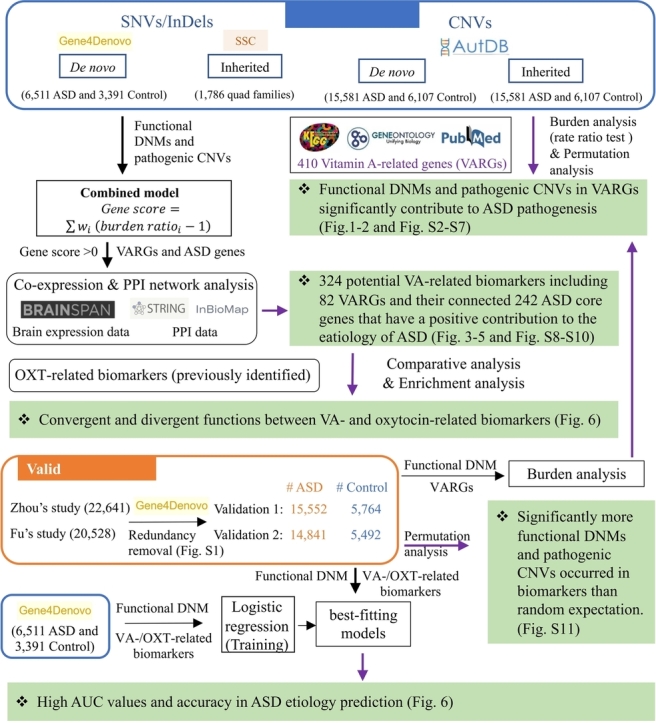
Highlights
-
•
Multilevel genomic ASD data were integrated to investigate whether VARG is associated with ASD pathogenesis.
-
•
Functional DNMs and pathogenic CNVs in VARGs play a major role in ASD pathogenesis, with strong supporting evidence.
-
•
Using a combined model, 324 potential VA-related biomarkers were identified in ∼8 % of patients with ASD.
-
•
VA- and oxytocin-related biomarkers show convergent and divergent functions.
-
•
VA- & oxytocin-related biomarkers strongly predict ASD etiology in individuals with functional DNMs in corresponding biomarkers.
1. Introduction
Autism spectrum disorder (ASD) is a common and highly heritable neurodevelopmental disorder characterised by impaired social interactions, restricted interests, and repetitive behaviours. According to the report of the U.S. Centers for Disease Control and Prevention in 2021, ∼2.27 % of children aged eight years were diagnosed with ASD, with boys presenting a 4.2-fold higher risk of ASD than girls [1]. Although genetic and environmental factors contributing to ASD pathology have been widely investigated [2], [3], effective treatment options are lacking.
Vitamin A (VA) is an essential lipid-soluble micronutrient, converted to retinol in human intestinal epithelial cells [4]. Retinol is then transported to the liver for storage, or delivered to certain tissues, such as the brain, where it is converted to retinoic acid (RA) to play a regulatory role by binding to nuclear receptors [5], [6]. It has been well-documented that RA is crucial for cortical neuron generation, synaptic function, and cognitive function [7], [8], [9], [10]. Furthermore, variations in certain RA-related genes have been revealed to contribute to ASD aetiology. For instance, common variants in RA-related orphan receptor (ROR) [11], and de novo mutations (DNMs) in RA induced 1 (RAI1) [12] and arginine-glutamic acid dipeptide repeats (RERE) [13], are associated with ASD aetiology. In addition, ubiquitin protein ligase E3A (UBE3A), an ASD risk gene mainly affected by copy number variations (CNVs), reportedly impairs synaptic plasticity in ASD by negatively regulating aldehyde dehydrogenase 1 family member A2 (ALDH1A2), a rate-limiting enzyme of RA synthesis [14]. Given that RA is the main active derivative of VA, we speculated that abnormalities in VA and its downstream RA system might be associated with ASD pathogenesis [15].
In China, ∼0.7 % of children aged 6–12 years suffer from ASD [16]. Patients with ASD in China have an overall lower VA level than typical controls, especially in boys with ASD [17], [18], [19], [20], [21], [22], [23]. Negative correlations between serum VA concentration in patients with ASD and the corresponding symptom severity were identified using the Childhood Autism Rating Scale, Autism Behaviour Checklist, and Social Responsiveness Scale [19], [21], [24]. Notably, VA supplementation ameliorates ASD symptoms, at least in a subset of patients [25], [26], [27]. Therefore, VA may have a potential role in ASD pathogenesis and may affect the clinical responses of patients with ASD with vitamin A deficiency (VAD) to VA intervention.
DNMs and rare inherited variants (RIVs) are important risk factors for ASD aetiology, especially for the disruptive variants in coding regions [28], [29]. In addition, CNVs have been strongly linked to ASD aetiology [30], [31] and occur in ∼4 % of patients with ASD [31]. Our previous studies identified several ASD genes [32], [33] and demonstrated the contribution of rare variations to ASD pathology in vitamin D-related genes [34], oxytocin-related genes (OTRGs) [35], and brain-size-related genes [36]. We also revealed the divergence and convergence in three ASD subcategories based on DNMs [37]. However, the complex relationships between rare variations in VARGs and ASD pathogenicity remain largely unexplored.
Since VAD may reduce brain oxytocin release by regulating CD38 expression, thereby affecting autistic beheaviour [38], [39], we speculated that there might be convergent and divergent molecular networks regulating the effects of VA and oxytocin intervention on ASD symptoms. To evaluate whether VA contributes to ASD aetiology, we compared the burden differences of rare variations in VARGs between ASD probands and controls, with respect to patient sex and non-verbal intelligence quotient (NVIQ). Additionally, we systematically compared the genetic similarities and differences between VA and oxytocin to ASD aetiology at the molecular levels. This study aimed to provide new insights into the pathophysiological mechanisms of ASD to help future clinical investigations on VA-, oxytocin-, or combination-based ASD treatments.
2. Materials and methods
2.1. VARG curation
In total, 410 VARGs (Table S1) were manually curated from three sources: 68 retinol metabolic (https://www.genome.jp/dbget-bin/www_bget?pathway:map00830) and 9 vitamin digestion and absorption pathway genes (https://www.genome.jp/dbget-bin/www_bget?pathway:map04977) from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [40], 223 genes from 19 VA-related functions in the Gene Ontology (GO) database [41], and 181 genes from 22 published studies searched in the PubMed database from January 2009 up to December 2018, including genes involved in VA digestion and absorption, retinol metabolism, and those significantly differentially expressed after VA intervention. Of the 22 studies searched, ten were clinic-based studies with sample sizes ranging from 8 to 171, while twelve were conducted using cell-line and mouse models.
2.2. Data collection and processing
DNMs (de novo single nucleotide variations [SNVs] and Insertion/Deletion [InDels]) from 6,511 patients with ASD and 3,391 controls were collected from the Gene4Denovo database [42]. The associated clinical information (sex and NVIQs) was retrieved from corresponding papers, if available (Table S2). Variant annotation was performed as described in our previous studies [43], [44]. Deleterious missense variants (Dmis) were predicted by ReVe [45] using scores>0.7. Dmis and loss-of-function (LoF) mutations were defined as functional mutations. Only coding variants with minor-allele frequencies<0.1 % were used for the downstream analysis. Inherited variants, sex and NVIQ information from 1,786 quad families (both parents unaffected, an affected child, and an unaffected sibling) were retrieved from Simons Simplex Collection (Table S2) [29]. The same variant annotation and filtration methods used for DNMs were applied to inherited variants. The remaining variants were considered as RIVs and were used for downstream analysis.
De novo CNVs (dnCNVs) and inherited CNVs (ihCNVs) from 15,581 patients with ASD and 6,107 controls were collated from the AutDB database [46] (Table S2). All these CNVs had a frequency below 0.5 %, and were therefore classified as rare CNVs. CNV annotation and pathogenicity prediction were performed as described in our previous study [35]. CNVs of different lengths may contain different numbers of VARGs in patients with ASD and controls. Therefore, the normalised CNV abundance for VARGs, defined by the scaled fraction of the total shared gene length between VARGs and CNVs in patients with ASD or controls to the total gene length in VARGs, was used for further CNV burden analysis with a scale factor of 1,000 base pairs.
2.3. Burden analysis and gene prioritisation
Burden analysis was performed between patients with ASD and controls using DNMs, RIVs, dnCNVs, and ihCNVs in VARGs. A similar burden analysis was also performed between subgroups of patients with ASD and controls stratified by sex or NVIQs.
