

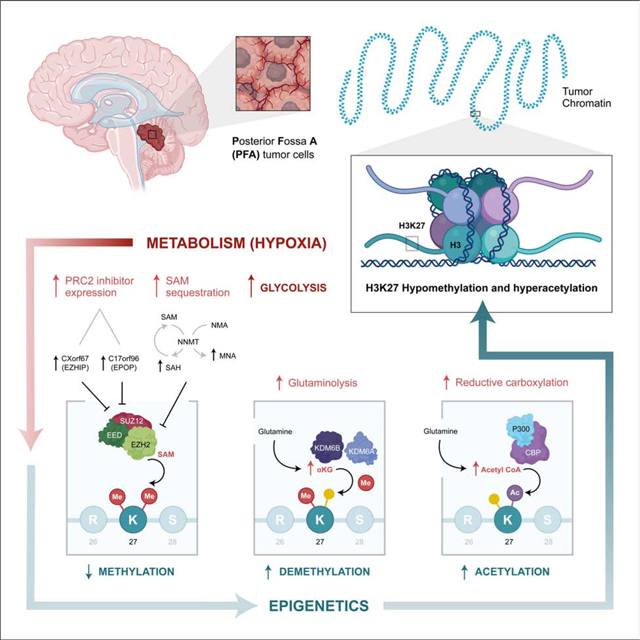
SUMMARY
Posterior fossa A (PFA) ependymomas are lethal malignancies of the hindbrain in infants and toddlers. Lacking highly recurrent somatic mutations, PFAs are proposed as an epigenetically driven tumor for which model systems are lacking. Here, we demonstrate that PFAs are maintained under hypoxia, associated with restricted availability of specific metabolites to diminish histone methylation and increase histone demethylation and acetylation at histone 3 lysine 27 (H3K27). PFAs initiate from a cell lineage in the first trimester of human development that resides in restricted oxygen. Unlike other ependymomas, transient exposure of PFA cells to ambient oxygen induces irreversible cellular toxicity. PFA tumors exhibit a low basal level of H3K27me3 and, paradoxically, inhibition of H3K27 methylation specifically disrupts PFA growth. Targeting metabolism and/or the epigenome presents a unique opportunity for rational therapy for infants with PFA ependymoma.
Graphical Abstract
INTRODUCTION
Ependymomas are malignant glial tumors that occur throughout the central nervous system (Thompson et al., 2015). Of the nine distinct molecular types of ependymoma, Posterior Fossa A (PFA) ependymomas, found in the hindbrain of infants and young children, are the most prevalent type (Ramaswamy and Taylor, 2016). Current standard-of-care remains neurosurgical resection followed by radiotherapy, but numerous clinical trials have failed to identify life-extending chemotherapies (Ramaswamy et al., 2016). Up to 40% of children with PFA ependymoma succumb to their disease and survivors are often left disabled due to toxicity from the tumor and treatment (Ramaswamy and Taylor, 2016).
Unlike most cancers, PFA ependymomas rarely harbor recurrent somatic single nucleotide variants (sSNVs) or copy number aberrations, but rather display epigenetic dysregulation, including abnormalities of DNA methylation and histones modifications (Mack et al., 2014; Pajtler et al., 2015a). A small minority of PFA tumors harbor somatic mutations in CXORF67 (EZHIP) or HF3A K27M mutations, which have previously been described in diffuse midline gliomas and are associated with H3K27 hypomethylation, which is also observed in PFA tumors (Hübner et al., 2019; Jain et al., 2019; Pajtler et al., 2018; Panwalkar et al., 2017; Ryall et al., 2017).
Research on PFA ependymoma has been hampered due to the lack of representative cell lines, xenografts, or animal models that recapitulate the disease, leading to a lack of therapeutics being brought forward to clinical trials (Aldape et al., 2019). We recently demonstrated that the lineage of origin for PFA ependymoma exists only very early in human development, during the first trimester when the microenvironment of the developing hindbrain is highly hypoxic (Vladoiu et al., 2019). Transcriptional analyses show that PFA ependymomas, but not other molecular ependymoma variants, have elevated hypoxic signaling (Pajtler et al., 2015b). Consequently, we hypothesized that the metabolic environment of the developing human fetal hindbrain contributes to PFA ependymomas through intermediary metabolism on the epigenome, offering an opportunity for novel targeted therapy.
RESULTS
Hypoxia is essential for PFA ependymoma maintenance
To generate patient-derived models, we interrogated the role of oxygen levels on the growth of surgical samples of PFA ependymomas. Dissociated single cells (1×106) from PFA ependymoma surgical biopsies were cultured under varying oxygen concentrations (21%, 10%, 5%, 1% and 0.2% O2). Immediate culture in 1% O2 allowed the establishment of PFA continuous primary cell cultures (9/10 specimens), while atmospheric O2(21%) largely failed (1/10, p = 0.0010) (Figure 1A). The single PFA culture that was established in 21% O2 displayed faster growth in 1% O2 (data not shown). After three generations in vitro, PFA cells in 1% vs. 21% O2 proliferated more with decreased markers of cellular senescence markers (p21WAF1) and apoptosis (Annexin V, propidium iodide) (Figures 1B-1G, Figure S1A-B). PFA cells, but not fetal neural stem cells (fNSCs) or supratentorial ependymoma (ST-EPN) cells, underwent G1 cell cycle arrest after exposure to 21% O2 (p=0.00008, Figure 1H). PFA cells cultured in 1% vs. 21% O2 displayed increased sphere formation in limiting dilution (Figure S1C). Even short-term exposure (4 days) of PFA cells to 21% O2 irreversibly inhibited proliferation, despite subsequent return to hypoxic conditions (Figure 1I). In a clinical cohort of PFA patients, a hypoxic gene signature informed poor prognosis (p=0.00134) (Figure 1J, Figure S1D). We conclude that maintenance of PFA ependymoma is highly dependent on a hypoxic microenvironment.
Figure 1 – PFA ependymoma mandates hypoxic conditions.

A) Patient derived PFA ependymoma primary cells undergo sustained growth in 1%, but not 21% oxygen. Cell lines were considered established if they could sustain 5 consecutive passages in culture, stalled if fewer than 5 passages, and failed if they never demonstrated growth in vitro. Two tailed Fisher’s exact test was used for statistical comparison.
B) Schematic representation of continuous culture in 1% versus 21% O2. A “generation” denotes a consecutive passage in each condition.
C) Proliferation of PFA cells cultured continuously in 1% O2 (blue) is significantly sustained and increased compared to 21% O2 (grey). Cell counts represent average viable cells remaining per plate at the end of the 3rd generation. Statistical comparison was performed using two-tailed t-test.
D) Crystal violet staining of 3rd generation PFA cells demonstrates high confluence of cells in 1% O2 compared to 21% O2. Each square is a representative image for the culture condition.
E) Senescence-associated β-Galactosidase staining of 3rd generation PFA cells demonstrates increased senescence in 21% O2, but not 1% O2. Blue colour indicates positive X-gal staining.
F) Immunoblot for p21Waf1 demonstrates increased expression in 21% compared to 1% O2.
G) Flow cytometry-based quantification of apoptosis using Annexin V and propidium iodide in PFA cell lines reveals a significant increase in apoptosis in 21% O2. Statistical comparisons were conducted using Welch’s t-test from technical triplicates for each sample.
H) Cell cycle analysis demonstrates an increase in G1 arrest of PFA cells, but not control STEPN or fetal neural stem cells (fNSCs), in 21% O2 versus 1% O2. Statistical comparison was performed using multiple t-testing for the G1 cell cycle phase between 1% vs. 21% oxygen of each cell type.
I) PFA cells cultured in 21% O2 for 4 days and then returned to 1% O2, display an irreversible decrease in proliferation. Statistical comparison was performed using twotailed t-test.
J) Poor outcome among PFA patients with a hypoxic gene signature (MSigDB). Kaplan-Meier survival curve stratified by hypoxia signature (high = red, low = blue) is correlated with worse overall survival in a cohort of 18 PFA patients. Statistical comparison was performed using the log-rank test after FDR correction.
PFA ependymomas exhibit a unique metabolism
Given the association of hypoxia with maintenance of tumorigenesis and the modulation of cancer metabolism in tumors, we interrogated six PFA cultures and their corresponding surgical specimens with whole genome and RNA sequencing (RNAseq). RNAseq and proteomics were also compared in three primary cultures grown in 1% and 21% O2 (Figures 2A, 2B, S2, S3A-E, Table S1-3). Confirming our prior observations (Mack et al., 2014), we did not observe any recurrent somatic SNVs or copy number aberrations (Figure S2). PFA cells cultured in 1% vs. 21% O2 displayed altered transcription of genes important in regulation of the epigenome (e.g., CXorf67) and metabolism (e.g., SLC16A3, AK4, and ALDOC) (Figure 2A). Mass spectrometry identified a broad set of differentially expressed metabolic enzymes, including LDHA, PGK1, and PFKL (Figure 2B). At the transcriptional and protein levels, hypoxia response and glycolytic metabolism pathways were upregulated in 1% O2, while cellular respiration and electron transport chain genes were increased in 21% O2. Comparison in the methylation profiles of five primary patient derived cultures and their parental tissue demonstrated a strong correlation, confirming that cell cultures recapitulate the original tissue (Figure S3F). Mass spectrometry profiling of metabolites from surgical biopsies of 15 PFA ependymomas, 4 ST-EPNs, and 5 normal human pediatric brains demonstrated increased glycolysis, gluconeogenesis and pyruvate metabolism, pentose phosphate metabolism, arginine and proline metabolism, polyamine metabolism and fatty acid beta oxidation in PFA tissues, but not supratentorial ependymoma or normal brain (Figures 2C and S4). Thus, PFA ependymomas have metabolic regulation distinct from other ependymal brain tumors and normal brain.
Figure 2 – PFA ependymomas harbor a unique metabolic and global histone hypomethylation profile.

A) Volcano plot of differential gene expression analysis for PFA cells cultured in 1% versus 21% oxygen. Significantly differentially expressed genes are indicated in colour (log2foldchange > 1, Padj < 0.05).
B) Volcano plot of differential proteomic expression analysis for PFA cells cultured in 1% O2 versus 21% O2. Significantly differentially expressed proteins are indicated in colour (FDR < 0.05).
C) Integrative metabologram of gene expression and metabolite abundance for selected KEGG pathways in PFA tissue relative to ST-EPN tissue (derived from (Hakimi et al., 2016a)). Log2FoldChange shown on a set scale from −3.0 to 3.0.
D) Relative levels of histone K27 methylation and acetylation in PFA (n=5) versus ST-EPN surgical (n=3) biopsies assessed by post-translational modification histone-tail mass spectrometry.
E) Relative abundance of H3.1 and H3.2 K27 methylation and acetylation in cultured PFA, ST and (fNSCs) under 1% and 21% O2 as assessed by post-translational modification histone-tail mass spectrometry.
F) Increased glucose consumption rate of cultured PFA, as compared to ST and fNSCs, and in 1% versus 21% O2.
G) Treatment of PFA cells with a GLUT1 specific inhibitor, BAY-876 (1μM), perturbs PFA, but not fNSCs survival in 1% O2. Bar plot shows relative survival.
H) Glucose (50mM) supplementation rescues the growth defect of third generation PFA cells grown in 21% O2.
Data are displayed as mean ± SEM. *p < 0.05, **p < 0.01 and ***p < 0.001. Statistical comparisons were conducted using Welch’s t-test for E-H.
Hypoxia regulates the epigenome of PFA ependymoma
Leveraging the epigenetic dependencies of PFA ependymomas, we interrogated the chromatin regulation of PFA ependymomas under different oxygen conditions by measuring the abundance of all known histone tail post-translational modifications from PFA surgical biopsies, as well as PFA cultures in both 1% and 21% O2, using mass spectrometry. PFA surgical biopsies exhibited global loss of H3K27me3 and H3K27me2, with a concomitant increase of H3K27me1 on both H3.1/H3.2 and H3.3 histones (Figure 2D). PFA tumors had an increase in H3K27 acetylation as compared to ST-EPNs (Figure 2D). PFA ependymoma, but not ST-EPNs or fNSCs, exhibited decreased trimethylation and increased acetylation of both histones H3.1 and H3.2 on K27 when grown in 1% O2 as compared to 21% (Figures 2E). To validate these findings, we interrogated surgical ependymoma biopsy specimens for the distribution of hypoxia (as measured by immunohistochemical staining of carbonic anhydrase 9, CA9) and H3K27me3, demonstrating lower levels of H3K27 trimethylation in the hypoxic regions of PFA ependymoma, but not in supratentorial ependymoma controls (p=0.0195, Figure S5A). To validate the impact of oxygen tension on H3K27 trimethylation Cut&Run sequencing was performed. PFA cultures grown in 21% O2 possessed increased H3K27me3 marks, with changes in promoter silencing corresponding to RNA expression observed above (Figure 2A, Figure S5BD). Collectively, these results show that hypoxia maintains aberrant post-translational modifications of H3K27.
