

Abstract
The first two mRNA vaccines against infection by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that were approved by regulators require a cold chain and were designed to elicit systemic immunity via intramuscular injection. Here we report the design and preclinical testing of an inhalable virus-like-particle as a COVID-19 vaccine that, after lyophilisation, is stable at room temperature for over three months. The vaccine consists of a recombinant SARS-CoV-2 receptor-binding domain (RBD) conjugated to lung-derived exosomes which, with respect to liposomes, enhance the retention of the RBD in both the mucus-lined respiratory airway and in lung parenchyma. In mice, the vaccine elicited RBD-specific IgG antibodies, mucosal IgA responses and CD4+ and CD8+ T cells with a Th1-like cytokine expression profile in the animals’ lungs, and cleared them of SARS-CoV-2 pseudovirus after a challenge. In hamsters, two doses of the vaccine attenuated severe pneumonia and reduced inflammatory infiltrates after a challenge with live SARS-CoV-2. Inhalable and room-temperature-stable virus-like particles may become promising vaccine candidates.
The Coronavirus disease 2019 (COVID-19) pandemic has severely impacted financial and social systems1,2. Globally, there are at least 36 vaccines against COVID-19 that have been approved by at least one country3. Most of them require intramuscular injection, producing antibodies that circulate in the blood but do not necessarily elicit potent mucosal immune responses4–6. Because the transmission of acute respiratory syndrome coronavirus 2 (SARS-CoV-2) primarily occurs via respiratory droplets and the respiratory mucosa is the primary route of viral entry, suboptimal mucosal immunity may limit the utility of intramuscularly administered COVID-19 vaccines. Additionally, some vaccines require deep-freezing for transportation and long-term storage (this is the case for the messenger RNA vaccines manufactured by Pfizer/BioNTech and Moderna). To circumvent such limitations, we sought to develop a vaccine candidate that provides efficient stimulation of mucosal immunity, allows for a non-invasive and needle-free delivery route, and is lyophilisable and stable at room temperature (r.t.) for months.
SARS-CoV-2 belongs to the coronavirus family of viruses. They are enveloped, positive-stranded RNA viruses with spike-protein complexes that recognize and bind to host-cell receptors7,8. Specifically, the receptor-binding domain (RBD) in the SARS-CoV and SARS-CoV-2 spike protein S1 subunit binds to the host airway epithelium angiotensin-converting enzyme 2 (ACE2) receptor and then fuses the viral and host membranes through the S2 subunit, making the RBD a specific target for neutralizing antibodies and vaccines9–12. Previous studies have demonstrated the efficacy of SARS-CoV RBD as the target of potently neutralizing antibodies13,14. In vitro studies of SARS-CoV-2 show host-antibody engagement with the RBD, binding to it and exerting a neutralizing effect15. It also blocked the entry of SARS-CoV-2 and SARS-CoV into ACE2-expressing host cells, suggesting its potential as a viral attachment inhibitor. However, the administration of the RBD alone does not allow for specific targeted delivery and does not evade degradation or rapid clearance. The RBD must be protected through a drug-delivery carrier that optimizes dosage to the antigen presenting cells (APCs).
Virus-like particles (VLPs) and nanoparticles (NPs) are powerful drug-delivery carriers16. In particular, exosomes are a type of naturally occurring extracellular vesicle found in the body, which makes them a native and ideal delivery vesicle for targeted drug delivery17,18. Because they carry and express their parent cell’s RNAs, proteins and lipids, and because they express surface proteins and receptors from the parent cell, they are superior at targeting same tissue-recipient cells19,20. They contain a cocktail of molecular components composed of proteins, lipids and nucleic acids with therapeutic properties21. Furthermore, exosomes can be engineered by creating surface modifications to express proteins or peptides to enhance targeting19,22.
We have derived lung spheroid cells (LSCs) from human lung donor samples23. Their regenerative abilities have been demonstrated in rodent models22,24 and are being tested in a human clinical trial (HALT-IPF, Human Autologous Lung stem cell Transplant for Idiopathic Pulmonary Fibrosis) (www.clinicaltrials.gov)25. We have previously studied the safety and biodistribution of LSC-derived exosomes (LSC-Exo) through nebulization treatments in rodent models of IPF26. LSC-Exo are native NPs for lung therapeutics, derived from heterogeneous populations of lung cells including type I and type II pneumocytes and mesenchymal cells24. We have also confirmed efficient exosome delivery throughout the bronchi and parenchyma of the rodent lung26. By leveraging the characteristics of LSC-Exo and RBD, we engineered an inhalable vaccine by conjugating RBD onto the surface of LSC-Exo (RBD-Exo), creating a VLP that emulates the morphology of the native virus. In contrast to reported intramuscular COVID-19 vaccines, inhaled RBD-Exo induced the production of neutralizing antibodies against SARS-CoV-2, and triggered the mucosal immune system to generate antigen-specific secretory IgA (SIgA) and T-cell responses in the lung to suppress viral uptake by the lung epithelium (Fig. 1).
Fig. 1 |. Inhalation of the RBD-Exo VLP vaccine induces SARS-CoV-2 neutralization in hamsters and protects their lungs.

Schematic representation of the fabrication of the RBD-Exo vaccine, which is delivered into the lungs via inhalation. RBD-Exo induces mucosal immunity and systemic immunity with the generation of RBD-specific IgA and IgG antibodies against SARS-CoV-2 infection in hamsters. This schematic was created with BioRender.com.
Results
Superior lung biodistribution of exosomes over liposomes.
Lipid NPs have been widely used for RNA vaccine delivery in response to the pandemic of COVID-19. For example, the mRNA-1273 (Moderna)27, BNT162b1 (BioNTech and Pfizer)28,29 and ARCoV mRNA vaccines30 (Walvax Biotechnology, Suzhou Abogen Biosciences and Academy of Military Medical Sciences) are all lipid-nanoparticle-formulated RNA vaccines. Here we sought to determine the biodistribution and retention of NPs (LSC-exosomes or liposomes) in the murine lung. Red fluorescent protein (RFP) was loaded into LSC-Exo (RFP-Exo) and commercially available liposomes (RFP-Lipo) via electroporation, for ex vivo imaging and microscopic visualization31,32. The average protein encapsulation efficiencies of RFP-Exo and RFP-Lipo were determined to be 18.69% and 24.83%, respectively (Supplementary Fig. 1). The corresponding loading efficiencies were calculated to be 2.08% in exosomes (2.08 μg RFP per 100 μg protein from exosome) and 0.41% in liposome (0.41 μg RFP per 100 μg liposome). RFP-Exo and RFP-Lipo were nebulized to healthy CD1 mice which were killed at 4 or 24 h post NPs administration (Fig. 2a). Ex vivo imaging (Fig. 2b) and analysis (Fig. 2c) of the whole lung showed the greatest integrated density of NPs in mice that received RFP-Exo and were killed after 24 h. Significantly more exosomes than liposomes were retained and distributed throughout the lung (Fig. 2d,e). Significantly more exosomes than liposomes reached the trachea, but both NPs diffused over time (Fig. 2f). Exosome biodistribution at 4 h post administration was the highest in the bronchioles (Fig. 2g), with the parenchyma starting to show exosome signal after 24 h (Fig. 2h). Significantly fewer liposomes reached the bronchioles (Fig. 2g) and diffused into the parenchyma (Fig. 2h), suggesting faster degradation and/or systemic clearance of liposomes in the lung. To verify whether antigen presenting cells (APCs) could take up these NPs, we performed immunofluorescence (Fig. 2i) on parenchymal lung sections and quantified RFP+ NP uptake by CD11b+ APCs. More APCs were present and took up exosomes than liposomes (Fig. 2j). Because of exosomes’ excellent retention in the lung, as well as enhanced targeting to APCs, we used exosomes as the backbone of our VLPs.
Fig. 2 |. Lung distribution of the RFP-loaded LSC-exosomes.
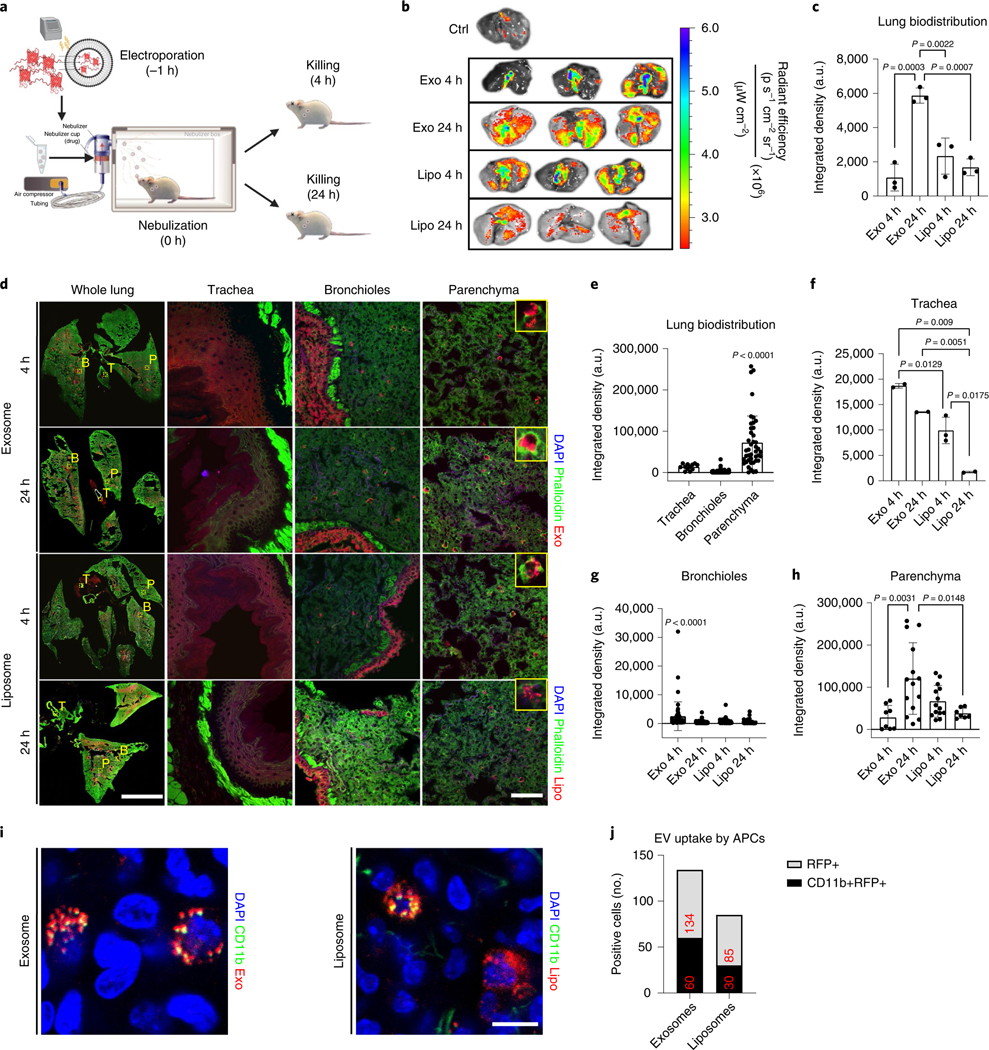
a, The experimental study schematic of RFP-loaded LSC-exosomes and RFP-loaded liposomes in healthy CD1 mice, n = 3 per group. b, Ex vivo imaging of mouse lungs after RFP-loaded LSC-exosome or RFP-loaded liposome delivery after 4 and 24 h. c, Quantification of the integrated density of RFP fluorescence in ex vivo mouse lungs; each dot represents data from one lung, n = 3 per group. d, Immunostaining of whole lung, tracheal, bronchial and parenchymal sections for DAPI (blue), phalloidin (green) and exosomes (red) or liposomes (red). These images were obtained under ×10 magnification. T, trachea; B, bronchioles; P, parenchyma. Scale bar, 1,000 μm for whole lung, 100 μm for parenchyma. e, Quantification of the integrated density of RFP fluorescence across all groups in tracheal, bronchial and parenchymal tiles from whole lung images; each dot represents data from one image tile, n = 12–276. f–h, Quantification of the integrated density of RFP fluorescence in tracheal (f), bronchial (g) and parenchyma (h) tiles from whole lung images; each dot represents data from one image tile, n = 2–82. i, Immunostaining of parenchymal sections for DAPI (blue), CD11b (green), and exosomes (red) or liposomes (red). Scale bar, 50 μm. j, Quantification of exosome or liposome uptake by CD11b+ APCs in ex vivo mouse lungs; numbers in red indicate total number of positive cells across all representative images, n = 6 images per group. EV, extracellular vesicle. Throughout, data are mean ± s.d. P values were calculated by one-way ANOVA with Bonferroni correction. The replicates in c are biological. Analysis in e–h, and j represents the technical replicates. The schematic in a was created with BioRender.com.
