

SUMMARY
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Omicron variant of concern, first identified in November 2021, rapidly spread worldwide and diversified into several subvariants. The Omicron spike (S) protein accumulated an unprecedented number of sequence changes relative to previous variants. In this review, we discuss how Omicron S protein structural features modulate host cell receptor binding, virus entry, and immune evasion and highlight how these structural features differentiate Omicron from previous variants. We also examine how key structural properties track across the still-evolving Omicron subvariants and the importance of continuing surveillance of the S protein sequence evolution over time.
INTRODUCTION
The Omicron variant has taken hold worldwide and is currently the predominant severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variant causing COVID-19. Increased infectivity and immune evasion and a substantially larger number of mutations compared to previous variants of concern (VOCs) characterize the Omicron variant (Figures 1A and 1B).1 The evolution of this SARS-CoV-2 VOC has lengthened the fallout from the COVID-19 pandemic, which has had social and financial impacts worldwide. Since its identification and classification as a VOC (https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern), scientists have been studying the Omicron variant to understand its structure and interactions with the human host, its differences from the previous VOCs, why these differences enabled it to outcompete other variants, and how its continued evolution is shaping the structure of the Omicron spike (S) protein. Several dominant subvariants of Omicron have been identified including BA.1-BA.5, BQ, XBB, and EG.5, with XBB, EG.5, and BA.2.86 currently prevalent across the globe2 (https://www.who.int/docs/default-source/coronaviruse/05062023xbb.1.16.pdf?sfvrsn=f1845468_3; https://www.who.int/docs/default-source/coronaviruse/09082023eg.5_ire_final.pdf) (Figure 1C). While multiple genes of SARS-CoV-2 can affect disease severity and are important to study, the S protein, which is the focus of this review, is the primary entity on the virus surface that is responsible for host receptor attachment during entry and is also the target for neutralizing antibodies. Omicron is the most immune-evasive variant to date (Figure 1D) and, through modifications in its S protein, has continued to improve its ability to evade the host immune system while retaining its ability to enter host cells.
Figure 1. Mutation accumulation and antibody neutralization of SARS-CoV-2 S protein.
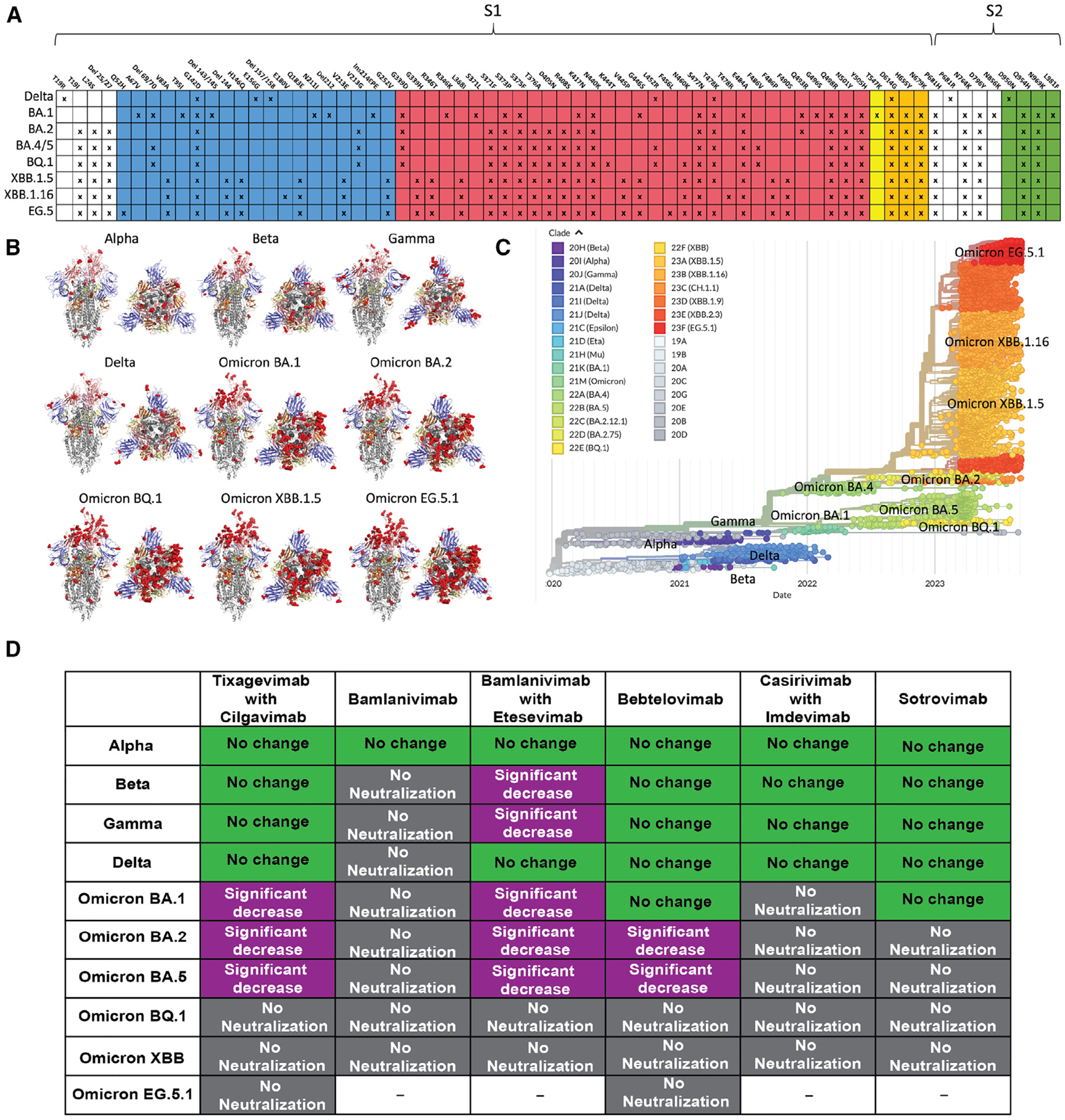
(A) Highest frequency Delta and Omicron SARS-CoV-2 variant mutations relative to the initial strain are indicated with Xs. Blue: NTD mutations; red: RBD mutations; yellow: SD1 mutations; orange: SD2 mutations. These diagrams were created using data from the Lineage Comparison tool from GISAID.
