

Abstract
Herein, we introduce a new methodology for designing transient organogels that offers tunability of the mechanical properties simply by matching the protective groups of the precursor to that of the solvent. We developed solvent-induced transient materials in which the solvent chemically participates in a set of reactions and actively supports the assembly event. The activation of a single precursor by an acid (accelerator) yields the formation of two distinct gelators and induces gelation. The interconversion cycle is supplied by the secondary solvent (originating from hydrolysis of the primary solvent by the accelerator), which then progressively solubilizes the gel network. We show that this gelation method offers a direct correlation between the mechanical and transient properties by modifying the chemical structure of the precursors and the presence of an accelerator in the system. Such a method paves the way for the design of self-abolishing and mechanically tunable materials for targeted purposes. The biocompatibility and versatility of amino acid-based gelators can offer a wide range of biomaterials for applications requiring a controllable and definite lifetime such as drug delivery platforms exhibiting a burst release or self-abolishing cell culture substrates.
Introduction
Biological supramolecular assemblies, which exist out of equilibrium, are under the constant exchange of energy and matter with their environment to sustain the transient state,1−3 in contrast to in-equilibrium biological assemblies. These systems often exhibit interesting properties, such as a triggerable response to external stimuli and self-healing.4,5 In contrast to biological assemblies, analogous artificial counterparts form under thermodynamic equilibrium, i.e., the intermolecular interactions, structure, and inner environment are kept stable when no external stimuli/disturbance is applied. In an effort to mimic naturally occurring systems and the unique properties of out-of-equilibrium systems, transient supramolecular materials have gained momentum in the past decades.4,6 Transient materials have been envisioned to be used as temporary delivery devices, such as drug delivery platforms7−9 and self-abolishing materials.4 Therefore, transient organogels can enrich the pool of materials with tunable properties such as finite and controllable release.
Dissipative (or dynamic) self-assembly (DSA) is an extensively studied representative of transient assembly mode, which relies on a reaction cycle,10−12 such as that exemplified in Scheme 1. Consumption of energy (i.e., fuel) by a gelator precursor forms the activated building blocks (activation reaction), which subsequently self-assemble.13−15 The transient assembled structure exhibits a limited lifetime governed by depletion of the fuel. Energy dissipation reverses the process, regenerating the non-assembling gelator precursor (deactivation reaction) and inducing the collapse of the network (disassembly). Thus, transient assemblies occur when the rate of energy consumption is higher than that of energy dissipation. Among DSA systems, several types of fuel have been previously reported, including chemical, light, and electrical fueling.16−18 Chemically fueled DSA systems intrinsically produce chemical waste during the DSA reaction cycle, that is, side products after activation and/or deactivation reactions, which can be problematic in some applications. Light-fueled DSA systems, which do not release chemical waste into the system, were developed to overcome this problem.16
Scheme 1. Schematic Description of DSA and Solvent-Induced Self-Assembly Systems.

The inset represents the set of reactions leading to self-assembly and gel-to-sol transition.
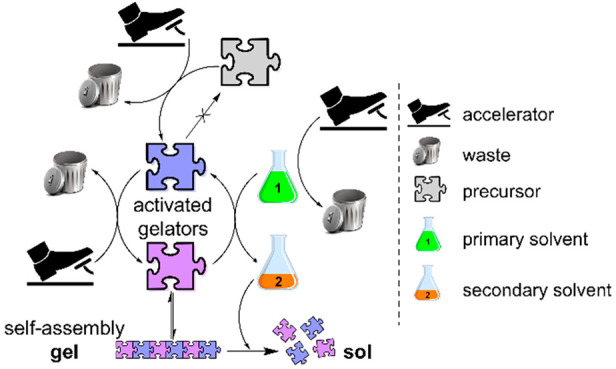
Recently, our group introduced the solvent as a new type of chemical agent inducing a transient assembly19 inspired by DSA. In both dissipative and non-dissipative self-assembly, solvents are encapsulated in the gel network and do not affect the assembly event via chemical reactions. Our “solvent-induced” supramolecular gel (Scheme 1) is a rare case in which the primary solvent, tert-butyl acetate (tBuOAc), actively controls a set of reactions promoting self-assembly. In contrast to typical DSA, the deprotection of the gelator precursor by an acid accelerator (activation reaction) gives rise to two distinct gelators. The N-Boc protective group is irreversibly deprotected, while the tBu ester group at the C-terminus is reversibly deprotected. Both gelators interconvert through a cycle of hydrolysis–esterification reactions supplied by the secondary solvent tert-butyl alcohol (tBuOH), a hydrolysis product of the primary solvent, reintroducing the tert-butyl group in the cycle. A key requirement for the proposed mechanism is that the primary solvent and the precursor gelator should have the same protective group, tBu ester, in our case. Interestingly, one of the gelators is soluble in tBuOH. Therefore, progressive dissolution of the network is observed over time. This new class of solvent-induced self-assembly is not to be defined as DSA. The transient assembly is induced by two acid-accelerated processes: the primary solvent hydrolysis generating the secondary solvent and the gelators’ interconversion. The transient nature of the system relies on the dual role adopted by the solvent, most importantly, the competing kinetics of the secondary solvent formation and the formation/dissolution of the gelators, dictating the assembly/disassembly.
Herein, we deepen and further optimize the solvent-induced transient assembly concept and prove its application to a set of phenylalanine-based peptide precursors bearing the same protective group (tBu) as that of the solvent. We report the transient assembly mechanism and assess the effect of the gelators’ chemical structure on the corresponding materials’ properties from the molecular to macroscopic length scale by varying the number of aromatic units (Figure 1). In addition, we decipher the assembly mode and the transitivity character of a multicomponent gel compared to its respective single-component gels. We show that the materials’ intrinsic and transitivity properties are tunable, depending on the gelation conditions and gelator structure, thus paving the way toward controllable materials.
Figure 1.

General structures of potential precursors 1–8 (upper table), monoprotected gelators 1a–3a, and deprotected gelators 1b–3b (lower table) as well as the corresponding gel systems I–IV.
Experimental Section
Materials
All chemicals were used as supplied without any further purification, unless stated otherwise. (S)-Phenylalanine tert-butyl ester HCl (Phe-OtBu) and N-(tert-butoxycarbonyl)-l-tyrosine (Boc-Tyr) were purchased from Carbosynth. N-(tert-Butoxycarbonyl)-l-phenylalanine (Boc-Phe), N-(tert-butoxycarbonyl)-l-valine (Boc-Val), and N-(tert-butoxycarbonyl)-alanine OH (Boc-Ala) were purchased from TCI. N-(tert-Butoxycarbonyl)-leucine OH (Boc-Leu), N-(tert-butoxycarbonyl)-isoleucine OH (Boc-Ile), and N-(tert-butoxycarbonyl)-glycine OH (Boc-Gly) were purchased from Sigma-Aldrich. 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethylammonium tetrafluoroborate (TBTU) and sodium hydrogen carbonate (NaHCO3) were purchased from Novabiochem and VWR Chemicals, respectively. tert-Butyl acetate (tBuOAc) was purchased from TCI and sulfuric acid from Fluka.