PFA ependymoma is addicted to glucose
The metabolic profiles of PFA ependymomas suggest that these cells have high levels of glycolysis compared to normal brain or other ependymal tumors (Figures 2C). Stable isotope tracing (U6-13C-glucose) gas chromatography-coupled mass spectrometry (GC-MS) demonstrated increased glucose consumption in 1% vs. 21% O2 for both PFA cells and other cell types (Figure 2F). However, relative glucose consumption was higher in PFA cells than in ST-EPN cells or fNSCs in 1% O2 (p = 0.0212 and 0.0039, respectively) (Figure 2F). Pharmacologic inhibition of glucose uptake by the glucose transporter GLUT1 using Bay-876 (1 μM) impaired growth of PFA cells compared to ST-EPN (Figure 2G). Glucose supplementation fully rescued the growth defect observed in PFA cells at 21% O2 (Figure 2H). Thus, high levels of glucose are both necessary and sufficient for growth of PFA cells, particularly at increased oxygen levels.
Intermediary products of PFA metabolism regulate histone tail methyltransferase activity
The hypomethylated and hyperacetylated epigenome observed in PFA ependymoma could be due to various mechanisms of histone modification. The methionine cycle is a key metabolic pathway that provides S-adenosyl methionine (SAM) as a substrate for methylating numerous proteins, including histones. Methionine cycle metabolites, including SAM, were found at similar levels in surgical biopsies of PFA and ST ependymoma (Figure 3A), but PFA cells cultured at 1% O2 display lower SAM levels compared to cells grown at 21% (p=0.0104), in contrast to ST-EPN, fNSCs or immortalized normal human astrocytes (iNHAs) (Figure 3B). Differences in levels of H3K27 trimethylation in PFA ependymoma between 1% and 21% O2 were not due to differences in the expression of core Polycomb Repressive Complex 2 (PRC2) complex genes (EZH2, EED, or SUZ12) (Figure 3C). However, two inhibitors of PRC2 activity, CXorf67 (EZHIP) and C17orf96 (EPOP), displayed elevated levels in PFA cells grown at 1% vs. 21% O2 (Figure 3C) (Beringer et al., 2016; Hübner et al., 2019; Jain et al., 2019). Decreased expression of EZHIP and EPOP in 21% O2 would be predicted to increase PRC2 activity with consequent increased trimethylation of H3K27. As SAM is a necessary substrate for the H3K27 methylase EZH2, we hypothesized that increased abundance of SAM in PFA cultures should result in growth inhibition (Figure 3D). Concordantly, exogenous treatment with SAM inhibited the growth of PFA cells, but not ST-EPN and iNHA cells, in a concentration-dependent manner (Figure 3E). Endogenous SAM is consumed by two alternate pathways: polyamine metabolism and the nicotinamide sink. SAM is consumed to generate polyamine metabolites through S-adenosylmethionine decarboxylase (SAMDC) activity or sequestered as 1-methylnicotinamide via the enzyme nicotinamide N-methyltransferase (NNMT) (Ulanovskaya et al., 2013). Inhibition of endogenous SAM consumption with Sardomozide (a SAMDC inhibitor) or JBSNF-000088 (an NNMT inhibitor) resulted in concentration-dependent growth inhibition of PFA ependymoma cells (Figures 3F and 3G). PFAs express high levels of the PRC2 inhibitors, EZHIP and EPOP, both in vitro and in vivo, with reduced levels of the EZH2 substrate SAM in vitro under hypoxic conditions, all contributing to maintaining an epigenome with low H3K27me3 levels.
Figure 3 – Hypoxia drives SAM deficiency, thereby perturbing PRC2-dependent methylation and maintaining H3K27 hypomethylation in PFA ependymoma.

A) Relative metabolite abundance of methionine cycle metabolites and intermediates, including the nicotinamide N-methyltransferase sink and polyamine metabolism, in PFA (n=15) versus ST-EPN (n=4) surgical biopsies. Significantly differentially abundant metabolites characterized by t-test with FDR correction (q-value < 0.05).
B) Hypoxia significantly decreases SAM abundance in PFA cells but not control ST-EPN, immortalized normal human astrocytes (iNHA) or fNSCs.
C) Hypoxia does not affect the transcript expression of PRC2 core subunits, but does increases the expression of PRC inhibitory genes, CXorf67 (EZHIP) and C17orf96 (EPOP), in 1% oxygen. Statistical comparison was performed using Wald test with Benjamini-Hochberg adjustment.
D) Cartoon depicting the relationship between metabolism of SAM (SAM consuming metabolic pathways) and the PRC2 complex activity during K27 methylation. Hypoxia mediated over expression of PRC2 complex inhibitory genes (blue) and targets/drugs (red) are indicated.
E) Supplementing exogenous SAM to PFA cells grown in 1% O2 diminishes survival in a dose dependent manner.
F) Dose dependent Sardomozide inhibition (7 days) of SAM decarboxylation (polyamine metabolism) diminishes PFA cell survival in 1% O2.
G) Dose dependent JBSNF-000088 inhibition (7 days) of the nicotinamide N-methyltransferase (methylation sink) perturbs PFA cell survival in 1% O2.
*p < 0.05 and **p < 0.01. Data are displayed as the mean ± SEM. Curves show logistic regression line of fit for E-G.
PFA metabolism fuels H3K27 histone demethylases
The activity of the H3K27 demethylases KDM6A and KDM6B are controlled, in part, by the concentration of the cofactor, α-ketoglutarate (αKG), and in particular by the ratio of αKG to succinate (Carey et al., 2015). The αKG/succinate ratio was much higher in PFA cells than in fNSCs, which decreased when PFA cells were shifted from 1% to 21% O2 (p = 0.0040, Figures 4A and 4B). No differences in the RNA expression levels of KDM6A or KDM6B were detected under 1% vs. 21% O2 (Figure 4C). 13C5-L-Glutamine tracing demonstrated that PFA cells preferentially underwent glutaminolysis under reduced Krebs cycle flux, producing αKG from glutamine and maintaining a high αKG/succinate ratio (p = 0.0023, Figures 4D and 4E), suggesting potential regulation of demethylase activity rather than total expression. In PFA cells, αKG produced from glutamate (m+5) was highly abundant in 1% O2, compared to m+3 or m+0 αKG which were abundant in 21%, consistent with the canonical forward activity of the Krebs cycle in 21% O2 (Figure S5).
Figure 4 – Hypoxia maintains histone lysine demethylase activity by increasing αKG abundance via glutamine catabolism in PFA ependymoma.

A) Snapshot of relative metabolite abundance of Krebs cycle intermediates in PFA (n=15) versus ST-EPN (n=4) surgical biopsies. Significantly differentially abundant metabolites characterized by t-test with FDR correction (q-value < 0.05).
B) PFA cells growing in 1% O2, exhibit significantly increased ratio of αKG/Succinate in PFA cells. Statistical comparison was performed using ratio paired t-test.
C) Histone lysine 27 demethylase gene expression is not differentially regulated by oxygen tension in PFA cells. Statistical comparison was performed using Wald test with Benjamini-Hochberg adjustment.
D) Cartoon depicting glutamine tracing in PFA cells growing in 1% O2. Labelled glutamine was used as the carbon source (13C5 light blue; unlabeled carbon dark blue). PFA cells preferentially undergo glutaminolysis and reductive carboxylation.
E) Labelled glutamine tracing of PFA cells reveals an elevated αKG:succinate ratio in 1% compared to 21% O2. Statistical analysis performed using two-tailed t-test with HolmSidak correction.
F) Cartoon depicting the roles of αKG and the KDMs in histone demethylation. Drugs which inhibit lysine 27 demethylase activity and their targets are in red, while exogenous αKG, which increases activity is in green.
G) Dose response of GSK-J4 inhibition (7 days) of KDM6A and KDM6B perturbs PFA cell survival in 1% O2.
H) Dose response of CB-839 inhibition (7 days) of GLS perturbs PFA cell survival in 1% O2.
I) Supplementing exogenous cell permeable dimethyl αKG rescues the growth defect of PFA cells cultured in 21% O2.
J) Supplementing exogenous cell permeable dimethyl αKG rescues PFA cell survival while under CB-839 inhibition in 1% O2.
*p < 0.05, **p < 0.01 and ****p < 0.0001. Data are displayed as the mean ± SEM. Curves show logistic regression line of fit for G-H.
We hypothesized that inhibiting either αKG production or KDM activity would diminish PFA growth, while exogenous αKG should rescue the effects of growth in 21% O2 (Figure 4F). CB839, an inhibitor of glutaminolysis, profoundly inhibited the growth of PFA cells, but not ST-EPN and iNHA cells (Figure 4G). GSK-J4, an inhibitor of the H3K27me3 demethylases KDM6A and KDM6B, demonstrated concentration-dependent growth inhibition of PFA cells, but not ST-EPN or iNHA controls (Figure 4H). Exogenous, αKG rescued the growth defect of PFA cells in 21% O2 (Figure 4I). To confirm that the inhibition of glutaminolysis by CB-839 was specific to αKG perturbation, we rescued PFA growth with exogenous αKG (Figure 4J). Thus, PFA cells are dependent on H3K27 demethylase activity maintained through a high αKG/succinate ratio generated by glutaminolysis and reduced Krebs cycle flux.
PFA undergoes reductive carboxylation to generate acetyl-CoA fueling H3K27 acetylation
Acetylation of H3K27 blocks the ability of EZH2 to methylate H3K27. Histone acetyltransferase (HAT) complexes consume acetyl-CoA, a product of intermediary metabolism, to acetylate histone tails. PFA surgical biopsies have very high levels of H3K27 acetylation, which diminishes upon exposure to 21% O2 (Figures 2D-E). The αKG/citrate ratio is a marker of reductive carboxylation, which results in the production of acetyl-CoA from citrate (Fendt et al., 2013; Lee et al., 2014). In PFA cells, but not fNSCs, the αKG/citrate ratio was higher in 1% vs. 21% O2 (Figures 5A and 5B). Expression of the HAT complex genes CREBBP and EP300 were unchanged between 1% and 21% O2 (Figure 5C).
Figure 5 – Hypoxia drives reverse TCA cycle activity to increase histone K27 acetylation in PFA ependymoma.

A) Relative metabolite abundance of Krebs cycle and glycolysis intermediates in PFA (n=15) versus ST-EPN (n=4) surgical biopsies. Significantly differentially abundant metabolites characterized by t-test with FDR correction (q-value < 0.05).
B) The αKG:citrate ratio is significantly lower in PFA cells in 21% versus 1% oxygen, which is not mirrored in fNSCs. Statistical comparison was performed using ratio paired t-test.
C) Expression of lysine acetyltransferase components CREBBP and p300 is not influenced by oxygen tension in PFA cells. Statistical comparison was performed using Wald test with Benjamini-Hochberg adjustment.
D) Cartoon depicting glutamine tracing in PFA cells growing in 1% O2. Labelled glutamine was used as the carbon source (13C5 light blue; unlabeled carbon dark blue). PFA cells preferentially undergo glutaminolysis and reductive carboxylation.
E) Labelled glutamine tracing of PFA cells demonstrates an increased aKG:citrate ratio, a marker of reductive carboxylation, in 1% O2. Statistical analysis performed using twotailed t-test with Holm-Sidak correction.
F) Cartoon depicting the roles of acetate and lysine 27 acetyltransferases in histone acetylation. Drugs and which inhibit lysine 27 acetyltransferase activity and their targets are in red, while exogenous acetate, which increases activity is in green.
G) Dose-dependent selective inhibition (7 days) of CREBBP/P300 by A485 diminishes PFA cell survival at 1% O2.
H) BMS-303141 (50μM) inhibition (7 days) of ACLY diminishes PFA cell survival in 1% O2. Statistical analysis was performed by t-test with Welch correction.
I) Addition of exogenous acetate (200μM and 2mM) rescues BMS-303141 inhibition of ACLY in PFA cells in 1% O2.
J) Addition of exogenous acetate (200μM and 2mM) rescues CB-839 inhibition of GLS in PFA cells in 1% O2.
****p < 0.0001. Data are displayed as the mean ± SEM. Curves show logistic regression line of fit for G,I,J.
We hypothesized that high levels of acetyl-CoA production could drive H3K27 acetylation in PFA cells. 13C5-L-Glutamine tracing demonstrated that in PFA cells, citrate was primarily produced by reductive carboxylation (m+5, m+3) in 1% O2, whereas citrate was consumed by forward activity of the Krebs cycle (m+4) in 21% (p < 0.0001, Figures 5D, 5E and S5). Production of cytosolic fumarate, malate, and aspartate (m+3) was detected in 1% O2 (Figure S6), which are produced when ATP citrate lyase (ACLY) converts citrate into acetyl-CoA. Thus, PFA ependymoma cells, but not ST-EPNs or fNSCs, consume glutamate and undergo reductive carboxylation, resulting in high levels of acetyl-CoA (Figures 5D, 5E, and S6). To test the functional relevance of our metabolic model, we determined the activity of P300/CREBBP acetyltransferase inhibitors and ACLY inhibitors against PFA ependymoma cells (Figure 5F). The P300-complex inhibitor A485 demonstrated a concentration-dependent inhibition of PFA cell growth, in contrast to ST-EPNs or iNHAs (Figure 5G). A485 treatment caused limited growth inhibition of diffuse intrinsic pontine glioma (DIPG) cells, a tumor type that similarly exhibits genome-wide H3K27 hypomethylation. In orthogonal experiments, pharmacologic inhibition of production of acetyl-CoA from citrate using the ACLY inhibitor BMS-303141 diminished the growth of PFA cells, but not ST-EPNs (Figure 5H). Blockade of ACLY can force cells to utilize external sources of acetate to create acetyl-coA via intracellular acetyl-CoA synthetases (Gao et al., 2016), exogenous acetate rescued the growth of PFA cells treated with the ACLY inhibitor in a concentration-dependent manner (Figure 5I). Consistent with our proposed model, supplementation of PFA cells with exogenous acetate rescued the growth inhibition mediated by the glutaminolysis inhibitor, CB-839 (Figure 5J). Collectively, we conclude that glutaminolysis, followed by reductive carboxylation, generates high levels of acetyl-CoA to maintain increased H3K27Ac.