(B) Locations of mutations relative to the initial strain mapped on SARS-CoV-2 variant S protein structures. Mutations are shown by red dots. Mutations shown are >60% prevalent in each variant. S structure coloring follows that of (A) and Figure 2A. All structures are in the 1-up-RBD conformation. Mutations shown on each structure are as follows: Alpha: N501Y, A570D, D614G, T716I, S982A, D1118H; not shown: P681H; Beta: D215G, K417N, E484K, N501Y, D614G, A701V; not shown: D80A; Gamma: L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I; and not shown: V1176F. Delta and Omicron mutations are those shown in (A). PDB IDs of S structures are Alpha, PDB: 7EDF; Beta, PDB: 7LYQ; Gamma, PDB: 8DLO; Delta, PDB: 7V7P; and Omicron BA.1, PDB: 7TL9. BA.2, BQ.1, XBB.1.5, and EG.5.1 mutations are all modeled on PDB: 7TL9.
(C) Phylogenetic tree showing the evolution of Omicron over time. Generated from GISAID.
(D) FDA-approved monoclonal antibodies for treatment of SARS-CoV-2 and their efficacy against VOCs and Omicron variants in comparison to the initial SARS-CoV-2 strain. Green: no change; purple: significant decrease; gray: no neutralization. Data are sourced from Qu et al.,20 Tao et al.,23 Imai et al.,30 and Wang et al.,31 as well as https://www.fda.gov/media/145611/download, https://www.fda.gov/media/145802/download, https://www.fda.gov/media/149534/download, https://www.fda.gov/media/154701/download, https://www.fda.gov/media/156152/download, https://covdb.stanford.edu/susceptibility-data/table-mab-susc/, and https://www.bv-brc.org/view/VariantLineage/.
On a molecular level, SARS-CoV-2 S proteins are upside-down, pyramid-shaped, trimeric structures displayed on the fatty acid membrane that forms the outer surface of the virus and encloses the viral RNA3 (Figures 2A and 2B). The pre-fusion SARS-CoV-2 S protein contains S1 and S2 subunits separated by a furin cleavage site (Figure 2C). S1 and S2 subunits facilitate receptor binding and fusion with the host cell membrane, respectively.4–8 The S1 subunit is dynamic and contains an N-terminal domain (NTD) and a receptor-binding domain (RBD) that interacts primarily with the angiotensin-converting enzyme 2 (ACE2) receptor expressed by a plethora of human cell types at varying levels9–11 as well as with a newly identified receptor, TMEM106B, which is reported to bind with lower affinity to S than ACE2 but allows viral entry into cells lacking expression of ACE2.12 Additional interactions have been reported between SARS-CoV-2 S and the co-receptors heparan sulfate13 and neuropilin-1.14 Upon receptor binding and proteolytic processing by host proteases, the S2 subunit undergoes conformational changes resulting in release of the fusion peptide (FP), which mediates fusion of viral and host cell membranes (Figure 2A).15–17
Figure 2. The SARS-CoV-2 S protein S1 subunit as an “arm” controlling the RBD down/up transition, a modulator of the propensity for pre- to postfusion conformation transition.