Methods
NMR Spectroscopy
The spectra of the synthesized compounds and xerogels were recorded on Bruker Advance III HD 300 and 500 MHz spectrometers in d6-DSMO and CDCl3 solvents. Chemical shifts (δ) are given in parts per million, and the coupling constant (J) is given in Hz. The spectra were referenced to the solvent signal (2.5 and 7.26 ppm in 1H NMR and 39.52 and 77.16 ppm in 13C NMR for d6-DSMO and CDCl3, respectively). 13C NMR spectra were recorded with a broadband 1H decoupling. Xerogels were obtained from fresh organogels dried in open air at room temperature (overnight).
FTIR Spectroscopy
The spectra of the synthesized compounds and xerogels were recorded on a Bruker Tensor 27 FT-IR in attenuated total reflectance (ATR) mode. Spectral width: 400–4000 cm–1; number of scans: 124; resolution: 4 cm–1. All spectra were baseline corrected. Xerogels were prepared as mentioned above in the NMR Spectroscopy section.
UV–Vis Spectroscopy
The UV–vis spectra of synthesized compounds in ACN and gels were recorded on a PerkinElmer Lambda 850 UV–vis spectrometer (spectral range: 200–500 nm; step: 1 nm; integration time: 0.2 s; slit width: 2 nm). The samples were measured in a 1 mm path length quartz cuvette at room temperature. Gel samples were prepared in situ in the quartz cuvettes one day before the measurements.
Rheology
Rheology measurements were performed on an Anton Paar MCR 302 modular compact rheometer with an upper geometry cylinder (cylinder-relative ST10-4V-8.8/97.5). Gel samples (1.0 mL) were prepared in glass vials (Fisherbrand Type III soda lime glass, 14 mm inner diameter) and allowed to rest for one day before measurements for gelation to occur. Frequency sweep measurements were performed within the linear viscoelastic region (LVR) as obtained by amplitude sweep measurements. All measurements were taken in triplicate at room temperature.
SEM Imaging
Microscopy images were obtained on a Zeiss EVO-50XVP microscope. Diluted gels (×10), with the organic solvent used for gelation, were pipetted (1 μL) onto carbon films (400 mesh copper grids) obtained from Agar Scientific and freeze-dried for 1 h before imaging.
Results and Discussion
Transient Assembly at the Molecular Level
Phenylalanine-derived peptide precursors 1–8 (Figure 1) bearing a phenylalanine unit and varying the second unit by using hydrophobic side-chained amino acids were synthesized (see the Supporting Information, Section 1, for synthesis details).
Under identical gelation conditions (tBuOAc solvent, 50 mM, 1 equiv of sulfuric acid), four self-supporting gels (SSG) were obtained (Figure 2a). Interestingly, no SSG formed from the precursors bearing tyrosine or an aliphatic side chain shorter than leucine. Although the gelation ability of specific molecules under certain conditions is not fully understood, common trends and hypotheses can be drawn. The presence of phenylalanine (Phe), tyrosine (Tyr), and leucine (Leu) motifs has been reported to play an important role in the self-assembly event. Because of the hydrophobicity, the π–π stacking tendency of Phe and Tyr, and the additional hydrogen-bonding donor site of Tyr, these aromatic amino acids are often present in amino acid-based gelators. Similarly, Leu is widely reported as an efficient amino acid for gelation due to its mobile aliphatic side chain.20−22 In our case, the length of the side chain and the electric charge affect the gelation process by electrostatic repulsion and/or steric hindrance23 which is in line with the existing literature.
Figure 2.

(a) Vial inversion test confirming the lack of flow and thus acting as supporting evidence of material formation for gel systems I, II, III, and IV. Gel IV is the multicomponent gel formed by precursors 1 and 3 with a 1:1 molar ratio. (b) Schematic depiction of the solvent-induced transient assembly.
The only precursors yielding SSGs (Figure 2a) contain either only phenylalanine or phenylalanine and leucine motifs (Boc-Phe-Phe-OtBu 1 (gel I), Boc-Phe-Phe-Phe-OtBu 2 (gel II), and Boc-Leu-Phe-OtBu 3 (gel III); Figure 1). In addition, the multicomponent gel (gel IV), consisting of precursors 1 and 3 with a 1:1 ratio, was prepared to study whether the mode of assembly is self-sorting (two distinct fibrous networks of each gelator individually) or co-assembly (a single fibrous network consisting of both gelators).24 The comparison of precursors 1, 2, and 3 also gives insight into the effect of the number of aromatic units on the gelation ability and gel properties.
For all systems (I, II, III, and IV), SSGs could be obtained at a concentration down to 25 mM (Tables S2–S5). The phase-transition temperatures (Table S1) show an increase proportional to the number of aromatic units, suggesting that the network stiffens by increasing the aromatic character of the precursors. Importantly, under similar gelation conditions (50 mM, 1.0 equiv of accelerator), each gel system exhibits a transient character with a different lifetime. Gel I is stable for 4–6 days and gel III for 20–22 days, while multicomponent gel IV collapses after 10 days, showing a lifetime approximately in between that of its individual components. For gel II, the gel-to-sol transition is observed after 8 days.
To assess whether precursors 2 and 3 follow the same transient assembly mechanism as for 1 (Figure 2b), nuclear magnetic resonance (NMR) studies were performed on the xerogels (dried gels) of each system a day after gelation (Figure 3). Gel I shows two amide signals at 8.83 and 8.89 ppm corresponding to gelators 1a and 1b and a singlet at 1.32 ppm attributed to the tBu ester group of 1a (Figure 3, black).19 Consequently, for gel III (Figure 3, blue), an identical trend is observed with the formation of 3a and 3b. As precursor 2 contains an additional amino acid unit compared to 1 and 3, two amide signals are expected per gelator present in gel II (Figure 3, red). The peaks marked with an asterisk correspond to a rotamer of 2a observed in d6-DMSO (Figure S5). Increasing temperature provides additional energy lifting the rotational restriction of the single bond. Therefore, 1H NMR spectra recorded at 30, 70, and 90 °C show the coalescence of the rotamer peaks (Figure S6). Additionally, the presence of the rotamer was confirmed by recording the spectrum in a different solvent, here, CDCl3, in which no additional signals were observed (Figure S3). HR-MS also confirmed the presence of a single molecule.
Figure 3.
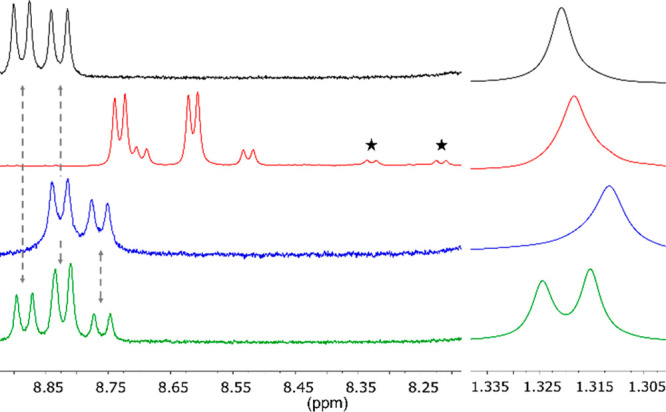
1H NMR (300 MHz, d6-DMSO) spectra of the dried gel systems I (black), II (red), III (blue), and IV (green) using 1.0 equiv of accelerator. Peaks belonging to the rotamer are marked by a star.