Basal PRC2 activity is essential for PFA survival – the PFA Goldilocks model
We leveraged the ability to culture PFA cells to perform a genome-wide CRISPR knockout screen to identify essential genes in three patient-derived PFA cell cultures, compared to another malignant brain cancer, glioblastoma (GBM) (Figure 6A) (MacLeod et al., 2019). PFA cultures had total of 272 context-specific fitness genes (Z-score < 0.05, average PFA Bayes Factor > 4, average GBM Bayes Factor < 4) (Table S4). Three of the top ten essential genes specific to PFA ependymoma were EED, SUZ12, and EZH2, all core components of PRC2 (Figure 6A). Essential genes specific to PFA ependymoma by pathway analysis focused on histone lysine methylation (Figure 6A). The essentiality of PRC2 genes in PFA seems paradoxical, as our results above suggest that increasing H3K27 trimethylation decreases the fitness of PFA ependymoma (Figures 3–5), but while PFA cells are hypomethylated at H3K27, they display basal H3K27 trimethylation. We compared our PFA CRISPR screens to previously published screens on human fNSCs (Table S5) (MacLeod et al., 2019). A total of 316 essential genes were observed, with 128 genes overlapping with the PFA vs. GBM comparison (Table S6). These results suggest that a minimal basal level of H3K27me3 is essential for PFA survival and further attenuation of H3K27me3 decreases the fitness of PFA ependymoma cells. PFA cells have a unique epigenome, suggesting a model in which they thrive in a narrow Goldilocks zone, with deviation to either increased or decreased H3K27me3 levels leading to diminished cellular fitness (Figure 6B).
Figure 6 – Paradoxically, inhibition of the PRC2 complex also shows activity against PFA ependymoma.

A) Pathway analysis of significantly essential genes by genome-wide CRISPR knockout essentiality screen in PFA cells compared to GBM cells (z-score, p-value < 0.05). The top 10 hits ranked by average difference of quantile normalized Bayes Factor between PFA and GBM screens are listed. Genes associated with the PRC2 complex are denoted in red.
B) Cartoon depicting several mechanisms of K27 modification active in PFA ependymoma. Targeted inhibition of MAT2A by PF9366 and the PRC2 complex components EED and EZH2 by A395 and UNC1999 respectively are depicted.
C) Dose-dependent inhibition (7 days) of MAT2A by PF9366 inhibits SAM production, resulting in diminished PFA cell survival, while simultaneously augmenting iNHA control cell survival in 1% O2. ST, DIPG and fNSC control lines showed no effect upon treatment.
D) Dose-dependent inhibition (7 days) of EED by A395 diminishes PFA cell survival in 1% O2. An inactive analog of the A395 probe (dashed) did not affect PFA cell survival. ST, DIPG and fNSC control lines showed no effect upon treatment.
E) Dose-dependent inhibition (10 days) of EZH2 by UNC1999 diminishes PFA cell survival in 1% O2. ST, DIPG and fNSC control lines showed no effect upon treatment.
Curves show logistic regression line of fit for C-E.
Our results demonstrated that interventions regulating H3K27 methylation, demethylation, or acetylation that increase H3K27me3 levels diminish the fitness of PFA cells. To test our Goldilocks model, we inhibited H3K27 trimethylation through inhibition of SAM production using PF9366, an inhibitor of MAT2A, the enzyme responsible for generating SAM. PFA cells, but not other brain cell types, treated with PF9366 demonstrated inhibition of cell growth (Figure 6C). Inhibition of the PRC2 component EED using A395, but not the inactive control probe, diminished the growth of PFA cells (Figure 6D). Similarly, EZH2 inhibition using the compound UNC1999 readily inhibited PFA growth compared to other control lines (Figure 6E). Therefore, inhibition of either production of the methyl donor SAM or the activity of the PRC2 complex (through EED or EZH2) support our Goldilocks model of the PFA epigenome.
Normal fetal hindbrain cells in the PFA lineage of origin have a hypoxic phenotype
PFA ependymoma is a glial tumor that arise very early in the fetal gliogenic lineage from gliogenic progenitors and roof plate-like stem cells (Vladoiu et al., 2019). Murine roof plate-like stem cells were almost exclusively present at E10, while gliogenic progenitors have a peak prevalence at E16 (Figure 7A). Single cell RNA-seq data from different time points across murine hindbrain development demonstrated that hypoxia gene expression signatures correspond to the peak prevalence of these two cell populations (Figure 7B). Pathway analysis revealed that E16 gliogenic progenitors displayed strong transcriptional signatures of glycolysis, gluconeogenesis and monosaccharide metabolism, similar to the transcriptome of PFA ependymoma, but postnatal P0 gliogenic progenitors upregulated genes associated with electron transport chain and cellular respiration pathways (Figure 7C). As PFA ependymomas have a very low mutational burden and resemble normal fetal hindbrain cells, we hypothesized hindbrain.
Figure 7 – The normal glial lineage of origin of PFA in the hindbrain displays early hypoxia during normal development.

A) Relative frequency of gliogenic progenitors and roof-plate like stem cells across several embryonic timepoints. Frequency is calculated as number of cells of that type at each timepoint, relative to total number of cells of that type across all timepoints.
B) Violin plot of the signature score for hypoxia genes across the embryonic timepoints. Hypoxia signature set based on 15 genes (Aldoa, Aldoc, Bnip3, Bnip3l, Car9, Ddit4, Gpi1, Ldha, P4ha1, P4ha2, Pdk1, Pgk1, Tpi1, Vegfa, Zfp395). Set scale from −1 to 1.
C) Gene set enrichment analysis of mouse cerebellar gliogenic progenitors across four developmental timepoints, generated from scRNAseq. Cytoscape maps generated using q < 0.15, P < 0.05.
D) Heatmap of PFA enriched metabolites demonstrates a higher correlation to E16.5 compared to P2 embryonic timepoints. A total of 19 significantly enriched metabolites in PFA ependymoma tissue (FDR < 0.05, foldchange > 0) were assessed in mouse hindbrain tissue isolated from at E16.5 and P2 embryonic timepoints. Normalized metabolite abundance was plotted with a set scale of 3 to −3. Annotations of metabolic KEGG pathway are indicated for reference.
DISCUSSION
Pediatric brain tumors, including lethal midline glioma, medulloblastoma, atypical teratoid rhabdoid tumor (ATRT), and ependymoma, reflect the epigenetic developmental states from their cells-of-origin (Mack et al., 2016). IDH-mutant astrocytomas in young adults have previously demonstrated a link between metabolism and the epigenome in neuro-oncology (Parsons et al., 2008). We now identify a metabolic-epigenomic link in PFA ependymomas that informs the phenotype of PFA ependymoma and possible development of novel therapeutic strategies for these lethal cancers.
A diverse array of normal stem cells and cancer cells preferentially grow in hypoxia, but PFA ependymoma cells appear unique in that they are permanently poisoned after only transient exposure to room air. Future work to isolate and study the lineage-of-origin will determine if the unique metabolism of PFA cells merely reflects their cells-of-origin in the early hindbrain or is an acquired phenotype. The specific roles of metabolic factors, including hypoxia, either acute or chronic, during fetal development in the development of PFA ependymoma awaits robust prospective in vivo investigation in model organisms.
The occurrence of PFA ependymoma in the hypoxic hindbrain during fetal and early post-natal life controls the availability of the intermediary products of metabolism, including SAM, which regulates histone methylation, α-ketoglutarate, which regulates histone demethylation, and acetyl-CoA, which regulates histone acetylation; collectively resulting in H3K27M hypomethylation. Comparisons of metabolite abundance from primary PFA tissues did not provide the resolution to identify the enrichment or depletion of key metabolites that we observed in primary cultures. This is likely a limitation of bulk PFA tissues, which contain contaminating immune cells and stroma, and highlights the importance of in vitro primary cell culture models to decipher such phenotypes (Vladoiu et al., 2019).
Pharmacologic agents targeting selected metabolites or the resulting histone post-translational modification displayed specific activity against PFAs. While the PFA ependymoma epigenome exhibits marked global H3K27 hypomethylation, the residual levels of H3K27 trimethylation were critical for cell survival, leading to the seemingly paradoxical finding that inhibition or potentiation of PRC2 complex activity profoundly reduces the fitness of PFA cells, suggesting broad utility as a targeted therapy.
PFA ependymoma represents a very unusual malignancy of infancy, with a paucity of known recurrent somatic mutations, in which microenvironmental regulation of the epigenome appears to play a major role in tumorigenesis. Targeting the highly tuned availability of metabolic intermediates, as well as the finely balanced epigenome should be evaluated for children with PFA ependymoma.
STAR* METHODS
RESOURCE AVAILABILITY
Lead Contact:
For all additional information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Michael Taylor (mdtaylor@sickkids.ca).
Materials Availability:
All patient derived cell lines generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement as per the policies of the Lead Contact’s institution. All unique materials and reagents generated in this study can be obtained from the Lead Contact.
Data and Code Availability:
All datasets generated in this study have been deposited and are available as follows: Methylation - GEO: GSE146426; RNAseq & WGS – EGAS00001004312; Cut&Run Seq – GEO: GSE146858; Proteomics – PRIDE: PXD018191. This study did not generate any unique code requiring deposition.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient derived PFA and ST Tissue:
All tissues (PFA, ST) used in this study were obtained with properly informed consent of patients. All experimental procedures were performed in accordance with the Research Ethics Boards (REB 1000055059) at The Hospital for Sick Children (Toronto, Canada). Primary tumour cultures used in this study are from patients that have not undergone radiotherapy or chemotherapy prior to surgical resection.
Patient derived PFA and ST Cell Cultures:
All samples used in this study were obtained with properly informed consent of patients. All experimental procedures were performed in accordance with the Research Ethics Boards at The Hospital for Sick Children (Toronto, Canada). Patient derived PFA-ependymoma cell lines (MDT-PFA1, MDT-PFA2, MDT-PFA3, MDT-PFA4, MDT-PFA5, MDT-PFA7 MDT-PFA8, MDT-PFA9, MDT-PFA13, MDT-PFA15) and supratentorial ependymoma cell lines (MDT-ST1, MDT-ST4) were established in this study. GBM and DIPG K27M cell cultures were obtained from Dr. Peter Dirks (The Hospital for sick Children, Canada) and Dr. Nada Jabado (McGil University, Canada) respectively. All cell cultures were confirmed to match original tumors by STR fingerprinting, where tumor tissues were available. The following PFA and ST cell cultures were derived from male patients: MDT-PFA1, MDT-PFA2, MDT-PFA3, MDT-PFA5, MDT-PFA7, MDT-PFA8, MDT-PFA9, MDT-PFA13, MDT-PFA15, MDT-ST4. The following PFA and ST cell cultures were derived from female patients: MDT-PFA4, MDT-ST1. Human fetal neural stem cells, fNSC (HF7450, HF6562) and immortalised normal human astrocytes (iNHA) were obtained from Dr. Peter Dirks (The Hospital for sick Children, Canada) and Dr. Nada Jabado (McGil Univeristy, Canada) respectively.
Mouse Housing and Husbandry:
All mouse breeding and procedures were performed as approved by The Centre for Phenogenomics (Toronto). Pairs of C57BL/6J mice were obtained from The Jackson laboratory for mouse breeding. Embryos of mated C57BL/6J female mice were dissected to collect hindbrain tissue from E10, E12, E14, E16 and E18 gestational time points. Hindbrain of C57BL/6J pups was dissected to collect tissue from P0, P5, P7 and P14 postnatal time points.