Fabrication and characterization of RBD-Exo VLPs.
To synthesize RBD-Exo VLP, RBD antigens were first conjugated with (1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-poly(ethy lene-glycol)-N-hydroxysuccinimide) (DSPE-PEG-NHS) to form RBD-PEG-DSPE according to our previous methods (Fig. 3a,b)33,34. Next, RBD-PEG-DSPE was conjugated on the surface of LSC-Exo and the binding capacity was quantified to be 0.52 μg RBD per 1010 exosomes by enzyme-linked immunosorbent assay (ELISA) analysis. Correspondingly, approximately 892 antibody molecules were bound to each individual VLP. Naive Exo and RBD-Exo were further characterized using transmission electron microscopy (TEM). Gold nanoparticles were conjugated to anti-RBD antibodies to demonstrate RBD presence on the exosome surface (Fig. 3c and Supplementary Fig. 2). Immunoblotting of RBD-Exo, RBD and Exo lysates further demonstrated RBD presented on RBD-Exo and RBD, but not on the Exo control (Fig. 3d). Likewise, exosomal marker CD63 was found to be present on RBD-Exo and Exo, but not on the free RBD control (Fig. 3d). Nanoparticle tracking analysis (NTA) revealed that RBD decoration slightly increased the average diameter of exosomes (Fig. 3e). These compound data confirmed the production of RBD-Exo VLPs. Likewise, RBD conjugated with a commercially available liposome (RBD-Lipo) was synthesized as a control and validated through TEM images with gold nanoparticle-labelled secondary antibodies (Supplementary Fig. 3).
Fig. 3 |. Characterization of RBD-Exo and its stability.

a, Schematic illustrating the preparation of LSC-Exo with RBD to generate RBD-Exo. b, SDS–PAGE gel of RBD and RBD-PEG-DSPE. c, TEM images of LSC-Exo and RBD-Exo. RBD was discovered using gold nanoparticle-labelled secondary antibodies with diameters of 15 nm. d, Immunoblots of RBD and CD63 in lysed RBD-Exo, RBD and Exo. e, Size measurements of LSC-Exo (left) and RBD-Exo (right) via NTA. f–i, TEM images (f), concentrations (g), size change (h) and RBD level change (i) of RBD-Exo after storing at −80 °C, 4 °C and r.t. for 3 weeks (3W) or 3 months (3M). RBD level was calculated using the ratio of the level in the treatment group to that in the pre-lyophilisation group (Pre-lyo), n = 5 per group in g and h, n = 4 per group in i. j, Summary of stability data of RBD-Exo over 3 weeks or 3 months. k, Left: representative immunostaining of C57BL/6 dendritic cells for DAPI (blue) and RBD-RhB (red) or RBD-RhB-Exo (red). Right: flow cytometry analysis of RBD and RBD-Exo internalization by C57BL/6 dendritic cells, n = 3. Scale bar, 50 μm. Data are mean ± s.d., P values calculated by two-tailed, unpaired Student’s t-test (k) or one-way ANOVA with Bonferroni correction (g–i). NS, not significant. All replicates are biological. The uncropped gel (b) and blots (d) are provided in Source Data 3. The schematic in a was created with BioRender.com.
Room-temperature stability of RBD-Exo VLPs.
We next tested whether RBD-Exo VLPs were stable during storage at r.t. (21 °C). RBD-Exo VLPs were lyophilised and stored at −80 °C, 4 °C or r.t. for 21 d (3 weeks) or 3 months. After rehydration, the morphology and structures of RBD-Exo were well preserved, as indicated by TEM images (Fig. 3f). These results indicated the structural integrity of RBD-Exo after r.t. storage. Their sizes and concentrations were determined by NTA. As shown in Fig. 3g,h, room temperature storage had minimal impact on VLPs’ size and concentration. Moreover, the level of RBD on RBD-Exo remained unchanged after storage (Fig. 3i). Additionally, stability testing of RBD-Exo was performed under 40 °C ± 2 °C/75% ± 5% relative humidity according to the ‘accelerated agent’ of the International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) guidelines. As illustrated in Supplementary Fig. 4a, RBD-Exo lysate under the ‘accelerated agent’ condition for 6 months exhibited similar binding activity with ACE2 relative to fresh RBD-Exo lysate, suggesting the conformational integrity of RBD in RBD-Exo. Immunoblotting and quantitative results in Supplementary Fig. 4b showed that RBD-Exo had good stability against degradation for 6 months under ICH conditions. Collectively, these data demonstrated that RBD-Exo has high physical and antigenic stability at all tested temperatures (Fig. 3j), potentially superior to other approved vaccines (Supplementary Table 1).
Internalization of RBD-Exo VLPs by dendritic cells.
To effectively elicit immune responses, VLPs need to be recognized and internalized by APCs. RBD and RBD-Exo were labelled using NHS-Rhodamine (NHS-RhB) to investigate the uptake of RBD-Exo by subsets of APCs in vivo. RBD-RhB or RBD-Exo-RhB were nebulized to mice, which were killed at 24 h post inhalation. Resident APCs in the lung were examined for RBD-Exo uptake using surface markers B220 (B cells), F4/80 (macrophages) and NLDC-155 (dendritic cells). As shown in Supplementary Fig. 5a,c, despite RBD-Exo appearing to be taken up by all the major types of APCs, populations of NLDC-155+ RBD-Exo+ cells were much higher than B220+ RBD-Exo+ and F4/80+ RBD-Exo+, indicating that RBD-Exo was preferentially captured and processed by dendritic cells (DCs) in the lung. In addition, we found minimal free RBD uptake by the APCs in lung compared with RBD-Exo (Supplementary Fig. 5b).
RBD-RhB or RBD-Exo-RhB were co-cultured with C57BL/6 dendritic cells. Confocal laser scanning microscopy (CLSM) exhibited enhanced RBD-Exo internalization by C57BL/6 dendritic cells as compared with free RBD (Fig. 3k). Flow cytometry confirmed the results of CLSM, which indicated that exosomes as a carrier enhanced cellular uptake of RBD by DCs.
RBD-Exo VLP vaccination induces systemic immune response.
CD1 mice were randomized to receive two vaccinations 2 weeks apart for placebo (PBS), control Exo, free RBD or RBD-Exo via either intravenous (IV) injection or inhalation (nebulization, N). At 7 d after the second vaccination, the mice were challenged with SARS-CoV-2 mimics (labelled by AF647) via intratracheal delivery (Fig. 4a). Mice were killed at 2 or 6 d post challenge and lung tissues were imaged using ex vivo imaging system to visualize SARS-CoV-2 mimics clearance (Fig. 4b). Lung sections were prepared and stained for CLSM imaging. Ex vivo imaging (Fig. 4b) and CLSM (Supplementary Fig. 6,7) indicated that RBD-Exo VLP vaccinations facilitated the rapid clearance of SARS-CoV-2 mimics. Furthermore, nebulization administration across all groups induced more rapid clearance of SARS-CoV-2 mimics than IV injection (Fig. 4c), suggesting that nebulization is a more targeted and potent delivery strategy for RBD-Exo vaccines. ELISA revealed that inhalation of RBD-Exo VLPs induced the most RBD-specific IgG antibodies against RBD (Fig. 4d) in the mouse sera. An IgG subclass profile of RBD-specific antibody responses in serum was further assessed. As shown in Fig. 4e, RBD-Exo IV injection induced a type 2 T helper (Th2)-biased immune response with a predominant production of IgG1 antibodies, whereas RBD-Exo nebulization elicited a type 1 T helper (Th1)-skewing immune response as shown by an IgG2a/IgG1 ratio >1. In addition to the IgG isotype, RBD-Exo VLP elicited significant production of RBD-specific IgA antibodies in serum (Extended Data Fig. 1a), which was higher than that from RBD-Exo IV treatment.
Fig. 4 |. Vaccination with RBD-Exo induces antibody production and enhances the clearance of SARS-CoV-2 mimics in mice.