At the amide region, the spectrum of multicomponent gel IV (Figure 3, green) is the superimposition of its individual component’s gel systems (Figure 3, black and blue). As precursors 1 and 3 are present, four gelator molecules are observed in the gel state. Indeed, three amide signals (including the overlapping signal at 8.83 ppm corresponding to two amide groups) and two tBu ester signals are seen. The results are supported by HR-MS measurements confirming four distinct gelators 1a, 1b, 3a, and 3b. In all gels’ NMR spectra, a broad peak at around 3–6 ppm corresponds to tBuOH, as supported by the control experiment of induced hydrolysis of tBuOAc (Figure S26). In addition, NMR spectra were measured on the xerogels of each system at half of their respective lifetimes. All systems follow an identical trend; that is, the expected gelators are present in the corresponding gels, although in a different ratio (Figures S35, S41, and S47). These results suggest that throughout the lifetime of the gels, the chemical interchange carries on until the materials collapse.
Surprisingly, all systems formed SSGs also when a lower amount of accelerator was used to trigger gelation (0.5 equiv instead of 1.0 equiv). Analysis of the 1H NMR spectra of these gels revealed that the same reactions occurred, as the spectra are quantitatively similar. However, since half the amount of triggering agent (acid accelerator) was used, the remains of the unreacted precursors were also present in the systems (Figures S29, S33, S39, and S45). Notably, because less accelerator was used to trigger the hydrolysis cycle (required water originates from the aqueous accelerator), a smaller amount of tBuOH formed during gelation. Therefore, gels made with 0.5 equiv of accelerator do not show a transient character, unlike those made with 1.0 equiv, and are stable to date. Respectively, the lifetime of the gels formed using 1.5 equiv of accelerator was shorter (Tables S2–S5). A higher concentration of accelerator produces more tBuOH in the medium, increasing the collapse rate of the materials from 4–10 days to 1–3 days.
Additionally, the effect of the precursor concentration on the dynamic properties (lifetime) of the materials was investigated (Tables S2–S5). It was found that gels with a high precursor concentration (100 mM) exhibited longer lifetimes than those with a lower concentration (50 mM). This difference is presumably due to the higher amount of gelator to be dissolved and stronger supramolecular interactions maintaining the assembly.
To verify the assumption that the gelation trigger and the subsequent gelation mechanism are related only to the identities of the C-terminus protective group and solvent, an attempt to induce gelation using the same trigger in dichloromethane (DCM) was made with precursors 1 and 3. No gelation was observed; instead, the product precipitated, and the analysis of the 1H NMR spectrum revealed the complete deprotection of the precursors (Figure S49). In addition, gelation was attempted using Boc-deprotected molecules 1a–3a as precursors using the same trigger. SSGs formed within the same time, and 1H NMR spectroscopy verified the presence of both monoprotected and fully deprotected gelators, along with tBuOH, in the medium. Therefore, both the solvent and the precursor’s C-terminus must bear the same functional group, in this case tBu ester, for the reaction cycle and gelation to occur, regardless of the peptide backbone and/or the presence of a Boc protective group on the N-terminus.
Higher-Order Assembly Determination
To investigate the effect of the gelators’ structure on the fibrous network, scanning electron microscopy (SEM) images shown in Figure 4 were recorded on diluted gels. All samples were freeze-dried for 1 h prior to imaging to minimize potential structural rearrangement of the network during drying at room temperature.25 Gel I (Figure 4a) contains densely packed, curved fibers, branching and entangling with one another, which support the high stiffness value measured by rheology (vide infra). In contrast, the fibers of gel III (Figure 4c), although similar in shape to gel I, appear less packed, verifying the lower stiffness than gel I. In addition, helical features are present, supporting the peak corresponding to the helical-type assemblies observed in IR measurements (vide infra). For multicomponent gel IV (Figure 4d), straightened fibers highly entangled into bigger bundles are observed. The resulting fibrous network exhibits a different shape than its individual components, suggesting that a new structure has formed and implying co-assembly of the gelators.26 In gel II, SEM image shows individual curved fibers branching around nucleation points (Figure 4b).
Figure 4.
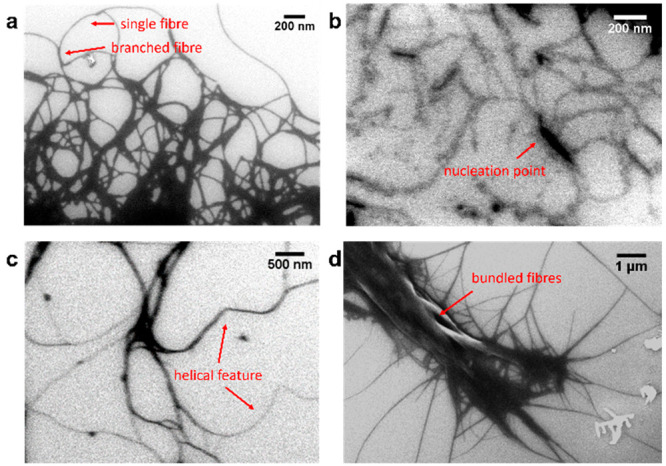
SEM images of (a) gel I, (b) gel II, (c) gel III, and (d) gel IV. The final concentration of all gels was 5 mM after dilution with tBuOAc. Samples were pipetted onto a silicon surface and freeze-dried for 1 h prior to imaging.
Infrared (IR) spectroscopy was then employed to assess the differences in supramolecular interactions leading to gelation and the structural differences of the gelators. We specifically emphasized on the secondary structures within the amide I region (1700–1600 cm–1). The ATR-FTIR spectra of the xerogels were measured one day after gelation (Figure 5a).
Figure 5.

(a) IR spectra of gels I–IV showing the amide I region (1600–1700 cm–1). (b) UV–vis absorption spectra. Dashed curves correspond to the spectra of precursors 1 (black), 2 (red), and 3 (blue) in solution and solid curves to their corresponding gels I, II, III, and IV (green). All measurements were performed on gels one day after gelation. For all samples, in solution and gel state, the concentration was 50 mM. The optical path length was 1 mm.