METHOD DETAILS
Primary Cell Culture Establishment:
Upon receiving patient tissue from the operating room, all posterior fossa and supratentorial ependymoma tissues were dissociated as quickly as possible. Tissues were minced small and dissociated into single cells with 1x collagenase/dispase (Roche) in PBS solution, followed by incubation at 37ºC for 30 minutes with gentle vortex every 10 minutes. After 30 minutes incubation, cells were passed through 70um cell strainer and pelleted by centrifugation. Pellets were dissolved in EPN culture media and seeded (1*106 cells/per plate) on to laminin-coated Primaria plates (Corning). EPN culture media consists of serum free Human Neurocult NS-A Basal media (StemCell Technologies) supplemented with N2 (20ng/mL, Life Technologies), B27 (Life Technologies), EGF (10ng/mL Life Technologies), FGF (10ng/mL Peprotech), heparin (2μg/mL), BSA (300 μg/mL, Sigma -Aldrich) and 2mM Glutamine (GlutaMAX, 75μg/ml, Life Technologies). Cells were seeded on pre-coated laminin plates (poly-l-ornithine treatment followed by laminin (Sigma-Aldrich). All plates were parallelly tested under different oxygen gradients using an oxygen restricted hypoxia chamber at the following partial pressures: 1% Hypoxia (1% O2 oxygen, 94% Nitrogen and 5% CO2), 5% Hypoxia (5% O2 oxygen, 90% Nitrogen and 5% CO2), 10% Hypoxia (10% O2 oxygen, 85% Nitrogen and 5% CO2) and 21% Normoxia (21% O2 oxygen, 74% Nitrogen and 5% CO2). Cells were replenished with fresh EPN cell culture media every 3 days. All PFA and ST cell cultures implicated in the study were established and maintained as continuous culture at 1% Hypoxia unless otherwise explicitly stated. DIPG K27M, GBM, fNSC and iNHA cell cultures were also continuously cultured in EPN culture media at 1% Hypoxia unless otherwise explicitly stated.
Growth Curve/Proliferation Analysis:
Master cell culture plates (maintained at 1% oxygen) were divided into two plates (1X 106 cells each) and each plate was incubated at 1% oxygen and 21% oxygen respectively. These plates were further passaged and maintained for 21 days as triplicates. At the end of 21 days, viable cell numbers of both conditions were calculated by trypan blue cell viability assay using automated Vi-CELL XR Cell Viability Analyzer. Additionally, growth curves of PFA cells exposed to 1% vs. 21% oxygen tension were performed. Three PFA cell lines (MDT-PFA2, MDT-PFA4, MDT-PFA5) were cultured continuously in either 1% or 21% oxygen for a total of 21 days. Cells were seeded at low density (40,000 cells) in multiple 12-well and 24-well plates. Replicates were counted at various time points throughout the time-course by trypan blue cell viability assay using automated Vi-CELL XR Cell Viability Analyzer and plots were fitted using an exponential growth non-linear regression model.
Cell Confluence Analysis:
Cells maintained at 1% and 21% oxygen for 3 generations as described above were subjected to crystal violet-based confluence analysis at the end of 21 days. All three replicate plates of each condition were washed once with PBS and stained with 0.1% crystal violet in 20% methanol. After 1-hour staining, plates were washed with cold water, dried, and images of all four quadrants of each plate were captured by Evos Microscope.
Senescence β-Galactosidase Staining:
Cells maintained at 1% and 21% oxygen for 3 generations as described before were assessed at the end of 21 days for senescence phenotype using Senescence β-Galactosidase Staining Kit (Cell Signaling Tech) per manufacturer’s instructions. Briefly, cells were washed with 1X PBS and fixed with 1X fixative solution for 15 mins at RT. After incubation, cells were washed again with 1X PBS followed by staining with β-Galactosidase staining solution, plates were sealed and incubated at 37C in dry incubator for overnight. Next day, plates were analyzed by development of blue colour. Images of all plates were captured by Evos Microscope and analyzed.
Apoptosis and Cell Cycle Analysis:
Apoptosis and cell death of PFA cell cultures and control cell cultures treated with different oxygen gradients (1% and 21% oxygen) were assessed by using Annexin V 568 kit (BD) as per manufacturer’s guidelines. Quantification of live cells against dead cells was done by using LIVE/DEAD 488 fluorescence dye from Invitrogen. Bromodeoxyuridine (BrdU) based proliferation and cell cycle analysis of PFA cell cultures and control cell cultures treated with different oxygen gradients (1% and 21% oxygen) were done by FITC BrdU Flow Kit from BD Biosciences following their recommended protocol. Live cell cycle analysis of PFA, ST, and fNSC cell cultures treated with different oxygen gradients were performed using Hoechst 33342 Ready Flow Reagent (Invitrogen) per manufacturer instructions. Samples were run on a BD Biosciences LSR II machine.
Western Blotting:
Total cell lysates were prepared using RIPA buffer with added protease inhibitors (Sigma-Aldrich) and lysate protein concentration estimated by Pierce™ BCA Protein Assay Kit (ThermoFisher). 15ug of total cell lysates was separated on 12% Mini-PROTEAN® TGX™ Precast Protein Gels (Bio-Rad) and got wet-transferred to a PVDF membrane (GE Healthcare). Post transfer, membrane was blocked using 5% skim milk in TBST (50 mM Tris, 150 mM NaCl, 0.1% Tween 20, pH 7.4) (TBST) for 1 hr. Membranes were incubated with primary antibodies for overnight washed with TBST more than 3 times and incubated again with secondary antibodies. Membranes were further developed for signal using SuperSignal West Pico Chemiluminescent solution and imaged on a ChemiDoc MP Imaging System (Bio-Rad).
Transient Growth in 21% Oxygen:
A total of 100,000 PFA cells were seeded in triplicate in 10cm laminin coated plates, and concurrently incubated in either 1% Hypoxia (12 days), 21% Normoxia (12 days) or 21% Normoxia (4 days) followed by 1% Hypoxia (8 days). Replicates were counted at 12-day endpoint by trypan blue cell viability assay using automated Vi-CELL XR Cell Viability Analyzer. Data are displayed as mean ± SEM.
In Vitro Limiting Dilution Analysis (LDAs):
LDAs were performed in a 96-well plate format, with one plate cultured in 1% O2 and one plate cultured in 21% O2. Serial dilutions of PFA cells were performed to achieve a cell seeding number of 2,000 cells per well at the highest dose and 4 cells per well at the lowest dose. A total of 10 cell doses were tested with 6 technical replicates per dose. Fresh media was added to the cells every 5 days. Wells were scored for sphere formation on day 7, 14 and 21. Spheres were imaged with an EVOS XL Core Imaging System using a 20x air objective. Statistical analysis was performed with the Extreme Limiting Dilution Analysis (ELDA) web-based software (Hu and Smyth, 2009).
Co-isolation of DNA and RNA:
DNA and total RNA was extracted from snap frozen tumour tissue and frozen cell pellets using the Qiagen AllPrep Universal kit. DNA was extracted from snap frozen whole blood samples using the Qiagen QiaAmp DNA Blood Midi kit. DNA was eluted into EB buffer, and RNA eluted into RNAse-free water. Quantification and purity were initially assessed using Picogreen or nanodrop spectrophotometer and RIN was subsequently confirmed to be >7.
WGS Library Preparation:
To minimize library bias and coverage gaps associated with PCR amplification of high GC or AT-rich regions we have implemented a version of the TruSeq DNA PCR-free kit (E6875–6877B-GSC, New England Biolabs), automated on a Microlab NIMBUS liquid handling robot (Hamilton Robotics). Briefly, 500 ng of genomic DNA was arrayed in a 96-well microtitre plate and subjected to shearing by sonication (Covaris LE220). Sheared DNA was end-repaired and size selected using paramagnetic PCRClean DX beads (C-1003–450, Aline Biosciences) targeting a 300–400 bp fraction. After 3’ A-tailing, full length TruSeq adapters were ligated. Libraries were purified using paramagnetic (Aline Biosciences) beads. PCR-free genome library concentrations were quantified using a qPCR Library Quantification kit (KAPA, KK4824) prior to sequencing with paired-end 150 base reads on the Illumina HiSeqX platform using V4 chemistry according to manufacturer recommendations.
RNAseq Library Preparation:
Qualities of total RNA samples were determined using an Agilent Bioanalyzer RNA Nanochip or Caliper RNA assay and arrayed into a 96-well plate (Thermo Fisher Scientific). Polyadenylated (PolyA+) RNA was purified using the NEBNext Poly(A) mRNA Magnetic Isolation Module (E7490L, NEB) from 500 ng total RNA normalized in 35 μL for DNase I-treatment (1 Unit, Invitrogen). DNase-treated RNA was purified using RNA MagClean DX beads (Aline Biosciences, USA) on a Microlab NIMBUS liquid handler (Hamilton Robotics, USA). Messenger RNA selection was performed using NEBNext Oligod(T)25 beads (NEB) with incubation at 65oC for 5 minutes followed by snap-chilling at 4oC to denature RNA and facilitate binding of poly(A) mRNA to the beads. mRNA was eluted in 36 μL of Tris Buffer. First-strand cDNA was synthesized from the purified polyadenylated messenger RNA using the Maxima H Minus First Strand cDNA Synthesis kit (Thermo-Fisher, USA) and random hexamer primers at a concentration of 8ng/μL along with a final concentration of 0.04μg/uL Actinomycin D, followed by PCR Clean DX bead purification on a Microlab NIMBUS robot (Hamilton Robotics, USA). The second strand cDNA was synthesized following the NEBNext Ultra Directional Second Strand cDNA Synthesis protocol (NEB) that incorporates dUTP in the dNTP mix, allowing the second strand to be digested using USERTM enzyme (NEB) in the post-adapter ligation reaction and thus achieving strand specificity. cDNA was fragmented by Covaris LE220 sonication for 130seconds (2×65seconds) at a “Duty cycle” of 30%, 450 Peak Incident Power (W) and 200 Cycles per Burst in a 96-well microTUBE Plate (P/N: 520078) to achieve 200–250 bp average fragment lengths. The paired-end sequencing library was prepared following the BC Cancer Agency Genome Sciences Centre strand-specific, plate-based library construction protocol on a Microlab NIMBUS robot (Hamilton Robotics, USA). Briefly, the sheared cDNA was subject to end-repair and phosphorylation in a single reaction using an enzyme premix (NEB) containing T4 DNA polymerase, Klenow DNA Polymerase and T4 polynucleotide kinase, incubated at 20oC for 30 minutes. Repaired cDNA was purified in 96-well format using PCR Clean DX beads (Aline Biosciences, USA), and 3’ A-tailed (adenylation) using Klenow fragment (3’ to 5’ exo minus) and incubation at 37oC for 30 minutes prior to enzyme heat inactivation. Illumina PE adapters were ligated at 20oC for 15 minutes. The adapter-ligated products were purified using PCR Clean DX beads, then digested with 3μL of USERTM enzyme (1U/μL, NEB) at 37oC for 15 minutes followed immediately by 13 cycles of indexed PCR using Phusion DNA Polymerase (Thermo Fisher Scientific Inc. USA) and Illumina’s PE primer set. PCR parameters: 98˚C for 1 minute followed by 13 cycles of 98˚C for 15 seconds, 65˚C for 30 seconds and 72˚C for 30 seconds, and then 72˚C for 5 minutes. The PCR products were purified and size-selected using a 1:1 PCR Clean DX beads-to-sample ratio (twice), and the eluted DNA quality was assessed and quantified using the Caliper LabChip GX DNA High Sensitivity assay (PerkinElmer) and the Quant-iT dsDNA high sensitivity assay (ThermoFisher Scientific). Libraries were normalized and pooled and the final concentration of the pooled library was double checked and determined by Qubit dsDNA HS Assay Kit using a Qubit fluorometer (ThermoFisher Scientific). Clusters were generated on the Illumina cluster station and sequence was run on the Illumina HiSeq2500 platform following the manufacturer’s instructions.
Proteomics:
Cells (2X 106) of three independent plates were exposed to 1% and 21% oxygen concentration for 7 days, 14 days and 21 days. At end of incubation, cells were scraped in ice cold PBS, snap frozen, and stored at −80C until further processing. For sample preparation, cells were lysed in denaturing urea buffer in the presence of protease and phosphatase inhibitors. Extracted proteins were digested into peptides with trypsin. After desalting, peptides were analyzed by mass spectrometry (MS, “1D separation”) using around 1 ug per injection.
Histone extraction and Histone tail PTM mass spectrometry analysis:
Histone extraction, LC/MS, and data analysis are performed as described previously (Sidoli et al., 2016). Briefly, tissue and cell pellets of continuous culture were lysed to isolate nuclei using 0.3% NP-40 alternative supplemented nuclear isolation buffer. To the isolated nuclei, 0.4 N H2SO4 was added and incubated for 3hrs with constant rotation at 4ºC. Samples were then centrifuged and supernatant was collected. Chilled 100% trichloroacetic acid (w/v) was added to the supernatant in the ratio of 1:3(v/v) to get 33% final concentration of TCA and incubated on ice overnight to precipitate histones. Histone pellets were further rinsed with ice cold acetone containing 0.1% HCl followed by ice-cold acetone and air dried to evaporate acetone. To the resulting histones, double distilled water added and protein concentration estimated by Bradford assay. Samples (Histone extract −20ug) were resuspended in NH₄HCO₃ and derivatized with propionic anhydride followed by trypsin digestion. 250 fmol/mg of isotopically-labeled synthetic histone peptides was spiked in the samples and got derivatized with propionic anhydride. C18 Stage tips was used to desalt of histone peptides before nanoLC-MS analysis. All the raw data files analyzed using EpiProfile 2.0 (Yuan et al., 2018).
