

Abstract
Background
Specific peripheral proteins have been implicated to play an important role in the development of Alzheimer’s disease (AD). However, the roles of additional novel protein biomarkers in AD etiology remains elusive. The availability of large-scale AD GWAS and plasma proteomic data provide the resources needed for the identification of causally relevant circulating proteins that may serve as risk factors for AD and potential therapeutic targets.
Methods
We established and validated genetic prediction models for protein levels in plasma as instruments to investigate the associations between genetically predicted protein levels and AD risk. We studied 71,880 (proxy) cases and 383,378 (proxy) controls of European descent.
Results
We identified 69 proteins with genetically predicted concentrations showing associations with AD risk. The drugs almitrine and ciclopirox targeting ATP1A1 were suggested to have a potential for being repositioned for AD treatment.
Conclusions
Our study provides additional insights into the underlying mechanisms of AD and potential therapeutic strategies.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13195-023-01378-4.
Keywords: Genetic instrument, Protein biomarker, Alzheimer’s disease, Risk
Summary box
What is already know on this topic
There is one study evaluating associations between genetically predicted protein levels in dorsolateral prefrontal cortex and risk of Alzheimer’s disease (AD); another study focuses on 38 dementia-associated proteins to determine associations of their genetically predicted levels in plasma with AD risk; a third study assesses 184 cerebrospinal fluid proteins, 100 plasma proteins, and 27 brain proteins using protein quantitative trait loci as instruments for their associations with AD risk. Existing studies did not systematically evaluate associations of predicted levels proteins across the proteome in plasma using genetic prediction models, findings of which may identify novel proteins to confer translational perspective for risk assessment and therapeutic strategies of AD.
What this study adds
Our study identifies 69 potential AD-associated proteins in plasma using comprehensive genetic prediction models as instruments. We also prioritize drugs almitrine and ciclopirox targeting ATP1A1 to have a potential for being repositioned for AD treatment.
How this study might affect research, practice, or policy
The promising proteins identified in our study could be further investigated for their roles in AD risk assessment and therapeutic strategies.
Introduction
Alzheimer’s disease (AD), the most common cause of dementia, has become a growing public health concern due to an unprecedented increase in life expectancy globally. In the USA, reported deaths from AD have increased 146.2% between 2000 and 2018, making it the sixth leading cause of death [1]. It is predicted that the annual cost of caring for AD patients will reach to a trillion dollars by 2050. AD is an irreversible and progressive disorder with neuropathological changes often occurring long before any symptom becomes apparent. The abnormal accumulation of amyloid-beta (Aβ) plaques, a hallmark of AD, is known to occur as early as two decades before the onset of clinical symptoms [2]. Abnormal phosphorylation of tau, the second canonical AD protein aggregate, is believed to occur shortly thereafter (15–20 years before symptom onset) [3]. While a great deal of research effort has focused on targeting pathological Aβ aggregates and tau neurofibrillary tangles, several drugs were approved by U.S. Food and Drug Administration (FDA), including Aduhelm® [4] and Leqembi® [5]. These approved drugs could relieve symptoms while whether they can cure AD relies on further analyses. As a result, it is critical to identify novel biomarkers and biological pathways that may contribute to AD risk.
Physiological changes that take place outside the brain (e.g., immune, vascular, and metabolic changes) have been shown to directly influence the function of neural cells and relate strongly to risk of developing AD [6, 7]. The identification of circulating peripheral proteins that drive the associations between peripheral biological changes and increased risk for AD may enhance our understanding of AD pathogenesis and thereby inform future therapeutic strategies. In addition to Aβ and tau, a number of proteins have also been recognized to be related to AD [8]. Translational and epidemiological research indicates that biological processes which operate outside of the central nervous system can contribute considerably to one’s risk of developing AD [6, 9]. These peripheral biological processes can be reflected in plasma and serum protein composition, i.e., secreted proteins. Identifying proteins that are causally associated with AD-relevant outcomes will deepen our understanding regarding how peripheral molecular changes, biological pathways, and regulatory mechanisms influence AD risk.
AD is highly heritable. Twin and family studies support that genetic factors could play a role in at least 80% of AD cases [10]. A recent genome-wide association study (GWAS) has identified 29 independent disease-associated risk loci by studying 71,880 (proxy) cases and 383,378 (proxy) controls of European ancestry [11]. The present study aimed at identifying novel protein biomarkers for AD through evaluating the associations between genetically predicted protein concentrations and AD risk, a design of proteome-wide association study (PWAS). Similar to the design of Mendelian randomization (MR) and transcriptome-wide association study (TWAS) [12–15], such a design can potentially reduce common biases imbedded in conventional epidemiological studies, such as selection biases, residual confounding, or reverse causality. We established and validated comprehensive protein genetic prediction models to fully capture the genetically regulated components of protein levels by using both cis- and trans-acting elements, thus providing higher statistical power than only using cis-acting elements alone (a common practice for related studies). We then related genetically predicted plasma concentrations to AD risk and, in doing so, causally implicated 69 circulating proteins in the AD pathogenesis, shedding light on the peripheral biology of AD.
Methods
The genome and plasma proteome data of European descendants included in the INTERVAL study (subcohort 1 and subcohort 2) was used to establish and validate protein genetic prediction models. Detailed information about the INTERVAL study dataset has been described elsewhere [16]. In brief, participants were aged 18–80 and were generally in good health. The SOMAscan assay was used to measure the relative concentrations of 3620 plasma proteins or protein complexes. Quality control (QC) was performed at the sample and SOMAmer level. After excluding eight non-human protein targets, a total of 3283 SOMAmers remained for further study. DNA was used to assay ~ 830,000 variants on the Affymetrix Axiom UK Biobank genotyping array. Standard sample and variant QC was conducted, as described in the original publication [16]. SNPs were further phased using SHAPEIT3 and imputed using a combined 1000 Genomes Phase 3-UK10K reference panel via the Sanger Imputation Server, resulting in over 87 million imputed variants. Such SNPs were filtered using criteria of (1) imputation quality of at least 0.7, (2) minor allele frequency (MAF) of at least 5%, (3) Hardy–Weinberg equilibrium (HWE) p ≥ 5 × 10−6, (4) missing rates < 5%, and (5) presenting in the 1000 Genome Project data for European populations. In total, there were 4,662,360 variants passing these criteria.