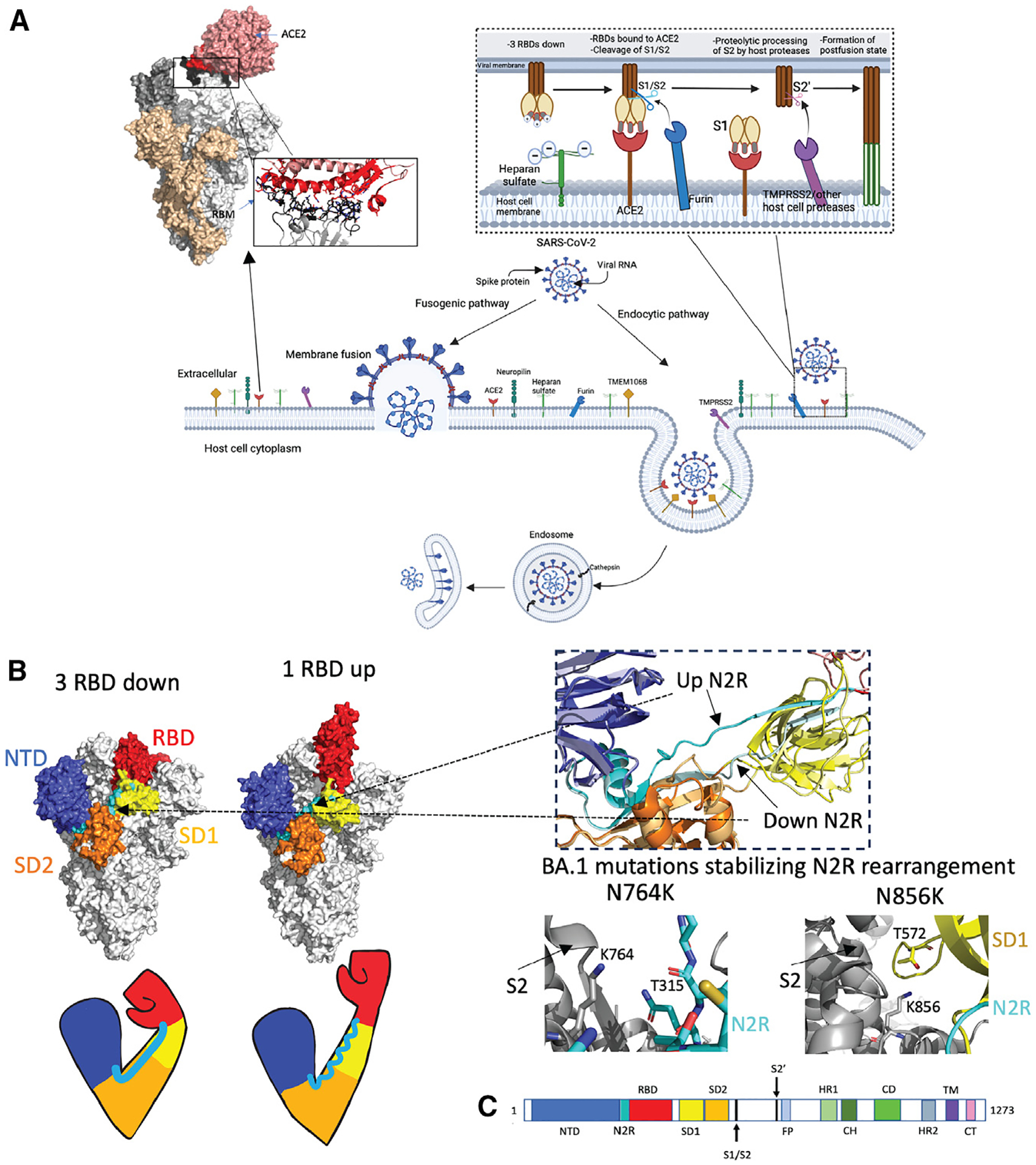
(A) Schematic representation of SARS-CoV-2 viral cell entry fusogenic and endocytic pathways. The top right inset shows interactions between SARS-CoV-2 S and heparan sulfate, ACE2, and proteases at the host cell surface, leading to the formation of the postfusion state. Top left: Omicron S bound to ACE2 (PDB: 7T9K) with a zoomed-in image of the receptor binding motif-ACE2 interaction. Interacting stretches in RBD 438–460 and 469–506 are colored black. Directly interacting residues in RBD: 444KVSGNY449, 453YRLF456, 473YQAGNK478, 484AGFNCYFPLRSYSFRPTYGVGHQ506: black sticks; ACE2 binding site (residues 18–56, 80–85, 325–333, and 347–360): red; Directly interacting residues on ACE2: 19STIEEQ24, 27TF28, 30DK31, 34HE35, 37ED38, 41YQ42, 45LA46, 48WN9, 52T, 55T, 80A, 82M, 83Y, 326G, 329EN330, 352GKGD FR357: red sticks. Figure made using BioRender.com and Pymol.
(B) Arm analogy of S protein S1 subunit. SARS-CoV-2 Omicron S in 3-RBD-down (left, PDB: 7TF8) and 1-RBD-up (right, PDB: 7TEI) conformations. NTD residues 24–293 (shoulder): blue; N2R residues 293–330: cyan; RBD residues 330–52813: red; SD1 (528–590): yellow; SD2 residues 590–680 (elbow): orange; S2: gray. Right top: zoom in on N2R region showing a comparison of the up RBD “rearranged” up N2R and the down N2R. Bottom right: BA.1 mutations N764K (PDB: 7TL9) and N856K (PDB: 7TF8), which stabilize the N2R rearrangement in Omicron BA.1 S.
(C) Schematic of the S sequence with visible S1 domains colored the same as in the above structure image. NTD, N-terminal domain; RBD, receptor-binding domain; SD1 and SD2, subdomains 1 and 2. The S2 subunit contains the fusion peptide (FP), heptad repeat 1 (HR1), central helix (CH), connector domain (CD), and HR2 subdomains. The transmembrane domain28 and cytoplasmic tail (CT) follow.