S-Adenosylmethionine (SAM) quantitation:
Total abundance of S-Adenosylmethionine (SAM) in the PFA cell culture maintained in different oxygen gradients was quantified using S-Adenosylmethionine (SAM) ELISA Kit (CELL BIOLABS, INC) following manufacturer’s assay protocol. Briefly, Cell pellets (5*106 cells) were homogenised on ice with cold PBS and centrifuged at 10,000g and supernatant got collected used for assay immediately. SAM conjugate coated assay plates were made on the previous day of experiment. 50 ul of sample and SAM-BSA (standard) and blank was added to assay plate and got incubated for 10mins at RT. After Incubation, 50ul of Anti-SAM was added to each testing well and incubated at RT for 1 hour on orbital shaker. After Incubation, each well got washed multiple times with 1X wash buffer. 100 ul of secondary HRP conjugate was added, again incubated again at RT for 1 hour on orbital shaker and washed multiple times with 1X wash buffer. Substrate solution was added into each testing well and incubated until color changes reasonably and reaction got stopped using stop solution and absorbance of each microwell was read using microplate reader (Tecan) using 450nm as primary wavelength.
Cell viability assay:
PFA cell cultures and control cells were seeded at a density of 5000 cells (Non-epigenetic target validation) and 1500 cells (Epigenetic target validation) in 96 well Primaria plates (Corning) for dose-response analysis. Twenty-four-hours post seeding, cells were treated with indicated compounds for 7 days and 14 days (epigenetic target validation) with varying compound concentrations. For longer assay period, media was placed every 4 days with indicated drug/compounds. All compounds, except BMS-303141 were dissolved in DMSO (Sigma-Aldrich) which was used as vehicle and serving as control to normalize cell indices. PFA cells and control cells were independently treated with BAY876, CB-839, BMS-303141, Sardomozide-HCl, JBSNF-0000088, SAM, GSK-J4, UNC1999, PF9366, GSK343, A395, A395N, and A485 in a dose-dependent manner and cell viability was assessed at the end of incubation. All the above listed drugs and compounds used in this study were procured from various vendors (See key resource table). Cell viability of PFA and control cells were either measured by Alamar Blue Cell Viability Reagent (ThermoFisher Scientific) or CyQUANT Direct Cell Proliferation Assay (ThermoFisher Scientific) as per manufacturer’s protocol at endpoint. Graphpad Prism software used to plot the dose-dependent response data using nonlinear regression of variable slope (four parameters), least squares (ordinary) fitting method. Each plotted data represents mean and SEM of three experimental replicates.
Immunohistochemistry (IHC) and analysis:
Formalin-fixed, paraffin-embedded serial sections of pediatric 8 ependymomas (4 supratentorial and 4 of the posterior fossa) were obtained from the BC Children’s Hospital (Vancouver, BC, Canada). PFA status for posterior fossa tumors had been previously determined by global reduction of H3K27me3, as described (PMID:28733933). All IHC was performed using the Ventana Discovery platform with CA9 (RD AF2188) and H3K27me3 (Cell Signalling 9733) antibodies, both with dilutions of 1:200. In brief, tissue sections were incubated in Tris EDTA buffer (cell conditioning 1; CC1 standard) at 95C for 1 hour to retrieve antigenicity, followed by incubation with the respective primary antibody for 1 hour. Bound primary antibodies were incubated with the respective secondary antibodies (Jackson Laboratories) with 1:500 dilution, followed by Ultramap HRP and Chromomap DAB detection. For IHC analysis, hypoxic tumor areas were initially identified by CA9 positivity. H3K27me3 positivity was analyzed for each tumor in 3 randomly selected hypoxic regions and three randomly selected non hypoxic areas (each region ~ 1 mm2). Average H3K27me3 scores for each tumor were subsequently calculated for hypoxic and non-hypoxic regions. H3K27me3 IHC positivity scores were automatically quantified in the regions of interest with Aperio ImageScope software. In order to ensure that H3K27me3 scores reflected the positivity of tumor cells, and not interstitial or normal brain cells, all IHC analyses was performed by an MD with training in pathology (AD).
Metabolite extraction, derivatization, gas chromatography/mass spectrometry:
For each technical replicate, 750,000 cells were plated on laminin-coated 6-well plates and cultured in serum-free EPN-complete media supplemented with B27, N2, EGF/FGF, heparin, BSA, and pen/strep antibiotics, and anti-mycoplasma prophylaxis (Plasmocin, InVivogen ant-mpp). After 24 h, polar metabolites and fatty acids were extracted using methanol/water/chloroform as previously described (Badur et al., 2018). Briefly, each technical replicate was cultured in growth-factor added serum-free EPN media lacking glucose or glutamine supplemented with [U13C6]-glucose (Cambridge Isotope) or [U13C5]-glutamine (Cambridge Isotope), respectively for 24h. After removing spent media, cells were rinsed with 0.9% (w/v) saline and quenched with 250 μl of −80°C methanol. 100 μl of ice-cold water containing 1 μg norvaline internal standard was added to each sample, after which both solution and cells were collected via scraping. Upon transferring to fresh sample tubes, cell lysates were mixed with 250 μl of −20°C chloroform containing 1 μg heptadecanoate (internal standard for fatty acids) and 1 μg coprostan-3-ol (internal standard for cholesterol). After vortexing and centrifugation, the top aqueous layer (polar metabolites) and bottom organic layer (lipids) were collected and dried under airflow. Polar metabolites and fatty acids were derived using MOX-tBDMCS derivatization as previously described (Lewis et al., 2014). Gas chromatography/mass spectrometry (GC/MS) analysis was performed as previously described using an Agilent 7890A with a 30m DB-35MS capillary column (Agilent Technologies) connected to an Agilent 5975C MS (Lewis et al., 2014; Zhang et al., 2016).
Extracellular Flux and Glucose/Glutamine Consumption Rate Measurements:
Extracellular fluxes, including glucose/glutamine uptake and lactate/glutamate secretion, were calculated by subtracting final spent medium from initial medium substrate concentrations measured using a Yellow Springs Instrument 2950 and normalized by the numbers of PFA-EPN, ST-EPN, or fNSC cells over 24h.
Glucose rescue:
Cells incubated at different oxygen tensions (1% and 21%) were challenged with varying concentrations of glucose and evaluated for proliferation efficiency. Glucose free Neurobasal media was purchased from ThermoFisher Scientific. Cells (1X106 cells) were seeded and incubated with serum-free complete media for 24h. After 24h incubation, replicates of each condition were challenged with complete media without Glucose and with increased glucose concentration (50mM) for 7days with intermittent media change maintaining varying glucose concentrations. Seven days later, viable cell numbers of both conditions were calculated by trypan blue cell viability assay using automated Vi-CELL XR Cell Viability Analyzer.
Dimethyl alpha-Ketoglutarate Rescue of Normoxia and GLS inhibition:
PFA cells were plated as replicates in laminin-coated six well Primaria plates and exposed to 1% and 21% oxygen concentration. Cells at 21% oxygen supplemented with and without cell permeable dimethyl alpha-Ketoglutarate in their growth media assessed for cell proliferation post 4days incubation using automated Vi-CELL XR Cell Viability Analyzer. PFA cells cultured on laminin-coated 6 well plate and maintained at 1% oxygen. Post 24hrs seeding, one set of replicate plates were treated with GLS inhibitor (CB839) and other sets were treated with GLS inhibitor (CB839) but supplemented with dimethyl alpha-Ketoglutarate. Cells that were not treated, with GLS inhibitor, treated but supplemented with exogenous dimethyl alpha-Ketoglutarate were subjected to cell proliferation analysis using automated Vi-CELL XR Cell Viability Analyzer.
Acetate rescue of ACLY or GLS inhibition:
Complete media was supplemented with sodium acetate (Sigma Aldrich) at 200μM and 2mM or PBS of equal volume. pH of each condition was measured and proper buffering at pH 7.35 was confirmed. In a laminin-coated 96-well plate, we plated 5000 cells per replicate and assayed 5 technical replicates per titration point per condition. For each inhibitor, we titrated the inhibitory effect on cell viability by 1:2 dilution series of the ACLY inhibitor BMS-303141 and the GLS inhibitor CB-839. After 72 h incubation, Alamar Blue (Thermo Fisher Scientific) was used to measure cell viability. Data were presented as box-whisker plot for each titration point.
Single Cell RNAseq:
Single cell data was used from a previously published manuscript (Vladoiu et al.). Methods for collection, library preparation, alignment and QC are according to the previously published article.
Genome-wide knockout screen:
We performed CRISPR-Cas9 based genome-wide knockout screening on three PFA cell cultures (MDT-PFA2, MDT- PFA4, and MDT-PFA5) using the 70K TKOv3 library (Addgene #90294) as described in previous study (Hart and Moffat, 2016a; MacLeod et al., 2019). Briefly, All PFA cell cultures (minimum 100 million cells) were independently transduced with a pooled gRNA library with 0.3–0.4 M.O.I range to obtain 500-fold coverage. On the following day, transduced cell cultures were replaced with fresh complete media with 1μg/mL puromycin. Post puromycin selection (48hrs), viable cells were collected and pooled from all screen plates. Twenty million of these pooled cells were pelleted and saved as T0 samples and remaining cells were (minimum twenty million cells) were continuously passaged for 14 doublings as two duplicate arms. From each arm of continuous doublings, twenty million cells were pelleted. Pellets from timepoints T0, T3, T5, and T7 were harvested for genomic DNA isolation, and PCR amplification of gRNAs was conducted using a two-step procedure (oligonucleotides listed in Key Resource Table) (MacLeod et al., 2019). Amplicons were barcoded, gel purified and sent for next generation sequencing using either Illumina HiSeq2500 of NextSeq500 instruments. Samples were sequenced at a depth of 500-fold (T0 reference) or 200-fold (all other timepoints).
Cut&Run Sequencing:
CUT&RUN was conducted following the published protocol (Skene and Henikoff, 2017) with slight modifications as follows. Cells were harvested and counted, with 250000 cells per sample reaction. Permeabilized cells, along with pre-activated Concanavalin A-coated beads were mixed with primary antibody (anti-histone H3K27me3, Diagenode, C15410069) overnight at 4°C, 1ng per 100ul volume, 900rpm on the Thermomixer. After several washes, CUTANA pAG-MNase (EpiCypher, 15–1016) was added to each sample and incubated for 10 minutes at room temperature as per manufacturing protocol. Chromatin digestion and release was done under the high calcium/low salt conditions, including a 2.5 min incubation step following the addition of the Incubation Buffer. Fragmented DNA samples were extracted using Zymo DNA-Clean/Concentrator kit (D4004). Library preparation was done with NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB, E7645S) and purified with NEBNext Sample Purification Beads (NEB, E7104S). Adaptor ligation was performed assuming input amount about 10ng, with working adaptor concentration at 1.5μM. PCR enrichment of adaptor-ligated DNA was done with the following cycling conditions: initial denaturation for 30 sec at 98°C, denaturation for 15 sec at 98°C and annealing/extension for 10 sec at 65°C (10 cycles). Samples were then sequenced using an Illumina Hi-Seq after BioAnalyzer Trace quality control as described above.
Metabolomics:
Global metabolite abundance profiling on snap frozen tissues of PFA ependymoma (n=15), ST ependymoma (n=4), normal fetal brain controls (n=5) and hindbrain of C57BL/6J mouse embryonic timepoint, E16 (n=10) and postnatal timepoint, P2 (n=10) tissues were performed in collaboration with Metabolon Inc (https://www.metabolon.com). Method details of sample preparation, metabolite identification and quantification were provided by Metabolon Inc as below.
Sample Preparation:
Upon receiving, all samples were maintained at a temperature of −80C until processed. Samples were prepared using the automated MicroLab STAR® system from Hamilton Company. Several recovery standards were added prior to the first step in the extraction process for QC purposes. To remove protein, dissociate small molecules bound to protein or trapped in the precipitated protein matrix, and to recover chemically diverse metabolites, proteins were precipitated with methanol under vigorous shaking for 2 min (Glen Mills GenoGrinder 2000) followed by centrifugation. The resulting extract was divided into five fractions: two for analysis by two separate reverse phase (RP)/UPLC-MS/MS methods with positive ion mode electrospray ionization (ESI), one for analysis by RP/UPLC-MS/MS with negative ion mode ESI, one for analysis by HILIC/UPLC-MS/MS with negative ion mode ESI, and one sample was reserved for backup. Samples were placed briefly on a TurboVap® (Zymark) to remove the organic solvent. The sample extracts were stored overnight under nitrogen before preparation for analysis.
QA/QC:
Several types of controls were analyzed in concert with the experimental samples: a pooled matrix sample generated by taking a small volume of each experimental sample (or alternatively, use of a pool of well-characterized human plasma) served as a technical replicate throughout the data set; extracted water samples served as process blanks; and a cocktail of QC standards that were carefully chosen not to interfere with the measurement of endogenous compounds were spiked into every analyzed sample, allowed instrument performance monitoring and aided chromatographic alignment. Instrument variability was determined by calculating the median relative standard deviation (RSD) for the standards that were added to each sample prior to injection into the mass spectrometers. Overall process variability was determined by calculating the median RSD for all endogenous metabolites (i.e., non-instrument standards) present in 100% of the pooled matrix samples. Experimental samples were randomized across the platform run with QC samples spaced evenly among the injections.
Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectroscopy (UPLC-MS/MS):
All methods utilized a Waters ACQUITY ultra-performance liquid chromatography (UPLC) and a Thermo Scientific Q-Exactive high resolution/accurate mass spectrometer interfaced with a heated electrospray ionization (HESI-II) source and Orbitrap mass analyzer operated at 35,000 mass resolution. The sample extract was dried then reconstituted in solvents compatible to each of the four methods. Each reconstitution solvent contained a series of standards at fixed concentrations to ensure injection and chromatographic consistency. One aliquot was analyzed using acidic positive ion conditions, chromatographically optimized for more hydrophilic compounds. In this method, the extract was gradient eluted from a C18 column (Waters UPLC BEH C18–2.1×100 mm, 1.7 μm) using water and methanol, containing 0.05% perfluoropentanoic acid (PFPA) and 0.1% formic acid (FA). Another aliquot was also analyzed using acidic positive ion conditions; however, it was chromatographically optimized for more hydrophobic compounds. In this method, the extract was gradient eluted from the same afore mentioned C18 column using methanol, acetonitrile, water, 0.05% PFPA and 0.01% FA and was operated at an overall higher organic content. Another aliquot was analyzed using basic negative ion optimized conditions using a separate dedicated C18 column. The basic extracts were gradient eluted from the column using methanol and water, however with 6.5mM Ammonium Bicarbonate at pH 8. The fourth aliquot was analyzed via negative ionization following elution from a HILIC column (Waters UPLC BEH Amide 2.1×150 mm, 1.7 μm) using a gradient consisting of water and acetonitrile with 10mM ammonium formate, pH 10.8. The MS analysis alternated between MS and data-dependent MSn scans using dynamic exclusion. The scan range varied slighted between methods but covered 70–1000 m/z.
QUANTIFICATION & STATISTICAL ANALYSIS
All statistical analyses methods, including dispersion of data, are indicated in the figure legends.
Survival Analysis Stratified by Hypoxia Gene Signature:
We derived transcriptional activity scores for every transcriptional signature gene set curated by the Molecular Signature Database (MSigDB; n = 21,450) in a PFA ependymoma patient cohort with clinically annotated transcriptional dataset (Liberzon et al., 2015; Witt et al., 2011). Then we stratified the cohort into high vs. low groups using the median transcriptional signature score as the cut-off and performed the log-rank test in Kaplan-Meier survival analysis with FDR adjustment. Among 735 gene sets associated with worse overall survival, 9 were hypoxia gene signatures derived from transcriptional and epigenetic profiles of a variety of cancer and stem cells (Fardin et al., 2009; Kondo et al., 2006; Maina et al., 2005; Manalo et al., 2005; Qi et al., 2010; Walmsley et al., 2011; Winter et al., 2007).
Bulk RNA-sequencing Data Pre-Processing:
After raw fastq files quality check, the strand-specific 75bp paired-end reads were aligned to hg38 (downloaded from iGenomes project) reference using STAR (STAR_2.4.2a) and the UCSC gene annotation (downloaded from the IGenome project) (Dobin et al., 2013). The “ReadsPerGene” raw counts from STAR were used for further analysis with the DESeq2 package (Love et al., 2014). Normalized gene expression matrix (size factors correction and vst transformed normalized matrix) and differential expression analyses were performed with the DESseq2 packages. Gene set enrichment analysis was performed using GSEA (Mootha et al., 2003; Subramanian et al., 2005). Genes were ranked using the sign of log2FC*-log10(pvalue) and analyzed under the pre-ranked option. Significantly enriched pathways defined by FDR (qval noted in individual figure legends) were visualized using Enrichment Map in Cytoscape (Merico et al., 2010; Shannon et al., 2003). Node sizes are proportional to the number of genes and edge weight (Jaccard and overlap coefficient set at 0.66 cut-off) represents the number of shared genes between each gene set.
Whole-Genome Sequencing Data Pre-Processing and Somatic Mutation Assessment:
First we performed quality check on the 150bp paired-end raw sequence fastq files (fastqc 0.11.5), then the reads were aligned to human genome hg38 (downloaded from iGenomes project) with bwa 0.7.15 and samtools 1.3.1 (Andrews, 2010; Li and Durbin, 2010; Li et al., 2009). We then applied the best practice from GATK to further process the bams (markduplicate, IndelRealignment, BQSR, with picard 2.6.0 and GATK 3.5). Those bams were used for CNV calling and somatic SNV detections. We ran the following 5 mutation callers to identify somatic mutations for both the tissue and line samples using the matched blood sample as reference: (1) Strelka 1.0.14, (2) varscan copynumber and snv 2.3.8, (3) MuSE 1.0rc, (4) mutect2 GATK 3.6, (5) multiSNV v2.3 (Cibulskis et al., 2013; Fan et al., 2016; Josephidou et al., 2015; Koboldt et al., 2012; Saunders et al., 2012). Common SNPs from the snp147Common file which contains dbsnp mutations that uniquely mapped variants that appear in at least 1% of the population or are 100% non-reference were removed. Somatic mutations called by 3 or more callers were kept. Annovar annotation was used to identify damaging mutations (nonsynonymous, stop gain, stop loss) and mutations close to spliced site (Wang et al., 2010). Copy number segments were obtained from the Sequenza algorithm (v2.1.2) (Favero et al., 2015). CNV segments were filtered out against common variants found in the Database of Genomic Variants using a reciprocal overlap of 50%. Following this, adjacent segments whose logR ratios were within 0.25 of one another were merged using a weighted average (based on the number of marks involved in each segment). CNV gene status were defined using the following rules: the largest segment overlapping the gene was chosen as the segment that would decide the state of that gene, and did not necessarily have to overlap the majority of the gene (in the case of if the remainder of the gene was overlapped by no segments). Neutral, loss, and deletion were assigned based on logR values alone, whereas gains and amplification would require that at least 50% of the gene was overlapped by that gain/amplification segment. logR values used to assign the states are as follows: Deletion: logR < log2(0.7/2), Loss: logR < −0.4, Gain: logR > 0.32, Amplification: logR > log2(5/2).
Analysis of 850K methylation data comparing PFA tissue & cell line:
Raw IDAT files were processed with the R package ChAMP v2.14.0 using default parameters (Tian et al., 2017). The final 732,084 probes across 10 samples were normalized in ChAMP using the BMIQ method. No batch effect linked to array slide or position was identified. The 10% most variable probes across all samples as determined by the median absolute deviation were used to calculate the correlation between each PFA tumor and line. The correlations were plotted using corrplot v0.84 (Wei and Simko, 2017).
Proteomics:
All analyses were performed on the latest generation of Orbitrap instrumentation (Orbitrap Fusion) using liquid chromatography (LC) gradients. MaxQuant was used for peptide identification, protein inference and label-free quantification (Cox and Mann, 2008). Subsequent data analysis steps were done in Perseus/R/STRING (Tyanova et al., 2016). Differential expression analysis between Hypoxia and Normoxia conditions was performed. Permutation-based FDR was calculated considering the entire dataset. Hits with FDR<5% were considered significant.
Single Cell RNAseq:
Single cell analysis was performed using Seurat v3.1.4, based on methods previously published (Stuart et al., 2019; Vladoiu et al., 2019). Briefly, 84 validated hypoxia genes were identified from previous publications (Buffa et al., 2010; Chi et al., 2006; Connor et al., 2019; Favaro et al., 2011; Harris et al., 2015; Koong et al., 2000; Sørensen et al., 2010; Toustrup et al., 2011). As these gene lists were compiled from various tumor types, we identified genes which were significantly upregulated in our PFA cell cultures grown in 1% oxygen (padj < 0.05, log2foldchange > 0). These 15 genes were converted to their mouse matched gene symbols (Aldoa, Aldoc, Bnip3, Bnip3l, Car9, Ddit4, Gpi1, Ldha, P4ha1, P4ha2, Pdk1, Pgk1, Tpi1, Vegfa, Zfp395) and a hypoxia gene signature was created. From our previously published mouse developmental cerebellar atlas, cells annotated as gliogenic progenitors or roof plate like stem cells from timepoints E10, E12, E14, E16, E18, and P0 were isolated and scored against the hypoxia gene signature. The hypoxia signature was built using the AddModuleScore built-in function from Seurat package (v.3.1.4).
Analysis of genome-wide CRISPR screen data:
Read counts for each guide RNA were generated by mapping the sequencing reads to the gRNA library using the MAGECK count function (Li et al., 2014). The BAGEL algorithm was used to determine the log2foldchange relative to the reference T0, and calculate a Bayes Factor (BF) for each gene (Hart and Moffat, 2016b). The BAGEL algorithm uses a reference set of 684 essential genes and 927 non-essential genes for standardizing calculations of BFs. Bayes factor scores for all genes across the 3 PFA samples were quantile normalized (qBF) with 2 previously published GBM lines (G583NS, G549NS) and fetal neural stem cell (fNSC) lines (HF6562, HF7450) which have been screened using the same TKOv3 CRISPR library (Macleod, 2019). To determine unique essential genes of high significance, average qBF scores were calculated for PFAs and GBMs, and the average difference was calculated. A z-score probability was calculated comparing PFA and GBM averages. A final candidate gene list was produced, sorted by largest difference between PFA and GBM qBF scores as well as several other thresholds (p-value < 0.05, average PFA qBF > 4, average GBM qBF < 4). An identical approach was used for comparing PFAs and HFs.
Analysis of Cut&Run Sequencing Data for H3K27 trimethylation:
Single-end Cut&Run reads were aligned to Hg38 using Hisat2 (v2.1.0) using parameters --no-splicedalignment and --nounal (Kim et al., 2015). Non-uniquely mapped reads, PCR duplicates and reads aligning in the Hg38 blacklist (Amemiya et al., 2019) regions were removed. All samples were normalized using spike-in (yeast) alignment counts. Signal tracks files were computed using bedtools. MACS2 function bdgpeakcall was used to call peaks from the bedgraph files (Gaspar, 2018). A custom script was used to calculate area under the peaks; comparison of peak area in different samples was utilized to call the inducible peaks. Genomic heatmaps were plotted using deeptools2 (Ramírez et al., 2016).
Global metabolite profiling by Metabolon Inc:
Data Extraction and Compound Identification:
Raw data was extracted, peak-identified and QC processed using Metabolon’s hardware and software. Compounds were identified by comparison to library entries of purified standards or recurrent unknown entities. Metabolon maintains a library based on authenticated standards that contains the retention time/index (RI), mass to charge ratio (m/z), and chromatographic data (including MS/MS spectral data) on all molecules present in the library. Furthermore, biochemical identifications are based on three criteria: retention index within a narrow RI window of the proposed identification, accurate mass match to the library +/− 10 ppm, and the MS/MS forward and reverse scores between the experimental data and authentic standards. The MS/MS scores are based on a comparison of the ions present in the experimental spectrum to the ions present in the library spectrum. While there may be similarities between these molecules based on one of these factors, the use of all three data points can be utilized to distinguish and differentiate biochemicals. More than 3300 commercially available purified standard compounds have been acquired and registered into LIMS for analysis on all platforms for determination of their analytical characteristics. Additional mass spectral entries have been created for structurally unnamed biochemicals, which have been identified by virtue of their recurrent nature (both chromatographic and mass spectral). These compounds have the potential to be identified by future acquisition of a matching purified standard or by classical structural analysis.
Curation:
A variety of curation procedures were carried out to ensure that a high-quality data set was made available for statistical analysis and data interpretation. The QC and curation processes were designed to ensure accurate and consistent identification of true chemical entities, and to remove those representing system artifacts, mis-assignments, and background noise. Metabolon data analysts use proprietary visualization and interpretation software to confirm the consistency of peak identification among the various samples. Library matches for each compound were checked for each sample and corrected if necessary.
Metabolite Quantification and Data Normalization:
Peaks were quantified using area-underthe-curve. For studies spanning multiple days, a data normalization step was performed to correct variation resulting from instrument inter-day tuning differences. Essentially, each compound was corrected in run-day blocks by registering the medians to equal one (1.00) and normalizing each data point proportionately. For studies that did not require more than one day of analysis, no normalization is necessary, other than for purposes of data visualization. In certain instances, biochemical data may have been normalized to an additional factor (e.g., cell counts, total protein as determined by Bradford assay, osmolality, etc.) to account for differences in metabolite levels due to differences in the amount of material present in each sample.
Metabolograms and Differential Metabolite Scores:
To represent each metabolic pathway, we used the metabologram representation described previously (Hakimi et al., 2016b). Briefly, the metabologram is composed of two components: the left half circle corresponds to the transcriptomics component while the right half circle corresponds to the metabolomics component. Genes involved in each metabolic superpathway were extracted from KEGG annotations. The outer circle represents the fold change for each gene and metabolites. The inner circle displays to the average fold change of all genes and metabolites of the metabolic pathway. Color scale has been set from −3 to 3. Normalized read counts were generated as described above in RNA sequencing anlaysis. Normalized metabolite scores were generated by normalizing each sample with the mass extracted, determining relative values for each sample compared to its median metabolite score (to set the median to 1).