In subcohort 1 (N = 2481), protein levels were log transformed and adjusted for age, sex, duration between blood draw and processing, and the first three principal components of ancestry. For the rank-inverse normalized residuals of each protein of interest, we followed the TWAS/FUSION framework [17] to develop genetic prediction models, using nearby SNPs (within 100 kb) of potentially associated SNPs as potential predictors. A false discovery rate (FDR) < 0.05 and P-value ≤ 5 × 10−8 were used to determine potentially associated SNPs in cis- and trans- regions, respectively. We defined cis-region as a region within 1 Mb of the transcriptional start site (TSS) of the gene encoding the target protein of interest. Subsequently, we extracted all SNPs located within 100 kb of the aforementioned potentially associated SNPs to serve as potential predictors for establishing protein prediction models, excluding any ambiguous SNPs. In order to include potential predictors from both cis and trans regions, we converted all the chromosome numbers to Z and combined them as a single pseudo chromosome. Four methods, namely, best linear unbiased predictor, elastic net, LASSO, and top1, were used for establishing the models. For developed protein prediction models with prediction performance (R2) of at least 0.01 [15, 18–23], which is a common threshold used in relevant studies, we further conducted external validation using subcohort 2 (N = 820) data. In brief, we generated predicted expression levels by applying the established protein prediction models to the genetic data, and then compared the predicted v.s. measured levels of each protein of interest. We selected proteins with a model prediction R2 of ≥ 0.01 in subcohort 1 and a correlation coefficient of ≥ 0.1 in subcohort 2 for the downstream association analysis.
To assess the associations between genetically predicted circulating protein levels and AD risk, we applied the validated protein prediction models to the summary statistics from a large GWAS meta-analysis of AD risk [24]. Instead of using the conventional approach of including clinically diagnosed AD alone, this GWAS combined clinically confirmed and parental diagnoses based by-proxy phenotypes, which has been demonstrated to confer great value in substantially increasing statistical power [25]. In brief, this study included a total of 85,934 cases (39,106 clinically diagnosed AD and 46,828 proxy AD) and 401,577 controls of European ancestry, which were obtained from various sources including The European Alzheimer & Dementia Biobank dataset (EADB), GR@ACE/DEGESCO study, The Rotterdam Study (RS1 and RS2), European Alzheimer’s Disease Initiative (EADI) Consortium, Genetic and Environmental Risk in AD (GERAD) Consortium/Defining Genetic, Polygenic, and Environmental Risk for Alzheimer’s Disease (PERADES) Consortium, The Norwegian DemGene Network, The Neocodex–Murcia study (NxC), The Copenhagen City Heart Study (CCHS), Bonn studies, and UK Biobank. Detailed information on study participants as well as genotyping and imputation methods for the samples from each of the included study can be found in the supplementary files of the original GWAS paper [24]. Risk estimates for the single marker association analyses were adjusted for sex, batch (if applicable), age (if applicable), and top principal components (PCs).
The TWAS/FUSION framework was used to determine the protein-AD associations, by leveraging correlation information between SNPs included in the prediction models from the phase 3, 1000 Genomes Project data of European ancestry [17]. We calculated the PWAS test statistic Z-score = w'Z/(w'Σs,sw)1/2, where the Z is a vector of standardized effect sizes of SNPs for a given protein (Wald z-scores), w is a vector of prediction weights for the abundance feature of the protein being tested, and the Σs,s is the LD matrix of the SNPs estimated from the 1000 Genomes Project as the LD reference panel. The Bonferroni correction P-value < 0.05 was used to determine significant associations between genetically predicted protein concentrations and AD risk.
Ingenuity Pathway Analysis (IPA, Ingenuity System Inc, USA)) and Protein–Protein Interaction analysis via STRING database (version 12.0) with 0.400 confidence level [26] was implemented to cluster and classify enriched pathways for the identified proteins using default interaction resources, including Textmining, Experiments, Databases, Co-expression, Neighborhood, Gene Fusion, and Co-occurrence. We also investigated potentially repositionable drugs targeting the genes encoding associated proteins, by using the GREP (Genome for REPositioning drugs) tool [27]. We further conducted molecular docking analysis considering ATP1A1 protein as the drug target protein and almitrine and ciclopirox as the drug agents [28].
Results
In this study, potential predictors were identified for 1870 proteins, and protein prediction models were successfully established for 1864 proteins. For the 1413 of the remaining proteins, there was no SNP showing an association at FDR < 0.05 for cis SNPs and P-value ≤ 5 × 10−8 for trans SNPs. After internal and external validation, there were 1389 proteins showing internal and external validation performance of R2 ≥ 0.01. The median external validation R2 was 0.06. There were 459, 189, and 38 proteins that showed external validation R2 ≥ 0.1, 0.2, and 0.5, respectively. Overall, proteins that could be predicted well in INTERVAL subcohort 1 also tended to be predicted well in subcohort 2 in external validation analyses (a correlation coefficient of 0.96 for R2 in two data sets; Fig. 1). Using the TWAS/FUSION framework, we examined the association for a total of 1340 proteins. For the remaining 49 proteins, more than half of the SNPs included in the models were not present in the AD GWAS summary; therefore, their associations with AD risk were not considered. We identified 69 proteins with genetically predicted concentrations showing associations with AD risk after Bonferroni correction (P-value < 3.01 × 10−5) (Table 1; Fig. 2). Of those 69 proteins, positive associations were observed for 45 of them, and inverse associations were observed for 24 (Table 1; Fig. 2).
Fig. 1.
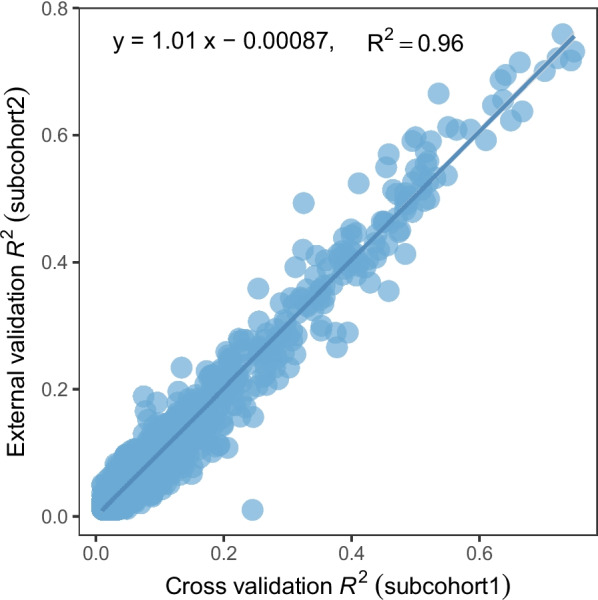
Performance of protein expression prediction models in INTERVAL subcohort1 and subcohort2 datasets for proteins showing internal and external validation performance of R2 ≥ 0.01
Table 1.