a, Schematic illustrating animal study design. b, Ex vivo imaging of the lungs of mice after intratracheal delivery of SARS-CoV-2 mimics at different time points. c, Semi-quantitative analysis of SARS-CoV-2 mimics labelled with AF647 from confocal images of lung tissues, n = 4. d, Anti-RBD antibody titre from murine serum detected by ELISA, n = 3. e, Ratio of RBD-specific IgG2a to IgG1 antibody generated, n = 3. f,g, RBD-specific SIgA antibody titres from NPLF (f) and BALF (g) detected by ELISA, n = 3. h,i, IFN-γ+, IL-4+ or IL-17a+ of CD4+ T cells (h) and CD8+ T cells (i) specific to the RBD peptide pool in the lung at week 1 after second vaccination. RBD-Exo LTS, RBD-Exo reconstitution after 3 weeks of storage at room temperature, n = 3. Throughout, data are mean ± s.d., statistical analysis by two-tailed, unpaired Student’s t-test (c–g) or two-way ANOVA with a Tukey post hoc test for multiple comparisons (h,i). All replicates are biological. The schematic in a was created with BioRender.com.
RBD-Exo VLP vaccination induces mucosal immune response.
Because the mucosa is the primary entry route of pathogens, the host immune system provides a dynamic immunologic barrier through antigen-specific SIgA responses that play a key role in preventing pathogen invasion5,35. To evaluate mucosal immune response, we measured SIgA antibodies against RBD from nasopharyngeal lavage fluid (NPLF) and bronchoalveolar lavage fluid (BALF) of the mice. ELISA revealed that inhalation of RBD-Exo VLPs produced the highest amount of SIgA antibodies in NPLF (Fig. 4f) and BALF (Fig. 4g). Comparatively, only negligible amounts of SIgA antibodies were found in animals that received vaccination via IV injection. Interestingly, a detectable IgG response against RBD was observed in the BALF, which will further protect the subject against SARS-CoV-2 infection (Extended Data Fig. 1b).
The immunogenicity of the RBD-Exo vaccine was compared to RBD-Lipo (nebulization) or RBD-Exo adjuvanted with aluminum hydroxide (Alum, intramuscular injection) (Extended Data Fig. 2). A lower level of RBD-specific IgG titres was observed in sera from mice with RBD-Exo nebulization as compared with RBD-Exo-Alum. Nevertheless, RBD-Exo elicited 10–100-fold higher RBD-specific SIgA antibody titres in both BALF and NPLF than in RBD-Exo-Alum, revealing the nebulization vaccine’s capability of triggering mucosal immunity. Notably, RBD-Exo VLP after 3 weeks (3W) of storage at r.t. was as effective as fresh RBD-Exo VLP (Extended Data Fig. 2). In addition, we found that nasopharyngeal-associated lymphoid tissue was involved in RBD-Exo VLP mediated immune responses that triggered comparable SIgA antibody responses in NPLF by intranasal administration (Extended Data Fig. 3). Taken together, our data demonstrated that inhalation of RBD-Exo produced both neutralizing antibodies and SIgA responses against RBD.
Having demonstrated that RBD-Exo is capable of inducing mucosal immunity in mice, SARS-CoV-2-specific CD4+ and CD8+ T-cell responses in pneumocytes were examined. After restimulation ex vivo with an RBD peptide pool, both RBD-Exo and RBD-Exo with long-term storage (LTS) (reconstitution after 3 weeks storage at room temperature) elicited potent high frequency of IFN-γ-producing CD4+ and CD8+ T cells, rather than IL-4 (Th2)- or IL-17a (Th17)-producing CD4+ and CD8+ T cells (Fig. 4h,i), suggesting induction of Th1-biased cellular immune responses in the lung. These results are valuable since the major safety premise of SARS-CoV-2 vaccine is eliciting a substantial Th1-biased immune response, rather than a Th2-biased response that might induce vaccine-associated enhanced respiratory disease36. The formation of inducible bronchus-associated lymphoid tissues (iBALT) induced by RBD-Exo vaccination was also observed in lungs of mice (Supplementary Fig. 8).
Cellular response of RBD-Exo VLP vaccination.
As the spleen is an important organ for immunity, we proceeded to assess whether the RBD-Exo VLP could be delivered into the spleen of mice via inhalation. Mice were killed at 24 h after inhalation of RBD-Exo-RhB and its distribution in major organs was evaluated through ex vivo imaging. In addition to the lungs, RBD-Exo was also distributed in the spleen, liver, kidneys and heart (Extended Data Fig. 4), which might be attributed to the translocation of RBD-Exo from the pulmonary tree to the bloodstream followed by delivery to other major organs through circulation37. Compared with the RBD-Exo nebulization group, lower RBD signal in major organs was detected in the RBD-RhB treatment group. This could possibly be attributed to the rapid clearance of free RBD.
Viral antigens presented to APCs such as DCs for further immunological response and protection against the pathogen are important38. We isolated murine splenocytes after two doses of immunizations and rechallenged these with RBD to assess DCs activation via flow cytometry. A greater percentage of DCs were CD86+ (Supplementary Fig. 9), CD40+ (Supplementary Fig. 10) and CD80+ (Supplementary Fig. 11) in splenocytes derived from RBD-Exo inhalation-treated mice, indicating that more DCs were activated.
We then evaluated cellular immune responses and systemic cytokines in vaccinated mice. Enzyme-linked immune absorbent spots (ELISpots) against IFN-γ were performed on splenocytes from animals in each treatment group (Fig. 5a). RBD and RBD-Exo vaccinations induced significantly greater IFN-γ secretion after restimulation with RBD (Fig. 5b). Specifically, RBD-Exo inhalation induced approximately 300 spot-forming units (s.f.u.) per 106 splenocytes, the highest among all groups (Fig. 5b). Furthermore, restimulation by RBD induced the highest levels of TNF-α (Fig. 5c) and IL-6 (Fig. 5d) secretions in animals vaccinated with RBD-Exo via inhalation. Of note, RBD-Exo resulted in robust Th1 T-cell immune responses in splenocytes by producing a higher frequency of CD4+ and CD8+ IFN-γ T cells, instead of Th2 T-cell responses or Th17 T-cell responses (Fig. 5e,f). Haematology parameters of mice with two doses of RBD-Exo immunizations indicated no significant change in immune cell populations as compared with PBS or free-RBD treatment (Supplementary Fig. 12). Furthermore, RBD-Exo vaccinations did not increase the production of IgE and RBD-specific IgE antibodies in mouse sera as compared with PBS or free RBD. These compound datasets indicated that RBD-Exo inhalation robustly induced a systemic T-cell immune response that can further protect the subjects from viral replication without allergic type responses.
Fig. 5 |. Induction of systemic cytokines in mice vaccinated with RBD-Exo.

a, IFN-γ release spots in a 96-well plate with 106 splenocytes per well after rechallenge with RBD. Splenocytes were derived from each treatment group that received IV or N administration. b, IFN-γ splenocytes shown as s.f.u. per 106 cells, n = 3. c, TNF-α levels from splenocytes supernatant restimulated by RBD, n = 4. d, IL-6 levels from splenocytes supernatant restimulated by RBD, n = 4. e,f, IFN-γ+, IL-4+ or IL-17a+ CD4+ T cells (e) and CD8+ T cells (f) specific to an RBD peptide pool in the splenocytes at week 1 after the last vaccination, n = 3. Throughout, data are mean ± s.d., statistical analysis by two-tailed, unpaired Student’s t-test (b–d) or two-way ANOVA with a Tukey post hoc test for multiple comparisons (e,f). All replicates are biological.
RBD-Exo VLP vaccination against live SARS-CoV-2 infection.
We then assessed the protective effects of the RBD-Exo VLP vaccine in high-dose SARS-CoV-2 infected hamsters, which can replicate severe clinical disease, accompanied by rapid weight loss and severe lung pathology39–42. After two doses of RBD-Exo vaccination, 15 Syrian golden hamsters (6–8 weeks old) were challenged with 1.99 × 104 50% tissue culture infective dose (TCID50) SARS-CoV-2 by intranasal and intratracheal routes (Fig. 6a). Viral loads in oral swabs (OS) and bronchoalveolar lavage (BAL) were determined by quantitative real-time PCR with reverse transcription (RT-qPCR). As shown in Fig. 6c, high levels of RNA copies were detected in all three groups, PBS, RBD and RBD-Exo, with a median peak of 6.632, 6.454 and 6.042 log10 RNA copies per ml in OS on day 2. However, RNA levels were substantially diminished in RBD-Exo-immunized animals, with 3.43 log10 reductions in median peak RNA in OS on day 7 following the challenge. Consistent with the OS results, BAL viral load was approximately 1.942 log10 RNA copies per ml in the RBD-Exo immunization group, which was much lower than that in the PBS (5.916) and RBD (5.548) treatment groups (Fig. 6b). Furthermore, RBD-Exo vaccinations elicited significantly higher antibody titres than PBS and free-RBD control at every time point (1 d before challenge, and 4 d and 7 d after challenge) (Fig. 6d), in line with the decline of viral loads in BAL (Fig. 6b) and OS (Fig. 6c) over time. Clinical chemistry and haematological parameters of hamsters vaccinated with RBD-Exo were in the normal range (Supplementary Fig. 13). Hamsters were assessed by histopathology on day 7 after the virus challenge. Hematoxylin and eosin (H&E) staining revealed severe pulmonary lesions with marked inflammatory infiltrates and multifocal dense nodules with alveolar wall thickening in hamsters that received PBS vaccinations (Fig. 6e). Conversely, the pulmonary alveolus was highly visible in the RBD-Exo vaccination group and the number of polymorphonuclear neutrophils was significantly reduced (Fig. 6e). Masson trichrome staining and Ashcroft score analysis showed that RBD-Exo immunization notably decreased resolution of fibrosis by preserving alveolar epithelial structures compared with PBS or RBD vaccination (Fig. 6f,g).
Fig. 6 |. Protective effect of the RBD-Exo vaccine in the Syrian hamster model of SARS-CoV-2 infection.

a, Schematic showing hamster study design. b, Impact of RBD-Exo on viral genomic RNA (gRNA) in BAL fluid at 7 d post challenge. c, Impact of RBD-Exo on viral gRNA in oral swabs at the indicated time points. d, RBD-specific binding antibody from hamster serum at week 2 (1 d before challenge), 4 d and 7 d after viral challenge as detected by ELISA. e,f, H&E (e) and Masson’s trichrome (f) images of lung tissues from hamsters at 7 d post challenge. Scale bars, 500 μm (top), 100 μm (bottom). g, Quantitation of lung fibrosis of challenged hamsters by Ashcroft scoring performed blindly. Throughout, each dot stands for data from one animal, n = 5. Data are mean ± s.d., P values calculated by one-way ANOVA with Bonferroni correction. All replicates are biological. The schematic in a was created with BioRender.com.