To comprehensively evaluate the contribution of each gene to ASD aetiology, a combined model integrating DNMs, dnCNVs and ihCNVs was used for gene prioritisation, as described in our previous study [35]. The equation is summarized as follows:
where i is one of the three types of rare disruptive variations, and bri and represent the burden ratio and the odds ratio (weight) for the corresponding rare disruptive variation between patients with ASD and controls, respectively. Burden ratio refers to the ratio of the mean count of disruptive variations in the genes of interest between the ASD and control groups. Odds ratio refers to the ratio of individuals affected by disruptive variations in all reference genes between the ASD and control groups. Finally, the contribution scores were calculated using the odds ratios estimated in our study (O-Gene scores) and the odds ratios calculated by Krumm et al. (K-Gene scores) [47]. These scores were used for gene prioritisation. Genes with a score>0 were considered to positively contribute to ASD aetiology.
2.4. Collection of publicly available data used for integration analysis and validation
Observed/expected (oe) metrics reflecting the tolerance of a given gene to LoF variants were downloaded from the Genome Aggregation Database (GnomAD database v2.1) [48]. ASD genes were gathered from the following sources: (1) ASD candidates curated in our previous study [49]; (2) genes retrieved from the SFARI database (https://gene.sfari.org/) [50]; and (3) genes collected from the AutDB database [46]. Additional cohorts with publicly available DNM data from 22,641 individuals in the study of Zhou et al. (validation 1) [51] and 20,528 individuals in the study of Fu et al. (validation 2) [52] were collected. The individuals with redundancies who were part of the original DNM cohort were removed, retaining a total of 21,316 individuals (nASD = 15,552 and ncontrol = 5,764) in validation 1 and 20,333 individuals (nASD = 14,841 and ncontrol = 5,492) in validation 2 (Fig. S1). The DNMs obtained from these additional cohorts were processed using the same criteria that were applied to our original DNM cohort.
2.5. Network construction
Gene expression data from the human brain were retrieved from BrainSpan (http://www.brainspan.org/). Protein–protein interactions (PPIs) were downloaded from STRING (https://string-db.org/) [53] and InWeb_IM50 [54]. Gene pairs co-expressed with |R|>0.8 in the brain data [55], or PPIs with score> 400 (STRING data) [55] or score>0.8 (InWeb_IM50) [56] were considered as connected and used for downstream analysis. All network figures were produced using Cytoscape v.3.8.0 (https://cytoscape.org) [57].
2.6. Permutation test
Functional network connectivity was evaluated with a permutation test. In 10,000 randomly simulated networks, we compared the number of genes and the number of connections within PC-VARGs, between PC-VARGs and known ASD genes, between potential biomarkers associated with VA only, and those associated only with oxytocin. P values were determined based on the position of the actual value in the 10,000 randomly simulated datasets.
2.7. Enrichment analysis
GO enrichment analyses for target gene sets were performed using clusterProfiler [58] (version 3.14.3) with default parameters in R (https://www.r-project.org), which employed the false discovery rate (FDR) method to correct the P-values for all GO terms. GO terms with p.adjust (FDR)< 0.01 and a gene count of at least three were defined as significantly enriched.
2.8. Statistical analysis
A one-sided rate ratio (RR) test using the ‘rateratio.test’ function in R (version 1.0–2) (https://CRAN.R-project.org/package=rateratio.test) [59] was used to estimate the burden difference in VARGs between ASD probands and the corresponding controls. The ‘p.adjust’ function employed the FDR method to calculate the corrected P-values for multiple comparisons. Significant differences were defined for all comparisons with p.adjust (FDR)<0.05.
The diagnostic accuracy of our identified potential biomarkers was assessed via receiver operating characteristic (ROC) analysis with constructed logistic regression models. All affected individuals (ASDs or controls) who carried functional DNMs in potential biomarkers and sample size-matched case/controls from the original discovery cohorts were used for model construction. In our constructed logistic regression model, the dependent variable was ASD status, with patients with ASD assigned a value of 1 (y = 1) and controls assigned a value of 0 (y = 0). The independent variable was the status of an individual carrying a functional DNM (status = 1) or not carrying a functional DNM (status = 0) in each set of potential biomarkers (VA- or oxytocin-related biomarkers). To account for potential covariance on model effectiveness, we considered all available covariates from the collected data (sex and NVIQ) in our logistic regression models. Due to the availability of sex data for both individuals with ASD and control individuals, sex was retained for model construction. We then performed internal validation using the bootstrap method with 2,000 iterations to construct two fitting models for each set of potential biomarkers, one adjusting for sex and the other not adjusting for sex. For the model fitted with adjustment for sex, we employed the variance inflation factor (VIF) to estimate the regression coefficients due to the correlation between the sex predictor and the individual’s DNM status predictor. VIF values ≥5 indicate a high correlation and possible multicollinearity. The results showed that sex significantly contributed to the model performance (P < 0.05) and had a low correlation with the individual’s DNM status (VIF<1.6). To obtain the best-fitting models, we used the Akaike Information Criterion (AIC) as a criterion for the goodness-of-fit of each logistic regression model, where a lower AIC value indicates a better fit of the model to the data. The results showed that the models adjusting for an individual’s sex had lower AIC values, suggesting that they were the best-fitting models. Furthermore, all affected individuals by functional DNM in potential biomarkers and sample size-matched case/controls from two validation cohorts were used to estimate the performance of the best-fitting models we constructed. Finally, ROC curves were produced using the ‘roc’ function in the pROC package.
3. Results
3.1. DNM in VARG contributes to ASD susceptibility
A total of 8,175 and 3,629 coding DNMs were collected from 6,511 ASD probands and 3,391 controls, respectively (Table S2). The DNM burdens in 410 manually curated VARGs (Table S1) were then compared between ASD probands and controls in all coding DNMs and six DNM subtypes, namely, LoF mutations, predicted deleterious missense mutations, predicted tolerant missense mutations, synonymous SNVs, non-frameshift InDels, and functional mutations (Table S3). The coding DNMs were significantly more enriched in patients with ASD compared to the controls (0.029 vs. 0.017 variants per subject [vps]; RR test: RR = 1.65, FDR = 2.24 × 10−3; Fig. 1a). Notably, this significant enrichment increased to 1.80-fold in the functional DNMs (0.012 vs. 0.006 vps; RR test: FDR = 2.19 × 10−2) and to 3.39-fold in the LoF DNMs (0.004 vs. 0.001 vps; RR test: FDR = 2.19 × 10−2; Fig. 1a). Compared to the controls, significantly higher burdens of functional and LoF DNMs were detected in patients with ASD in two validation DNM cohorts (Fig. 1b). Additionally, 10,000 negative control gene sets were randomly generated, with each gene set containing 410 protein-coding genes to compare with the VARG set. Functional DNMs carried by patients with ASD were more prevalent in the VARG set compared with the negative control (Permutation test: P = 5.71 × 10−3; Fig. 1c), which was supported by two validation cohorts (Fig. 1d, e). Considering the association between DNM frequency and paternal age [60], [61], we compared the DNM burden in VARGs between ASD probands and matched unaffected siblings after adjusting for the father’s age at child conception using a linear regression model. Similarly, a significantly higher number of both functional DNMs (RR test: RR = 2.27, FDR = 2.16 × 10−2) and LoF DNMs (0.005 vs. 0 vps; RR test: RR = Inf, FDR = 5.86 × 10−3) were dectected in patients with ASD compared to the unaffected siblings (Fig. S2), supporting the findings in the original discovery cohort.
Fig. 1.

Mutational burden differences in VARGs between patients with ASD and controls. (a) Differences in DNM frequency (burden) between patients with ASD (n = 6,511) and controls (n = 3,391) for each mutation type in VARGs. (b) Differences in DNM frequency (burden) between ASD probands and controls for functional and LoF DNMs in VARGs based on the data from the study of Zhou et al. (nASD = 15,552 and ncontrol = 5,764) and that of Fu et al. (nASD = 14,841 and ncontrol = 5,492). (c–e) Permutation figures for functional DNM count in ASD probands between the VARG set and the negative control sets (10,000 permutations) for the discovery DNM cohort (c), and two validation cohorts (d, e). (f) Differences in RIV frequency (burden) between patients with ASD (n = 1,786) and controls (n = 1,786) for each mutation type in VARGs. DNM, de novo mutation; RIV, rare inherited variants. VARGs, vitamin A-related genes; LoF, loss-of-function; P values were calculated by the rate ratio test and corrected by the function of “p.adjust” with FDR method. *FDR< 0.05; * *FDR< 0.01; * **FDR< 0.001.