Proteins showing a significant association with Alzheimer’s disease risk for their genetically predicted concentrations in plasma
| Protein | Protein full name | SOMAMER ID | Encoding gene ID | Modeling methoda | Num of predicting SNPs in model | Num of predicting SNPs-Cisb | Num of predicting SNPs-Trans | Model internal cross validation R2 | Model external validation R2 | Z scorec | P-value | Distance of gene to closest risk SNP (kb)d |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TBCA | Tubulin-specific chaperone A | TBCA.12501.10.3 | TBCA | blup | 470 | 0 | 470 | 0.54 | 0.51 | − 50.90 | < 1.09 × 10−298 | 9,236.20 |
| S100A13 | Protein S100-A13 | S100A13.7223.60.3 | S100A13 | blup | 528 | 0 | 528 | 0.56 | 0.54 | − 39.10 | < 1.09 × 10−298 | 7,564.12 |
| DCK | Deoxycytidine kinase | DCK.9836.20.3 | DCK | lasso | 5 | 0 | 5 | 0.04 | 0.02 | 19.80 | 7.23 × 10−87 | 31,660.52 |
| PSME1 | Proteasome activator complex subunit 1 | PSME1.5918.5.3 | PSME1 | lasso | 13 | 0 | 13 | 0.19 | 0.11 | − 19.00 | 8.12 × 10−81 | 28,693.45 |
| SYVC | Valine–tRNA ligase | VARS.13083.18.3 | VARS | enet | 69 | 0 | 69 | 0.38 | 0.27 | 16.90 | 2.71 × 10−64 | 833.24 |
| PSG5 | Pregnancy-specific beta-1-glycoprotein 5 | PSG5.9314.9.3 | PSG5 | enet | 50 | 16 | 34 | 0.07 | 0.05 | 16.90 | 8.68 × 10−64 | 1,740.05 |
| UBP21 | Ubiquitin carboxyl-terminal hydrolase 21 | USP21.12681.63.3 | USP21 | enet | 39 | 0 | 39 | 0.14 | 0.08 | 15.90 | 3.69 × 10−57 | 26.10 |
| SURF1 | Surfeit locus protein 1 | SURF1.8009.121.3 | SURF1 | enet | 68 | 0 | 68 | 0.06 | 0.05 | 15.80 | 1.62 × 10−56 | 28,552.64 |
| RPAC1 | DNA-directed RNA polymerases I and III subunit RPAC1 | POLR1C.12939.1.3 | POLR1C | enet | 44 | 0 | 44 | 0.03 | 0.01 | 15.30 | 6.67 × 10−53 | 2,542.63 |
| P5I11 | Tumor protein p53-inducible protein 11 | TP53I11.13022.20.3 | TP53I11 | enet | 19 | 0 | 19 | 0.06 | 0.05 | 15.20 | 2.25 × 10−52 | 2,426.44 |
| NRBP | Nuclear receptor-binding protein | NRBP1.12616.45.3 | NRBP1 | enet | 40 | 11 | 29 | 0.07 | 0.02 | 14.90 | 3.56 × 10−50 | 9,881.28 |
| MO2R2 | Cell surface glycoprotein CD200 receptor 2 | CD200R1L.8980.19.3 | CD200R1L | top1 | 1 | 0 | 1 | 0.09 | 0.03 | − 13.30 | 4.44 × 10−40 | 42,252.95 |
| APBB2 | Amyloid beta A4 precursor protein-binding family B member 2 | APBB2.12753.6.3 | APBB2 | top1 | 1 | 0 | 1 | 0.05 | 0.03 | − 13.20 | 8.33 × 10−40 | 613.20 |
| MUCDL | Cadherin-related family member 5 | CDHR5.9962.1.3 | CDHR5 | top1 | 1 | 0 | 1 | 0.03 | 0.02 | 13.10 | 2.66 × 10−39 | 46,763.76 |
| TM59L | Transmembrane protein 59-like | TMEM59L.9959.60.3 | TMEM59L | enet | 47 | 0 | 47 | 0.05 | 0.01 | 12.40 | 2.48 × 10−35 | 16,869.43 |
| EMAP-2 | Endothelial monocyte-activating polypeptide 2 | AIMP1.2714.78.2 | AIMP1 | lasso | 4 | 0 | 4 | 0.06 | 0.03 | 11.50 | 1.51 × 10−30 | 67,037.86 |
| MED4 | Mediator of RNA polymerase II transcription subunit 4 | MED4.14021.81.3 | MED4 | top1 | 1 | 0 | 1 | 0.09 | 0.05 | 11.10 | 7.48 × 10−29 | NAd |
| QORL1 | Quinone oxidoreductase-like protein 1 | CRYZL1.9207.60.3 | CRYZL1 | enet | 18 | 0 | 18 | 0.16 | 0.13 | 11.10 | 7.76 × 10−29 | 7,440.72 |
| GST M1-1 | Glutathione S-transferase Mu 1 | GSTM1.7239.9.3 | GSTM1 | enet | 20 | 6 | 14 | 0.03 | 0.05 | 10 | 1.03 × 10−23 | 342.01 |
| XRCC4 | DNA repair protein XRCC4 | XRCC4.9886.28.3 | XRCC4 | lasso | 4 | 0 | 4 | 0.05 | 0.03 | 9.43 | 3.98 × 10−21 | 3,849.83 |
| Cofilin-1 | Cofilin-1 | CFL1.4203.50.2 | CFL1 | lasso | 6 | 0 | 6 | 0.11 | 0.08 | 9.04 | 1.59 × 10−19 | 5,600.20 |
| MAPK5 | MAP kinase-activated protein kinase 5 | MAPKAPK5.8382.47.3 | MAPKAPK5 | lasso | 21 | 0 | 21 | 0.48 | 0.49 | 8.79 | 1.48 × 10−18 | 1,439.76 |
| SULT 1E | Estrogen sulfotransferase | SULT1E1.9878.3.3 | SULT1E1 | lasso | 10 | 0 | 10 | 0.11 | 0.05 | − 8.08 | 6.54 × 10−16 | 30,508.08 |
| Testican-1 | Testican-1 | SPOCK1.5490.53.3 | SPOCK1 | enet | 24 | 0 | 24 | 0.05 | 0.01 | 7.38 | 1.63 × 10−13 | 14,121.40 |
| LRTM2 | Leucine-rich repeat and transmembrane domain-containing protein 2 | LRTM2.8906.60.3 | LRTM2 | enet | 49 | 0 | 49 | 0.04 | 0.01 | 6.70 | 2.03 × 10−11 | 111,790.06 |
| APRV1 | Retroviral-like aspartic protease 1 | ASPRV1.13023.8.3 | ASPRV1 | enet | 42 | 0 | 42 | 0.14 | 0.12 | 6.59 | 4.26 × 10−11 | 32,627.91 |
| PRPC | Salivary acidic proline-rich phosphoprotein 1/2 | PRH1.10502.15.3 | PRH1 | enet | 38 | 21 | 17 | 0.05 | 0.04 | 6.53 | 6.52 × 10−11 | 102,686.22 |
| Furin | Furin | FURIN.6276.16.3 | FURIN | enet | 43 | 0 | 43 | 0.07 | 0.05 | 6.51 | 7.76 × 10−11 | 12,182.62 |
| SL9B2 | Mitochondrial sodium/hydrogen exchanger 9B2 | SLC9B2.9088.20.3 | SLC9B2 | lasso | 4 | 0 | 4 | 0.02 | 0.02 | 6.48 | 9.02 × 10−11 | 63,740.34 |
| Gc-Globulin, Mixed Type | Vitamin D-binding protein | GC.6581.50.3 | GC | enet | 30 | 0 | 30 | 0.17 | 0.16 | 6.40 | 1.53 × 10−10 | 32,408.56 |
| PRB4 | Basic salivary proline-rich protein 4 | PRB4.12590.67.3 | PRB4 | top1 | 1 | 0 | 1 | 0.02 | 0.03 | − 6.29 | 3.21 × 10−10 | 102,259.78 |