To visualize SARS-CoV-2 in lung tissues, the expression and distribution of SARS nucleocapsid protein (SARS-N) were observed in the multifocal pneumocytes and alveolar septa for PBS-treated animals (Fig. 7a). These viral antigen positive cells frequently co-stained with pan-cytokeratin (pan-CK, epithelial cells marker), further confirming that they were alveolar epithelial cells (Fig. 7b). Of special note, the level of SARS-N protein in the lung was decreased significantly with RBD-Exo vaccinations (Fig. 7f). Furthermore, SARS-CoV-2 viral RNA (vRNA) in the lung was determined by in situ RNA hybridization (RNAscope). Compared with PBS or RBD vaccination, the RBD-Exo immunization group had dramatically decreased levels of both positive-sense and negative-sense vRNA (Fig. 7c), indicating that RBD-specific antibodies could reduce viral replication. Furthermore, we found that foci of virus-infected cells were frequently associated with large inflammatory infiltrates of ionized calcium binding adapter (Iba-1+) and CD206+ (Fig. 7d). Many neutrophils were also detected throughout the lung, with high expression of neutrophil myeloperoxidase (MPO) in challenged hamsters (Fig. 7e). Comparatively, RBD-Exo vaccinations showed the least MPO positive cells in lung (Fig. 7g). Diffuse expression of CD3-positive T lymphocytes was discovered in challenged hamsters (Fig. 7e,h), which were able to accelerate the clearance of the infected cells. Importantly, expression of the antiviral protein type 1 interferon response gene MX1, which possesses broad antiviral activity against RNA viruses, was significantly decreased in hamsters that received RBD-Exo vaccinations as compared with PBS or RBD vaccinations (Fig. 7e,i), further validating decreased virus replication owing to the highly potent antibodies induced by the RBD-Exo vaccine. Taken together, these histology results strongly indicated that RBD-Exo vaccination could effectively protect hamster lungs from SARS-CoV-2 infection.
Fig. 7 |. Histopathological changes and RNAscope analysis in Syrian hamsters vaccinated with RBD-Exo.
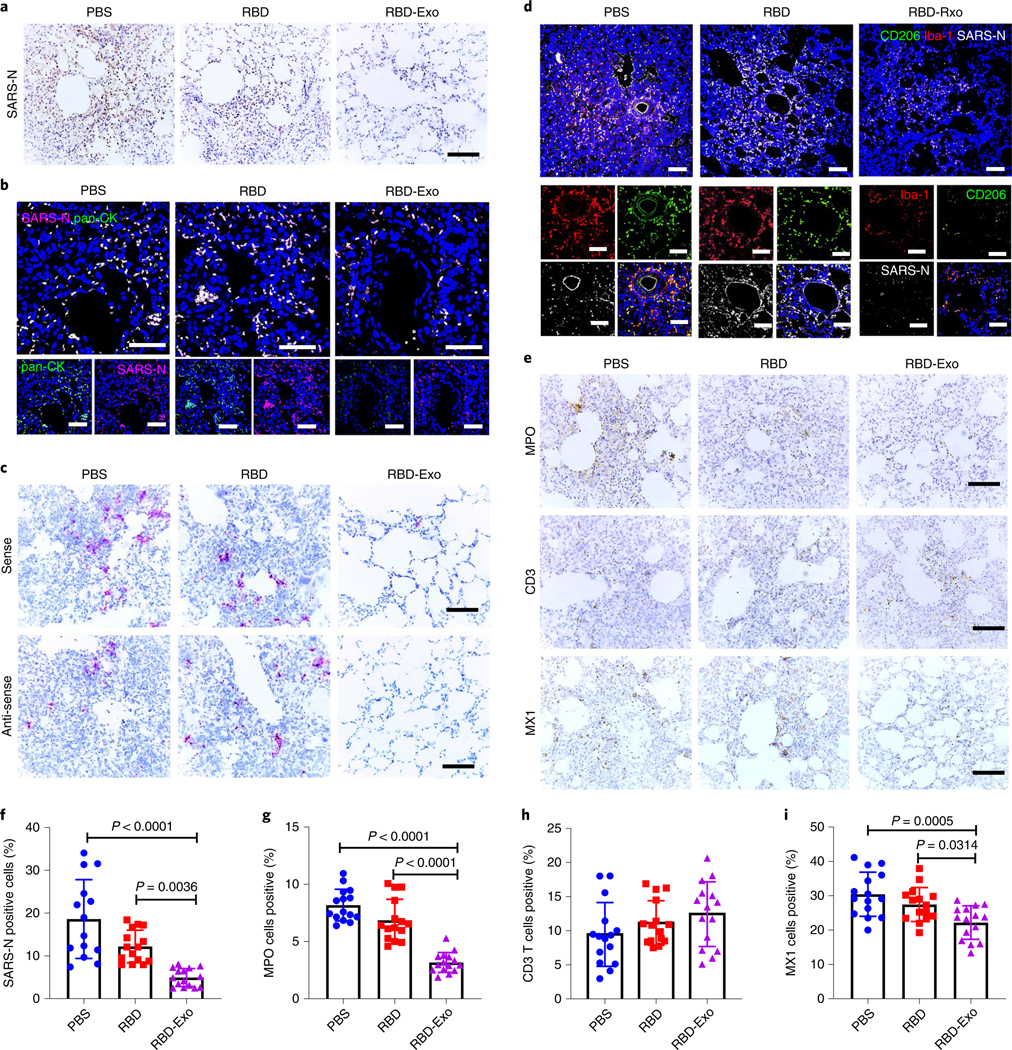
a, SARS-N IHC staining of lung tissues in hamsters vaccinated with PBS, RBD or RBD-Exo at 7 d post viral challenge. Scale bar, 100 μm. b, Immunofluorescence images of pan-CK (green), SARS-N (magenta) and DAPI (blue) of lung tissues in hamsters to study the distribution of SARS-N. Scale bar, 50 μm. c, RNAscope in situ hybridization detection of vRNA in lung tissues of hamsters at 7 d post challenge. Scale bar, 100 μm. d, Immunofluorescence images of Iba-1 (red), CD206 (green), SARS-N (greyscale) and DAPI (blue) of lung tissues in hamsters at 7 d post challenge. Scale bar, 50 μm. e, IHC staining of MPO, CD3 T lymphocytes and interferon inducible gene MX1 of hamsters at 7 d post challenge. Scale bar, 100 μm. f, Quantitation of SARS-N positive cells of lung tissues in hamsters. Each dot represents data from one image file, n = 15. g–i, Quantitation of positive MPO (g), CD3 (h) and MX1 (i) cell numbers in lung tissues of hamsters. Each dot stands for data from one image file, n = 15. Throughout, data are mean ± s.d., P values calculated by one-way ANOVA with Bonferroni correction. Analysis in f–i represents technical replicates from 5 independent biological samples.
Cross-protection activity of RBD-Exo VLP.
SARS-CoV-2 D614G variant, a more infectious variant43, was used to test the cross reactivity of RBD-Exo VLP vaccination. At 7 d after immunizations, the mice were challenged with green fluorescent protein (GFP)-expressing SARS-CoV-2 D614G pseudovirus via intranasal delivery. Ex vivo imaging results of lung showed that substantial GFP signal was detected in the mice vaccinated with free RBD. In comparison, RBD-Exo vaccination remarkably diminished the GFP signal (Fig. 8a,b), exhibiting a cross-variant neutralization ability, even after being stored for 3 weeks at r.t. (RBD-Exo LTS). Confocal imaging of the whole lung demonstrated that less SARS-CoV-2 D614G pseudovirus was distributed in both the trachea/bronchioles and parenchyma with RBD-Exo and RBD-Exo LTS than with free-RBD nebulization (Fig. 8c–e). Immunohistochemistry (IHC) analysis of viral spike protein showed that RBD-Exo and RBD-Exo LTS accelerated the clearance of pseudotyped SARS-CoV-2 D614G in the lung (Fig. 8f).
Fig. 8 |. RBD-Exo VLP efficiently neutralizes the SARS-CoV-2 D614g pseudovirus.

a, Ex vivo imaging of lungs of immunized mice after inoculation with SARS-CoV-2 D614G pseudovirus with GFP expression for 24 h. b, Quantification of the integrated density of GFP fluorescence in ex vivo mouse lungs; each dot represents data from one lung, n = 3 per group. c, Immunostaining imaging of whole lung (top row), trachea/bronchioles (middle row) and parenchyma (bottom row) of mice with different vaccinations for DAPI (blue), phalloidin (red) and SARS-CoV-2 D614G pseudovirus (green), and immunohistochemistry staining of spike protein of SARS-CoV-2 D614G pseudovirus (SARS-S, bottom row) in lung tissue after different vaccinations. Whole lung images were taken under ×10 magnification. Scale bar, 50 μm. d,e Quantification of the integrated density of GFP fluorescence in tracheal/bronchial (d) and parenchymal tiles (e) from whole lung images. Each dot represents data from one image tile, n = 43–55. f, Quantitation of positive spike protein cell numbers in lung tissues. Each dot stands for data from one image, n = 6. g, SARS-CoV-2 D614G pseudovirus-infected primary bronchial/tracheal epithelial cells with GFP expression, inhibited by IgA antibodies purified from vaccinated mice. Scale bar, 50 μm. h, Flow cytometry plots of SARS-CoV-2 D614G pseudovirus-infected primary small airway epithelial cells, which were inhibited by IgA antibodies purified from vaccinated mice, n = 3 per group. i,j, Purified IgG and IgA pseudovirus neutralization assay (i) and IC50 values (j), n = 3 per group in i; n = 9 per group in j. Data are mean ± s.d., statistical analysis by one-way ANOVA with Bonferroni correction (b, d–f, h) or two-tailed, unpaired Student’s t-test (j). The replicates in b, h and i are biological. Analysis in d–f and j represents technical replicates from 3 independent biological samples.
To investigate the mechanism of neutralization of RBD-Exo against SARS-CoV-2 variant invasion, the IgA isotype was purified from BALF and NPLF of mice as previously described44. An in vitro SARS-CoV-2 infection model, where primary small airway epithelial cells were co-cultured with primary bronchial/tracheal epithelial cells (lower chamber) via a transwell, was constructed to assess the blockage activity of IgA. As expected, IgA antibodies purified from the mice vaccinated with RBD-Exo VLP efficiently inhibited the invasion of SARS-CoV-2 D614G pseudovirus into primary bronchial/tracheal cells (Fig. 8g). We also found that the IgA antibodies obtained from RBD-Exo or RBD-Exo LTS vaccinated mice exhibited significant blockage activity against SARS-CoV-2 infection of primary small airway epithelial cells compared with PBS or free-RBD vaccinations (Fig. 8h). The protection activity of IgA and IgG produced by RBD-Exo against SARS-CoV-2 D614G variants was compared, where the IgG isotype was purified from mice sera. Both IgA and IgG antibodies from mice vaccinated with PBS could not inhibit the entry of SARS-CoV-2 D614G pseudovirus into A549 cells expressing ACE2 receptors (Fig. 8i). In contrast, IgA and IgG obtained from the mice immunized with RBD-Exo VLP significantly neutralized the SARS-CoV-2 D614G pseudovirus in the ng ml−1 range, with respective IC50 of 53.6 ng ml−1 and 65.4 ng ml−1 (Fig. 8j). These results indicated that IgA antibodies show a higher cross-activity than IgG antibodies.