Previously reported diphenylalanine (Phe-Phe)-based supramolecular gels mainly exhibit a β-sheet secondary structure.27−31 In our case, for gel I consisting of protected diphenylalanine gelator molecules 1a and 1b, a strong band corresponding to a helical-shaped assembly in the higher organization is observed at 1660 cm–1,32 in addition to the expected β-sheet band at 1691 cm–1 (Figure 5a, black).33 The discrepancy with the literature possibly arises from the presence of two different gelators instead of one. The IR spectrum of gel III (Figure 5a, blue) is qualitatively similar to that of gel I and exhibits both a helical assembly (1655 cm–1) and a β-sheet motif (1682 cm–1). IR spectra recorded on xerogels and non-dried “wet” gels present similar secondary structures with minor shifts (Figures S63b and S65b). Therefore, the drying conditions do not significantly affect the secondary structures of the gels. Surprisingly, the IR spectrum of gel II only shows a band corresponding to a helical assembly at 1651 cm–1 (Figure 5a, red). Although previous literature reports that tripeptide (Phe-Phe-Phe)-based amphiphiles form organogels exhibiting β-sheet structures,34 this is not observed in our case. Similarly to gel I, the discrepancy is possibly due to the presence of a second gelator in the system. Systematic experimental and computational studies report the role and importance of Leu and Phe amino acids in the formation of secondary structures. While Leu is considered a helix former due to the rotational freedom of the γ-branched side chain,35 Phe is reported to favor the formation of β-sheets. However, some reports also highlight the importance of aromatic interactions for helix stabilization36 and that Phe is not necessarily a helix breaker. Additionally, in our systems, the bulky tert-butyl group might also disturb the system as a whole and force it to yield divergent arrangements of the building blocks such as helices.
As gel IV is formed by both precursors 1 and 3 with a 1:1 ratio, the IR profile of its xerogel within the amide I region should provide insight into whether self-sorting or co-assembly occurred. If self-sorting occurred, then the mere superimposition of the individual components’ spectra would be observed. On the contrary, a different spectrum would be observed in the case of co-assembly. A peak at 1658 cm–1 corresponding to a helical-shaped assembly is observed (Figure 5a, green plot), and unlike the gels of its individual components, no β-sheet motif is clearly visible. This suggests that a new structure has formed by the co-assembly of the different gelators.37
In addition to the amide I region, the shifts of specific vibrational bands provide important information on the intermolecular interactions occurring during gelation, i.e., π–π stacking and hydrogen bonding (Table 1). The bands corresponding to the C=C stretching of the phenyl rings appear in the gelators (neat powder) at 1495 cm–1 (1a), 1496 cm–1 (1b), 1495 cm–1 (2a, 2b), 1497 cm–1 (3a), and 1496 cm–1 (3b).38 In the gel phase, these bands shift toward shorter wavenumbers to 1493 cm–1 (gel I), 1494 cm–1 (gel II), and 1495 cm–1 (gel III and IV), which suggests the π–π stacking of the phenyl rings. Although the small shifts of the C=C bands are of the same magnitude as the resolution limit of the instrument, their presence still indicates the involvement of π–π stacking in intermolecular interactions. Additionally, the bands corresponding to the terminal amine are merged and downshifted (gel I: 3328 cm–1; gel II: 3349 cm–1; gel III: 3323 cm–1; gel IV: 3320 cm–1), suggesting hydrogen bonding.
Table 1. Phenyl Ring C=C Stretching and N-Terminal N–H Stretching Wavenumber Peaks (cm–1) of the Synthesized Gelators (Neat Powders) and Gel Samples.
| C=C
stretching phenyl rings |
N–H
stretching terminal amine |
|||||
|---|---|---|---|---|---|---|
| gel system | gelator a | gelator b | gel | gelator a | gelator b | gel |
| gel I | 1495 | 1496 | 1493 | 3392, 3346, 3288 | 3249, 3171 | 3328 |
| gel II | 1495 | 1495 | 1494 | 3370, 3266 | 3375, 3363 | 3349 |
| gel III | 1497 | 1496 | 1495 | 3364, 3319 | 3392, 3340 | 3323 |
| gel IV | 1495, 1497 | 1496, 1496 | 1495 | 3392, 3364, 3346, 3319, 3288 | 3392, 3340, 3249, 3171 | 3320 |
Complementarily to IR, UV–vis spectroscopy provides insights into the molecular packing of the gelators and has been extensively used to elucidate the interactions leading to self-assembly.39 More specifically, information about the π–π stacking of the phenylalanine moiety can be obtained, such as the type of aggregates (H or J), by comparison of the band shift between the spectrum of monomers in solution (interaction free) and the spectrum of the gel (aggregated monomers).40 The absorption spectra of the gelators for each system were obtained at the same concentration as those of the gels (50 mM) in acetonitrile to enhance solubility (Figure 5b, dashed curves). Solvatochromism (the shift of the absorption maxima depending on the solvent)41 and the concentration effect were verified by recording the UV spectra in tBuOAc at 50 and 5 mM concentrations.
However, no significant shifts were observed in either case (Figure S50). The center of the absorption band (λmax) at 258 nm for precursors 1–3 corresponds to the π → π* transition of the phenyl rings. As seen in Figure 5b, the λmax is slightly red-shifted upon gelation to 259 nm for gels I and III and 260 nm for gel II (plain curves). Because of the weakly absorbing character of the phenylalanine residues, compared to, for example, naphthalene and anthracene, the observed shifts are minor (1–2 nm).42 However, these results still suggest the π system overlapping in J-aggregate. Additionally, the loss of fine vibronic structure in the spectra of gel I and II is attributed to charge transfer over molecular aggregation.43−45 In our case, in contrast to most of the gel aggregation studies reported,43,46,47 we observed an absorption intensity increase over gelation, even after normalization due to scattering. This behavior could be explained by the aggregation-induced enhanced emission also related to the absorption profile.45,48−50 The red-edge tails observed for gels I and II most probably arise from the scattered light from the aggregates (opaque gels). No broadening is observed for gels III and IV containing just one phenylalanine unit due to its weak π → π* transition. These results correlate with the rheological profile of the gels; the weaker the π–π interactions, the softer the gels. Interestingly, the intensity of the band of gel IV is observed between those of its individual component’s gels. Red-shifts of the weaker, longer wavelength n → π* transitions corresponding to the C=O carbonyl and N–H amide groups,51 respectively, are observed to be significant as the precursors’ aromaticity increases (3 < 1 < 2). This suggests hydrogen-bonding-type interactions from these groups, consistent with the literature.28,52−54
Macroscopic Properties Assessment
Rheology experiments were performed on the gel systems to assess the correlation between the gelators’ structures and the materials’ mechanical properties. A material exhibiting a greater storage modulus G′ than the loss modulus G″ upon shear strain is considered “solid”. At the point this inverses, the material is considered “liquid”.55,56 For all gel systems, the measured elastic modulus (or storage modulus), G′, is greater than the viscous modulus (or loss modulus), G″, verifying the viscoelastic nature of the gels. For comparison purposes, Figure 6a presents the oscillatory frequency sweep (FS) experiments performed on gel specimens within the linear viscoelastic region (LVR) one day after gelation at a constant concentration of 50 mM and 1.0 equiv of accelerator. The G′ of both gels I (black) and II (red) is 420 and 320 kPa (Table 2), respectively, which is in the range of the bladder and gut tissue stiffness.57,58 These quantitatively similar values suggest that an additional aromatic moiety (in this case, phenylalanine) does not significantly alter the stiffness of the material.
Figure 6.