Supplementary Material
A) Three PFA cell lines (MDT-PFA2, MDT-PFA4, MDT-PFA5) were cultured continuously in either 1% or 21% oxygen for a total of 21 days. Cells were seeded at low density (40,000 cells) in multiple 12-well and 24-well plates. Replicates were counted at various time points throughout the time-course, and plots were fitted using an exponential growth non-linear regression model.
B) PFA cells from the primary tumour were cultured at a variety of oxygen conditions (10%, 5%, 1% and 0.2%) for 200 hours of continuous culture. A seeding density of 100,000 cells was used and cells were counted at endpoint.
C) Limited dilution assay of PFA cells cultured in 1% (blue) or 21% (grey) oxygen. Graph depicts fraction of replicates negative for sphere formation at different dilutions of cell seeding. Images demonstrate relative sphere formation under each oxygen condition.
D) Kaplan-Meier survival analysis of PFA ependymoma patients stratified by the median level of the respective hypoxia transcriptional signature score. The log-rank test was used with the calculated hazard ratio ± 95% CI bolded. The hazard ratios for worse overall survival with high transcriptional activity ± 95% CI for all signatures are also plotted.
Oncoprint of damaging single nucleotide variants (SNVs) or focal gain/loss mutations in 6 matched PFA ependymoma tissue-line pairs. SNVs were called by 3 or more mutation callers and focal gains or losses called by GISTIC. Mutation frequency indicated by scale bar on right.
A) Volcano plot of differential gene expression analysis between PFA-EPN (blue) and STEPN (green) surgical biopsies (DEseq2). Significantly differentially expressed genes are indicated in colour (log2foldchange > 1, Padj < 0.05).
B) Volcano plot of differential gene expression analysis between PFA-EPN (blue) and normal brain (maroon) surgical biopsies (DEseq2). Significantly differentially expressed genes are indicated in colour (log2foldchange > 1, Padj < 0.05).
C) Cytoscape map visualization of upregulated pathways between PFA-EPN (blue) and STEPN (green) surgical biopsies. Gene set enrichment analysis (Padj < 0.05, qval <0.05) was used as input.
D) Cytoscape map visualization of upregulated pathways between PFA-EPN (blue) and normal brain (maroon) surgical biopsies. Gene set enrichment analysis (Padj < 0.05, qval <0.01) was used as input.
E) Cytoscape map visualization of upregulated pathways between PFA primary cells cultured in 1% oxygen (blue) or 21% oxygen (grey). Gene set enrichment analysis (Padj < 0.05, qval <0.01) was used as input.
F) Heatmap demonstrating the Pearson correlation scores of normalized and corrected Beta values determined from 850K methylation arrays, between matched PFA primary tissue and cell cultures. A high correlation score (blue) is seen between matched samples. Scale or correlation score normalized between −1 and 1.
A) Volcano plot of differential metabolite abundance between PFA and ST-EPN surgical biopsies. Metabolites are annotated by KEGG super pathway designation. Significant metabolites are highlighted in colour (FDR < 0.05, Fold Change > 1.5).
B) Differential abundance of metabolites grouped by KEGG pathway annotation in PFA-EPN (blue) compared to normal brain (red). Differential abundance score plotted with setscale of −1 to 1.
C) Volcano plot of differential metabolite abundance between PFA and normal brain surgical biopsies. Metabolites are annotated by KEGG super pathway designation. Significant metabolites are highlighted in colour (FDR < 0.05, Fold Change > 1.5).
D) Integrative metabologram of gene expression and metabolite abundance for selected KEGG pathways in PFA tissue relative to normal brain tissue. The schematic illustrates the format of the metabologram plots (derived from Hakmi et al). Log2FoldChange shown on a set scale from −3.0 to 3.0.
A) Representative immunohistochemistry images of PFA and ST ependymoma surgical biopsies stained for H3K27me3 and CA9, demonstrating that hypoxic regions of PFA-EPN tumors have lower level of H3K27me3. Scale bar: 100μM. Quantification of H3K27me3 positivity in CA9 low and high regions (as a surrogate for hypoxia), in PFA (n=4) and ST (n=4) surgical biopsies. Scoring was performed blinded by a trained pathologist. Statistical comparisons were conducted using Welch’s t-test.
B) Heatmap of genome wide H3K27me3 scores (0–100 set scale) in MDT-PFA2 and MDTPFA5 grown at 1% or 21% oxygen. Histone trimethylation marks are determined within +/− 10kb of annotated transcriptional start sites.
C) Average H3K27me3 score for all genes and their respective transcriptional start sites in 1% and 21% oxygen.
D) Plot of H3K27 trimethylation marks for highly differentially expressed candidate genes in PFA ependymoma cells cultured in 1% and 21% oxygen.
A) Relative abundance of metabolites in the urea cycle, TCA cycle, glycolysis and amino acid pools in 1% versus 21% oxygen. PFA cells (orange), ST-EPN cells (purple), fNSCs (blue).
B) Abundance of labeled αKG in 1% (blue) versus 21% (grey) oxygen growth conditions for fNSCs, PFA and ST-EPN cells. All possible isotope compositions (m+) are reported.
C) Abundance of labeled citrate in 1% (blue) versus 21% (grey) oxygen growth conditions for fNSCs, PFA and ST-EPN cells. All possible isotope compositions (m+) are reported.
A) Schematic of labelled [13C-glutamine] tracing in the cell. Isotope configurations are shown throughout the TCA cycle, in forward and reverse direction, and cytosolic acetyl-CoA production. Colours depict labeled carbons (light blue) and unlabelled carbons (navy blue).
B) Abundance of labeled malate, aspartate, and fumarate in 1% (blue) versus 21% (grey) oxygen growth conditions for fNSC, PFA and ST-EPN cells. Cytosolic (m+3, red) and mitochondrial (m+4, green) are indicated.
S1 – List of significantly differentially expressed genes (RNAseq) comparing PFA cells grown in 1% compared to 21% oxygen (Related to Figure 2, Figure S3).
S2 – List of significantly differentially expressed genes (RNAseq) comparing PFA-EPN and ST-EPN surgical biopsies (Related to Figure 2, Figure S3).
S3 – List of significantly differentially expressed genes (RNAseq) comparing PFA-EPN and normal control brain surgical biopsies (Related to Figure 2, Figure S3).
S4 – List of significant essential genes as determined by whole genome CRISPR knockout screen comparing PFA ependymoma to glioblastoma cells (Related to Figure 6).
S5 – List of significant essential genes as determined by whole genome CRISPR knockout screen comparing PFA ependymoma to fetal neural stem cells (Related to Figure 6).
S6 – List of significant essential genes as determined by whole genome CRISPR knockout screen common to both comparisons in S1 and S2 above (Related to Figure 6b).
S7 – List of significantly differential metabolites comparing PFA-EPN and ST-EPN surgical biopsies (Related to Figure 7).
Highlights.
Hypoxic microenvironment is essential for the propagation and growth of PFA ependymoma
Hypoxia controls metabolic intermediates that maintain an H3K27 hypomethylated genome
Inhibition or potentiation of histone lysine methylation diminishes PFA survival
Gliogenic lineage of developing fetal hindbrain mirrors PFA metabolic alterations
ACKNOWLEDGEMENTS
Finally, we would like to thank our funding sources: The National Institutes of Health grants CA203101 (L.J.Y.K.), CA197718, CA154130, CA169117, CA171652, NS087913, and NS089272 (J.N.R.), CA148699 and CA159859 (M.D.T.). M.D.T. is also supported by a Stand Up To Cancer (SU2C) St. Baldrick’s Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT1113) and SU2C Canada Cancer Stem Cell Dream Team Research Funding (SU2C-AACR-DT-19–15) provided by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research, with supplementary support from the Ontario Institute for Cancer Research through funding provided by the Government of Ontario. Stand Up to Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research. M.D.T is also supported by The Pediatric Brain Tumour Foundation, The Terry Fox Research Institute, The Canadian Institutes of Health Research, The Cure Search Foundation, b.r.a.i.n.child, Meagan’s Walk, SWIFTY Foundation, The Brain Tumour Charity, Genome Canada, Genome BC, Genome Quebec, the Ontario Research Fund, Worldwide Cancer Research, V-Foundation for Cancer Research, and the Ontario Institute for Cancer Research through funding provided by the Government of Ontario. M.D.T. is also supported by a Canadian Cancer Society Research Institute Impact grant and a Cancer Research UK Brain Tumour Award. M.D.T. is also supported by the Garron Family Chair in Childhood Cancer Research at the Hospital for Sick Children and the University of Toronto. The authors would also like to thank Meera Kamal (Administrative assistant), James Loukides (Tumour Bank Manager) and recognize the Labatt Brain Tumour Research Centre and the Michael and Amira Dan Brain Tumour Bank Network. We would also like to acknowledge the Isla Kerr fund for their continued support of ependymoma research at SickKids. K.A.M is supported by MITACS ELEVATE Postdoctoral fellowship. S.A.K is supported by ResTraComp doctoral scholarship, Hospital for Sick Children, the McLaughlin Centre, University of Toronto, and the Ruggles Family Foundation, University of Toronto. SCM is supported by Cancer Prevention Research Institute of Texas (CPRIT) scholar award (RR170023), Alex’s Lemonade Stand Foundation (ALSF) A award and Young investigator award, Pediatric Brain Tumor Foundation Award, Chad Tough Young Investigator Award, Cookies for Cancer Research Grant, RALLY research grant, BEAR Necessities Pediatric Cancer Foundation Grant, Children’s Cancer Research Fund award, Children’s Brain Tumor Foundation Award, and Baylor College of Medicine Junior Faculty Award. The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Eshelman Institute for Innovation, Genome Canada through Ontario Genomics Institute [OGI055], Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA, Darmstadt, Germany, MSD, Novartis Pharma AG, Pfizer, São Paulo Research FoundationFAPESP, Takeda, and Wellcome.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aldape K, Brindle KM, Chesler L, Chopra R, Gajjar A, Gilbert MR, Gottardo N, Gutmann DH, Hargrave D, Holland EC, et al. (2019). Challenges to curing primary brain tumours. Nat Rev Clin Oncol 16, 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep 9, 9354–9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. (2010). FastQC: a quality control tool for high throughput sequence data. [Google Scholar]
- Badur MG, Muthusamy T, Parker SJ, Ma S, McBrayer SK, Cordes T, Magana JH, Guan K-L, and Metallo CM (2018). Oncogenic R132 IDH1 Mutations Limit NADPH for De Novo Lipogenesis through (D)2-Hydroxyglutarate Production in Fibrosarcoma Sells. Cell Rep 25, 1018–1026.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beringer M, Pisano P, Di Carlo V, Blanco E, Chammas P, Vizán P, Gutiérrez A, Aranda S, Payer B, Wierer M, et al. (2016). EPOP Functionally Links Elongin and Polycomb in Pluripotent Stem Cells. Mol. Cell 64, 645–658. [DOI] [PubMed] [Google Scholar]
- Buffa FM, Harris AL, West CM, and Miller CJ (2010). Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br. J. Cancer 102, 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Finley LWS, Cross JR, Allis CD, and Thompson CB (2015). Intracellular αketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi J-T, Wang Z, Nuyten DSA, Rodriguez EH, Schaner ME, Salim A, Wang Y, Kristensen GB, Helland A, Børresen-Dale A-L, et al. (2006). Gene expression programs in response to hypoxia: cell type specificity and prognostic significance in human cancers. PLoS Med. 3, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G. (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor AA, Denroche RE, Jang GH, Lemire M, Zhang A, Chan-Seng-Yue M, Wilson G, Grant RC, Merico D, Lungu I, et al. (2019). Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell 35, 267–282.e267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Xi L, Hughes DST, Zhang J, Zhang J, Futreal PA, Wheeler DA, and Wang W. (2016). MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 17, 178–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaro E, Lord S, Harris AL, and Buffa FM (2011). Gene expression and hypoxia in breast cancer. Genome Med 3, 55–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, Szallasi Z, and Eklund AC (2015). Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol 26, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendt S-M, Bell EL, Keibler MA, Olenchock BA, Mayers JR, Wasylenko TM, Vokes NI, Guarente L, Vander Heiden MG, and Stephanopoulos G. (2013). Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat Commun 4, 2236–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Lin S-H, Ren F, Li J-T, Chen J-J, Yao C-B, Yang H-B, Jiang S-X, Yan G-Q, Wang D, et al. (2016). Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat Commun 7, 11960–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar JM (2018). Improved peak-calling with MACS2. BioRXiv 496521. [Google Scholar]
- Hakimi AA, Reznik E, Lee C-H, Creighton CJ, Brannon AR, Luna A, Aksoy BA, Liu EM, Shen R, Lee W, et al. (2016a). An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 29, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi AA, Reznik E, Lee C-H, Creighton CJ, Brannon AR, Luna A, Aksoy BA, Liu EM, Shen R, Lee W, et al. (2016b). An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 29, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris BHL, Barberis A, West CML, and Buffa FM (2015). Gene Expression Signatures as Biomarkers of Tumour Hypoxia. Clin Oncol (R Coll Radiol) 27, 547–560. [DOI] [PubMed] [Google Scholar]
- Hart T, and Moffat J. (2016a). BAGEL: a computational framework for identifying essential genes from pooled library screens. BMC Bioinformatics 17, 164–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, and Moffat J. (2016b). BAGEL: a computational framework for identifying essential genes from pooled library screens. BMC Bioinformatics 17, 164–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, and Smyth GK (2009). ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 347, 70–78. [DOI] [PubMed] [Google Scholar]