| CR026 | Dynactin-associated protein | DYNAP.10692.48.3 | DYNAP | top1 | 1 | 0 | 1 | 0.02 | 0.02 | 6.29 | 3.21 × 10−10 | 3,934.47 |
| CCNC | Cyclin-C | CCNC.7817.36.3 | CCNC | top1 | 1 | 0 | 1 | 0.02 | 0.03 | 6.11 | 1.01 × 10−9 | 14,622.63 |
| CLC4E | C-type lectin domain family 4 member E | CLEC4E.7077.9.4 | CLEC4E | top1 | 1 | 0 | 1 | 0.02 | 0.02 | 6.11 | 1.01 × 10−9 | 105,033.89 |
| CEACAM1 | Carcinoembryonic antigen-related cell adhesion molecule 1 | CEACAM1.8031.11.3 | CEACAM1 | enet | 21 | 0 | 21 | 0.23 | 0.26 | 5.94 | 2.91 × 10−9 | 2,400.48 |
| AT1A1 | Sodium/potassium-transporting ATPase subunit alpha-1 | ATP1A1.11993.227.3 | ATP1A1 | enet | 15 | 0 | 15 | 0.06 | 0.07 | 5.91 | 3.52 × 10−9 | 7,027.43 |
| CF226 | Uncharacterized protein C6orf226 | C6orf226.8078.15.3 | C6orf226 | lasso | 7 | 0 | 7 | 0.07 | 0.07 | 5.74 | 9.61 × 10−9 | 1,915.81 |
| AXIN2 | Axin-2 | AXIN2.8429.16.3 | AXIN2 | enet | 18 | 0 | 18 | 0.40 | 0.44 | 5.57 | 2.48 × 10−8 | 1,978.90 |
| TRML4 | Trem-like transcript 4 protein | TREML4.11139.4.3 | TREML4 | lasso | 9 | 0 | 9 | 0.21 | 0.20 | − 5.57 | 2.51 × 10−8 | 253.89 |
| DAF | Complement decay-accelerating factor | CD55.5069.9.3 | CD55 | lasso | 11 | 11 | 0 | 0.11 | 0.07 | − 5.57 | 2.55 × 10−8 | 197.03 |
| EVI2B | Protein EVI2B | EVI2B.13028.2.3 | EVI2B | enet | 11 | 0 | 11 | 0.07 | 0.05 | − 5.52 | 3.30 × 10−8 | 11,571.33 |
| TMIE | Transmembrane inner ear expressed protein | TMIE.7992.3.3 | TMIE | enet | 7 | 0 | 7 | 0.04 | 0.05 | 5.47 | 4.46 × 10−8 | 10,490.88 |
| AMBN | Ameloblastin | AMBN.6522.57.3 | AMBN | lasso | 11 | 0 | 11 | 0.15 | 0.07 | − 5.47 | 4.52 × 10−8 | 31,259.13 |
| SLAF5 | SLAM family member 5 | CD84.8770.136.3 | CD84 | blup | 46 | 0 | 46 | 0.05 | 0.08 | 5.46 | 4.65 × 10−8 | 644.50 |
| SG1C1 | Secretoglobin family 1C member 1 | SCGB1C1.5960.49.3 | SCGB1C1 | enet | 25 | 0 | 25 | 0.18 | 0.10 | − 5.42 | 5.84 × 10−8 | 47,187.26 |
| KCNE2 | Potassium voltage-gated channel subfamily E member 2 | KCNE2.10427.2.3 | KCNE2 | enet | 33 | 0 | 33 | 0.26 | 0.26 | − 5.38 | 7.55 × 10−8 | 8,215.37 |
| FA20A | Pseudokinase FAM20A | FAM20A.6433.57.3 | FAM20A | enet | 30 | 17 | 13 | 0.04 | 0.03 | − 5.36 | 8.43 × 10−8 | 4,985.48 |
| SHSA3 | Protein shisa-3 homolog | SHISA3.7057.18.3 | SHISA3 | enet | 22 | 0 | 22 | 0.23 | 0.16 | − 5.20 | 2.01 × 10−7 | 2,200.66 |
| NPTX2 | Neuronal pentraxin-2 | NPTX2.6521.35.3 | NPTX2 | enet | 30 | 0 | 30 | 0.04 | 0.05 | 5.20 | 2.01 × 10−7 | 1,685.45 |
| ILT-4 | Leukocyte immunoglobulin-like receptor subfamily B member 2 | LILRB2.5633.65.3 | LILRB2 | lasso | 8 | 6 | 2 | 0.21 | 0.22 | 5.19 | 2.15 × 10−7 | 6.22 |
| DIAC | Di-N-acetylchitobiase | CTBS.6115.40.3 | CTBS | enet | 20 | 0 | 20 | 0.14 | 0.09 | − 5.11 | 3.23 × 10−7 | 24,873.14 |
| IGFL4 | Insulin growth factor-like family member 4 | IGFL4.6353.60.3 | IGFL4 | enet | 18 | 0 | 18 | 0.10 | 0.05 | − 4.95 | 7.47 × 10−7 | 300.60 |
| NAP-2 | Neutrophil-activating peptide 2 | PPBP.2790.54.2 | PPBP | enet | 22 | 0 | 22 | 0.03 | 0.01 | − 4.93 | 8.04 × 10−7 | 34,653.31 |
| MCFD2 | Multiple coagulation factor deficiency protein 2 | MCFD2.10476.23.3 | MCFD2 | blup | 744 | 0 | 744 | 0.53 | 0.60 | 4.92 | 8.62 × 10−7 | 9,597.07 |
| SLUR1 | Secreted Ly-6/uPAR-related protein 1 | SLURP1.6401.73.3 | SLURP1 | lasso | 4 | 0 | 4 | 0.06 | 0.03 | − 4.86 | 1.17 × 10−6 | 1,285.78 |
| SGCB | Beta-sarcoglycan | SGCB.7034.4.3 | SGCB | enet | 16 | 0 | 16 | 0.10 | 0.06 | − 4.84 | 1.30 × 10−6 | 12,688.03 |
| RN165 | RING finger protein 165 | RNF165.11561.32.3 | RNF165 | enet | 18 | 0 | 18 | 0.15 | 0.11 | − 4.84 | 1.32 × 10−6 | 12,275.48 |
| PSD2 | PH and SEC7 domain-containing protein 2 | PSD2.9118.7.3 | PSD2 | enet | 43 | 0 | 43 | 0.15 | 0.16 | − 4.74 | 2.14 × 10−6 | 11,257.00 |
| ICOS | Inducible T-cell costimulator | ICOS.14084.191.3 | ICOS | enet | 27 | 0 | 27 | 0.17 | 0.12 | − 4.72 | 2.41 × 10−6 | 1,058.05 |
| SHPS1 | Tyrosine-protein phosphatase non-receptor type substrate 1 | SIRPA.5430.66.3 | SIRPA | blup | 2,563 | 2,506 | 57 | 0.63 | 0.62 | 4.63 | 3.71 × 10−06 | 1,480.84 |
| FKBP6 | Inactive peptidyl-prolyl cis–trans isomerase FKBP6 | FKBP6.12529.32.3 | FKBP6 | blup | 1,593 | 0 | 1,593 | 0.49 | 0.53 | 4.54 | 5.53 × 10−6 | 17,800.84 |
| Cathepsin H | Cathepsin H | CTSH.8465.52.3 | CTSH | lasso | 13 | 12 | 1 | 0.30 | 0.28 | 4.51 | 6.58 × 10−6 | 15.80 |
| Siglec-3 | Myeloid cell surface antigen CD33 | CD33.3166.92.1 | CD33 | enet | 98 | 92 | 6 | 0.41 | 0.45 | 4.47 | 7.78 × 10−6 | 0.37 |
| SPA9 | Serpin A9 | SERPINA9.7266.4.3 | SERPINA9 | lasso | 4 | 0 | 4 | 0.02 | 0.02 | 4.46 | 8.02 × 10−6 | 1,990.20 |
| B3GN1 | Beta-1,4-glucuronyltransferase 1 | B3GNT1.8259.25.3 | B3GNT1 | enet | 50 | 0 | 50 | 0.15 | 0.17 | 4.40 | 1.09 × 10−5 | 6,090.90 |
| APBB2 | Amyloid beta A4 precursor protein-binding family B member 2 | APBB2.12761.12.3 | APBB2 | lasso | 9 | 6 | 3 | 0.03 | 0.01 | 4.24 | 2.26 × 10−5 | 613.20 |