Considering that ASD has a strong male bias and results in various levels of intellectual disability [1], participants were further stratified into subgroups based on sex and NVIQ to compare the differences in VARG DNM burden between male and female ASD subgroups, and between each ASD subgroup and the corresponding control group. A 2.82-fold enrichment of coding DNMs in VARGs was observed in females with ASD compared with female controls (0.039 vs. 0.014 vps; RR test: FDR = 5.06 × 10−3; Fig. S3a). A significantly higher burden was also observed in males with ASD compared to male controls (RR test: RR = 1.72, FDR = 3.50 × 10−2; Fig. S3a). These results are consistent with our previous findings in OTRGs [35]. However, no burden difference was observed between females and males with ASD (Fig. S3a). Additionally, a significantly higher DNM burden was observed in the VARGs of patients with ASD and moderate intellectual disability (NVIQ, 50–80) in all coding DNMs (0.036 vs. 0.017 vps; RR test: RR = 2.05, FDR = 2.21 × 10−2) and in LoF DNMs (0.008 vs. 0.001 vps; RR test: RR = 6.87, FDR = 2.21 × 10−2; Fig. S3b). Compared with controls, patients with ASD and severe (NVIQ<50) or mild (NVIQ>80) intellectual disability exhibited 1.90-fold and 1.55-fold higher DNM burdens, respectively; however, this result was insignificant (Fig. S3b). These results suggest that ASD probands have a significantly higher DNM burden in VARGs than controls, particularly for functional and LoF DNMs. Additionally, the DNM burden in VARGs is particularly biased toward female patients and those with a relatively lower NVIQ. Similar findings were observed in OTRGs [35].
To explore the RIV burden differences in VARGs between patients with ASD and controls, 94,418 and 93,895 coding RIVs from ASD probands and unaffected siblings were catalogued, respectively, from 1786 quad families (Table S2). The coding RIVs in VARGs were then retained for mutation burden analysis (Table S4). No significant differences were observed between ASD probands and unaffected siblings (Fig. 1f), even when the RIVs were stratified by different inheritance patterns (Fig. S4a, S4b), sex, or NVIQ (Fig. S4c). These results suggest that VARG RIVs do not contribute significantly contribution to ASD aetiology, similar to our previous findings in OTRGs [35].
3.2. VARG-associated CNVs are significantly associated with ASD aetiology
Owing to the significant contribution made by CNVs to ASD aetiology [30], [31], we investigated whether VARG-associated CNVs contribute to ASD pathogenesis. To this end, 16,902 CNVs, comprising 697 dnCNVs and 16,205 ihCNVs, were collected from 15,581 patients with ASD, and 11,921 CNVs, comprising 212 dnCNVs and 11,709 ihCNVs, were collected from 6,107 controls (Table S2). Only CNVs associated with VARGs were used for burden analysis (Table S5). Since a specific CNV may contain several VARGs, potential bias may be directly derived from the VARG-associated CNVs for burden analysis. To reduce this bias, normalised CNV abundances in VARGs were applied for burden analysis (see Methods) in all CNVs and five CNV subtypes, namely ‘deletion,’ ‘duplication,’ ‘pathogenic,’ ‘likely pathogenic,’ and ‘uncertain significance’ (Table S6). ‘Benign’ and ‘likely benign’ CNVs were excluded from burden analysis due to their relatively low contribution to ASD aetiology and because they are rarely annotated with VARGs.
Compared with the controls, patients with ASD exhibited significant dnCNV enrichments in all VARG CNVs (0.020 vs. 0.004 normalised CNVs per subject [ncps]; RR test: RR = 5.36, FDR = 2.53 × 10−22; Fig. 2a). Significant enrichment was observed for most CNV subtypes, particularly for the top ranked ‘pathogenic’ dnCNVs, which showed a 9.65-fold enrichment in patients with ASD compared to the controls (0.01 vs. 0.001 ncps; RR test: FDR = 1.51 × 10−14; Fig. 2a). In patients with ASD, dnCNVs occurred more frequently in the VARG set than that in negative control genes (Permutation test with 10,000 iterations: P = 2.78 × 10−2; Fig. 2b). Additionally, analysis of a subset of the CNV data from same source (quad families) revealed similar results in all CNVs from different platforms by comparing the dnCNV burden between the ASD proband and their sibling controls (RR test: FDR = 1.48 × 10−22 for microarrays; FDR = 3.62 × 10−8 for WES; FDR = 3.36 × 10−18 for WGS; Fig. S5a, S5b, S5c), even after adjusting for paternal age at child conception using linear regression (RR test: FDR = 1.48 × 10−22; Fig. S5d). However, insignificant differences in the ihCNV burden of VARGs were observed between patients with ASD and controls, except for a modest yet significant, difference in ‘pathogenic’ ihCNVs (0.002 vs. 0.0002 ncps; RR test: RR = 13.13, FDR = 9.48 × 10−4; Fig. 2c). Similarly, no significant difference in the ihCNV burden of VARGs was observed between ASD probands and their sibling controls using data on quad families from the same source (Fig. S6). Finally, all ihCNVs in VARGs were stratified based on inheritance patterns to compare the differences in burden between patients with ASD and controls. No significant differences in ihCNV burden were observed (Fig. S7).
Fig. 2.

CNV burden differences in VARGs between patients with ASD and controls. (a) Differences in normalized dnCNV abundance (burden) between patients with ASD (n = 15,581) and controls (n = 6,017) for each CNV type in VARGs. (b) Permutation figure of normalized pathogenic dnCNV count in ASD probands between the VARG set and negative control set (10,000 permutations). (c) Differences in normalized ihCNVs abundance (burden between patients with ASD (n = 15,581) and controls (n = 6,017) for each CNV type in VARGs. (d) Forest plots for normalized dnCNV burden differences between female patients with ASD (nfemale = 757) and male patients with ASD (nmale = 3,764), between sex-stratified patients with ASD (nfemale = 757; nmale = 3,764) and controls (nfemale = 874; nmale = 750). (e) Forest plots for normalized dnCNV burden differences between NVIQ-stratified patients with ASD (nNVIQ ≤ 50 = 101, n50<NVIQ ≤ 80 = 273, nNVIQ>80 = 750) and controls (n = 6,017) for each CNV type in VARGs. Rate ratio is defined by the CNV rate (number per subject) in patients with ASD divided by that in controls. NVIQ, non-verbal intelligence quotient. CNV, copy number variation; dnCNV, de novo CNV; ihCNVs, inherited CNV; P values were calculated by the rate ratio test and corrected by the function of “p.adjust” with FDR method. *FDR< 0.05; * *FDR< 0.01; * **FDR< 0.001.
To determine whether the sex and NVIQ bias observed in DNMs (Fig. S3) was also present in the CNV data, CNV burden analysis was performed between patients with ASD and controls within the same sex, between male and female patients with ASD, and between ASD probands with different NVIQs (≤ 50, 50–80, and>80) and controls. Compared with same-sex controls, all dnCNVs in VARGs exhibited a 10.36-fold enrichment in males (0.014 vs. 0.001 ncps; RR test: FDR = 2.28 × 10−3; Fig. 2d) and a 1.74-fold enrichment in females (0.021 vs. 0.001 ncps; RR test: FDR = 2.66 × 10−4; Fig. 2d) with ASD. Additionally, female patients with ASD exhibited significant VARG enrichment in almost dnCNV subtypes compared with female controls, which were rarely observed in male patients with ASD compared with male controls (Fig. 2d). Insignificant differences in VARG CNV burden were observed between male and female patients with ASD for all dnCNV subtypes. Furthermore, CNV burden analysis of NVIQ bias revealed that patients with relatively lower NVIQ had a relatively higher dnCNV burden in VARGs compared to the controls (Fig. 2e). Hence, similar to our previous OTRG findings, the VARG-related CNV burden is significantly higher in patients with ASD compared with controls, particularly for pathogenic CNVs.
3.3. VARG prioritisation and relationships with known ASD risk factors
By employing the gene prioritisation system, a significant positive correlation between the O-Gene and K-Gene scores for VARGs was observed (Pearson correlation, R = 0.80, P < 2.20 × 10−16; Fig. S8), indicating that the estimated weight was reliable (Methods). Based on a score>0, 82 VARGs were found to positively contribute to ASD pathology (PC-VARGs) (Fig. 3a). The top three genes were UBE3A, MAPK3, and PPP4C (Fig. 3b and Table S7).
Fig. 3.

Relationship between PC-VARGs and ASD risk. (a) Workflow for PC-VARG identification. PC-VARG, VARG with a score> 0. (b) Gene score distribution of PC-VARGs. (c) Differences in the oe score distribution between PC-VARGs and each of the other five groups including VARGs, non-PC-VARGs, ASD genes, ASD core genes, and ASD associated genes. Wilcoxon test was used to compare the differences between any two groups. (d) Enrichment of PC-VARGs (n = 82) or non-PC-VARGs (n = 328) in known ASD core genes (n = 559) and (e) DEGs (n = 7,101) identified from the ASD and control cortex. DEGs, differentially expressed genes. P values were calculated by hypergeometric test and corrected by the function of “p.adjust” with FDR method. *FDR< 0.05; * *FDR< 0.01; * **FDR< 0.001.