The Omicron S mutations are most densely clustered in the RBD and the NTD, which are immunodominant regions targeted by neutralizing antibodies (Figures 1A and 1B).4,18 Several studies have noted decreased binding of antibodies from SARS-CoV-2-infected individuals to the Omicron S protein,18–29 with neutralization titers of plasma specimens obtained from individuals infected with the Omicron variant up to ~50 times lower than for the wild-type (WT) S protein.18 The Omicron variants are also significantly more resistant to S-protein-targeting monoclonal antibodies that were previously approved as therapeutics for SARS-CoV-2-infected individuals20,23,30,31 (https://www.fda.gov/media/145611/download; https://www.fda.gov/media/145802/download; https://www.fda.gov/media/149534/download; https://www.fda.gov/media/154701/download; https://www.fda.gov/media/156152/download; https://covdb.stanford.edu/susceptibility-data/table-mab-susc/; https://www.bv-brc.org/view/variantlineage/) (Figure 1D). The Omicron variant’s rapid accumulation of immune-evasive amino acid substitutions makes it necessary to continue to improve viral genome sequence detection and identification in populations throughout the world. Early detection of emerging SARS-CoV-2 variants with S mutations that may contribute to immune evasion will allow health officials to make informed decisions concerning population health.
The overall topology of the Omicron S protein resembles that of previous variants, with the RBD from each monomer folding over on SD1 and SD2 subdomains and touching the NTD of the adjacent protomer.10,17,32 The Omicron S protein has no mutations to any of its 22 N-linked glycosylation sites33 and no additional glycosylation sites compared to previous variants. Several structures of Omicron S variants available in literature incorporated the engineered 2P or Hexapro stabilization mutations34 that were designed to stabilize the S protein in its pre-fusion conformation. For the Omicron BA.1 S protein, the 2P mutations decreased antibody binding at key epitopes10 and restricted the S conformational landscape to predominantly present the 1-RBD-up state,35–38 whereas without the engineered proline mutations, the Omicron BA.1 and BA.2 S proteins accessed a wider range of conformational states including the 3-RBD-down state and multiple RBD-up states.10,32,39,40 In this review, we explore the major structural differences in the S protein of Omicron and previous variants. These differences include a more compact architecture, rearrangements in several domains, and changes to structural elements that control the RBD up/down transition. We further explore the epistatic interactions between mutations that, combined, cause a different effect than they would individually and note the increased accumulation of positively charged residues at the host-interacting surface of the S.
Omicron S protein RBD mutations stabilize the RBD-down state and retain ACE2 binding
A notable difference observed between the Omicron BA.1 S protein compared to previous SARS-CoV-2 variants is its tighter interprotomer packing and more compact architecture.10,32,38,39,41 Omicron BA.1 had 15 RBD mutations compared to the initial SARS-CoV-2 S protein (Figure 1A), The S371L, S373P, and S375F substitutions occur in an interfacial RBD loop, contributing, along with Y505H, to interactions between the down-state RBDs. The S373P substitution also shifts a short RBD helix, forcing a rotation of the N-linked glycan at N343 and allowing S371L (in BA.1) and S371F (in BA.2 and all later Omicron variants) to point toward the glycan to stabilize its new conformation.40 N440K, G446S, S477N, T478K, E484A, G496S, Q498R, N501Y, and Y505H substitutions are situated at the receptor binding motif (RBM), causing substantial remodeling of the receptor binding interface. In the RBD-down S protein structure, the rest of the mutations as well as some of the RBM mutations are situated in the regions involved in interprotomer packing42,43 and contribute to interactions between the down-state RBDs (Figure 3A). In a structure of the XBB.1 S ectodomain,44 despite the presence of the Hexapro stabilization mutations,34 the 3-RBD-down state dominated the S population, highlighting that the RBD-RBD down-state contacts are reinforced through Omicron evolution. The mutations acquired at the RBD-RBD interface of the 3-RBD-down state are retained throughout successive Omicron variants, including in EG.5 and BA.2.86, and are likely an immune-evasion mechanism by the virus to shield immunodominant regions that would be exposed in an RBD presenting its “up” conformation.
Figure 3. The compact nature of the Omicron S protein compared to previous SARS-CoV-2 variants.