| TMM85 | ER membrane protein complex subunit 4 | EMC4.13516.46.3 | EMC4 | enet | 73 | 0 | 73 | 0.21 | 0.20 | − 4.20 | 2.63 × 10−5 | 24,505.40 |
| Siglec-9 | Sialic acid-binding Ig-like lectin 9 | SIGLEC9.3007.7.2 | SIGLEC9 | blup | 618 | 580 | 38 | 0.72 | 0.73 | 4.19 | 2.75 × 10−5 | 99.83 |
| ADSV | Adseverin | SCIN.12684.5.3 | SCIN | lasso | 4 | 0 | 4 | 0.03 | 0.02 | 4.17 | 3.01 × 10−5 | 340.75 |
aenet: elastic net; blup: best linear unbiased predictor
bSNPs within 1 MB of the protein-encoding gene
cZ score represents the direction of the association between genetically predicted protein levels and AD risk
dRisk SNPs identified in previous GWAS or fine-mapping studies. The SNP list is included in Table S1
eNA indicates no risk SNP was reported on the chromosome
Fig. 2.

Associations Z scores for proteins showing an association at Bonferroni corrected P-value ≤ 0.05 with AD risk
For those proteins associated with AD risk, the Core Analysis was performed in Ingenuity Pathway Analysis. Assembly of RNA Polymerase I Complex and DNA Double-Strand Break Repair by Non-Homologous End Joining were two canonical pathways showing significant enrichments at P < 0.05 (Table S2; Figure S1). In the Network Analysis, Cell-To-Cell Signaling and Interaction, Hematological System Development and Function, Immune Cell Trafficking was identified which involved 19 associated proteins (Table S3; Figure S2). Based on the Disease and Biological Functions analysis, the top disease functional categories identified were shown in Table S4.
Protein interactions of 69 associated proteins were investigated using the STRING database (Figure S3). In the network, five proteins (ILT-4, PRPC, SHPS1, Siglec-3, and Siglec-9) had three or more interactions with other proteins. Among them, Siglec-3 (known as CD33) was reported as a risk factor for AD and both the mRNA level and protein abundance were found to be increased in AD patients compared to the age-matched controls [29]. This finding is consistent with our current study (Z-score = 4.47, P-value = 7.78 × 10−6).
Based on The Anatomical Therapeutic Chemical (ATC) test using GREP, the drugs almitrine and ciclopirox targeting ATP1A1 were suggested to have a potential for being repositioned for AD treatment (odds ratio (OR) = 63.0; P = 0.022 for almitrine; OR = 35.9, P = 0.035 for ciclopirox).
For molecular docking analysis, we downloaded the 3D structure of ATP1A1 protein from Protein Data Bank (PDB) with source code 3KDP and almitrine and ciclopirox drug from the PubChem database [30, 31]. AutoDock-Vina produced − 7.6 kcal/mol binding energy for ATP1A1 protein with almitrine drug agent and − 6.2 kcal/mol binding energy for ATP1A1 protein with ciclopirox drug agent. Figure 3 showed the 3D structure (left) and 2D schematic diagram (right) of the ATP1A1 potential target and almitrine drug with interacting amino acids: Leu80, Thr81, Met164, Arg198, Phe245, Ala271, Thr272, Ala274, Ser275, Asp740, Val741, Gln744, and Ala745. Figure 4 showed the 3D structure (left) and 2D schematic diagram (right) of the ATP1A1 potential target and ciclopirox drug.
Fig. 3.

The 3D structure (left) and 2D schematic diagram (right) of the ATP1A1 potential target and almitrine drug
Fig. 4.

The 3D structure (left) and 2D schematic diagram (right) of the ATP1A1 potential target and ciclopirox
Discussion
To our knowledge, the present study is the first large population-based study to systematically investigate the associations between genetically predicted circulating protein concentrations in plasma and AD risk using genetic instruments of comprehensive protein prediction models. Overall, we identified 69 proteins that were significantly associated with AD risk after Bonferroni correction. If validated in future studies, our findings could add substantial new knowledge to the etiology of AD and provide a list of protein markers to facilitate precision preventive or therapeutic trials of AD.
Recently, plasma proteins including Aß42 and phosphorylated tau (p-tau217, p-tau181, and others) have been identified as promising plasma biomarkers for clinically and pathologically defined AD [32–34]. While these biomarkers will be incredibly useful for participant risk stratification, it remains vitally important to identify additional AD biomarkers to further understand the pathophysiological processes leading to AD. By examining associations of genetically predicted protein levels in plasma with AD risk, we are able to go beyond a traditional examination of protein-AD association and begin to understand whether proteins may be causally relevant. For example, although plasma levels of YKL-40 [35] have been associated with AD, we did not observe evidence of an association for genetically predicted levels of YKL-40 (Z = 1.50; P = 0.13). This finding seems to support that although specific proteins such as YKL-40 could be strong biomarkers, they may not be causally relevant.