Discussion
Regardless of the availability of treatment strategies for COVID-1945,46, effective vaccination will remain essential. RBD-based vaccines have shown clinical promise in producing an antibody response capable of protecting against and neutralizing SARS-CoV-213,47,48. The airway mucosal immune response plays an integral role in early pathogen invasion, triggering both humoral and cell-mediated immune reactions that trigger systemic responses35,38. To that end, we conjugated the SARS-CoV-2 RBD onto lung-derived exosomes as an inhalable VLP vaccine. The VLPs trigger robust production of RBD-specific IgG and IgA to neutralize SARS-CoV-2. LSC-Exo is an ideal carrier for a SARS-CoV-2 VLP. Being native to the lung, LSCs and their derived LSC-Exo share surface proteins and receptors with membrane features found among airway epithelia. For this reason, exosomes are more distributed and longer retained in the lung and have enhanced internalization by APCs in the lung, providing a more targeted delivery carrier than commonly used liposomes. Our findings show that RBD-Exo vaccination led to both humoral and cellular immune responses, protecting against a SARS-CoV-2 mimic in mice and live SARS-CoV-2 infection in a hamster model. In addition to generating robust IgG antibodies, RBD-Exo vaccination produced high titres of RBD-specific IgA as well as CD4+ and CD8+ T cells in lung, which play key roles in protecting the lungs against viral invasion in the airway mucosa and inhibiting infection of both wild-type and D614G variant viruses.
Although the majority of vaccines are administered intramuscularly, establishing a mucosal immune protection is critical, as COVID-19 is primarily a respiratory disease49. Our data indicate that inhalation is a safe and effective administration route to elicit a robust mucosal immunity when exosomes are used as the vaccine carrier. Compared with intramuscular vaccines, inhaled vaccines provide two additional layers of protection50: vaccine-elicited IgA and resident T cells in the respiratory mucosa that provide an effective barrier to sites where viruses normally enter and propagate, and cross-reactive resident T cells that impede viral replication and reduce viral shedding and transmission. It is established that these resident T cells encounter antigens earlier and respond more quickly than systemic memory cells, providing further protection against the virus51,52. More importantly, mucosal vaccination via the inhalation route has the potential to confer sterilizing immunity in the respiratory tract relative to intramuscular injection, the former being preferable against emerging variants, and to stop virus transmission4. Another advantage of RBD-Exo nebulization is non-invasiveness. This can potentially encourage people who fear needles to get vaccinated.
There are some limitations to our strategy. The large-scale production and purification of exosomes is challenging. The cargos and surface ligands of exosomes vary from batch to batch. Owing to the instability of NHS ester in aqueous conditions, the attachment of DSPE-PEG-NHS to RBD will compete with ester hydrolysis in the reaction buffer53, which would result in a modest conjugation yield. Additionally, the spatial hindrance effect between DSPE-PEG-NHS and RBD further limits their conjugation efficiency. Moreover, inhalation vaccines are usually not recommended for people who have baseline respiratory complications. Nevertheless, room-temperature-stable and inhalable vaccines such as RBD-Exo may represent a promising vaccine candidate and warrant future research and development.
Current vaccine products require storage temperatures as low as −20 °C or −70 °C to ensure stability and preservation. However, maintaining such temperatures in transit is costly and requires specialized containers with temperature control. Also, the vaccines must be stored in deep freezers to maintain efficacy and shelf life, but many hospitals and vaccine-administration sites do not have the suitable facilities or space to accommodate these freezers, constraining vaccine distribution. Room-temperature and lyophilisable vaccines such as RBD-Exo VLPs have extended shelf life and could thus reduce transportation costs, facilitating vaccine distribution and accessibility.
Methods
Cell culture.
LSCs were constructed from whole lung samples of healthy human from the University of North Carolina at Chapel Hill Cystic Fibrosis and Pulmonary Diseases Research and Treatment Center and expanded as previously described22–24. LSCs were plated on a fibronectin-coated (Corning) flask and maintained in Iscove’s Modified Dulbecco’s Media (IMDM; Thermo Fisher) containing 20% fetal bovine serum (FBS; Corning). Media changes were performed every other day. LSCs were allowed to reach 70–80% confluence before generating serum-free secretome (LSC-secretome) as previously described26. LSC-secretome was collected and filtered with a 0.22 μm filter to remove cellular debris. C57BL/6 dendritic cells (1129–4807AU20) were purchased from Cellero and cultured in IMDM medium (Thermo Fisher) with 10% FBS. They are of the myeloid type and derive from bone marrow using murine recombinant granulocyte-macrophage colony-stimulating factor (GM-CSF) stimulation. Primary small airway epithelial cells (PCS-301–010) and primary bronchial/tracheal epithelial cells (PCS-300–010) were obtained from ATCC and cultured with airway epithelial cell basal medium. Splenocytes and pneumocytes were isolated from vaccinated mice as previously described54. All procedures in this study were in accordance with the ethical standard of the institutional research committee and with the guidelines set by the Declaration of Helsinki.
Exosome isolation and characterization.
Exosomes were collected and isolated from human LSC-secretome via ultrafiltration55. Filtered secretome was pipetted into a 100 kDa Amicon centrifugal filter unit (Millipore Sigma) and centrifuged at 400 RCF at 10 °C. Once all medium was filtered, the remaining exosomes were detached from the filter and resuspended using Dulbecco’s phosphate-buffered saline (DPBS; Thermo Fisher) with 25 mM trehalose (Millipore Sigma) for further analysis56. LSC-Exo, RFP-Exo, RFP-Lipo and RBD-Exo were analysed by nanoparticle tracking analysis (NTA; NanoSight NS3000, Malvern Panalytical). All samples were fixed onto copper grids and stained with vanadium negative staining for TEM (JEOL JEM-2000FX) to analyse exosomal composition and morphology before and after RBD binding. To determine the presence of RBD through TEM, RBD-Exo were incubated with anti-RBD primary antibody (40592-T62; Sino Biological) overnight at 4 °C. Unbound antibodies were removed via ultracentrifugation at 100,000 g for 30 min. Gold nanoparticles (15 nm) labelled with goat anti-rabbit IgG secondary antibody were added and incubated at room temperature for 2 h.
RFP loading.
RFP (ab268535; Abcam) was loaded into exosomes and commercial liposome particles (300205, Avanti Polar Lipids) via electroporation, yielding RFP-Exo and RFP-Lipo32. The liposome consisted of fully hydrogenated soy phosphatidylcholine (HSPC), N-(carbonyl-ethoxypolyethylene glycol 2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine sodium salt (DSPE-mPEG2000) and cholesterol. The mass ratio of DSPE-mPEG2000, HSPC and cholesterol was 18.8:61.6:19.6. One billion nanoparticles from each sample were diluted in Gene Pulser electroporation buffer (Bio-Rad) at a 1:9 ratio of nanoparticles to buffer. RFP (10 μg) was added to the nanoparticle-electroporation buffer solution, which was transferred into the ice-cold 0.4 cm Gene Pulser/MicroPulser electroporation cuvette (Bio-Rad). The electroporation cuvette was inserted into the Gene Pulser Xcell Total System (Bio-Rad) and electroporated under the following conditions: pulse type, square waveforms; voltage, 200 V; pulse length, 10 ms; number of pulses, 5; pulse interval, 1 s. Electroporation buffer and unloaded RFP were removed from the fluorescently labelled nanoparticles by ultrafiltration via an Amicon centrifugal filter (Millipore, UFC510096, 100 kDa molecular weight cut-off), followed by three washes with DPBS buffer (10 mM, pH 7.4, 13,000 g, 10 min per wash, 4 °C). RFP encapsulation efficiency and loading efficiency were determined by RFP fluorescence at 595 nm, with excitation at 547 nm. The encapsulation efficiency (EE) was calculated using the following formula: . The loading efficiency (LE) was calculated as: (ref. 57).
RBD conjugation on LSC-Exosomes.
Recombinant SARS-CoV-2 RBD protein (40592-V08B, Sino Biological) was purchased and reconstituted in DPBS. RBD was conjugated to LSC-Exo using a DSPE-PEG-NHS linker. Briefly, RBD (100 μg) was conjugated with DSPE-(PEG)5000-NHS (10 mg) in 2 ml PBS (10 mM, pH 7.4) overnight at 4 °C. Unconjugated DSPE-(PEG)5000-NHS was removed by centrifugation and washing using an Amicon centrifugal filter unit with 10 kDa molecular weight cut-off (Millipore, UFC801096, triple wash with 10 mM PBS, 4 °C, 4,500 g, 10 min per wash). To demonstrate the conjugation of RBD-PEG-DSPE, RBD and RBD-PEG-DSPE were analysed by blue native SDS–PAGE (4–20% acrylamide precast Tris-glycine gel) and stained with Coomassie blue. Then, the resultant RBD-PEG-DSPE was conjugated with LSC-Exo (2 × 1011 particles) for 24 h at 4 °C. Unbound RBD-DSPE-NHS and free RBD were concentrated and washed three times with PBS buffer (10 mM, pH 7.4, 4,500 g, 10 min per wash) in an Amicon centrifugal filter (Millipore, UFC810096, 100 kDa molecular weight cut-off) at 4 °C. To further purify the RBD-Exo, the resulting RBD-Exo solution was ultracentrifugated at 100,000 g at 4 °C for 2 h, followed by washing with PBS buffer once (10 mM, pH 7.4) and resuspension in 2 ml PBS buffer (10 mM, pH 7.4).
To quantify the RBD moiety on LSC-Exo, 106 RBD-Exo particles were taken from the 2 × 1011 particles stock solution and diluted into RIPA lysis buffer and lysed on ice for 30 min. The amount of released RBD was quantified via ELISA (EH492RB, Thermo Fisher).
The conjugation efficiency of RBD with LSC-Exo via DSPE-PEG-NHS linker was determined to be 10.5%.