Rheological studies of the gels. (a) Frequency sweep measurements, under a constant shear strain γ (%) of gel I (black, γ = 0.02%), gel II (red, γ = 0.04%), gel III (blue, γ = 0.04%), and gel IV (green, γ = 0.1%). The concentration of the gel specimens was 50 mM. (b) Frequency sweep measurements of gel systems III (blue curves) and IV (green curves) at 25 mM (dashes), 50 mM (solid), and 100 mM (dots). Gelation was triggered with 1.0 equiv of H2SO4. (c) Frequency sweep measurements of gel I (black), II (red) III (blue), and IV (green) triggered by 1.0 equiv (solid) and 0.5 equiv (dashes) of H2SO4. All gels were diluted to a concentration of 50 mM. (d) Plot of the elastic moduli (black) against the lifetime (red) of the gels as a function of the number of aromatic units in the precursor. Data markers refer to numerical values from experiments. The point at 1.5 aromatic units corresponds to the average amount of aromatic rings in the system per mole of precursor in gel IV, where the ratio between 1 and 3 is 1:1. Error bars are calculated by standard deviation. Details of the experimental conditions and amplitude sweep measurements are given in the Experimental Section.
Table 2. Comparison of the Stiffness (G′) and Elasticity (G′/G″ Cross Point) Values of Gels I–IV.
|
G′ (kPa) |
G′/G″ cross point (%) |
||||
|---|---|---|---|---|---|
| concn (M) | 0.5 equiv | 1.0 equiv | 0.5 equiv | 1.0 equiv | |
| gel I | 0.05 | 180 | 420 | 4.2 | 1.5 |
| gel II | 0.05 | 147 | 320 | no crossing points | |
| gel III | 0.025 | n.m.a | 10 | n.m. | 6.5 |
| 0.05 | 11 | 6.5 | 9 | 3 | |
| 0.1 | n.m. | 330 | n.m. | 2 | |
| gel IV | 0.05 | 52 | 20 | 4.5 | 3.5 |
| 0.1 | n.m. | 155 | n.m. | 1.2 | |
Not measured.
However, for gel III (blue) bearing an aliphatic leucine unit instead of an aromatic phenylalanine unit, the G′ value of 6.5 kPa indicates that lower aromaticity of the gelators yields a softer material (approximately 60-fold), which is in the range of the lung and liver tissue stiffness.57,58 Previous studies show that a difference in the number of hydrophobic aliphatic carbons in the side chains of the gelators also affects the stiffness of the corresponding materials. However, this effect is minor (2–10-fold increase of G′ depending on the side chain length) to the observed 60-fold increase when adding three carbons and the aromatic character between leucine and phenylalanine.59,60 Despite a lower stiffness, gel III exhibits 2-fold more elastic behavior than gel I (Table 2). The elasticity was assessed by comparison of the G′/G″ cross points of the amplitude sweep measurements. A higher elasticity is indicated by the materials’ resistance to shear strain, as the cross point is shifted toward higher shear strain (γ%) values. Interestingly, the amplitude sweep measurements of gel II exhibit no crossing points between G′ and G″, although the curves seem to merge at the maximum shear strain γ of 100%, indicating that a crossing point would be observed over this value, as reported earlier for highly elastic gels.61 The additional phenylalanine motif in precursor 2 provides a higher hydrophobicity and more available sites for π–π stacking than that in precursor 1 (two phenylalanine motifs), giving more stability to the molecular aggregation during gelation. Although more intense π–π stacking yields stiffer materials, the G′ values are quantitatively similar for gels I and II, while the elasticity is higher for gel II than for gel I. This suggests that the fiber arrangement (microenvironment) has more impact on the material properties than the molecular interactions. In addition, the G′ value of 20 kPa for the multicomponent gel IV (green), although quantitatively closer to gel III than gel I, suggests that a new structure has formed, implying co-assembly of the gelators.24
To further investigate the effect of gelation conditions on the stiffness of the materials, we measured the elastic modulus G′ at different concentrations of gel III (monocomponent) and gel IV (multicomponent, Figure 6b). An increase in the precursor concentration yields a stiffer material in both cases (approximately 10–40-fold higher G′ value), along with a decrease in the elastic behavior (Table 2).
Similar experiments were performed by changing the amount of accelerator (0.5 or 1.0 equiv) to determine whether the precursor concentration of the precursor or that of the accelerator has a more significant effect on the materials’ properties. For all four gel systems, although a quantitatively similar G′ value was observed (approximately 3-fold, Figure 6c), the elasticity was found to increase by 1.5–2-fold in gels formed with 0.5 equiv of accelerator. Therefore, we suggest that the mechanical properties of the gels are tunable within a wide range of stiffness by modifying the precursor structure, i.e., the number of aromatic rings, concentration, and the amount of accelerator used to trigger gelation.
The phase-transition temperature (Tgel–sol) indicates the thermal stability of the different materials. Gel samples (viscoelastic gel behavior initially confirmed by rheological measurements) were gradually heated by 5 °C increments at 10 min intervals. The vial inversion method was used to verify the gel-to-sol (i.e., the complete transition from self-supporting gel to free-flowing solution) and gel-to-sol-to-gel transitions (Supporting Information, Section 2.4), assuming that after thermal breaking the viscoelastic nature of the gels remains identical with that of the gels initially assessed by oscillatory frequency sweep measurements. The observed Tgel–sol was found to be proportional to the number of aromatic units within the gelator structure (gel III: 40 °C; gel I: 45 °C; and gel II: 55 °C at 50 mM) and to the gel concentration (10 °C increase from 25 to 100 mM, Table S1 and Figure S51). These results suggest that the fibrous network and consequently the bulk material stiffen with the increase in the aromatic character of the gelators. However, the observed temperature for gel IV is lower than that of its individual components, in contrast to the rheological data. This behavior may arise from differences between the fibrous network (related to the Tgel–sol) and the π–π interactions (related to the gel stiffness).42
Numerical values of the lifetime (Figure 6d, red y-axis) compared to the mechanical properties (Figure 6d, black y-axis) of the gels, when plotted as a function of the number of aromatic units in the precursor structure, highlight a common trend. The evolution of the stiffness is found to be inversely proportionally to that of the lifetime of the materials, that is, stiff materials have a short lifetime. To ensure the reproducibility of the measurements and the reliability of the results, lifetime monitoring and rheological measurements were performed in triplicate. Systematic studies will be beneficial in the future to corroborate these findings for an extended set of data (gelation conditions, precursors, and acid equivalent). This observation can be assessed based on two factors: the gelators’ solubility in the secondary solvent, tBuOH, and the material’s elasticity. Indeed, control experiments showed that gelators 1a, 2a, and 3a are soluble in tBuOH, whereas their deprotected counterparts 1b, 2b, and 3b are insoluble. Therefore, with time, the in situ formed tBuOH will progressively dissolve the monoprotected gelator 1a, 2a, and 3a until the collapse of the material, as verified and described above. Considering this and the identical reaction cycle for all gel systems depicted in Figure 2b, the mechanical properties of the gels are potential reasons for the different lifetimes. As the fibrous network is progressively dissolved by tBuOH, a more elastic material remains self-supporting for a longer period of time before collapsing.