We identified multiple AD-associated proteins using proteomic and genetic methods that were reported for the first time (Table 1). For some of them, there is already existing evidence from functional work supporting their potential links with AD. For example, cofilin-1, as a major actin depolymerizer in the central nervous system, plays a crucial role in maintaining the structure and proper function of neurons [36]. Cofilin rods, which are primarily composed of actin and cofilin-1 and form in response to stressing conditions, have been suggested to be associated with neurodegenerative diseases such as AD by disrupting dendritic transportation and inducing synaptic dysfunction [36, 37]. Additional research is warranted to understand the identified associations for the other proteins.
By using GREP, the drugs almitrine and ciclopirox were suggested to be potentially repositionable for AD treatment. A double-blind controlled study involving patients with memory loss, lack of concentration, impaired mental alertness, and emotional instability supported that almitrine-raubasine could improve cognitive impairments [35]. Another controlled multicenter study investigating patients with cognitive decline (assessed by MMSE, SCAG) again suggested almitrine-raubasine significantly improved symptomatology compared with placebo [38]. Three other trails conducted in China involving 206 patients with vascular dementia also supported significant beneficial effect of almitrine-raubasine combination on the improvement of cognitive function measured by MMSE [39], although high risk of bias was observed. Other research supported that ciclopirox could protect neuronal cells from cell death and astrocytes from peroxynitrate toxicity [40, 41]. Future work may be warranted to further investigate whether almitrine and ciclopirox can indeed treat AD.
The strengths of our study include a high statistical power to identify AD-associated proteins given the large sample size in the main association analysis. Instead of merely using individual protein quantitative trait loci (pQTL) as instruments, we developed comprehensive protein genetic prediction models using a state-of-the-art method and externally validated their performance before applying them to downstream association tests. Our previous work has supported that compared with individual QTLs, comprehensive prediction models can better capture genetically regulated components of molecular levels and thus further increase statistical power [42]. In two recently published studies, pQTLs in plasma were used to assess proteins potentially associated with AD risk [43, 44]. It is expected that the current work should have improved power as well as scope compared with these two existing studies. Particularly, in Walker et al. [44], only proteins showing an association for the directly measured levels were tested. In Yang et al. [43], a relatively smaller dataset (n = 636) was used to determine plasma pQTLs. Correspondingly, a smaller number of pQTLs for 127 proteins were identified for association analyses. In Wingo et al. [45], prediction models for 376 proteins in brain tissue were established, and 13 proteins were identified to be associated with AD risk. It is also worth noting that in the previous studies, AD GWAS summaries involving a less number of cases and controls were employed. Walker and Yang utilized the GWAS summary data from the Kunkle study [46], comprising 21,982 clinically diagnosed AD cases and 41,944 cognitively normal controls, while Wingo employed the AD GWAS summary data from the Jansen study [11], encompassing 71,880 cases (clinically diagnosed AD and AD-by-proxy) and 383,378 controls. In the present study, we utilized a more comprehensive GWAS summary data from a more recent study, including 85,934 cases (comprising 39,106 clinically diagnosed AD and 46,828 proxy AD) and 401,577 controls. We checked the associations of the proteins reported in these previous studies in the current work. Interestingly, only three of the reported proteins showed consistent associations (same effect direction and nominal P-value < 0.05) in the current work (Table S5). To further examine the robustness of these results, we extended our examination by using two independent protein genetic prediction models established by others using independent methods, namely Atherosclerosis Risk in Communities (ARIC) European ancestry models [47] and INTERVAL cis-models [48]. Notably, when we focused only on plasma, a majority of the examined proteins did not exhibit significant associations with the risk of AD when using either ARIC European ancestry or INTERVAL cis-models. This observation that aligns well with results based on our developed models suggests that these prior findings could potentially be false positives. Again, such a discrepancy could be potentially attributed to the relatively limited utility of individual pQTL SNPs in fully elucidating the genetically regulated components of protein levels. Further studies are warranted to better characterize the other previously reported proteins.
Several limitations of the current work also need to be acknowledged. First, our findings may be subject to potential pleiotropic effects, limiting the ability to draw causal insights. Second, given the nature of our study of using genetic instruments to predict plasma protein levels, we are only able to capture the genetically regulated components of the protein concentrations, without incorporating the components influenced by exogenous exposures. Like the concept of transcriptome-wide association studies (TWAS), our proteome-wide association study (PWAS) aims to investigate the relationship between the genetically determined components of protein levels and disease risk. Further prospective studies with measured protein levels in pre-disease plasma samples are needed to better evaluate the relationship. Finally, when we establish genetic models to estimate such genetically determined components of protein levels, we carefully controlled for age, sex, duration between blood draw and processing, and top genetic principal components. However, we acknowledge that specific factors such as smoking and body mass index (BMI) were not controlled for during model construction using the INTERVAL dataset due to a lack of relevant data available to us [49]. Future studies are in need to validate our findings.
In conclusion, in this large association study using genetic instruments, we identified multiple novel AD risk-associated proteins. If validated with further investigations, our study may add additional knowledge to the underlying mechanisms of AD.
Supplementary Information
Additional file 1: Figure S1. Enriched canonical pathways for the identified associated proteins. The and p-value below each term indicates the significance level of each pathway. Figure S2. The network was identified by Ingenuity Pathway Analysis (IPA). A solid line represents a direct interaction between two nodes and a dotted line indicates an indirect interaction. Figure S3. Network nodes represent proteins and edges represent protein-protein associations.
Additional file 2: Table S1. Risk SNPs identified to be associated with AD risk in previous GWAS or fine-mapping studies. Table S2. Ingenuity Canonical Pathways. Table S3. Network analysis. Table S4. Disease and Biological Functions analysis. Table S5. Association results of the proteins reported in previous studies.
Acknowledgements
We would like to acknowledge authors of the GWAS meta-analysis of AD risk by Jansen et al. to make their GWAS summary statistics to be available. Participants in the INTERVAL randomized controlled trial were recruited with the active collaboration of NHS Blood and Transplant England (www.nhsbt.nhs.uk), which has supported field work and other elements of the trial. DNA extraction and genotyping were co-funded by the National Institute for Health Research (NIHR), the NIHR BioResource (http://bioresource.nihr.ac.uk), and the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014) [*]. The academic coordinating center for INTERVAL was supported by core funding from the: NIHR Blood and Transplant Research Unit in Donor Health and Genomics (NIHR BTRU-2014-10024), UK Medical Research Council (MR/L003120/1), British Heart Foundation (SP/09/002; RG/13/13/30194; RG/18/13/33946), and NIHR Cambridge BRC (BRC-1215-20014) [*]. A complete list of the investigators and contributors to the INTERVAL trial is provided in reference [**]. The academic coordinating center would like to thank blood donor center staff and blood donors for participating in the INTERVAL trial.
*The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
**Di Angelantonio E, Thompson SG, Kaptoge SK, Moore C, Walker M, Armitage J, Ouwehand WH, Roberts DJ, Danesh J, INTERVAL Trial Group. Efficiency and safety of varying the frequency of whole blood donation (INTERVAL): a randomized trial of 45 000 donors. Lancet. 2017 Nov 25;390(10110):2360-2371.