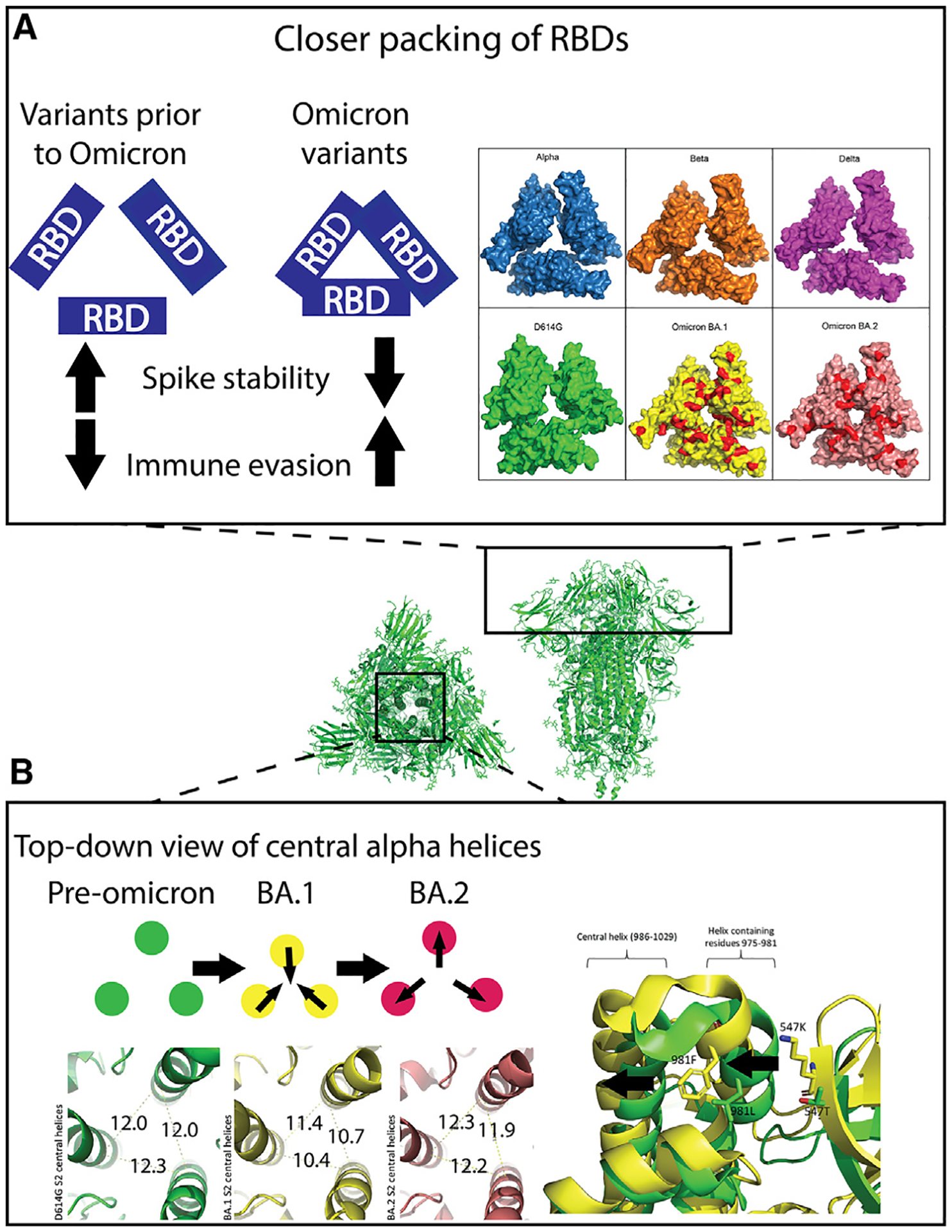
Center: top and side views of the 3-RBD-down D614G S ectodomain (PDB: 7KE8).
(A) Left: trend in Omicron variants to optimize inter-RBD contacts leading to stabilization of the RBD-down state, resulting in higher immune evasion. Right: map of RBD mutations on solved closed RBD structures in surface view. PDB IDs: Alpha, PDB: 7R13; Beta, PDB: 7LYL; Delta B.1.617.2, PDB: 7TOU; D614G, PDB: 7KDK; BA.1, PDB: 7TL1; BA.2, PDB: 7UB6. Red patches on BA.1 and BA.2: sites of mutation in Omicron variants relative to Delta variant.
(B) Top left: schematic showing the inward and outward movements of the S S2 central helices in pre-Omicron, BA.1 and BA.2. Bottom left: green, D614G S2 central helix (PDB: 7KE8) distances measured from C alpha-atom of residue R995 on each monomer; yellow: Omicron BA.1 S2 central helix (PDB: 7TF8) distances measured the same as D614G; red: Omicron BA.2 S2 central helix (PDB: 7UB0) distances measured the same as above. Right: aligned structures of D614G (green) PDB: 7KE8 and BA.1 (yellow) PDB: 7TF8. Residues 547 and 981 are shown as sticks. The arrows indicate the movements of the two helices due to these two mutations.
ACE2 receptor binding to the Omicron S protein has been studied extensively and compared with previous variants. The literature presents mixed results, with some studies conducted using surface plasmon resonance (SPR) and bio-layer interferometry (BLI) reporting no significant change between ACE2 binding for the S proteins of Omicron BA.1 and of the Delta variant that preceded Omicron in global dominance4,45–47 However, other studies using SPR, BLI, and microscale thermophoresis (MST) analysis have reported 2- to 6-fold increased S-ACE2 affinity for Omicron BA.1 compared to the Delta and WT variants,25,37,46,48 while still others employing MST and enzyme-linked immunosorbent assay49 analysis reported 1.25-to 72-fold decreased binding of ACE2 to the S protein of Omicron BA.1 compared to the Delta variant50,51 According to deep mutational scanning, ELISA, and SPR analysis, some of these mutations, when tested individually, increased affinity for ACE2 (N440K, S477N, T478K,52 G496S, Q498R,4 N501Y53), while others decreased ACE2 affinity (R346K through long-range interactions, K417N,4 G496S,54 E484A, G496S, Y505H53), possibly leaving Omicron BA.1 with a similar binding affinity for ACE2 as the preceding Delta variant.4 The collective effect of the Omicron RBD mutations to receptor interactions therefore are minimal despite large mutational changes, while the contributions by these mutations to immune evasion are substantial. Indeed, many of these RBD residue substitutions lead to decreased antibody binding either caused by individual epitope residue mutations or by the occlusion of the antibody epitope due to the improved RBD-RBD packing in the RBD-down state.4,39,55 Thus, the same mutations that remodeled the S protein architecture may have contributed to immune escape by the Omicron variant.