Conclusion
We developed solvent-induced transient self-assembly for the formation of a transient gel materials. A diprotected precursor molecule forms in situ two gelator building blocks (N-deprotected/C-protected and N-/C-deprotected) through activation by an accelerator, while the primary solvent chemically participates in the interconversion cycle between the two activated gelators through the generation of the secondary solvent. Additionally, the secondary solvent progressively solubilizes the fibrous network, leading to the collapse of the material over time. The gelation trigger and subsequent mechanism are related only to the precursor’s C-terminus protective group and the solvent, therefore offering a wide range of potential gelator candidates for the gelation of peptide-based systems. UV–vis spectroscopic data correlate the strength of the intermolecular interactions to the mechanical properties of the materials. Gels exhibiting greater stiffness showed greater absorption intensity increase and loss of vibronic structure, and vice versa, which can further be attributed to the prevalence of the π–π stacking interactions. IR spectroscopy, rheology, thermal stability, and lifetime studies highlighted the co-assembly of the gelators in the multicomponent gel IV. Additionally, the lifetimes of the gels are found to be inversely proportional to their mechanical properties. This work provides important insights into the formation of transient peptide-based supramolecular gels. The biocompatibility and versatility of modified amino acid-based gelators can be exploited, along with gelation in water-based solutions, to serve targeted medical purposes. The hydrolysis/esterification cycles proposed in this work could be used for developing hydrogels with a wide range of mechanical and transient properties, using, for instance, external tert-butylated compounds as a supplier of the tBu group to support the out-of-equilibrium self-assembly. The solvent-induced self-assembly can, therefore, provide interesting alternatives for responsive gelation and self-abolishing materials requiring a definite lifetime, such as delivery systems exhibiting a burst release. Alternatively, the self-abolishing character of these materials could be used for designing temporary ink devices. Thorough dynamic studies on the kinetics of gelator and secondary solvent formation, for instance, by high-pressure liquid chromatography14 would be beneficial to get insights into the in-depth acid-accelerated processes.
Acknowledgments
The authors acknowledge the Jane and Aatos Erkko Foundation for supporting the current work. J.R.H. thanks the UKRI Future Leaders Fellowship for funding (MR/T020415/1).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemmater.3c02327.
Materials, synthesis and characterization data of precursors and gels, gelation protocol, 1H and 13C NMR, IR, and UV–vis spectra, rheology amplitude sweep plots, Tgel–sol, gelation concentration screening (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Singh N.; Formon G. J. M.; De Piccoli S.; Hermans T. M. Devising Synthetic Reaction Cycles for Dissipative Nonequilibrium Self-Assembly. Adv. Mater. 2020, 32, 1–6. 10.1002/adma.201906834. [DOI] [PubMed] [Google Scholar]
- Van Rossum S. A. P.; Tena-Solsona M.; Van Esch J. H.; Eelkema R.; Boekhoven J. Dissipative Out-of-Equilibrium Assembly of Man-Made Supramolecular Materials. Chem. Soc. Rev. 2017, 46, 5519–5535. 10.1039/C7CS00246G. [DOI] [PubMed] [Google Scholar]
- Ragazzon G.; Prins L. J. Energy Consumption in Chemical Fuel-Driven Self-Assembly. Nat. Nanotechnol. 2018, 13, 882–889. 10.1038/s41565-018-0250-8. [DOI] [PubMed] [Google Scholar]
- Rieß B.; Boekhoven J. Applications of Dissipative Supramolecular Materials with a Tunable Lifetime. ChemNanoMat 2018, 4, 710–719. 10.1002/cnma.201800169. [DOI] [Google Scholar]
- Heuser T.; Weyandt E.; Walther A. Biocatalytic Feedback-Driven Temporal Programming of Self-Regulating Peptide Hydrogels. Angew. Chem., Int. Ed. 2015, 54, 13258–13262. 10.1002/anie.201505013. [DOI] [PubMed] [Google Scholar]
- Wang G.; Liu S. Strategies to Construct a Chemical-Fuel-Driven Self-Assembly. ChemSystemsChem 2020, 2, e1900046 10.1002/syst.201900046. [DOI] [Google Scholar]
- Vintiloiu A.; Leroux J. C. Organogels and Their Use in Drug Delivery - A Review. J. Controlled Release 2008, 125, 179–192. 10.1016/j.jconrel.2007.09.014. [DOI] [PubMed] [Google Scholar]
- Hu B.; Yan H.; Sun Y.; Chen X.; Sun Y.; Li S.; Jing Y.; Li H. Organogels Based on Amino Acid Derivatives and Their Optimization for Drug Release Using Response Surface Methodology. Artificial Cells, Nanomed. Biotechnol. 2020, 48, 266–275. 10.1080/21691401.2019.1699833. [DOI] [PubMed] [Google Scholar]
- Esposito C. L.; Kirilov P.; Roullin V. G. Organogels, Promising Drug Delivery Systems: An Update of State-of-the-Art and Recent Applications. J. Controlled Release 2018, 271, 1–20. 10.1016/j.jconrel.2017.12.019. [DOI] [PubMed] [Google Scholar]
- Boekhoven J.; Brizard A. M.; Kowlgi K. N. K.; Koper G. J. M.; Eelkema R.; Van Esch J. H. Dissipative Self-Assembly of a Molecular Gelator by Using a Chemical Fuel. Angew. Chem., Int. Ed. 2010, 49, 4825–4828. 10.1002/anie.201001511. [DOI] [PubMed] [Google Scholar]
- De S.; Klajn R. Dissipative Self-Assembly Driven by the Consumption of Chemical Fuels. Adv. Mater. 2018, 30, 1–6. 10.1002/adma.201706750. [DOI] [PubMed] [Google Scholar]
- Singh N.; Lainer B.; Formon G. J. M.; De Piccoli S.; Hermans T. M. Re-Programming Hydrogel Properties Using a Fuel-Driven Reaction Cycle. J. Am. Chem. Soc. 2020, 142, 4083–4087. 10.1021/jacs.9b11503. [DOI] [PubMed] [Google Scholar]
- Kriebisch B. A. K.; Jussupow A.; Bergmann A. M.; Kohler F.; Dietz H.; Kaila V. R. I.; Boekhoven J. Reciprocal Coupling in Chemically Fueled Assembly: A Reaction Cycle Regulates Self-Assembly and Vice Versa. J. Am. Chem. Soc. 2020, 142, 20837–20844. 10.1021/jacs.0c10486. [DOI] [PubMed] [Google Scholar]