Stability studies on RBD-Exo VLPs.
RBD-Exo lyophilisates were stored at −80 °C, 4 °C and room temperature (21 °C) for 21 d (3 weeks) or 3 months. Then, RBD-Exo lyophilisates were dispersed in PBS and their size and concentration were detected using NTA. In addition, the concentration of RBD on Exo was quantified using an ELISA kit.
An accelerated stability testing study of RBD-Exo lyophilisates was performed under the ICH guidelines at 40 ± 2 °C and 75 ± 5% relative humidity to further verify the potential of RBD-Exo as a vaccine58. The samples were evaluated periodically at 0, 3 weeks, 3 months and 6 months for stability analysis. Western blot and ELISA analyses were used for evaluating the stability and conformation integrity of RBD.
RBD-Exo internalization by APCs.
RBD was labelled using NHS-Rhodamine (Thermo Fisher) according to the manufacturer’s protocol. RBD-RhB and RBD-RhB-Exo were co-cultured with C57BL/6 dendritic cells for 4 h with the same amounts of RBD (1 μg). The free RBD-RhB and RBD-RhB-Exo were removed, and cells were washed with three times with DPBS buffer. Cells were imaged with an Olympus FLUOVIEW CLSM (Olympus; FV3000).
Mouse studies using SARS-CoV-2 mimics.
All studies complied with the requirements of the Institutional Animal Care and Use Committee (IACUC, 19–806-B). Male CD1 mice (7–8 weeks old) (Crl:CD1(ICR)) were purchased from Charles River Laboratory (Wilmington, MA, USA). RFP-Exo and RFP-Lipo were administered via nebulization (Pari Trek S Portable 459 Compressor Nebulizer Aerosol System; 047F45-LCS, PARI). PBS, Exo, RBD, RBD-Exo treatments (1010 per kg of mouse weight) and RBD-Lipo treatments (1.26 × 1010 per kg of mouse weight) were given in two doses once a week for 2 weeks via nebulization or IV injection. Spike protein (Sino Biological) was modified onto the surface of lentivirus (Cellomics Technology) and the SARS-CoV-2 mimics were synthesized according to our previous report59. Mice were challenged with SARS-CoV-2 mimics labelled with AF647 (106 particles per kg of body weight) by nebulization 1 week after the second treatment dose. Lungs were excised and imaged at days 2 and 6 post vaccination with a Xenogen live imager (PerkinElmer). Blood and all other major organs were collected for further analysis.
IgG antibody titre.
Microtitre plates (Nunc Cell Culture, Thermo Fisher) were coated with 10 μg ml−1 RBD in 100 μl coating buffer (R&D Systems) and incubated overnight at 4 °C. Wells were then blocked with 1% (w/v) bovine serum albumin (BSA; Sigma-Aldrich) in 200 μl PBS-T for 1 h at 37 °C. After washing three times with PBS-T, serial dilutions (1:100, 1:1,000, 1:5,000, 1:10,000, 1:50,000, 1:100,000, 1:200,000) of sera samples were added and control sera samples with 1:100 dilution were added into wells for incubation for 1.5 h at 37 °C. Then, samples were washed with PBS-T three times and then incubated with HRP-labelled anti-mouse IgG secondary antibody at a 1/2,000 dilution (100 μl per well) or HRP-labelled anti-hamster IgG secondary antibody at a 1/20,000 dilution (100 μl per well) for 1 h at 37 °C. After washing four times with PBS-T, 3,3’,5,5’-tetramethylbenzidine soluble substrate (TMB; Thermo Fisher) was added to each well (100 μl per well). After incubation of 30 min at room temperature, 50 μl of stop solution (2 M H2SO4, Sigma-Aldrich) was added and optical absorption at 450 nm was determined by a plate reader. The end-point titre of IgG was quantified by the reciprocal of maximal serum dilution that exceeded twice the s.d. above the mean readout of the control group. The individual antibody titres are shown as , calculated as the reciprocal of maximal serum dilution.
For IgG2a and IgG1 ELISA analyses, a similar protocol with minor modifications was followed as described above; goat anti-mouse IgG2a-HRP (A-10685, Thermo Fisher) and goat anti-mouse IgG1-HRP (ab97240, Abcam) were respectively used as secondary antibodies.
IgA antibody titre.
RBD-specific IgA from NPLF and BALF was measured using ELISA. To collect NPLF, the trachea was cut in the middle and the nasopharynx was rinsed upwards from the incision with 200 μl DPBS. The fluid was collected and rinsing was repeated three times for a total of 600 μl wash fluid. To collect BALF, the trachea was exposed by thoracotomy and a transverse incision was made at the top of the bronchial bifurcation. A needle was inserted into the trachea to wash the lungs with 200 μl DPBS. The wash fluid was collected and rinsing was repeated three times for a total of 600 μl wash fluid. Microtitre plates (Nunc Cell Culture, Thermo Fisher) were coated with 10 μg ml−1 RBD in 100 μl coating buffer (R&D Systems) and incubated overnight at 4 °C. Then, RBD solution was removed and wells were blocked with 1% (w/v) BSA (Sigma-Aldrich) dissolved in 200 μl PBS-T for 1 h at 37 °C. After washing with PBS-T three times, serial dilutions (1:50, 1:100, 1:1,000, 1:5,000, 1:10,000, 1:50,000) of NPLF and BALF, and control samples (1:50 dilution) were added and incubated for 1.5 h at 37 °C. After washing three times with PBS-T, HRP-labelled anti-mouse IgA secondary antibody at a 1/2,000 dilution (100 μl per well) was added and incubated for 1 h at 37 °C. Samples were removed and washed with PBS-T four times and TMB soluble substrate (Thermo Fisher) was added to each well (100 μl per well) for 30 min incubation at room temperature. Stop solution (50 μl) (2 M H2SO4, Sigma-Aldrich) was added and optical absorption at 450 nm was measured on a plate reader. Calculation for the end-point titre of IgA is the same as that for IgG as described above.
Cytokine measurement in splenocytes.
Splenocytes from each vaccinated mouse were challenged with 1 μg ml−1 RBD and plated into ELISpot wells (106 per well) (R&D Systems) that were coated with anti-mouse IFN-γ capture antibody. RBD-specific cells secreting IFN-γ were measured using an ELISpot assay according to the manufacturers’ protocol. Spot-forming units (s.f.u) were analysed using an anatomical microscope (Nikon) and the spots were counted using ImageJ software (NIH; https://imagej.nih.gov/ij/). Splenocytes from each vaccinated mouse were cultured in 6-well plates (5 × 106 cells per well) and restimulated with 5 μg ml−1 RBD. After a 48 h incubation, antigen-specific cytokine amounts of IL-6 and TNF-α from culture medium were detected by ELISA using a mouse IL-6 ELISA kit (RAB0308, Sigma-Aldrich) and mouse tumour necrosis factor α ELISA kit (RAB0477, Sigma-Aldrich) following the manufacturer’s protocols.
T-cell immunity in pneumocytes and splenocytes.
Mouse pneumocytes and splenocytes were obtained at day 7 post vaccinations and plated at a 1 × 106 per well concentration. A peptide pool composed of 15-mers (overlapping by 15 amino acids) spanning the SARS-CoV-2-S RBD (PP002-A, Sino Biological) was employed to stimulate pneumocytes and splenocytes for 12 h at a cell incubator. Brefeldin A (1 μg ml−1) was then added into pneumocytes and splenocytes for a 4 h incubation, followed by collection and staining of cells with anti-CD4-FITC (100406, Biolegend) or anti-CD8-FITC (ab22504, Abcam). Then, cells were washed, fixed using 4% paraformaldehyde and permeabilized with saponin for intracellular staining of anti-IFN-γ-PE (507806, Biolegend), anti-IL-14-PE (504103, Biolegend) or anti-IL-17a-APC (17–7177-81, Invitrogen). After a 1 h incubation, cells were collected, washed and tested with a CytoFLEX flow cytometer (Beckman Coulter) and analysed using FCS Express V6 (De Novo software; https://denovosoftware.com).
Hamster studies with live SARS-CoV-2.
Fifteen male and female Syrian golden hamsters (6–8 weeks old; Envigo) were randomly divided into three treatment groups. All hamsters were housed at Bioqual. Hamsters were administered with two doses of PBS (placebo), RBD or RBD-Exo 1 week apart by nebulization (n = 5 per group, 3 females/2 males). At 1 week after the vaccinations, the hamsters were challenged with 1.99 × 104 TCID50 of SARS-CoV-2 using the intranasal and intratracheal routes (50 μl in each nare). BAL, OS and blood were collected at the indicated time. Hamsters were necropsied at day 7 post challenge. All immunologic and virologic analyses were conducted blindly. All hamster studies were performed in compliance with all relevant local, state and federal regulations and were approved by the Bioqual Institutional Animal Care and Use Committee (20–091P).
Histopathology and immunohistochemistry in infected hamsters.
Tissues were fixed with 4% paraformaldehyde for 24 h and transferred to 70% ethanol. The samples were paraffin embedded within 7 d of fixation and blocks were sectioned at 5 μm. Slides were then baked for 1 h at 65 °C, deparaffinized in xylene and rehydrated by a series of graded ethanol to distilled water. Subsequently, the slides were stained with hematoxylin (HSS16, Sigma-Aldrich) and eosin Y (318906, Sigma-Aldrich). Trichrome (HT10516, Sigma-Aldrich) assay was conducted according to the manufacturer’s instruction. Optical microscopy was performed to analyse these slides. For SARS-N, CD3, MPO and MX1 for IHC staining, dewaxing and rehydration were first performed, followed by retrieval in citrate buffer (AP9003125, Thermo Fisher) and treatment with 3% H2O2 in methanol for 10 min. Slides were permeabilized and blocked with Dako Protein blocking solution (X0909, DAKO) containing 0.1% saponin (47036, Sigma-Aldrich). Then, slides were incubated with primary rabbit anti-SARS-N antibody (Novus, NB100–56576, 1:200), rabbit anti-CD3 (Abcam, ab16669, 1:200), rabbit anti-MPO (Thermo Fisher, PA5–16672, 1:200) and anti-MX1 (Millipore Sigma, MABF938, 1:200) overnight at 4 °C, followed by goat anti-rabbit HRP secondary antibody (Abcam, ab6721, 1:1,000) or goat anti-mouse HRP secondary antibody (Abcam, ab6789, 1:1,000) for 1 h at r.t., counterstaining with hematoxylin and then bluing with 0.25% ammonia water.
SARS-CoV-2 genomic RT-qPCR assay.