Authors’ contributions
L.W., C.W. and J. Z. conceived and designed the study. S.L. and D.G. developed the protein genetic prediction models. S.L. and J.Z. performed statistical analyses. H.Z. performed IPA and PPI analyses. M.A.A. performed molecular docking analysis. J.Z. wrote the first version of manuscript. L.W. and K.W. helped with drafting the manuscript. C.W. contributed to statistical analyses. Z.Z., P.S., S.F., A.S.B., M.A.A., and H-W. D. contributed to interpretation of results and/or manuscript revision. All authors have reviewed and approved the final manuscript.
Funding
This research is supported by University of Hawaii Cancer Center and Florida State University. L.W. is also supported by U54 HG013243. Keenan Walker was supported by the Intramural Research Program of the National Institutes of Health (National Institute on Aging).
Availability of data and materials
Summary statistics of the GWAS meta-analysis of AD risk by Bellenguez et al. are available at GWAS Catalog (https://www.ebi.ac.uk/gwas/) under accession no. GCST90027158. For the INTERVAL SomaLogic study, the individual-level genotype and protein data, and full summary association results from the genetic analysis, are available through the European Genotype Archive (accession number EGAS00001002555). Summary association results are also publicly available at http://www.phpc.cam.ac.uk/ceu/proteins/, through PhenoScanner (http://www.phenoscanner.medschl.cam.ac.uk) and from the NHGRI-EBI GWAS Catalog (https://www.ebi.ac.uk/gwas/downloads/summary-statistics). The scripts and protein genetic prediction models are available at https://github.com/Arthur1021/Protein-prediction-models.
Declarations
Ethics approval and consent to participate
This study was approved by the University of Hawaii Institutional Review Board (2019–00402).
Consent for publication
Not applicable.
Competing interests
L.W. provided consulting service to Pupil Bio Inc. and received honorarium. No potential conflicts of interest were disclosed by other authors.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jingjing Zhu and Shuai Liu contributed equally to this work and are co-first authors.
Contributor Information
Chong Wu, Email: cwu18@mdanderson.org.
Lang Wu, Email: lwu@cc.hawaii.edu.
References
- 1.2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020;16(3):391–460.
- 2.Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 3.Barthélemy NR, Li Y, Joseph-Mathurin N, Gordon BA, Hassenstab J, Benzinger TLS, et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat Med. 2020;26(3):398–407. doi: 10.1038/s41591-020-0781-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haddad HW, Malone GW, Comardelle NJ, Degueure AE, Poliwoda S, Kaye RJ, et al. Aduhelm, a novel anti-amyloid monoclonal antibody, for the treatment of Alzheimer’s Disease: a comprehensive review. Health Psychol Res. 2022;10(3):37023. doi: 10.52965/001c.37023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2023;388(1):9–21. doi: 10.1056/NEJMoa2212948. [DOI] [PubMed] [Google Scholar]
- 6.Yousef H, Czupalla CJ, Lee D, Chen MB, Burke AN, Zera KA, et al. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat Med. 2019;25(6):988–1000. doi: 10.1038/s41591-019-0440-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen X-N, Niu L-D, Wang Y-J, Cao X-P, Liu Q, Tan L, et al. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: a meta-analysis and systematic review of 170 studies. J Neurol Neurosurg Psychiatry. 2019;90(5):590–598. doi: 10.1136/jnnp-2018-319148. [DOI] [PubMed] [Google Scholar]
- 8.Mantzavinos V, Alexiou A. Biomarkers for Alzheimer’s disease diagnosis. Curr Alzheimer Res. 2017;14(11):1149–1154. doi: 10.2174/1567205014666170203125942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.James ED, Niblett PG. Provisional study to quantify and compare parameters of urine leakage in stress and instability incontinence. Br J Urol. 1988;62(3):223–227. doi: 10.1111/j.1464-410X.1988.tb04323.x. [DOI] [PubMed] [Google Scholar]
- 10.Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(10):a006296. doi: 10.1101/cshperspect.a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51(3):404–413. doi: 10.1038/s41588-018-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Birney E. Mendelian randomization. Cold Spring Harb Perspect Med. 2022;12(4):a041302. doi: 10.1101/cshperspect.a041302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47(9):1091–1098. doi: 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu D, Zhu J, Zhou D, Nikas EG, Mitanis NT, Sun Y, et al. A transcriptome-wide association study identifies novel candidate susceptibility genes for prostate cancer risk. Int J Cancer. 2022;150(1):80–90. doi: 10.1002/ijc.33808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong H, Liu S, Zhu J, Wu L. Associations between genetically predicted levels of blood metabolites and pancreatic cancer risk. Int J Cancer. 2023;153(1):103–110. doi: 10.1002/ijc.34466. [DOI] [PubMed] [Google Scholar]
- 16.Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558(7708):73–79. doi: 10.1038/s41586-018-0175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BWJH, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48(3):245–252. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu D, Zhou D, Sun Y, Zhu J, Ghoneim D, Wu C, et al. A transcriptome-wide association study identifies candidate susceptibility genes for pancreatic cancer risk. Cancer Res. 2020;80(20):4346–4354. doi: 10.1158/0008-5472.CAN-20-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu J, O’Mara TA, Liu D, Setiawan VW, Glubb D, Spurdle AB, et al. Associations between genetically predicted circulating protein concentrations and endometrial cancer risk. Cancers. 2021;13(9):2088. doi: 10.3390/cancers13092088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu J, Yang Y, Kisiel JB, Mahoney DW, Michaud DS, Guo X, et al. Integrating genome and methylome data to identify candidate DNA methylation biomarkers for pancreatic cancer risk. Cancer Epidemiol Biomark Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol. 2021;30(11):2079–2087. doi: 10.1158/1055-9965.EPI-21-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu D, Zhu J, Zhao T, Sharapov S, Tiys E, Wu L. Associations between genetically predicted plasma N-glycans and prostate cancer risk: analysis of over 140,000 European descendants. Pharmacogenomics Pers Med. 2021;14:1211–1220. doi: 10.2147/PGPM.S319308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Y, Zhu J, Zhou D, Canchi S, Wu C, Cox NJ, et al. A transcriptome-wide association study of Alzheimer’s disease using prediction models of relevant tissues identifies novel candidate susceptibility genes. Genome Med. 2021;13(1):141. doi: 10.1186/s13073-021-00959-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhong H, Zhu J, Liu S, Ghoneim DH, Surendran P, Liu T, et al. Identification of blood protein biomarkers associated with prostate cancer risk using genetic prediction models: analysis of over 140, 000 subjects. Hum Mol Genet. 2023;32:ddad139. doi: 10.1093/hmg/ddad139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet. 2022;54(4):412–436. doi: 10.1038/s41588-022-01024-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu JZ, Erlich Y, Pickrell JK. Case-control association mapping by proxy using family history of disease. Nat Genet. 2017;49(3):325–331. doi: 10.1038/ng.3766. [DOI] [PubMed] [Google Scholar]
- 26.Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, et al. The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49(D1):D605–D612. doi: 10.1093/nar/gkaa1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakaue S, Okada Y. GREP: genome for REPositioning drugs. Bioinforma Oxf Engl. 2019;35(19):3821–3823. doi: 10.1093/bioinformatics/btz166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alam MA, Shen H, Deng H-W. A robust kernel machine regression towards biomarker selection in multi-omics datasets of osteoporosis for drug discovery. 2022. 10.48550/ARXIV.2201.05060.