To investigate the functional impact of these PC-VARGs, gene-level tolerance to LoF variants represented by the observed/expected (oe) score, was used for comparative analysis among VARGs and ASD genes. Genes with low oe values are indicative of a strong intolerance to LoF variants, meaning those genes have a high probability of leading to pathogenesis. Specifically, the VARGs and ASD genes were classified into six gene sets including: (1) all 410 VARGs; (2) 82 PC-VARGs; (3) 328 non-PC-VARGs; (4) 1,229 known ASD genes identified in our previous ASD study [55]; (5) 559 ASD core genes defined in our previous ASD study (ASD core) [55]; and (6) 670 ASD-associated genes excluding the ASD core genes (ASD-associated). As expected, PC-VARGs exhibited significantly lower oe scores than VARGs (P = 0.026) and non-PC-VARGs (P = 0.006, Wilcoxon test; Fig. 3c), indicating stronger intolerance to LoF variants in PC-VARGs. Notably, PC-VARGs showed a stronger intolerance to LoF variants when compared to ASD-associated genes (P = 0.018) and a weaker intolerance to LoF variants compared to ASD core genes (P = 0.005, Wilcoxon test; Fig. 3c). This suggests that dysfunctions in PC-VARGs have an overall higher probability of leading to ASD pathogenesis in comparison to ASD-associated genes, however the overall probability is lower when compared to ASD core genes.
Next, PC-VARG enrichment was estimated in both ASD core genes and differentially expressed genes (DEGs) between ASD and control cortex samples. For comparison, non-PC-VARGs were also used in this analysis. ASD core genes exhibited significant overlap with PC-VARGs (P = 4.72 × 10−7), not with non-PC-VARGs (P = 0.98, hypergeometric test; Fig. 3d). In addition, DEGs were more significantly overlapped with PC-VARGs (P = 6.1 × 10−3) than non-PC-VARGs (P = 4.0 × 10−2, hypergeometric test; Fig. 3e). Together, these comparisons demonstrated that PC-VARGs are significantly associated with known ASD risk factors.
3.4. PC-VARGs are functionally associated with ASD aetiology
To further explore how PC-VARGs contribute to ASD aetiology, PPI and co-expression networks were constructed with these genes. Their interconnectivities were then evaluated using a permutation test with 10,000 iterations. The constructed PPI network contained significantly more genes (61/82 PC-VARGs; Permutation test: P < 1 × 10−4) and more internal connections (n = 121; Permutation test: P < 1 × 10−4; Fig. 4a) than those in a randomly permuted network. Similarly, the constructed co-expression network contained significantly more genes (20/82 PC-VARGs; Permutation test: P = 0.02) and more internal connections (n = 28; Permutation test: P = 0.01; Fig. 4b) than a randomly permuted network. These findings demonstrate the functional relevance of PC-VARGs.
Fig. 4.

PC-VARGs participate in known ASD-related pathways. (a) Interconnectedness of PC-VARGs based on gene co-expression and (b) PPI data are shown. The histograms present the distribution of genes and connections of 10,000 iterations, and the vertical black lines represented the number of observed genes and connections. P values and the observed numbers are shown in each of the figures. (c) PC-VARGs are significantly enriched in four major GO functional groups. (d) PC-VARGs enriched in the three major functional groups (including steroid hormone related function, transcription and chromatin and neuron development) are shown in the heatmap. Genes involved in vitamin A-related pathways and those that carry recurrent variants in the ASD cohort are also labeled.
Subsequently, the clusterProfiler [58] software was employed to annotate the 82 PC-VARGs using gene ontology (GO) terms (Table S8). The PC-VARGs were highly enriched in VA-related functions with 38 genes (Fig. 4c). In addition, three ASD-related functions exhibited enrichment, including functions related to steroid hormones [62], transcription and chromatin, and neuron development (Fig. 4c) [63]. These ASD-related functions were associated with ∼56.10 % (46/82) of the PC-VARGs. Notably, 25/46 ASD-related PC-VARGs also participated in VA-related functions (Fig. 4d). Specifically, seven genes (PNPLA4, AWAT2, AKR1B15, CYP26B1, CYP27C1, DGAT1, and PPARD) were involved in the ‘Retinoid metabolic process’, while three (CNOT1, CYP26B1, and RARA) were involved in ‘Retinoic acid receptor signalling pathway’ (Fig. 4d). Furthermore, connections (co-expression and/or PPI) between PC-VARGs and ASD core genes were significantly higher than in randomly constructed networks based on 10,000 iterations (Fig. S9). These findings suggest that PC-VARGs may contribute to ASD aetiology via direct involvement in VA-/ASD-related functions or by indirectly regulating known ASD genes.
3.5. VA-related molecular biomarkers contribute to ASD aetiology
Given the observed large number of connections between PC-VARGs and ASD core genes, 82 PC-VARGs and their connected 242 PC-ASD core genes were considered as VA-related biomarkers (324 biomarkers). Among these, 72 of the 82 PC-VARGs and 239 of their connected PC-ASD core genes were present in the PPI and/or brain co-expression data. Functional networks were constructed based on the 72 PC-VARGs and the 239 PC-ASD core genes (Fig. 5a). Approximately 59.72 % (43/72) of the PC-VARGs and 54.81 % (131/239) of their connected ASD core genes were involved in known ASD-related networks, such as transcriptional regulation and chromatin organization [63], neuronal development [63], steroid hormone-related function [62], [64], [65], and synaptic function [63]. Additionally, 26.39 % (19/72) of PC-VARGs and 15.90 % (38/239) of their connected ASD core genes participated in multiple ASD-related networks (Fig. 5a, Table S10). Based on the connections observed in the functional networks, the top 10 PC-VARGs directly involved in ASD-related networks and connected to at least 30 ASD core genes, were identified including NSD1, CTNNB1, CNOT1, UBE3A, MAPK3, DNMT3A, ATM, PIK3CA, NCOA6, and SMARCA5 (Table S9).
Fig. 5.

Functional network of potential VA-related biomarkers. (a) Connection among 324 potential VA-related biomarkers based on protein-protein interaction (PPI) and co-expression data in the human brain. Potential biomarkers implicated in these four functional networks are displayed. The protein-protein interactions are indicated with solid lines; gene co-expression connections are shown as dash lines and the connection by both protein-protein interaction and co-expression are indicated with parallel lines. (b) Summary of patients with ASD affected by each type of DNMs or CNVs in VA-related biomarkers.
The functional DNMs and pathogenic CNVs in 324 VA-related potential biomarkers accounted for ∼7.39 % (481/6511) of patients with ASD in the DNM cohort and ∼0.87 % (136/15,581) of patients with ASD in the CNV cohorts (Fig. 5b). These data suggest that ∼8 % of ASD probands carrying VA-related rare disruptive genetic variations might be affected by VA intervention.
3.6. Comparing VA-related and oxytocin-related biomarkers in ASD aetiology
Leveraging the 963 OTRGs, 172 PC-OTRGs, and 458 oxytocin-related biomarkers identified in our previous study [35], we found that only 9.51 % (39/410) of VARGs were shared with OTRGs (Fig. 6a). Of these, eight genes positively contributed to ASD aetiology (Fig. 6b). Among the remaining unshared PC-VARGs and PC-OTRGs (74 and 164, respectively), UBE3A and SHANK3 were ranked as the top risk genes associated with ASD aetiology, when exclusively associated with VA and oxytocin, respectively (Fig. 6b). After incorporating PC-VARG/-OTRG-connected PC-ASD core genes, 243 genes were found to be shared between potential VA- and oxytocin-related biomarkers; however, most of these were PC-ASD core genes (Fig. 6c, Table S10). These results indicate that the potential convergent contributions of VA and oxytocin to ASD aetiology may lie in their modulation in ASD core genes.
Fig. 6.

Potential functions and clinical applications of VA- and oxytocin-related biomarkers in ASD. (a–c) Comparison between VA-related biomarkers and oxytocin-related biomarkers in ASD. (d) Comparison between patients affected by DNMs and (e) CNVs in VA-related biomarkers, and oxytocin-related biomarkers (f) Evaluation of gene connections within or between biomarkers only associated with VA, or only associated with oxytocin (permutation test with 10,000 iterations). (g) ROC curves in the modelling cohort, (h) ROC curves in the processed validation cohort from the study of Zhou et al. (PMID:35982159), (i) ROC curves in the processed validation cohort from the study of Fu et al. (PMID:35982160). The best-fitting logistic regression models were constructed using the functional DNMs in VA- and oxytocin-related biomarkers, respectively, from the discovery cohort. Prediction of the ASD state in individuals for the discovery and validation cohorts. (Please see Methods). AUC values (95 % CIs) and accuracies were obtained from the corresponding logistic regression models for prediction. ROC, receiver operating characteristic; AUC, area under the curves; VA, Vitamin a; OXT, oxytocin;.