A QIAsymphony SP (Qiagen) automated sample preparation platform along with a virus/pathogen DSP midi kit and the complex800 protocol were used to extract viral RNA from 800 μl of oral swabs or bronchoalveolar lavage fluid. A reverse primer specific to the orf1a sequence of SARS-CoV-2 (5′-CGTGCCTACAGTACTCAGAATC-3′) was annealed to the extracted RNA and then reverse transcribed into complementary DNA using SuperScript III Reverse Transcriptase (Thermo Fisher) along with RNase Out (Thermo Fisher). The resulting cDNA was then treated with RNase H (Thermo Fisher) and added to a custom 4x TaqMan gene expression master mix (Thermo Fisher) containing primers and a fluorescently labelled hydrolysis probe specific for the orf1a sequence of SARS-CoV-2 (forward primer 5′-GTGCTCATGGATGGCTCTATTA-3′, reverse primer 5′-CGTGCCTACAGTACTCAGAATC-3′, probe 5′-/56-FAM/ACCTACCTT/ZEN/GAAGGTTCTGTTAGAGTG GT/3IABkFQ/−3). All PCR setup steps were performed using QIAgility instruments (Qiagen). The qPCR was then carried out on a QuantStudio 3 real-time PCR system (Thermo Fisher). SARS-CoV-2 genomic (orf1a) RNA copies per reaction were interpolated using quantification cycle data and a serial dilution of a highly characterized custom RNA transcript containing the SARS-CoV-2 orf1a sequence. Mean RNA copies per ml were then calculated by applying the assay dilution factor (11.7). The limit of quantification for this assay is approximately 31 RNA copies per ml (1.49 log10) with 800 μl of sample.
RNAscope in situ hybridization in hamsters.
SARS-CoV-2 antisense specific probe v-nCoV2019-S (ACD, 848561) and SARS-CoV-2 v-nCoV2019-S-sense (ACD, 845701) were used to respectively target the positive-sense and the negative-antisense of the Spike sequence. Before performing RNAscope assay, slides were first deparaffinized in xylene, rehydrated and incubated with RNAscope H2O2 (ACD, 322335) for 10 min at room temperature, followed by treatment with retrieval in ACD P2 retrieval buffer (ACD, 322000) for 15 min at 98 °C. Then, slides were incubated with protease plus (ACD, 322331) for 30 min at 40 °C. Probe hybridization and detection were performed through the RNAscope 2.5 HD Detection Reagents-RED (ACD, 322360) according to the manufacturer’s instructions.
Immunofluorescence staining of hamster lung sections.
In brief, the pretreatment of slides was the same as for IHC, including dewaxing, rehydration, retrieval and 3% H2O2 treatment. Then, slides were blocked with 5 % BSA for 30 min, followed by 3 rinses with DPBS. Subsequently, slides were incubated with primary rabbit anti-SARS-N antibody (1:200) overnight at 4 °C and goat anti-rabbit Alexa Fluor 647 (Abcam, ab150079, 1:500), AF-488-CD206 (Santa Cruz Biotechnologies,sc-376108 AF488, 1:150) and Alexa Fluor 568-Iba-1 (Abcam, ab221003, 1:200) were incubated at r.t. for 1 h, or goat anti-rabbit Alexa Fluor 647 (Abcam, ab150079, 1:500) and FITC-pan-CK (Abcam, ab78478, 1:200) were incubated at r.t. for 1 h. Finally, slides were mounted with ProLong Gold antifade mountant with 4′,6-diamidino-2-phenylindole (Invitrogen) and imaged on an Olympus FLUOVIEW CLSM (Olympus, FV3000).
SARS-CoV-2 D614G pseudovirus neutralization assay.
Female CD1 mice (7 weeks old) were immunized with PBS, RBD, RBD-Exo or RBD-Exo LTS in two doses once a week via nebulization. At 7 d after vaccinations, each mouse was challenged with 8 × 108 genomic copies (GC) of SARS-CoV2 D614G pseudovirus carrying the GFP reporter (C1120G, Montana Molecular). Lungs were collected and imaged at 24 h post vaccination with a Xenogen live imager (PerkinElmer) and an Olympus FLUOVIEW CLSM (Olympus, FV3000).
IgA and IgG purification.
IgA antibodies in BALF and NPLF of immunized mice were collected and purified. IgG antibodies were collected from serum of mice and purified according to previous reports44. Briefly, serum, BALF or NPLF (500 μl) were diluted with DPBS, heat-inactivated at 56 °C for 1 h and mixed with protein L/agarose beads (gel-protl-2; InvivoGen) overnight at 4 °C. The above solution was transferred to chromatography columns and washed with PBS (10 mM, pH 7.4) at a volume of 10 column. IgA and IgG were then collected by eluting with pH 3.0 glycine buffer (0.1 M). Then, pH was immediately adjusted to 7.5 using 1 M tris (pH 8.0). The collected IgA and IgG were transferred to PBS (1×) buffer using Amicon ultra centrifugal filters with a 10 kDa membrane. IgA and IgG concentrations were determined by measuring the absorbance at 280 nm with a NanoDrop (Thermo Fisher) instrument.
Blockage activity of IgA antibodies.
Primary small airway epithelial cells were co-cultured with primary bronchial/tracheal epithelial cells (lower chamber) using transwells for 24 h. Purified IgA antibodies from mice vaccinated with PBS, free RBD, RBD-Exo or RBD-Exo LTS were incubated with SARS-CoV-2 D614G pseudovirus for 1 h at 37 °C. The concentration of SARS-CoV-2 D614G pseudovirus used was 6.6 × 108 GC ml−1. After incubation, the mixture was added to the transwell and incubated for another 24 h. The primary small airway epithelial cells in the upper chamber were collected for flow cytometry assay, while the primary bronchial/tracheal cells in the lower chamber were imaged by microscopy.
IgA and IgG neutralization assay.
To compare their protection activity against SARS-CoV-2 variant, purified IgA and IgG antibodies from PBS or RBD-Exo treatments were incubated with SARS-CoV-2 D614G pseudotyped virus for 1 h at 37 °C. Subsequently, they were incubated with ACE2-expressing A549 cells (a549-hace2tpsa, Invivogen) for 24 h. After incubation, cells were washed with PBS and the GFP positive cells were counted by microscopy.
Statistical analysis.
All quantitative experiments were conducted in triplicate independently. Data are shown as mean ± s.d. Two-tailed unpaired Student’s t-test was used to analyse differences between any two groups. Comparisons of more than two groups were conducted using one-way analysis of variance (ANOVA) followed by post hoc Bonferroni test. Grouped data were analysed by two-way ANOVA followed by Tukey post hoc test for multiple comparisons. P < 0.05 was considered statistically significant.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. RBD-specific IgA and Igg responses.

a,b, RBD-specific IgA antibody titers in serum (a) of mice and RBD-specific IgG antibody titers in BALF (b) of mice detected by ELISA after two doses of immunizations with different treatments. Data are mean ± s.d. n = 3. Statistical analysis was performed by one-way ANOVA test with Bonferroni correction. ns indicates no significance. All replicates are biological.
Extended Data Fig. 2 |. Immunogenicity stability test of RBD-Exo after storage.

a-c, RBD-specific IgG antibody titer from murine serum (a) and RBD-specific secretory IgA antibody titer from bronchoalveolar lavage fluid (BALF) (b) and nasopharyngeal lavage fluid (NPLF) (c), detected by ELISA after two doses of immunizations with different treatments. Data are mean ± s.d. n = 3. Statistical analysis was performed by one-way ANOVA test with Bonferroni correction. ns indicates no significance. All replicates are biological.
Extended Data Fig. 3 |. Comparison of intranasal and inhalation immunization.

RBD-specific IgG antibodies in serum of mice and secretory IgA antibodies from BALF and NPLF of mice detected by ELISA, in which the mice were vaccinated with RBD-Exo VLP by intranasal administration or inhalation. Data are mean ± s.d. n = 3. Statistical analysis was performed by two-way ANOVA tests with a Tukey post hoc test for multiple comparisons. ns indicates no significance. All replicates are biological.
Extended Data Fig. 4 |. Biodistribution of RBD-Exo in mice after inhalation.
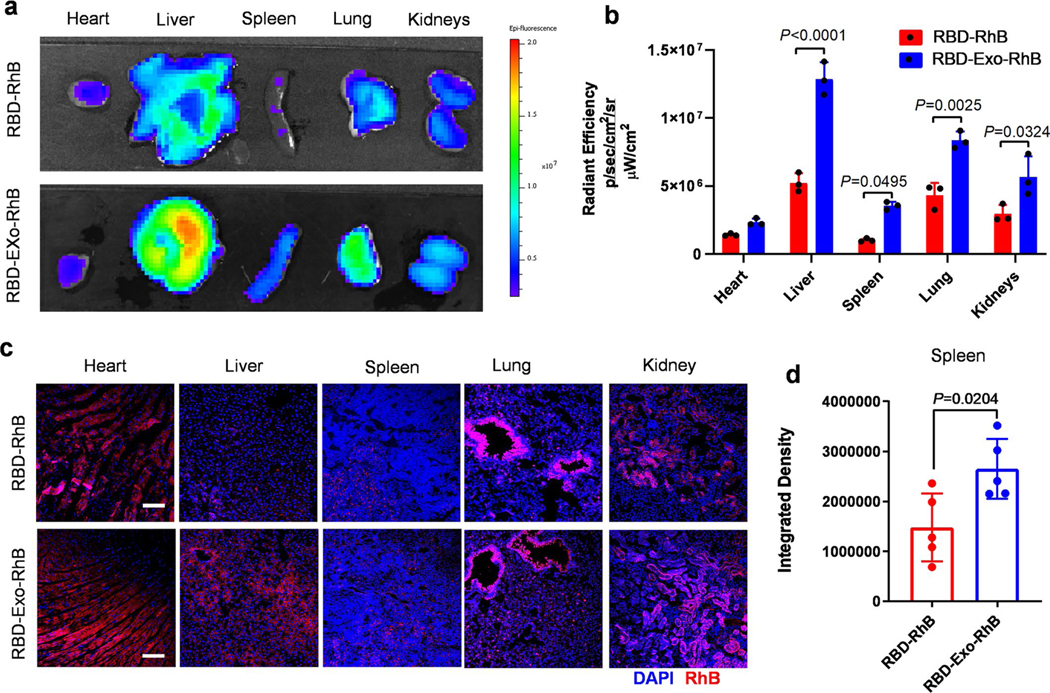
a, Ex vivo imaging of major organs of mice 24 hours after RBD-Exo-RhB or RBD-RhB inhalation. b, Quantification of the integrated density of RhB fluorescence in the major organs; n = 3 per group. c, Confocal images showing the biodistribution of RBD-Exo-RhB or RBD-RhB in heart, liver, spleen, lung, and kidney tissues. Scale bar, 50 μm. d, Quantitative results from in spleen tissues. n = 5. Data are mean ± s.d. Statistical analysis was performed by two-tailed, unpaired Student’s t-test. The replicates in b are biological. Analysis in d represents the technical replicates from three independent biological samples.