- 29.Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78(4):631–643. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019;47(D1):D1102–D1109. doi: 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SY, Jeong H-H, Kim J, Moon J-H, Sohn K-A. Robust pathway-based multi-omics data integration using directed random walks for survival prediction in multiple cancer studies. Biol Direct. 2019;14(1):8. doi: 10.1186/s13062-019-0239-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palmqvist S, Janelidze S, Quiroz YT, Zetterberg H, Lopera F, Stomrud E, et al. Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA. 2020;324(8):772–781. doi: 10.1001/jama.2020.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palmqvist S, Janelidze S, Stomrud E, Zetterberg H, Karl J, Zink K, et al. Performance of fully automated plasma assays as screening tests for Alzheimer disease-related β-amyloid status. JAMA Neurol. 2019;76(9):1060–1069. doi: 10.1001/jamaneurol.2019.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Janelidze S, Berron D, Smith R, Strandberg O, Proctor NK, Dage JL, et al. Associations of plasma phospho-tau217 levels with tau positron emission tomography in early Alzheimer disease. JAMA Neurol. 2021;78(2):149–156. doi: 10.1001/jamaneurol.2020.4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Craig-Schapiro R, Perrin RJ, Roe CM, Xiong C, Carter D, Cairns NJ, et al. YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol Psychiatry. 2010;68(10):903–912. doi: 10.1016/j.biopsych.2010.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen B, Wang Y. Cofilin rod formation in neurons impairs neuronal structure and function. CNS Neurol Disord Drug Targets. 2015;14(4):554–560. doi: 10.2174/1871527314666150225144052. [DOI] [PubMed] [Google Scholar]
- 37.Schönhofen P, de Medeiros LM, Chatain CP, Bristot IJ, Klamt F. Cofilin/actin rod formation by dysregulation of cofilin-1 activity as a central initial step in neurodegeneration. Mini Rev Med Chem. 2014;14(5):393–400. doi: 10.2174/1389557514666140506161458. [DOI] [PubMed] [Google Scholar]
- 38.Allain H, Bentué-Ferrer D. Clinical efficacy of almitrine-raubasine. An overview Eur Neurol. 1998;39(Suppl 1):39–44. doi: 10.1159/000052069. [DOI] [PubMed] [Google Scholar]
- 39.Alzheimer’s Association 2016 Alzheimer’s disease facts and figures. Alzheimers Dement J Alzheimers Assoc. 2016;12(4):459–509. doi: 10.1016/j.jalz.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 40.Farinelli SE, Greene LA. Cell cycle blockers mimosine, ciclopirox, and deferoxamine prevent the death of PC12 cells and postmitotic sympathetic neurons after removal of trophic support. J Neurosci Off J Soc Neurosci. 1996;16(3):1150–1162. doi: 10.1523/JNEUROSCI.16-03-01150.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lublin A, Isoda F, Patel H, Yen K, Nguyen L, Hajje D, et al. FDA-approved drugs that protect mammalian neurons from glucose toxicity slow aging dependent on cbp and protect against proteotoxicity. PLoS ONE. 2011;6(11):e27762. doi: 10.1371/journal.pone.0027762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Y, Wu L, Shu X-O, Cai Q, Shu X, Li B, et al. Genetically predicted levels of DNA methylation biomarkers and breast cancer risk: data from 228 951 women of European descent. J Natl Cancer Inst. 2020;112(3):295–304. doi: 10.1093/jnci/djz109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang C, Farias FHG, Ibanez L, Suhy A, Sadler B, Fernandez MV, et al. Genomic atlas of the proteome from brain, CSF and plasma prioritizes proteins implicated in neurological disorders. Nat Neurosci. 2021;24(9):1302–1312. doi: 10.1038/s41593-021-00886-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walker KA, Chen J, Zhang J, Fornage M, Yang Y, Zhou L, et al. Large-scale plasma proteomic analysis identifies proteins and pathways associated with dementia risk. Nat Aging. 2021;1(5):473–489. doi: 10.1038/s43587-021-00064-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wingo AP, Liu Y, Gerasimov ES, Gockley J, Logsdon BA, Duong DM, et al. Integrating human brain proteomes with genome-wide association data implicates new proteins in Alzheimer’s disease pathogenesis. Nat Genet. 2021;53(2):143–146. doi: 10.1038/s41588-020-00773-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Author Correction: Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51(9):1423–1424. doi: 10.1038/s41588-019-0495-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang J, Dutta D, Köttgen A, Tin A, Schlosser P, Grams ME, et al. Plasma proteome analyses in individuals of European and African ancestry identify cis-pQTLs and models for proteome-wide association studies. Nat Genet. 2022;54(5):593–602. doi: 10.1038/s41588-022-01051-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pathak GA, Singh K, Miller-Fleming TW, Wendt FR, Ehsan N, Hou K, et al. Integrative genomic analyses identify susceptibility genes underlying COVID-19 hospitalization. Nat Commun. 2021;12(1):4569. doi: 10.1038/s41467-021-24824-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wingerd J, Sponzilli EE. Concentrations of serum protein fractions in white women: effects of age, weight, smoking, tonsillectomy, and other factors. Clin Chem. 1977;23(7):1310–1317. doi: 10.1093/clinchem/23.7.1310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Enriched canonical pathways for the identified associated proteins. The and p-value below each term indicates the significance level of each pathway. Figure S2. The network was identified by Ingenuity Pathway Analysis (IPA). A solid line represents a direct interaction between two nodes and a dotted line indicates an indirect interaction. Figure S3. Network nodes represent proteins and edges represent protein-protein associations.
Additional file 2: Table S1. Risk SNPs identified to be associated with AD risk in previous GWAS or fine-mapping studies. Table S2. Ingenuity Canonical Pathways. Table S3. Network analysis. Table S4. Disease and Biological Functions analysis. Table S5. Association results of the proteins reported in previous studies.
Data Availability Statement
Summary statistics of the GWAS meta-analysis of AD risk by Bellenguez et al. are available at GWAS Catalog (https://www.ebi.ac.uk/gwas/) under accession no. GCST90027158. For the INTERVAL SomaLogic study, the individual-level genotype and protein data, and full summary association results from the genetic analysis, are available through the European Genotype Archive (accession number EGAS00001002555). Summary association results are also publicly available at http://www.phpc.cam.ac.uk/ceu/proteins/, through PhenoScanner (http://www.phenoscanner.medschl.cam.ac.uk) and from the NHGRI-EBI GWAS Catalog (https://www.ebi.ac.uk/gwas/downloads/summary-statistics). The scripts and protein genetic prediction models are available at https://github.com/Arthur1021/Protein-prediction-models.