Using the data from our original discovery cohorts, two groups of 10,000 negative control gene sets were randomly generated, with each gene set containing 324 or 458 protein-coding genes to compare with the identified potential VA- (n = 324) and oxytocin-related biomarkers (n = 458), respectively. Functional DNMs and normalised pathogenic CNVs carried by patients with ASD were more prevalent in VA- and oxytocin-related biomarkers compared with the negative control gene sets (Permutation test: P < 1 × 10−4 for functional DNMs in VA- and oxytocin-related biomarkers; P = 4.20 × 10−4 and P = 3.00 × 10−5 for normalized pathogenic dnCNVs in VA- and oxytocin-related biomarkers, respectively; P = 2.93 × 10−2 and P = 2.84 × 10−2 for normalized pathogenic ihCNVs in VA- and oxytocin-related biomarkers, respectively; Fig. S10). These significant patterns between potential biomarkers and negative controls were also observed in the validation cohorts (Fig. S11).
By comparing the patients affected by the potential biomarkers, ∼7 % of the ASD probands in our original discovery cohorts were determined to carry both VA- and oxytocin-related rare disruptive variations, including 6.31 % (411/6,511) of patients carrying functional DNMs (Fig. 6d) and 0.86 % (134/15,581) carrying pathogenic CNVs (Fig. 6e). In the two validation cohorts, approximately 4.94 % (768/15,552) and 4.51 % (669/14,841) of patients with ASD carried functional DNMs in both VA- and oxytocin-related biomarkers (Fig. S12). These results imply that this subset of patients may benefit from VA and oxytocin intervention. Furthermore, functional DNMs and pathogenic CNVs in VA-related or oxytocin-related biomarkers alone accounted for 1.09 % and 2.84 % of patients with ASD in our discovery cohorts, respectively.
Notably, internal connections were observed with only VA-related (Permutation test: P = 2 × 10-4; Fig. 6f) or oxytocin-related biomarkers (Permutation test: P < 1 × 10-4; Fig. 6f) at significantly higher frequencies than those observed in random connections. However, this high connection frequency was not observed between these two subset biomarker subsets (Permutation test: P = 0.62; Fig. 6f). Moreover, potential biomarkers related only to VA tended to be involved in steroid-mediated processes (Table S10), whereas those associated with oxytocin only were enriched in calcium-related functions (Table S10). Among these shared biomarkers, ∼60.49 % of genes (147/243) were involved in ASD-related functions, such as chromatin organisation and nervous system function (Table S10). These findings suggest that VA and oxytocin impact ASD aetiology by affecting similar and differential functions.
To facilitate the application of our findings to a clinical setting, two best-fitting logistic regression models were constructed using individuals carrying functional DNMs in VA- and oxytocin-related biomarkers and sample size-matched case/controls from the discovery cohort. Using the constructed models, high AUC values and accuracies were observed for VA- and oxytocin-related biomarkers in predicting ASD aetiology in the modelling cohort (AUC = 98.23 %, accuracy = 97.76 % for VA-related biomarkers; AUC = 98.16 %, accuracy = 97.34 % for oxytocin-related biomarkers; binomial test: P < 10−16; Fig. 6g). A significantly high predictive performance was also observed for individuals carrying functional DNMs in corresponding biomarkers in two validation cohorts (precision = 93.8–94.07 %, AUC = 94.68–94.79 %, accuracy = 93.80–94.07 % for VA-related biomarkers; precision = 93.05–93.5 %, AUC = 93.54–94.25 %, accuracy = 93.05–93.50 % for oxytocin-related biomarkers; binomial test: P < 10−16; Fig. 6h, i).
4. Discussion
Rare mutations are major contributors to ASD aetiology and pathophysiological disturbances. These mutations may affect the response of patients to treatment, such as VA and oxytocin [2], [66]. We systematically evaluated the contribution of rare variations in VARGs to ASD aetiology and compared the similarities and differences between VA- and oxytocin-related biomarkers. Functional DNMs and pathogenic CNVs (including dnCNVs and ihCNVs) in VARGs were found to contribute to ASD pathogenesis. We then prioritised 324 potential VA-related biomarkers that positively contribute to ASD aetiology, consisting of 82 PC-VARGs and 242 of their connected ASD core genes. Eighty-two PC-VARGs directly or indirectly interact with known ASD core genes and are thus involved in ASD-related functions, contributing to ASD aetiology. Notably, among the ∼8 % of patients with ASD affected by rare disruptive variations in VA-related biomarkers, ∼87.5 % (∼7 %/8 % of total patients) were simultaneously affected by oxytocin-related biomarkers. In addition, ∼3 % of total patients were affected by rare disruptive variations in biomarkers related to oxytocin only. In addition, we found 26 and 27 shared genes for VA- and oxytocin-related biomarkers, respectively, showing consistently significant association with ASD from both DNM and CNV burden analyses (Table S11). It is interesting to note that both VA- and oxytocin-related biomarkers demonstrated high performance in predicting ASD aetiology, but only for individuals with functional DNM in the corresponding biomarkers. For other individuals, there should be more investigations focused on resolving the aetiology prediction for these individuals.
Notably, potential biomarkers related to VA only are involved in steroid-mediated processes contributing to ASD aetiology by affecting epigenetic foetal programming during early brain development [62], [64], [65]. Although elevated steroid hormone levels and steroidogenic activity have reportedly been associated with autism [62], [65], and steroid hormones and VA metabolites can regulate early brain development by binding to homologous nuclear receptors [67], how VA is involved in steroid-mediated processes and affects ASD phenotypes warrants further exploration. In contrast, potential biomarkers associated with oxytocin only were enriched in the calcium-related functions, which strongly affect central oxytocin release [68] and are associated with ASD-like behaviours [69]. Among the shared VA and oxytocin-related genes, mitogen-activated protein kinase 3 (MAPK3) was the top one gene associated with ASD pathology. The MAPK pathway is reportedly regulated by oxytocin [35], [70] and is activated by RA in an RA receptor (RAR)-independent manner [71]. Activating this pathway inhibits neural stem cell proliferation and ultimately impacts functions such as learning and memory [71]. Therefore, we speculate that both oxytocin and VA may affect the MAPK pathway, indicating the potential of combined interventions in patients with ASD with MAPK3 mutations.
Furthermore, we observed two known ASD core genes belonging exclusively to PC-VARGs, including UBE3A and NSD1. Out of 32 pathogenic CNVs, UBE3A was the top mutated gene in patients with ASD, whereas no disruptive variant was observed in the controls. Genetic variation in UBE3A has been linked to distinct neurodevelopmental disorders, such as the Angelman and Dup15q syndromes [72]. NSD1 was highly connected with ASD core genes; it carried two functional DNMs and two pathogenic dnCNVs in patients with ASD, but only one functional DNM in controls. UBE3A and NSD1 reportedly function as coactivators that interact with RAR to regulate gene transcription [73], [74]. Notably, NCOA6 (nuclear receptor coactivator 6), a potential new VA-related risk gene for ASD, carries two recurrent stop-gain DNMs (p.R303X) in patients with ASD and is connected to 37 PC-ASD core genes. Similarly, NCOA6 functions as a coactivator for different nuclear receptors, including steroids, retinoids and vitamin D3 receptors [75], contributing to ASD aetiology. Given that disruptive rare variants in these genes may affect RA-mediated transcriptional regulation, we speculate that patients with these genetic variants may be more sensitive to VA supplementation. However, the intricate RA-mediated regulatory network in ASD needs further exploration.
ASD risk is complex, encompassing genetic factors [2], [76], [77] and environmental factors [78], [79]. Approximately 65.12 % (211/324) of potential VA-related molecular biomarkers and 57.62 % (189/328) of non-PC-VARGs (Table S12) were found to be significantly differentially expressed through DNA methylation and/or modification of histone in patients with ASD than the controls [78], [79], [80]. Among them, most genes (∼89.57 % [189/211] of VA-related molecular biomarkers; ∼84.13 % [159/189] of non-PC-VARGs) were located in the differentially methylated regions, which were detected in cord blood DNA from foetuses that were later diagnosed with ASD (Table S12). This finding suggests that the regulatory function of VA metabolites in early brain development [67] may be associated with epigenetic alterations in VA-related genes [80].