The RBD-RBD interfacial packing, first observed in the Omicron BA.1 S protein, was further optimized in the Omicron BA.2 S protein through the acquisition of additional mutations.10,32 The Omicron BA.2 S contains S371F instead of S371L in BA.1 and the additional substitutions T376A, D405N, R408S, and L452Q. In the Omicron BA.2 S protein, the G446S and G496S substitutions that appeared in BA.1 were reverted back to G. The BA.2 mutations drove closer RBD-RBD interprotomer packing while also stabilizing the RBD internally.32 The T376A substitution in the interfacial RBD loop contributed to enhanced RBD-RBD packing. The S371F substitution results in closer packing of two helices in the RBD as well as the N343 glycan stabilization described above and could have contributed to overall RBD stability. The R408S substitution breaks an interprotomer hydrogen bond, which is compensated for by the D405N substitution creating a new interprotomer H-bond.32 One study has reported the BA.2 S protein to have improved binding affinity for ACE2 than the BA.1 variant54 via SPR analysis, while another study reported similar binding to ACE2 for the BA.1 and BA.2 S proteins32 via ELISA. Taken together, these data show that despite the improved RBD-RBD down-state contacts in BA.2, the S protein retains robust binding to the ACE2 receptor. The absence of the G496S mutation is the only change in the BA.2 RBM compared to BA.1 and has been linked to the improved ACE2 interaction, although the increased stability of the BA.2 RBD may have also contributed to improving its interaction with the ACE2 receptor.53,54
The majority of RBD differences as Omicron evolved past BA.2 involve mutations to the RBM residues between residues 440 and 506. The amino acid sequences of the BA.4 and BA.5 S proteins are identical. The BA.4 and BA.5 S proteins lack the Q493R substitution seen in BA.1 and BA.2 while incorporating 2 additional substitutions, L452R (previously seen in Delta) and F486V. While no major structural changes were noted in RBD crystal structures due to these mutations,56 they have been shown to be important for antibody escape in neutralization assays against vaccine and naturally immune serum as well as monoclonal antibodies.56,57 In addition, F486V was found to decrease ACE2 binding to the RBD, while the R493Q reversion mutation compensated for this by increasing ACE2 binding.58 XBB.1.5 and XBB.1.16 variants contained RBM mutations V445P, G446S, F486P, and F490S and demonstrated similar ACE2 binding affinity to their predecessors, BA.2 and BQ.1.31 Omicron EG.5 has an additional F456L mutation that, as measured by deep mutation scanning, decreases ACE2-binding affinity in the BA.2 background but enhances ACE2-binding affinity in the XBB.1.5 background.59 Convergent evolution of XBB lineages has been noted in the appearance of RBM mutations L445F and F456L, nicknamed in a bioRxiv pre-print60 as the “FLip” mutations due to the flipping of L and F residues at these positions in the RBM of variants such as FL.1.5.1, XBB.1.5.7, and HK.3. Together, they were shown to enhance ACE2 binding and immune evasion in another bioRxiv pre-print.61 The residues in non-RBM regions of the RBD remained the same through BQ.1 and did not change until G339H, R346T, and L368I appeared in the XBB variants.
Changes in the stability of the Omicron S
Stability of the pre-fusion S protein is an important determinant of S fitness. While the S protein should be stable enough in its pre-fusion conformation to prevent premature triggering, it must also have the ability undergo the conformational changes required to transition to the postfusion form when engaged by the right triggers, which include host receptor binding and proteolytic processing. There are some apparent disagreements in the literature about the stability of the Omicron S protein relative to previous variants. Using Omicron S ectodomains that contain the Hexapro and 2P mutations, differential scanning calorimetry (DSC) and differential scanning fluorimetry (DSF) studies have shown increased thermostability of the Omicron S trimeric ectodomain compared to previous variants.21,62 Other studies using both Hexapro-stabilized and non-proline-stabilized S have found decreased thermostability in the Omicron trimeric ectodomain and the RBD compared to previous variants.32,63,64 However, there seems to be agreement in the literature about BA.1 being less thermostable than BA.2. While the reinforced RBD-RBD packing in the down state may have enabled the Omicron BA.1 variant to evade many previously effective antibodies, the large number of accumulated mutations resulted in decreased stability of both the S and the RBD structure. The Omicron BA.1 RBD was also more susceptible to digestion by trypsin and chymotrypsin than WT in protease-digestion assays,64 indicating decreased stability. The inflection temperature of the Omicron BA.1 RBD measured by DSF decreased by ~7°C compared to the WT strain, indicating less stability compared to WT.32,64 Additional mutations acquired in BA.2 partially overcame the decreased stability of the S and the RBD observed in BA.1 and could have contributed to BA.2 overtaking BA.1.32
Omicron mutations remodel NTD loops and alter packing of S2 helices