- Hübner J-M, Müller T, Papageorgiou DN, Mauermann M, Krijgsveld J, Russell RB, Ellison DW, Pfister SM, Pajtler KW, and Kool M. (2019). EZHIP / CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro-Oncology 21, 878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain SU, Do TJ, Lund PJ, Rashoff AQ, Diehl KL, Cieslik M, Bajic A, Juretic N, Deshmukh S, Venneti S, et al. (2019). PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat Commun 10, 2146–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephidou M, Lynch AG, and Tavaré S. (2015). multiSNV: a probabilistic approach for improving detection of somatic point mutations from multiple related tumour samples. Nucleic Acids Res. 43, e61–e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koong AC, Denko NC, Hudson KM, Schindler C, Swiersz L, Koch C, Evans S, Ibrahim H, Le QT, Terris DJ, et al. (2000). Candidate genes for the hypoxic tumor phenotype. Cancer Res. 60, 883–887. [PubMed] [Google Scholar]
- Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan Z-F, Lim HW, Liu S, et al. (2014). Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 20, 306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis CA, Parker SJ, Fiske BP, McCloskey D, Gui DY, Green CR, Vokes NI, Feist AM, Vander Heiden MG, and Metallo CM (2014). Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 55, 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stütz AM, Wang X, Gallo M, Garzia L, Zayne K, et al. (2014). Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506, 445–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack SC, Hubert CG, Miller TE, Taylor MD, and Rich JN (2016). An epigenetic gateway to brain tumor cell identity. Nat. Neurosci 19, 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod G, Bozek DA, Rajakulendran N, Monteiro V, Ahmadi M, Steinhart Z, Kushida MM, Yu H, Coutinho FJ, Cavalli FMG, et al. (2019). Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep 27, 971–986.e979. [DOI] [PubMed] [Google Scholar]
- Merico D, Isserlin R, Stueker O, Emili A, and Bader GD (2010). Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 5, e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Pajtler KW, Wen J, Sill M, Lin T, Orisme W, Tang B, Hübner J-M, Ramaswamy V, Jia S, Dalton JD, et al. (2018). Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 136, 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajtler KW, Witt H, Sill M, Jones DTW, Hovestadt V, Kratochwil F, Wani K, Tatevossian R, Punchihewa C, Johann P, et al. (2015a). Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 27, 728–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajtler KW, Witt H, Sill M, Jones DTW, Hovestadt V, Kratochwil F, Wani K, Tatevossian R, Punchihewa C, Johann P, et al. (2015b). Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 27, 728–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panwalkar P, Clark J, Ramaswamy V, Hawes D, Yang F, Dunham C, Yip S, Hukin J, Sun Y, Schipper MJ, et al. (2017). Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol. 134, 705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC-H, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu I-M, Gallia GL, et al. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy V, and Taylor MD (2016). Treatment implications of posterior fossa ependymoma subgroups. Chin J Cancer 35, 93–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy V, Hielscher T, Mack SC, Lassaletta A, Lin T, Pajtler KW, Jones DTW, Luu B, Cavalli FMG, Aldape K, et al. (2016). Therapeutic Impact of Cytoreductive Surgery and Irradiation of Posterior Fossa Ependymoma in the Molecular Era: A Retrospective Multicohort Analysis. J. Clin. Oncol 34, 2468–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T. (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryall S, Guzman M, Elbabaa SK, Luu B, Mack SC, Zapotocky M, Taylor MD, Hawkins C, and Ramaswamy V. (2017). H3 K27M mutations are extremely rare in posterior fossa group A ependymoma. Childs Nerv Syst 33, 1047–1051. [DOI] [PubMed] [Google Scholar]
- Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, and Cheetham RK (2012). Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S, Bhanu NV, Karch KR, Wang X, and Garcia BA (2016). Complete Workflow for Analysis of Histone Post-translational Modifications Using Bottom-up Mass Spectrometry: From Histone Extraction to Data Analysis. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, and Henikoff S. (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R. (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.a 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen BS, Toustrup K, Horsman MR, Overgaard J, and Alsner J. (2010). Identifying pH independent hypoxia induced genes in human squamous cell carcinomas in vitro. Acta Oncol 49, 895–905. [DOI] [PubMed] [Google Scholar]
- Thompson YY, Ramaswamy V, Diamandis P, Daniels C, and Taylor MD (2015). Posterior fossa ependymoma: current insights. Childs Nerv Syst 31, 1699–1706. [DOI] [PubMed] [Google Scholar]
- Tian Y, Morris TJ, Webster AP, Yang Z, Beck S, Feber A, and Teschendorff AE (2017). ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 33, 3982–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toustrup K, Sørensen BS, Nordsmark M, Busk M, Wiuf C, Alsner J, and Overgaard J. (2011). Development of a hypoxia gene expression classifier with predictive impact for hypoxic modification of radiotherapy in head and neck cancer. Cancer Res. 71, 5923–5931. [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J. (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- Ulanovskaya OA, Zuhl AM, and Cravatt BF (2013). NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol 9, 300–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vladoiu MC, El-Hamamy I, Donovan LK, Farooq H, Holgado BL, Sundaravadanam Y, Ramaswamy V, Hendrikse LD, Kumar S, Mack SC, et al. (2019). Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 572, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, and Hakonarson H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164–e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei T, and Simko V. (2017). R package “corrplot”: Visualization of a Correlation Matrix (Version 0.84). [Google Scholar]
- Yuan Z-F, Sidoli S, Marchione DM, Simithy J, Janssen KA, Szurgot MR, and Garcia BA (2018). EpiProfile 2.0: A Computational Platform for Processing Epi-Proteomics Mass Spectrometry Data. J. Proteome Res 17, 2533–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Badur MG, Divakaruni AS, Parker SJ, Jäger C, Hiller K, Murphy AN, and Metallo CM (2016). Distinct Metabolic States Can Support Self-Renewal and Lipogenesis in Human Pluripotent Stem Cells under Different Culture Conditions. Cell Rep 16, 1536–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A) Three PFA cell lines (MDT-PFA2, MDT-PFA4, MDT-PFA5) were cultured continuously in either 1% or 21% oxygen for a total of 21 days. Cells were seeded at low density (40,000 cells) in multiple 12-well and 24-well plates. Replicates were counted at various time points throughout the time-course, and plots were fitted using an exponential growth non-linear regression model.
B) PFA cells from the primary tumour were cultured at a variety of oxygen conditions (10%, 5%, 1% and 0.2%) for 200 hours of continuous culture. A seeding density of 100,000 cells was used and cells were counted at endpoint.
C) Limited dilution assay of PFA cells cultured in 1% (blue) or 21% (grey) oxygen. Graph depicts fraction of replicates negative for sphere formation at different dilutions of cell seeding. Images demonstrate relative sphere formation under each oxygen condition.
D) Kaplan-Meier survival analysis of PFA ependymoma patients stratified by the median level of the respective hypoxia transcriptional signature score. The log-rank test was used with the calculated hazard ratio ± 95% CI bolded. The hazard ratios for worse overall survival with high transcriptional activity ± 95% CI for all signatures are also plotted.
Oncoprint of damaging single nucleotide variants (SNVs) or focal gain/loss mutations in 6 matched PFA ependymoma tissue-line pairs. SNVs were called by 3 or more mutation callers and focal gains or losses called by GISTIC. Mutation frequency indicated by scale bar on right.
A) Volcano plot of differential gene expression analysis between PFA-EPN (blue) and STEPN (green) surgical biopsies (DEseq2). Significantly differentially expressed genes are indicated in colour (log2foldchange > 1, Padj < 0.05).
B) Volcano plot of differential gene expression analysis between PFA-EPN (blue) and normal brain (maroon) surgical biopsies (DEseq2). Significantly differentially expressed genes are indicated in colour (log2foldchange > 1, Padj < 0.05).
C) Cytoscape map visualization of upregulated pathways between PFA-EPN (blue) and STEPN (green) surgical biopsies. Gene set enrichment analysis (Padj < 0.05, qval <0.05) was used as input.
D) Cytoscape map visualization of upregulated pathways between PFA-EPN (blue) and normal brain (maroon) surgical biopsies. Gene set enrichment analysis (Padj < 0.05, qval <0.01) was used as input.
E) Cytoscape map visualization of upregulated pathways between PFA primary cells cultured in 1% oxygen (blue) or 21% oxygen (grey). Gene set enrichment analysis (Padj < 0.05, qval <0.01) was used as input.
F) Heatmap demonstrating the Pearson correlation scores of normalized and corrected Beta values determined from 850K methylation arrays, between matched PFA primary tissue and cell cultures. A high correlation score (blue) is seen between matched samples. Scale or correlation score normalized between −1 and 1.
A) Volcano plot of differential metabolite abundance between PFA and ST-EPN surgical biopsies. Metabolites are annotated by KEGG super pathway designation. Significant metabolites are highlighted in colour (FDR < 0.05, Fold Change > 1.5).
B) Differential abundance of metabolites grouped by KEGG pathway annotation in PFA-EPN (blue) compared to normal brain (red). Differential abundance score plotted with setscale of −1 to 1.
C) Volcano plot of differential metabolite abundance between PFA and normal brain surgical biopsies. Metabolites are annotated by KEGG super pathway designation. Significant metabolites are highlighted in colour (FDR < 0.05, Fold Change > 1.5).
D) Integrative metabologram of gene expression and metabolite abundance for selected KEGG pathways in PFA tissue relative to normal brain tissue. The schematic illustrates the format of the metabologram plots (derived from Hakmi et al). Log2FoldChange shown on a set scale from −3.0 to 3.0.
A) Representative immunohistochemistry images of PFA and ST ependymoma surgical biopsies stained for H3K27me3 and CA9, demonstrating that hypoxic regions of PFA-EPN tumors have lower level of H3K27me3. Scale bar: 100μM. Quantification of H3K27me3 positivity in CA9 low and high regions (as a surrogate for hypoxia), in PFA (n=4) and ST (n=4) surgical biopsies. Scoring was performed blinded by a trained pathologist. Statistical comparisons were conducted using Welch’s t-test.
B) Heatmap of genome wide H3K27me3 scores (0–100 set scale) in MDT-PFA2 and MDTPFA5 grown at 1% or 21% oxygen. Histone trimethylation marks are determined within +/− 10kb of annotated transcriptional start sites.
C) Average H3K27me3 score for all genes and their respective transcriptional start sites in 1% and 21% oxygen.
D) Plot of H3K27 trimethylation marks for highly differentially expressed candidate genes in PFA ependymoma cells cultured in 1% and 21% oxygen.
A) Relative abundance of metabolites in the urea cycle, TCA cycle, glycolysis and amino acid pools in 1% versus 21% oxygen. PFA cells (orange), ST-EPN cells (purple), fNSCs (blue).
B) Abundance of labeled αKG in 1% (blue) versus 21% (grey) oxygen growth conditions for fNSCs, PFA and ST-EPN cells. All possible isotope compositions (m+) are reported.
C) Abundance of labeled citrate in 1% (blue) versus 21% (grey) oxygen growth conditions for fNSCs, PFA and ST-EPN cells. All possible isotope compositions (m+) are reported.
A) Schematic of labelled [13C-glutamine] tracing in the cell. Isotope configurations are shown throughout the TCA cycle, in forward and reverse direction, and cytosolic acetyl-CoA production. Colours depict labeled carbons (light blue) and unlabelled carbons (navy blue).
B) Abundance of labeled malate, aspartate, and fumarate in 1% (blue) versus 21% (grey) oxygen growth conditions for fNSC, PFA and ST-EPN cells. Cytosolic (m+3, red) and mitochondrial (m+4, green) are indicated.
S1 – List of significantly differentially expressed genes (RNAseq) comparing PFA cells grown in 1% compared to 21% oxygen (Related to Figure 2, Figure S3).
S2 – List of significantly differentially expressed genes (RNAseq) comparing PFA-EPN and ST-EPN surgical biopsies (Related to Figure 2, Figure S3).
S3 – List of significantly differentially expressed genes (RNAseq) comparing PFA-EPN and normal control brain surgical biopsies (Related to Figure 2, Figure S3).
S4 – List of significant essential genes as determined by whole genome CRISPR knockout screen comparing PFA ependymoma to glioblastoma cells (Related to Figure 6).
S5 – List of significant essential genes as determined by whole genome CRISPR knockout screen comparing PFA ependymoma to fetal neural stem cells (Related to Figure 6).
S6 – List of significant essential genes as determined by whole genome CRISPR knockout screen common to both comparisons in S1 and S2 above (Related to Figure 6b).
S7 – List of significantly differential metabolites comparing PFA-EPN and ST-EPN surgical biopsies (Related to Figure 7).
Data Availability Statement
All datasets generated in this study have been deposited and are available as follows: Methylation - GEO: GSE146426; RNAseq & WGS – EGAS00001004312; Cut&Run Seq – GEO: GSE146858; Proteomics – PRIDE: PXD018191. This study did not generate any unique code requiring deposition.