- Schnitter F.; Bergmann A. M.; Winkeljann B.; Rodon Fores J.; Lieleg O.; Boekhoven J. Synthesis and Characterization of Chemically Fueled Supramolecular Materials Driven by Carbodiimide-Based Fuels. Nat. Protoc. 2021, 16, 3901–3932. 10.1038/s41596-021-00563-9. [DOI] [PubMed] [Google Scholar]
- Boekhoven J.; Hendriksen W. E.; Koper G. J. M.; Eelkema R.; Van Esch J. H. Transient Assembly of Active Materials Fueled by a Chemical Reaction. Science 2015, 349, 1075–1080. 10.1126/science.aac6103. [DOI] [PubMed] [Google Scholar]
- Liu M.; Creemer C. N.; Reardon T. J.; Parquette J. R. Light-Driven Dissipative Self-Assembly of a Peptide Hydrogel. Chem. Commun. 2021, 57, 13776–13779. 10.1039/D1CC04971B. [DOI] [PubMed] [Google Scholar]
- Deng J.; Bezold D.; Jessen H. J.; Walther A.. Light-Powered, yet Chemically Fueled, Dissipative DNA Systems with Transient Lifecycles. 2019. 10.26434/chemrxiv.10453769.v1. [DOI]
- Selmani S.; Schwartz E.; Mulvey J. T.; Wei H.; Grosvirt-Dramen A.; Gibson W.; Hochbaum A. I.; Patterson J. P.; Ragan R.; Guan Z. Electrically Fueled Active Supramolecular Materials. J. Am. Chem. Soc. 2022, 144, 7844–7851. 10.1021/jacs.2c01884. [DOI] [PubMed] [Google Scholar]
- Chevigny R.; Schirmer J.; Piras C. C.; Johansson A.; Kalenius E.; Smith D. K.; Pettersson M.; Sitsanidis E. D.; Nissinen M. Triggering a Transient Organo-Gelation System in a Chemically Active Solvent. Chem. Commun. 2021, 57, 10375–10378. 10.1039/D1CC04021A. [DOI] [PubMed] [Google Scholar]
- Hamley I. W. Self-Assembly, Bioactivity, and Nanomaterials Applications of Peptide Conjugates with Bulky Aromatic Terminal Groups. ACS Appl. Bio Mater. 2023, 6, 384–409. 10.1021/acsabm.2c01041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Chen L.; Zhang M.; Yi T. Low-Molecular-Mass Gels Responding to Ultrasound and Mechanical Stress: Towards Self-Healing Materials. Chem. Soc. Rev. 2014, 43, 5346–5371. 10.1039/C4CS00066H. [DOI] [PubMed] [Google Scholar]
- Maity M.; Maitra U. Supramolecular Gels from Conjugates of Bile Acids and Amino Acids and Their Applications. Eur. J. Org. Chem. 2017, 2017, 1713–1720. 10.1002/ejoc.201601616. [DOI] [Google Scholar]
- Tang J. D.; Mura C.; Lampe K. J. Stimuli-Responsive, Pentapeptide, Nanofiber Hydrogel for Tissue Engineering. J. Am. Chem. Soc. 2019, 141, 4886–4899. 10.1021/jacs.8b13363. [DOI] [PubMed] [Google Scholar]
- Draper E. R.; Adams D. J. How Should Multicomponent Supramolecular Gels Be Characterised?. Chem. Soc. Rev. 2018, 47, 3395–3405. 10.1039/C7CS00804J. [DOI] [PubMed] [Google Scholar]
- Mears L. L. E.; Draper E. R.; Castilla A. M.; Su H.; Zhuola; Dietrich B.; Nolan M. C.; Smith G. N.; Doutch J.; Rogers S.; Akhtar R.; Cui H.; Adams D. J. Drying Affects the Fiber Network in Low Molecular Weight Hydrogels. Biomacromolecules 2017, 18, 3531–3540. 10.1021/acs.biomac.7b00823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raeburn J.; Adams D. J. Multicomponent Low Molecular Weight Gelators. Chem. Commun. 2015, 51, 5170–5180. 10.1039/C4CC08626K. [DOI] [PubMed] [Google Scholar]
- Chevigny R.; Sitsanidis E. D.; Schirmer J.; Hulkko E.; Myllyperkiö P.; Nissinen M.; Pettersson M. Nanoscale Probing of the Supramolecular Assembly in a Two-Component Gel by Near-Field Infrared Spectroscopy. Chem.—Eur. J. 2023, 29, e202300155 10.1002/chem.202300155. [DOI] [PubMed] [Google Scholar]
- Huang R.; Su R.; Qi W.; Zhao J.; He Z. Hierarchical, Interface-Induced Self-Assembly of Diphenylalanine: Formation of Peptide Nanofibers and Microvesicles. Nanotechnology 2011, 22, 245609 10.1088/0957-4484/22/24/245609. [DOI] [PubMed] [Google Scholar]
- Yan X.; Zhu P.; Li J. Self-Assembly and Application of Diphenylalanine-Based Nanostructures. Chem. Soc. Rev. 2010, 39, 1877–1890. 10.1039/b915765b. [DOI] [PubMed] [Google Scholar]
- Marchesan S.; Vargiu A. V.; Styan K. E. The Phe-Phe Motif for Peptide Self-Assembly in Nanomedicine. Molecules 2015, 20, 19775–19788. 10.3390/molecules201119658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X.; Cui Y.; He Q.; Wang K.; Li J. Organogels Based on Self-Assembly of Diphenylalanine Peptide and Their Application to Immobilize Quantum Dots. Chem. Mater. 2008, 20, 1522–1526. 10.1021/cm702931b. [DOI] [Google Scholar]
- Barth A. Infrared Spectroscopy of Proteins. Biochimic. Biophys. Acta Bioenerg. 2007, 1767, 1073–1101. 10.1016/j.bbabio.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Barth A.; Zscherp C. What Vibrations Tell Us about Proteins. Q. Rev. Biophys. 2002, 35, 369–430. 10.1017/S0033583502003815. [DOI] [PubMed] [Google Scholar]
- Ariga K.; Kikuchi J. I.; Naito M.; Yamada N. FT-IR, TEM, and AFM Studies of Supramolecular Architecture Formed by Tripeptide-Containing Monoalkyl Amphiphiles. Polym. Adv. Technol. 2000, 11, 856–864. . [DOI] [Google Scholar]
- Lyu P. C.; Sherman J. C.; Chen A.; Kallenbach N. R. Alpha-Helix Stabilization by Natural and Unnatural Amino Acids with Alkyl Side Chains. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 5317–5320. 10.1073/pnas.88.12.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield S. M.; Patel P. R.; Waters M. L. Contribution of Aromatic Interactions to α-Helix Stability. J. Am. Chem. Soc. 2002, 124, 9751–9755. 10.1021/ja026668q. [DOI] [PubMed] [Google Scholar]
- Abul-Haija Y. M.; Roy S.; Frederix P. W. J. M.; Javid N.; Jayawarna V.; Ulijn R. V. Biocatalytically Triggered Co-Assembly of Two-Component Core/Shell Nanofibers. Small 2014, 10, 973–979. 10.1002/smll.201301668. [DOI] [PubMed] [Google Scholar]
- Barth A. The Infrared Absorption of Amino Acid Side Chains. Prog. Biophys. Mol. Biol. 2000, 74, 141–173. 10.1016/S0079-6107(00)00021-3. [DOI] [PubMed] [Google Scholar]