The SARS-CoV-2 NTD is made up of b-strands connected by several loops. The role of the NTD in viral infectivity is less well studied than that of the RBD. The SARS-CoV-2 S protein has evolved to have longer loops than SARS-CoV and other betacoronaviruses,65 and these loops have been hotspots for mutations since the emergence of SARS-CoV-2. The NTD mutations have predominantly appeared in 3 loops (N1: residues 14–26, N3: residues 141–156, and N5: residues 246–260)66 that make up the “NTD neutralization supersite.” The SARS-CoV-2 NTD is predicted to interact with sialosides67,68 and additional accessory receptors69,70 as it does in other coronaviruses, although the details and structural consequences of these interactions are not well understood. The large number of mutations and deletions that have accumulated in the NTD over time (Figure 1A) indicates the importance of this region as a target for the host immune response. In addition, the hypervariable NTD loops have been identified as a locus of control for S protein metastability. Allosteric coordination between the NTD and RBD domains has been reported,16 with the composition of the NTD loops having an effect on the RBD moving into the up position.71 In contrast to the RBD mutations, which do not substantially change the shape of the domain while still contributing to immune evasion and modulating receptor binding, the mutations in the NTD remodel the NTD antigenic loops.39
The Omicron variant was able to extensively remodel the NTD antigenic loops without losing S function.66 Deletion of NTD residue 144 led to increased immune evasion, while a deletion at residues 69/70 did not substantially alter recognition of serum antibodies.17,72 Residues 69/70 were deleted in the Omicron variants BA.1, BA.4/5, and BQ.1, while residue 144 was deleted in BA.2, XBB, and EG.5 Omicron subvariants. Sequence optimization at these NTD loops is likely a strategy for immune evasion. In BA.1, the 5 mutations, 3 deletions, and 3-residue insertion reconfigure the N-terminal segment and the surface-exposed loops.32,39 Most of the mutations in the BA.1 NTD are located at an antigenic supersite, causing a loss of neutralization by antibodies that target this epitope.73 Omicron BA.1 and BA.2 differ greatly in their NTD mutational content with 12 amino acid differences (including point mutations and deletions), and BA.2 has its own unique remodeling of the NTD N-terminal segment and loops.10,40 The BA.2 NTD antigenic supersite N1 loop displays a structural rearrangement relative to BA.1 due to the loss of 2 proline residues caused by the deletion at 25–27.74 This deletion at 25–27 appeared in BA.2 and is maintained throughout in subsequent Omicron variants.74 The BA.4/5 NTDs remain the same as BA.2, without any major changes emerging until XBB.1, where 5 additional point mutations are introduced.
Aside from the RBD and the NTD, where the majority of the mutations are concentrated, other regions of the S protein undergo structural changes as well, including the S2 subunit of pre-fusion S that contains 3 central α-helices, with each of the 3 monomers contributing one α-helix (Figure 3B). In the Omicron BA.1 S, the helices were shifted closer together,10,32 with two key BA.1 mutations, L981F and T547K, likely contributing to this structural shift. The elongation of the side chain upon mutation of T to K at residue 547 causes steric clashes with the helix formed at residues 975–981 and pushes this helix toward the central helix containing residues 986–1029. F981 interactions enabled by the L981F mutation also facilitate the movement of the 975–981 helix, pushing the central helices closer together by ~1–2Å compared to the D614G S (Figure 3B). Interestingly, in Omicron BA.2, the L981F and T547K mutations are reversed, and the three central helices relax to ~12Å apart, comparable to the D614G central helices.32 These observations support the idea that the BA.1 mutations were beneficial for immune evasion but contributed to destabilizing the S. Thus, the virus evolved to have a less strained structure by reversing some of the mutations that contributed to the strain in the Omicron BA.1 S.
Allosteric elements control S conformations
A single S protein protomer S1 subunit can be visualized as a bent arm, with the NTD as the shoulder, SD2 as the elbow, and SD1 as the wrist that rotates open the RBD “hand,” which extends upward to engage in a “fist bump” with the host receptor (Figure 2B). The NTD-to-RBD (N2R) linker connects the NTD and the RBD while contributing a β-strand to both SD1 and SD2.16 In an RBD-up protomer, the N2R linker becomes disordered, causing a decrease in the angle between the “shoulder” (NTD) and the “forearm” (N2R), which hinge at SD2 (the “elbow”). The 3-RBD-down pre-fusion Omicron BA.1 S protein shows a conformational rearrangement (Figure 2B) of the N2R linker in one of its down-state protomers.10,38 This rearrangement was also observed in the Delta variant S.10 When the N2R rearrangement takes place, the angle of the bend in the “elbow” becomes smaller, bringing the NTD and the RBD closer together, making the length of the affected side of the S protein trimer shorter between the two NTDs38 (Figure 2B). Since substantial reorganization of the N2R region is observed during the RBD up/down transition, this observation of N2R rearrangement in a down-state RBD in BA.1 suggested that the BA.1 RBD may be primed to transition into the 1-RBD-up state. The rearrangement of the N2R is stabilized by the N764K and N856K mutations on the S2 subunit of BA.1. Residue K764 interacts with the backbone oxygen of T315 at the SD2 end of the N2R, while residue K856 interacts with T572 and D568, which are part of SD1 (Figure 2B).10,38 This N2R rearrangement, which disrupted the β-strand arrangement of this region with SD1 and SD2, was not observed in BA.2 cryoelectron microscopy (cryo-EM) structures, consistent with the overall increased structural stability observed for the BA.2 S.