Supplementary Material
Acknowledgements
This work was supported by grants from the National Institutes of Health (HL123920, HL144002, HL146153, HL147357, HL149940 and HL154154 to K.C.; HL164998 to P.-U.C.D.) and the American Heart Association (18TPA34230092 and 19EIA34660286 to K.C.). We thank S. Kar, M. Porto, M. Lok and B. Spence at BIOQUAL. Inc for performing the hamster studies.
K.C. is a co-founder and equity holder of Xsome Biotech Inc. Xsome Biotech Inc. is a licensee of the intellectual property covering exosome delivery technologies and antiviral therapies from North Carolina State University.
Footnotes
Competing interests
The other authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41551-022-00902-5.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41551-022-00902-5.
Peer review information Nature Biomedical Engineering thanks Hiroshi Kiyono and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw and analysed datasets generated during the study are too large to be publicly shared, yet they are available for research purposes from the corresponding authors on reasonable request. Source data are provided with this paper.
References
- 1.Wang C, Horby PW, Hayden FG & Gao GF A novel coronavirus outbreak of global health concern. Lancet 395, 470–473 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guan W-j et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med 382, 1708–1720 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma K. et al. Vaccines for COVID-19: where do we stand in 2021? Paediatr. Respir. Rev 39, 22–31 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.King RG et al. Single-dose intranasal administration of AdCOVID elicits systemic and mucosal immunity against SARS-CoV-2 and fully protects mice from lethal challenge. Vaccines 9, 881 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hassan AO et al. A single-dose intranasal ChAd vaccine protects upper and lower respiratory tracts against SARS-CoV-2. Cell 183, 169–184 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kyriakidis NC, Lopez-Cortes A, Gonzalez EV, Grimaldos AB & Prado EO SARS-CoV-2 vaccines strategies: a comprehensive review of phase 3 candidates. NPJ Vaccines 6, 28 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walls AC et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gorbalenya AE et al. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol 5, 536–544 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Monteil V. et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 181, 905–913 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wrapp D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mercurio I, Tragni V, Busto F, De Grassi A. & Pierri CL Protein structure analysis of the interactions between SARS-CoV-2 spike protein and the human ACE2 receptor: from conformational changes to novel neutralizing antibodies. Cell. Mol. Life Sci 78, 1501–1522 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lan J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Chen W-H, Hotez PJ & Bottazzi ME Potential for developing a SARS-CoV receptor-binding domain (RBD) recombinant protein as a heterologous human vaccine against coronavirus infectious disease (COVID)-19. Hum. Vaccines Immunother 16, 1239–1242 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tai W. et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol 17, 613–620 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tai W, Zhang X, He Y, Jiang S. & Du L. Identification of SARS-CoV RBD-targeting monoclonal antibodies with cross-reactive or neutralizing activity against SARS-CoV-2. Antiviral Res. 179, 104820 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parodi A. et al. Bio-inspired engineering of cell- and virus-like nanoparticles for drug delivery. Biomaterials 147, 155–168 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Kahroba H, Hejazi MS & Samadi N. Exosomes: from carcinogenesis and metastasis to diagnosis and treatment of gastric cancer. Cell. Mol. Life Sci 76, 1747–1758 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Popowski K. et al. Exosome therapeutics for lung regenerative medicine. J. Extracell. Vesicles 9, 1785161 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kao C-Y & Papoutsakis ET Extracellular vesicles: exosomes, microparticles, their parts, and their targets to enable their biomanufacturing and clinical applications. Curr. Opin. Biotechnol 60, 89–98 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Antimisiaris SG, Mourtas S. & Marazioti A. Exosomes and exosome-inspired vesicles for targeted drug delivery. Pharmaceutics 10, 218 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simons M. & Raposo G. Exosomes – vesicular carriers for intercellular communication. Curr. Opin. Cell Biol 21, 575–581 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Henry E. et al. Adult lung spheroid cells contain progenitor cells and mediate regeneration in rodents with bleomycin-induced pulmonary fibrosis. Stem Cells Transl. Med 4, 1265–1274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dinh P-UC et al. Derivation of therapeutic lung spheroid cells from minimally invasive transbronchial pulmonary biopsies. Respir. Res 18, 132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cores J. et al. Safety and efficacy of allogeneic lung spheroid cells in a mismatched rat model of pulmonary fibrosis. Stem Cells Transl. Med 6, 1905–1916 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cores J. et al. A pre-investigational new drug study of lung spheroid cell therapy for treating pulmonary fibrosis. Stem Cells Transl. Med 9, 786–798 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dinh P-UC et al. Inhalation of lung spheroid cell secretome and exosomes promotes lung repair in pulmonary fibrosis. Nat. Commun 11, 1064 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corbett KS et al. Evaluation of the mRNA-1273 vaccine against SARS-CoV-2 in nonhuman primates. N. Engl. J. Med 383, 1544–1555 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sahin U. et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature 586, 594–599 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Mulligan MJ et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 586, 589–593 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Zhang N-N et al. A thermostable mRNA vaccine against COVID-19. Cell 182, 1271–1283 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naseri Z, Oskuee RK, Jaafari MR & Forouzandeh Moghadam M. Exosome-mediated delivery of functionally active miRNA-142–3p inhibitor reduces tumorigenicity of breast cancer in vitro and in vivo. Int. J. Nanomed 13, 7727–7747 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakase I. et al. Arginine-rich cell-penetrating peptide-modified extracellular vesicles for active macropinocytosis induction and efficient intracellular delivery. Sci. Rep 7, 1991 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Z. et al. Pretargeting and bioorthogonal click chemistry-mediated endogenous stem cell homing for heart repair. ACS Nano 12, 12193–12200 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen D. et al. Antibody-armed platelets for the regenerative targeting of endogenous stem cells. Nano Lett. 19, 1883–1891 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kiyono H. & Azegami T. The mucosal immune system: from dentistry to vaccine development. Proc. Jpn. Acad. B 91, 423–439 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laczko D. et al. A single immunization with nucleoside-modified mRNA vaccines elicits strong cellular and humoral immune responses against SARS-CoV-2 in mice. Immunity 53, 724–732 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miragoli M. et al. Inhalation of peptide-loaded nanoparticles improves heart failure. Sci. Transl. Med 10, eaan6205 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Lycke N. Recent progress in mucosal vaccine development: potential and limitations. Nat. Rev. Immunol 12, 592–605 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Sia SF et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 583, 834–838 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tostanoski LH et al. Ad26 vaccine protects against SARS-CoV-2 severe clinical disease in hamsters. Nat. Med 26, 1694–1700 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imai M. et al. Syrian hamsters as a small animal model for SARS-CoV-2 infection and countermeasure development. Proc. Natl Acad. Sci. USA 117, 16587–16595 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baum A. et al. REGN-COV2 antibodies prevent and treat SARS-CoV-2 infection in rhesus macaques and hamsters. Science 370, 1110–1115 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hou YJ et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 370, 1464–1468 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Z. et al. Enhanced SARS-CoV-2 neutralization by dimeric IgA. Sci. Transl. Med 13, eabf1555 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Q. et al. Cellular nanosponges inhibit SARS-CoV-2 infectivity. Nano Lett. 20, 5570–5574 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rao L. et al. Decoy nanoparticles protect against COVID-19 by concurrently adsorbing viruses and inflammatory cytokines. Proc. Natl Acad. Sci. USA 117, 27141–27147 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeng C. et al. Leveraging mRNA sequences and nanoparticles to deliver SARS-CoV-2 antigens in vivo. Adv. Mater 32, 2004452 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou X. et al. Engineering antiviral vaccines. ACS Nano 14, 12370–12389 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doremalen NV et al. Intranasal ChAdOx1 nCoV-19/AZD1222 vaccination reduces viral shedding after SARS-CoV-2 D614G challenge in preclinical models. Sci. Transl. Med 13, eabh0755 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lund FE & Randall TD VIEWPOINT: COVID-19 scent of a vaccine. Science 373, 397 (2021). [DOI] [PubMed] [Google Scholar]
- 51.Hassan AO et al. A single intranasal dose of chimpanzee adenovirus-vectored vaccine protects against SARS-CoV-2 infection in rhesus macaques. Cell Rep. Med 2, 100230 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu S. et al. Safety, tolerability, and immunogenicity of an aerosolised adenovirus type-5 vector-based COVID-19 vaccine (Ad5-nCoV) in adults: preliminary report of an open-label and randomised phase 1 clinical trial. Lancet Infect. Dis 21, 1654–1664 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wong LS, Khan F. & Micklefield J. Selective covalent protein immobilization: strategies and applications. Chem. Rev 109, 4025–4053 (2009). [DOI] [PubMed] [Google Scholar]
- 54.Dong K. et al. Pathogen-mimicking nanocomplexes: self-stimulating oxidative stress in tumor microenvironment for chemo-immunotherapy. Mater. Today 20, 346–353 (2017). [Google Scholar]
- 55.Greening DW, Xu R, Ji H, Tauro BJ & Simpson RJ A protocol for exosome isolation and characterization: evaluation of ultracentrifugation, density-gradient separation, and immunoaffinity capture methods. Methods Mol. Biol 1295, 179–209 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Bosch S. et al. Trehalose prevents aggregation of exosomes and cryodamage. Sci. Rep 6, 36162 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saxena A, Sachin K, Bohidar HB & Verma AK Effect of molecular weight heterogeneity on drug encapsulation efficiency of gelatin nano-particles. Colloids Surf. B 45, 42–48 (2005). [DOI] [PubMed] [Google Scholar]
- 58.Bajaj S, Singla D. & Sakhuja N. Stability testing of pharmaceutical products. J. Appl. Pharm. Sci 2, 129–138 (2012). [Google Scholar]
- 59.Li Z. et al. Cell-mimicking nanodecoys neutralize SARS-CoV-2 and mitigate lung injury in a non-human primate model of COVID-19. Nat. Nanotechnol 16, 942–951 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw and analysed datasets generated during the study are too large to be publicly shared, yet they are available for research purposes from the corresponding authors on reasonable request. Source data are provided with this paper.