- Yu G.; Yan X.; Han C.; Huang F. Characterization of Supramolecular Gels. Chem. Soc. Rev. 2013, 42, 6697–6722. 10.1039/c3cs60080g. [DOI] [PubMed] [Google Scholar]
- Hestand N. J.; Spano F. C. Expanded Theory of H- and J-Molecular Aggregates: The Effects of Vibronic Coupling and Intermolecular Charge Transfer. Chem. Rev. 2018, 118, 7069–7163. 10.1021/acs.chemrev.7b00581. [DOI] [PubMed] [Google Scholar]
- Mayerhöfer T. G.; Popp J. Beyond Beer’s Law: Revisiting the Lorentz-Lorenz Equation. ChemPhysChem 2020, 21, 1218–1223. 10.1002/cphc.202000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairi P.; Roy B.; Routh P.; Sen K.; Nandi A. K. Self-Sustaining, Fluorescent and Semi-Conducting Co-Assembled Organogel of Fmoc Protected Phenylalanine with Aromatic Amines. Soft Matter 2012, 8, 7436–7445. 10.1039/c2sm25673h. [DOI] [Google Scholar]
- Ghosh S.; Li X.-Q.; Stepanenko V.; Würthner F. Control of H- and J-Type π Stacking by Peripheral Alkyl Chains and Self-Sorting Phenomena in Perylene Bisimide Homo- and Heteroaggregates. Chem.—Eur. J. 2008, 14, 11343–11357. 10.1002/chem.200801454. [DOI] [PubMed] [Google Scholar]
- Hisaki I.; Shigemitsu H.; Sakamoto Y.; Hasegawa Y.; Okajima Y.; Nakano K.; Tohnai N.; Miyata M. Octadehydrodibenzo[12]Annulene-Based Organogels: Two Methyl Ester Groups Prevent Crystallization and Promote Gelation. Angew. Chem., Int. Ed. 2009, 48, 5465–5469. 10.1002/anie.200900501. [DOI] [PubMed] [Google Scholar]
- Pacheco-Liñán P. J.; Martín C.; Alonso-Moreno C.; Juan A.; Hermida-Merino D.; Garzón-Ruíz A.; Albaladejo J.; Van Der Auweraer M.; Cohen B.; Bravo I. The Role of Water and Influence of Hydrogen Bonding on the Self-Assembly Aggregation Induced Emission of an Anthracene-Guanidine-Derivative. Chem. Commun. 2020, 56, 4102–4105. 10.1039/D0CC00990C. [DOI] [PubMed] [Google Scholar]
- Ishi-i T.; Murakami K.; Imai Y.; Mataka S. Self-Assembled Fluorescent Hexaazatriphenylenes That Act as a Light-Harvesting Antenna. J. Org. Chem. 2006, 71, 5752–5760. 10.1021/jo060768n. [DOI] [PubMed] [Google Scholar]
- Wagner W.; Wehner M.; Stepanenko V.; Ogi S.; Würthner F. Living Supramolecular Polymerization of a Perylene Bisimide Dye into Fluorescent J-Aggregates. Angew. Chem., Int. Ed. 2017, 129, 16224–16228. 10.1002/ange.201709307. [DOI] [PubMed] [Google Scholar]
- Dawn A.; Shiraki T.; Haraguchi S.; Sato H.; Sada K.; Shinkai S. Transcription of Chirality in the Organogel Systems Dictates the Enantiodifferentiating Photodimerization of Substituted Anthracene. Chem.—Eur. J. 2010, 16, 3676–3689. 10.1002/chem.200902936. [DOI] [PubMed] [Google Scholar]
- Hong Y.; Lam J. W. Y.; Tang B. Z. Aggregation-Induced Emission: Phenomenon, Mechanism and Applications. Chem. Commun. 2009, 29, 4332–4353. 10.1039/b904665h. [DOI] [PubMed] [Google Scholar]
- An B.-K.; Lee D.-S.; Lee J.-S.; Park Y.-S.; Song H.-S.; Park S. Y. Strongly Fluorescent Organogel System Comprising Fibrillar Self-Assembly of a Trifluoromethyl-Based Cyanostilbene Derivative. J. Am. Chem. Soc. 2004, 126, 10232–10233. 10.1021/ja046215g. [DOI] [PubMed] [Google Scholar]
- Skrabania K.; Miasnikova A.; Bivigou-Koumba A. M.; Zehm D.; Laschewsky A. Examining the UV-Vis Absorption of RAFT Chain Transfer Agents and Their Use for Polymer Analysis. Polym. Chem. 2011, 2, 2074–2083. 10.1039/c1py00173f. [DOI] [Google Scholar]
- Ravarino P.; Di Domenico N.; Barbalinardo M.; Faccio D.; Falini G.; Giuri D.; Tomasini C. Fluorine Effect in the Gelation Ability of Low Molecular Weight Gelators. Gels 2022, 8, 98. 10.3390/gels8020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Hu X.; Chen J.; Wei Z.; Song C.; Huang R. N-(9-Fluorenylmethoxycarbonyl)-L-Phenylalanine/Nano-Hydroxyapatite Hybrid Supramolecular Hydrogels as Drug Delivery Vehicles with Antibacterial Property and Cytocompatibility. J. Mater. Sci.: Mater. Med. 2020, 31, 73. 10.1007/s10856-020-06410-9. [DOI] [PubMed] [Google Scholar]
- Das T.; Häring M.; Haldar D.; Díaz Díaz D. Phenylalanine and Derivatives as Versatile Low-Molecular-Weight Gelators: Design, Structure and Tailored Function. Biomater. Sci. 2018, 6, 38–59. 10.1039/C7BM00882A. [DOI] [PubMed] [Google Scholar]
- Aufderhorst-Roberts A.; Frith W. J.; Kirkland M.; Donald A. M. Microrheology and Microstructure of Fmoc-Derivative Hydrogels. Langmuir 2014, 30, 4483–4492. 10.1021/la5005819. [DOI] [PubMed] [Google Scholar]
- Arnedo-Sánchez L.; Nonappa; Bhowmik S.; Hietala S.; Puttreddy R.; Lahtinen M.; De Cola L.; Rissanen K. Rapid Self-Healing and Anion Selectivity in Metallosupramolecular Gels Assisted by Fluorine-Fluorine Interactions. Dalton Trans. 2017, 46, 7309–7316. 10.1039/C7DT00983F. [DOI] [PubMed] [Google Scholar]
- Guimarães C. F.; Gasperini L.; Marques A. P.; Reis R. L. The Stiffness of Living Tissues and Its Implications for Tissue Engineering. Nat. Rev. Mater. 2020, 5, 351–370. 10.1038/s41578-019-0169-1. [DOI] [Google Scholar]
- Handorf A. M.; Zhou Y.; Halanski M. A.; Li W. J. Tissue Stiffness Dictates Development, Homeostasis, and Disease Progression. Organogenesis 2015, 11, 1–15. 10.1080/15476278.2015.1019687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azyat K.; Makeiff D.; Smith B.; Wiebe M.; Launspach S.; Wagner A.; Kulka M.; Godbert N. The Effect of Branched Alkyl Chain Length on the Properties of Supramolecular Organogels from Mono-N-Alkylated Primary Oxalamides. Gels 2023, 9, 5. 10.3390/gels9010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama T.; Harada Y.; Hase T.; Mori S.; Kimura S.; Yokoya M.; Yamanaka M. Effect of Alkyl Chain Length of N -Alkyl- N′ -(2-benzylphenyl)Ureas on Gelation. Chem.—Asian J. 2021, 16, 1750–1755. 10.1002/asia.202100433. [DOI] [PubMed] [Google Scholar]
- Öhrlund Å. Evaluation of Rheometry Amplitude Sweep Cross-Over Point as an Index of Flexibility for HA Fillers. J. Cosmetics. Dermatological Sciences and Applications 2018, 8, 47–54. 10.4236/jcdsa.2018.82008. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.