In addition, maternal multivitamin supplementation during pregnancy has been associated with the risk of ASD in the foetus. Previous studies have demonstrated that maternal folic acid and multivitamin supplementation were positively associated with the VA, vitamin D (VD), and folate levels in ASD offspring and negatively associated with the risk of ASD in the offspring [81], [82]. Both ASD probands and their mothers reportedly exhibited low levels of plasma oxytocin, and the maternal oxytocin levels were negatively associated with the symptom severity in offspring [83]. Given the previously identified positive contribution of VD to ASD pathogenesis [84], [85], a comprehensive evaluation of the contribution of oxytocin and multivitamins to ASD pathogenesis, considering genetic and environmental factors and their interactions, should be performed in future investigations.
5. Conclusions
The findings of this study provided a basis for future studies aimed at understanding the pathophysiological mechanisms in ASD and a set of potential molecular targets for VA-, oxytocin-, or combination-based ASD treatments. Future clinical studies on VA and/or oxytocin intervention should simultaneously consider treatment outcomes, genetic and environmental factors, and clinical conditions to facilitate the provision of precise interventions in ASD with VA and/or oxytocin.
Funding
This work was supported by the Guangdong Key Project "Early diagnosis and treatment of autism spectrum disorders" (#202007030002 to ZSS), "Development of new tools for diagnosis and treatment of Autism" (#2018B030335001 to ZSS), National Natural Science Foundation of China (#31871191 to ZSS), Strategic Priority Research Program of Chinese Academy of Sciences (XDPB16 to ZSS), Science and Technology Major Project of Hunan Provincial Science and Technology Department (#2021SK1010 to Kun Xia), National Natural Science Foundation of China (#82130043 and #81730036 to Kun Xia).
CRediT authorship contribution statement
Zhongsheng Sun, Kun Xia, and Tao Wang conceived, designed, and supervised the study. Tao Wang and Liqiu Liu acquired, analyzed and verified the underlying data. Tianda Fan organized the article tables. Tao Wang and Liqiu Liu organized the figures and interpreted the data. Zhongsheng Sun, Tao Wang, and Liqiu Liu drafted and revised the manuscript.
Declaration of Competing Interest
We declare no competing interests.
Acknowledgements
We thank the members of the Beijing Institute of Life Science, Chinese Academy of Sciences for their valuable discussion regarding this work.
Ethics approval and consent to participate
All data used in this study are from public available dataset.
Consent for publication
All authors read and approved the final version of the manuscript and consent to its publication.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.csbj.2023.05.015.
Contributor Information
Kun Xia, Email: xiakun@sklmg.edu.cn.
Zhongsheng Sun, Email: sunzs@biols.ac.cn.
Appendix A. Supplementary material
Supplementary material
.
Supplementary material
.
Supplementary material
.
Supplementary material
.
Supplementary material
.
Supplementary material
.
Supplementary material
.
Supplementary material
.
Supplementary material
.
Supplementary material
.
Supplementary material
.
Supplementary material
.
Supplementary material
.
Data Availability
The datasets supporting the conclusions of this article were retrieved from Simons Simplex Collection (DOI: 10.1038/ng.3303), AutDB database (http://autism.mindspec.org/autdb), Gene4Denovo database (http://www.genemed.tech/gene4denovo/download), and studies of Zhou et al. [51] and Fu et al. [52].
References
- 1.Maenner M.J., Shaw K.A., Bakian A.V., Bilder D.A., Durkin M.S., Esler A., et al. Prevalence and characteristics of autism spectrum disorder among children aged 8 years - Autism and developmental disabilities monitoring network, 11 sites, United States, 2018. MMWR Surveill Summ. 2021;70:1–16. doi: 10.15585/mmwr.ss7011a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iakoucheva L.M., Muotri A.R., Sebat J. Getting to the cores of autism. Cell. 2019;178:1287–1298. doi: 10.1016/j.cell.2019.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim J.Y., Son M.J., Son C.Y., Radua J., Eisenhut M., Gressier F., et al. Environmental risk factors and biomarkers for autism spectrum disorder: an umbrella review of the evidence. Lancet Psychiatry. 2019;6:590–600. doi: 10.1016/S2215-0366(19)30181-6. [DOI] [PubMed] [Google Scholar]
- 4.Bang Y.J., Hu Z., Li Y., Gattu S., Ruhn K.A., Raj P., et al. Serum amyloid A delivers retinol to intestinal myeloid cells to promote adaptive immunity. Science. 2021;373:eabf9232. doi: 10.1126/science.abf9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shibata M., Pattabiraman K., Lorente-Galdos B., Andrijevic D., Kim S.K., Kaur N., et al. Regulation of prefrontal patterning and connectivity by retinoic acid. Nature. 2021;598:483–488. doi: 10.1038/s41586-021-03953-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Upadhyay J., Patra J., Tiwari N., Salankar N., Ansari M.N., Ahmad W. Dysregulation of multiple signaling neurodevelopmental pathways during embryogenesis: a possible cause of autism spectrum disorder. Cells. 2021:10. doi: 10.3390/cells10040958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siegenthaler J.A., Ashique A.M., Zarbalis K., Patterson K.P., Hecht J.H., Kane M.A., et al. Retinoic acid from the meninges regulates cortical neuron generation. Cell. 2009;139:597–609. doi: 10.1016/j.cell.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Z., Marro S.G., Zhang Y., Arendt K.L., Patzke C., Zhou B., et al. The fragile X mutation impairs homeostatic plasticity in human neurons by blocking synaptic retinoic acid signaling. Sci Transl Med. 2018:10. doi: 10.1126/scitranslmed.aar4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dumetz F., Ginieis R., Bure C., Marie A., Alfos S., Pallet V., et al. Neuronal morphology and synaptic plasticity in the hippocampus of vitamin A deficient rats. Nutr Neurosci. 2022;25:779–790. doi: 10.1080/1028415X.2020.1809877. [DOI] [PubMed] [Google Scholar]
- 10.Wołoszynowska-Fraser M.U., Kouchmeshky A., McCaffery P. Vitamin A and retinoic acid in cognition and cognitive disease. Annu Rev Nutr. 2020;40:247–272. doi: 10.1146/annurev-nutr-122319-034227. [DOI] [PubMed] [Google Scholar]
- 11.Sayad A., Noroozi R., Omrani M.D., Taheri M., Ghafouri-Fard S. Retinoic acid-related orphan receptor alpha (RORA) variants are associated with autism spectrum disorder. Metab Brain Dis. 2017;32:1595–1601. doi: 10.1007/s11011-017-0049-6. [DOI] [PubMed] [Google Scholar]
- 12.Abad C., Cook M.M., Cao L., Jones J.R., Rao N.R., Dukes-Rimsky L., et al. A rare De Novo RAI1 gene mutation affecting BDNF-enhancer-driven transcription activity associated with autism and atypical Smith-Magenis syndrome presentation. Biology (Basel) 2018:7. doi: 10.3390/biology7020031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fregeau B., Kim B.J., Hernandez-Garcia A., Jordan V.K., Cho M.T., Schnur R.E., et al. De Novo mutations of RERE cause a genetic syndrome with features that overlap those associated with proximal 1p36 deletions. Am J Hum Genet. 2016;98:963–970. doi: 10.1016/j.ajhg.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu X., Li C., Gao X., Xia K., Guo H., Li Y., et al. Excessive UBE3A dosage impairs retinoic acid signaling and synaptic plasticity in autism spectrum disorders. Cell Res. 2018;28:48–68. doi: 10.1038/cr.2017.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar S., Reynolds K., Ji Y., Gu R., Rai S., Zhou C.J. Impaired neurodevelopmental pathways in autism spectrum disorder: a review of signaling mechanisms and crosstalk. J Neurodev Disord. 2019;11:10. doi: 10.1186/s11689-019-9268-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou H., Xu X., Yan W., Zou X., Wu L., Luo X., et al. Prevalence of Autism spectrum disorder in china: a nationwide multi-center population-based study among children aged 6 to 12 years. Neurosci Bull. 2020;36:961–971. doi: 10.1007/s12264-020-00530-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang N., Zhao Y., Gao J. Association between peripheral blood levels of vitamin A and Autism spectrum disorder in children: a meta-analysis. Front Psychiatry. 2021;12 doi: 10.3389/fpsyt.2021.742937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo M., Zhu J., Yang T., Lai X., Lei Y., Chen J., et al. Vitamin A and vitamin D deficiencies exacerbate symptoms in children with autism spectrum disorders. Nutr Neurosci. 2019;22:637–647. doi: 10.1080/1028415X.2017.1423268. [DOI] [PubMed] [Google Scholar]
- 19.Liu X., Liu J., Xiong X., Yang T., Hou N., Liang X., et al. Correlation between nutrition and symptoms: nutritional survey of children with autism spectrum disorder in chongqing, China. Nutrients. 2016:8. doi: 10.3390/nu8050294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng B., Zhu J., Yang T., Guo M., Lai X., Li Q., et al. Vitamin A deficiency increases the risk of gastrointestinal comorbidity and exacerbates core symptoms in children with autism spectrum disorder. Pedia Res. 2021;89:211–216. doi: 10.1038/s41390-020-0865-y. [DOI] [PubMed] [Google Scholar]
- 21.Guo M., Li L., Zhang Q., Chen L., Dai Y., Liu L., et al. Vitamin and mineral status of children with autism spectrum disorder in Hainan Province of China: associations with symptoms. Nutr Neurosci. 2020;23:803–810. doi: 10.1080/1028415X.2018.1558762. [DOI] [PubMed] [Google Scholar]
- 22.Zhu J., Guo M., Yang T., Lai X., Tang T., Chen J., et al. Nutritional status and symptoms in preschool children with Autism spectrum disorder: a two-center comparative study in Chongqing and Hainan Province, China. Front Pedia. 2020;8:469. doi: 10.3389/fped.2020.00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang T., Chen L., Dai Y., Jia F., Hao Y., Li L., et al. Vitamin A status is more commonly associated with symptoms and neurodevelopment in boys with autism spectrum disorders-a multicenter study in China. Front Nutr. 2022;9 doi: 10.3389/fnut.2022.851980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou W., Li S. Decreased levels of serum retinoic acid in chinese children with autism spectrum disorder. Psychiatry Res. 2018;269:469–473. doi: 10.1016/j.psychres.2018.08.091. [DOI] [PubMed] [Google Scholar]
- 25.Guo M., Zhu J., Yang T., Lai X., Liu X., Liu J., et al. Vitamin A improves the symptoms of autism spectrum disorders and decreases 5-hydroxytryptamine (5-HT): a pilot study. Brain Res Bull. 2018;137:35–40. doi: 10.1016/j.brainresbull.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Lai X., Zhang Q., Zhu J., Yang T., Guo M., Li Q., et al. A weekly vitamin A supplementary program alleviates social impairment in Chinese children with autism spectrum disorders and vitamin A deficiency. Eur J Clin Nutr. 2021;75:1118–1125. doi: 10.1038/s41430-020-00827-9. [DOI] [PubMed] [Google Scholar]
- 27.Freedman R., Hunter S.K., Hoffman M.C. Prenatal primary prevention of mental illness by micronutrient supplements in pregnancy. Am J Psychiatry. 2018;175:607–619. doi: 10.1176/appi.ajp.2018.17070836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iossifov I., O'Roak B.J., Sanders S.J., Ronemus M., Krumm N., Levy D., et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krumm N., Turner T.N., Baker C., Vives L., Mohajeri K., Witherspoon K., et al. Excess of rare, inherited truncating mutations in autism. Nat Genet. 2015;47:582–588. doi: 10.1038/ng.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo H., Peng Y., Hu Z., Li Y., Xun G., Ou J., et al. Genome-wide copy number variation analysis in a Chinese autism spectrum disorder cohort. Sci Rep. 2017;7:44155. doi: 10.1038/srep44155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanders S.J., He X., Willsey A.J., Ercan-Sencicek A.G., Samocha K.E., Cicek A.E., et al. Insights into Autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87:1215–1233. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y., Zeng C., Li J., Zhou Z., Ju X., Xia S., et al. PAK2 haploinsufficiency results in synaptic cytoskeleton impairment and Autism-related behavior. Cell Rep. 2018;24:2029–2041. doi: 10.1016/j.celrep.2018.07.061. [DOI] [PubMed] [Google Scholar]
- 33.Song W., Li Q., Wang T., Li Y., Fan T., Zhang J., et al. Putative complement control protein CSMD3 dysfunction impairs synaptogenesis and induces neurodevelopmental disorders. Brain Behav Immun. 2022;102:237–250. doi: 10.1016/j.bbi.2022.02.027. [DOI] [PubMed] [Google Scholar]
- 34.Li J., Wang L., Yu P., Shi L., Zhang K., Sun Z.S., et al. Vitamin D-related genes are subjected to significant de novo mutation burdens in autism spectrum disorder. Am J Med Genet B Neuropsychiatr Genet. 2017 doi: 10.1002/ajmg.b.32543. [DOI] [PubMed] [Google Scholar]
- 35.Wang T., Zhao T., Liu L., Teng H., Fan T., Li Y., et al. Integrative analysis prioritised oxytocin-related biomarkers associated with the aetiology of autism spectrum disorder. EBioMedicine. 2022;81 doi: 10.1016/j.ebiom.2022.104091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J., Wang L., Guo H., Shi L., Zhang K., Tang M., et al. Targeted sequencing and functional analysis reveal brain-size-related genes and their networks in autism spectrum disorders. Mol Psychiatry. 2017;22:1282–1290. doi: 10.1038/mp.2017.140. [DOI] [PubMed] [Google Scholar]
- 37.Li J., Hu S., Zhang K., Shi L., Zhang Y., Zhao T.et al., : A comparative study of the genetic components of three subcategories of autism spectrum disorder. Mol Psychiatry 2018. [DOI] [PubMed]
- 38.Lai X., Wu X., Hou N., Liu S., Li Q., Yang T., et al. Vitamin A deficiency induces autistic-like behaviors in rats by regulating the RARβ-CD38-oxytocin axis in the hypothalamus. Mol Nutr Food Res. 2018:62. doi: 10.1002/mnfr.201700754. [DOI] [PubMed] [Google Scholar]
- 39.Tolomeo S., Chiao B., Lei Z., Chew S.H., Ebstein R.P., Novel A. Role of CD38 and oxytocin as tandem molecular moderators of human social behavior. Neurosci Biobehav Rev. 2020;115:251–272. doi: 10.1016/j.neubiorev.2020.04.013. [DOI] [PubMed] [Google Scholar]
- 40.Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gene Ontology C. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021;49:D325–D334. doi: 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao G., Li K., Li B., Wang Z., Fang Z., Wang X., et al. Gene4Denovo: an integrated database and analytic platform for de novo mutations in humans. Nucleic Acids Res. 2020;48:D913–D926. doi: 10.1093/nar/gkz923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang H., Wang T., Zhao X., Wu H., You M., Sun Z., et al. AI-Driver: an ensemble method for identifying driver mutations in personal cancer genomes. NAR Genom Bioinform. 2020;2:lqaa084. doi: 10.1093/nargab/lqaa084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang T., Ruan S., Zhao X., Shi X., Teng H., Zhong J., et al. OncoVar: an integrated database and analysis platform for oncogenic driver variants in cancers. Nucleic Acids Res. 2021;49:D1289–D1301. doi: 10.1093/nar/gkaa1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J., Zhao T., Zhang Y., Zhang K., Shi L., Chen Y., et al. Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 2018;46:7793–7804. doi: 10.1093/nar/gky678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Basu S.N., Kollu R., Banerjee-Basu S. AutDB: a gene reference resource for autism research. Nucleic Acids Res. 2009;37:D832–D836. doi: 10.1093/nar/gkn835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krumm N., O'Roak B.J., Karakoc E., Mohajeri K., Nelson B., Vives L., et al. Transmission disequilibrium of small CNVs in simplex autism. Am J Hum Genet. 2013;93:595–606. doi: 10.1016/j.ajhg.2013.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alfoldi J., Wang Q., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y., Wang T., Wang Y., Xia K., Li J., Sun Z. Targeted sequencing and integrative analysis to prioritize candidate genes in neurodevelopmental disorders. Mol Neurobiol. 2021;58:3863–3873. doi: 10.1007/s12035-021-02377-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abrahams B.S., Arking D.E., Campbell D.B., Mefford H.C., Morrow E.M., Weiss L.A., et al. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs) Mol Autism. 2013;4:36. doi: 10.1186/2040-2392-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou X., Feliciano P., Shu C., Wang T., Astrovskaya I., Hall J.B., et al. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat Genet. 2022;54:1305–1319. doi: 10.1038/s41588-022-01148-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fu J.M., Satterstrom F.K., Peng M., Brand H., Collins R.L., Dong S., et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat Genet. 2022;54:1320–1331. doi: 10.1038/s41588-022-01104-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Szklarczyk D., Gable A.L., Nastou K.C., Lyon D., Kirsch R., Pyysalo S., et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49:D605–D612. doi: 10.1093/nar/gkaa1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li T., Wernersson R., Hansen R.B., Horn H., Mercer J., Slodkowicz G., et al. A scored human protein-protein interaction network to catalyze genomic interpretation. Nat Methods. 2017;14:61–64. doi: 10.1038/nmeth.4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang T., Zhang Y., Liu L., Wang Y., Chen H., Fan T., et al. Targeted sequencing and integrative analysis of 3,195 Chinese patients with neurodevelopmental disorders prioritized 26 novel candidate genes. J Genet Genom. 2021;48:312–323. doi: 10.1016/j.jgg.2021.03.002. [DOI] [PubMed] [Google Scholar]
- 56.Li T., Wernersson R., Hansen R.B., Horn H., Mercer J., Slodkowicz G., et al. A scored human protein–protein interaction network to catalyze genomic interpretation. Nat Methods. 2016;14:61–64. doi: 10.1038/nmeth.4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fay M.: rateratio.test: Exact Rate Ratio Test. 2014.
- 60.Wu J., Yu P., Jin X., Xu X., Li J., Li Z., et al. Genomic landscapes of Chinese sporadic autism spectrum disorders revealed by whole-genome sequencing. J Genet Genom. 2018;45:527–538. doi: 10.1016/j.jgg.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 61.Kong A., Frigge M.L., Masson G., Besenbacher S., Sulem P., Magnusson G., et al. Rate of de novo mutations and the importance of father's age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baron-Cohen S., Tsompanidis A., Auyeung B., Norgaard-Pedersen B., Hougaard D.M., Abdallah M., et al. Foetal oestrogens and autism. Mol Psychiatry. 2020;25:2970–2978. doi: 10.1038/s41380-019-0454-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.De Rubeis S., He X., Goldberg A.P., Poultney C.S., Samocha K., Cicek A.E., et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malkki H. Neurodevelopmental disorders: elevated fetal sex steroids might confer risk for autism. Nat Rev Neurol. 2014;10:366. doi: 10.1038/nrneurol.2014.107. [DOI] [PubMed] [Google Scholar]
- 65.Baron-Cohen S., Auyeung B., Norgaard-Pedersen B., Hougaard D.M., Abdallah M.W., Melgaard L., et al. Elevated fetal steroidogenic activity in autism. Mol Psychiatry. 2015;20:369–376. doi: 10.1038/mp.2014.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Geschwind D.H. Oxytocin for Autism spectrum disorder - down, but not out. N Engl J Med. 2021;385:1524–1525. doi: 10.1056/NEJMe2110158. [DOI] [PubMed] [Google Scholar]
- 67.Petkovich M., Brand N.J., Krust A., Chambon P. A human retinoic acid receptor which belongs to the family of nuclear receptors. Nature. 1987;330:444–450. doi: 10.1038/330444a0. [DOI] [PubMed] [Google Scholar]
- 68.Sheng W., Harden S.W., Tan Y., Krause E.G., Frazier C.J. Dendritic osmosensors modulate activity-induced calcium influx in oxytocinergic magnocellular neurons of the mouse PVN. Elife. 2021:10. doi: 10.7554/eLife.63486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Q., Kong Y., Wu D.Y., Liu J.H., Jie W., You Q.L., et al. Impaired calcium signaling in astrocytes modulates autism spectrum disorder-like behaviors in mice. Nat Commun. 2021;12:3321. doi: 10.1038/s41467-021-23843-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grinevich V., Neumann I.D. Brain oxytocin: how puzzle stones from animal studies translate into psychiatry. Mol Psychiatry. 2021;26:265–279. doi: 10.1038/s41380-020-0802-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lin Y.L., Persaud S.D., Nhieu J., Wei L.N. Cellular retinoic acid-binding protein 1 modulates stem cell proliferation to affect learning and memory in male mice. Endocrinology. 2017;158:3004–3014. doi: 10.1210/en.2017-00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lopez S.J., Segal D.J., LaSalle J.M. UBE3A: an E3 ubiquitin ligase with genome-wide impact in neurodevelopmental disease. Front Mol Neurosci. 2018;11:476. doi: 10.3389/fnmol.2018.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fang M., Li Y., Ren J., Hu R., Gao X., Chen L. Epilepsy-associated UBE3A deficiency downregulates retinoic acid signalling pathway. Front Genet. 2021;12 doi: 10.3389/fgene.2021.681295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang N., vom Baur E., Garnier J.M., Lerouge T., Vonesch J.L., Lutz Y., et al. Two distinct nuclear receptor interaction domains in NSD1, a novel SET protein that exhibits characteristics of both corepressors and coactivators. Embo J. 1998;17:3398–3412. doi: 10.1093/emboj/17.12.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee S.K., Anzick S.L., Choi J.E., Bubendorf L., Guan X.Y., Jung Y.K., et al. A nuclear factor, ASC-2, as a cancer-amplified transcriptional coactivator essential for ligand-dependent transactivation by nuclear receptors in vivo. J Biol Chem. 1999;274:34283–34293. doi: 10.1074/jbc.274.48.34283. [DOI] [PubMed] [Google Scholar]
- 76.Xia L., Ou J., Li K., Guo H., Hu Z., Bai T., et al. Genome-wide association analysis of autism identified multiple loci that have been reported as strong signals for neuropsychiatric disorders. Autism Res. 2020;13:382–396. doi: 10.1002/aur.2229. [DOI] [PubMed] [Google Scholar]
- 77.Xia K., Guo H., Hu Z., Xun G., Zuo L., Peng Y., et al. Common genetic variants on 1p13.2 associate with risk of autism. Mol Psychiatry. 2014;19:1212–1219. doi: 10.1038/mp.2013.146. [DOI] [PubMed] [Google Scholar]
- 78.Sun W., Poschmann J., Cruz-Herrera Del Rosario R., Parikshak N.N., Hajan H.S., Kumar V., et al. Histone acetylome-wide association study of Autism spectrum disorder. Cell. 2016;167:1385–1397 e1311. doi: 10.1016/j.cell.2016.10.031. [DOI] [PubMed] [Google Scholar]
- 79.Ramaswami G., Won H., Gandal M.J., Haney J., Wang J.C., Wong C.C.Y., et al. Integrative genomics identifies a convergent molecular subtype that links epigenomic with transcriptomic differences in autism. Nat Commun. 2020;11:4873. doi: 10.1038/s41467-020-18526-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mordaunt C.E., Jianu J.M., Laufer B.I., Zhu Y., Hwang H., Dunaway K.W., et al. Cord blood DNA methylome in newborns later diagnosed with autism spectrum disorder reflects early dysregulation of neurodevelopmental and X-linked genes. Genome Med. 2020;12:88. doi: 10.1186/s13073-020-00785-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tan M., Yang T., Zhu J., Li Q., Lai X., Li Y., et al. Maternal folic acid and micronutrient supplementation is associated with vitamin levels and symptoms in children with autism spectrum disorders. Reprod Toxicol. 2020;91:109–115. doi: 10.1016/j.reprotox.2019.11.009. [DOI] [PubMed] [Google Scholar]
- 82.Levine S.Z., Kodesh A., Viktorin A., Smith L., Uher R., Reichenberg A., et al. Association of maternal use of folic acid and multivitamin supplements in the periods before and during pregnancy with the risk of autism spectrum disorder in offspring. JAMA Psychiatry. 2018;75:176–184. doi: 10.1001/jamapsychiatry.2017.4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu X.J., Shou X.J., Li J., Jia M.X., Zhang J.S., Guo Y., et al. Mothers of autistic children: lower plasma levels of oxytocin and Arg-vasopressin and a higher level of testosterone. PLoS One. 2013;8 doi: 10.1371/journal.pone.0074849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li J., Wang L., Yu P., Shi L., Zhang K., Sun Z.S., et al. Vitamin D-related genes are subjected to significant de novo mutation burdens in autism spectrum disorder. Am J Med Genet B Neuropsychiatr Genet. 2017;174:568–577. doi: 10.1002/ajmg.b.32543. [DOI] [PubMed] [Google Scholar]
- 85.Lee B.K., Eyles D.W., Magnusson C., Newschaffer C.J., McGrath J.J., Kvaskoff D., et al. Developmental vitamin D and autism spectrum disorders: findings from the Stockholm Youth Cohort. Mol Psychiatry. 2021;26:1578–1588. doi: 10.1038/s41380-019-0578-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Supplementary material
Supplementary material
Supplementary material
Supplementary material
Supplementary material
Supplementary material
Supplementary material
Supplementary material
Supplementary material
Supplementary material
Supplementary material
Supplementary material
Data Availability Statement
The datasets supporting the conclusions of this article were retrieved from Simons Simplex Collection (DOI: 10.1038/ng.3303), AutDB database (http://autism.mindspec.org/autdb), Gene4Denovo database (http://www.genemed.tech/gene4denovo/download), and studies of Zhou et al. [51] and Fu et al. [52].