The FP proximal region (FPPR) includes residues 823–862 and is another control element of the RBD up/down transition. In previous variants, this region is disordered in the RBD-up protomer. Zhang et al.40 noted that in BA.1, the salt bridge created between SD1 and the FPPR residue N856K caused the FPPR to remain locked in its ordered conformation even after the RBD was raised upward. They noted that the RBD needed to rise higher than in previous variants to accommodate the ordered FPPR region in BA.139 and that the N856K interaction likely created steric hinderance to the RBD raising up. Interestingly, BA.2, which lacks the N856K mutation, was found to have more order in its FPPR loops than in the D614G variant, indicating that N856K may not be the only contributor to this FPPR ordering.
It is possible that increased interactions with mutated residues on S2 could have allosteric effects on the conformation and interactions of the S1 subunit, including antibody binding by S1. Increased S1–S2 interactions could lead to less efficient detachment of S1 from S2, hindering transition to the postfusion conformation. This idea is supported in a study by Kumar et al.75 that explores the 6 Omicron mutations (N764K, D796Y, N856K, Q954H, N969K, and L981F) found in the S2 subunit of S and found that introduction of S2 mutation N856K greatly reduced syncytia formation and therefore use of the fusogenic cell entry pathway. The same study also found that the Omicron S2 mutations generally decreased antibody neutralization, perhaps by keeping the receptor-binding epitopes folded over the adjacent protomer for larger amounts of time,75 thus decreasing the exposure of antibody-binding epitopes. Further investigation into the interprotomer interactions between the S1 and S2 subunits of SARS-CoV-2 S are needed to understand the structural determinants of the dynamics of S1 detachment during host cell entry.
Omicron subvariants retain receptor tropism but show altered utilization of TMPRSS2
The fusogenic pathway of cell entry by SARS-CoV-2 involves cleavage by the transmembrane serine protease 2 (TMPRSS2) at the S2′ site. Through S-pseudotyped virus assays, the Omicron BA.1 variant has been shown to have a shift in its preferred cell entry method compared to previous VOCs.28 Several studies have reported decreased utilization of TMPRSS2, decreased fusogenicity, and increased reliance on the endocytic pathway for BA.128,76,77 One study, using site-directed mutagenesis, pseudovirus infection, and fusogenicity assays, attributed this change to the S2 subunit, particularly the N856K and N969K substitutions.75,76 This could be explained by added interactions by N856K in the FPPR and/or N969K’s location in the HR1,78 which rearranges during the pre- to postfusion transition. Additional studies using similar methods79,80 attributed the reduced TMPRSS2 usage and less efficient S cleavage primarily to S375F, T547K, and H655Y substitutions in BA.1. While the S375F and H655Y substitutions were maintained throughout the Omicron variants, T547Y was lost after BA.1. Yet another study found that S371F, S375F, and T376A (all present throughout Omicron), either in combination or individually, nearly abrogated syncytia formation, while L981F and N764K increased syncytia formation.78
The combination of these mutations enabling syncytia formation highlights Omicron’s ability to compensate for mutations that promote immune evasion (such as S371F, S375F, and T376A) but decrease cell entry abilities, with cell entry enhancing mutations that occur in a completely different domain of the S protein (S2; through the L981F and N764K mutations). BA.2, by reversing T547K and N856K (which decrease syncytia formation75) and L981F (which increases syncytia formation), perhaps created a net-zero change in the preference for the fusogenic pathway and showed similar fusogenic abilities to BA.1.40 Interestingly, BA.5 and later Omicron variants seem to have adapted back to use the TMPRSS2 cleavage pathway,81 although which mutations contributed to this change is not definitely known.
Despite extensive mutations in the RBD, the Omicron variant retained its ACE2 binding as described above. Recently, another S receptor, TMEM106B, was identified and demonstrated to bind every major S variant tested to varying degrees of affinity.12 Using hydrogen-deuterium exchange mass spectrometry (HDXMS) and cryo-EM, the S binding site for TMEM106B was identified to overlap with the ACE2 binding site.12 However, TMEM106B binding is optimized by the E484D mutation, which is not present in the Omicron variants, indicating that Omicron variants may prefer ACE2 to TMEM106B.12 While some Omicron subvariants shifted away from using TMPRSS2 proteolysis in their fusogenic pathways, there have not seemed to be any shifts away from using the RBM for binding to receptors such as ACE2 and TMEM106B.
Evolving electrostatics of the Omicron S RBD and implications for interactions with the host membrane
When studying the evolution of viral surface proteins, it is useful to pay attention to trends in amino acid mutations that crop up over time, as these can be predictors of future mutations and their effects on virus infectivity. One such trend was noticed by Kim et al.,82 who compared the electrostatic maps of RBD-down structures of SARS-CoV-2 S proteins from earlier variants to the newer Omicron variants. Kim et al. noticed a trend toward an increase in positively charged amino acids accumulating at a central patch on the host cell interaction side of the Omicron S protein (Figure 4A). This accumulation of positive charges contributes to increased interaction with the negatively charged glycans, such as heparan sulfate, in the glycocalyx on the outside of the host cell membrane.13,83–85 Heparan sulfate has also been shown in vitro to induce RBD-up states of the S protein. These electrostatic interactions between the S and host cell membrane could lead to an increased number of viruses accumulating at the cell surface, thus resulting in a higher probability of encountering the host cell surface receptors and infecting the cell.
Figure 4. Electrostatics, pre- to postfusion S conformation changes, and other factors influencing cell entry.
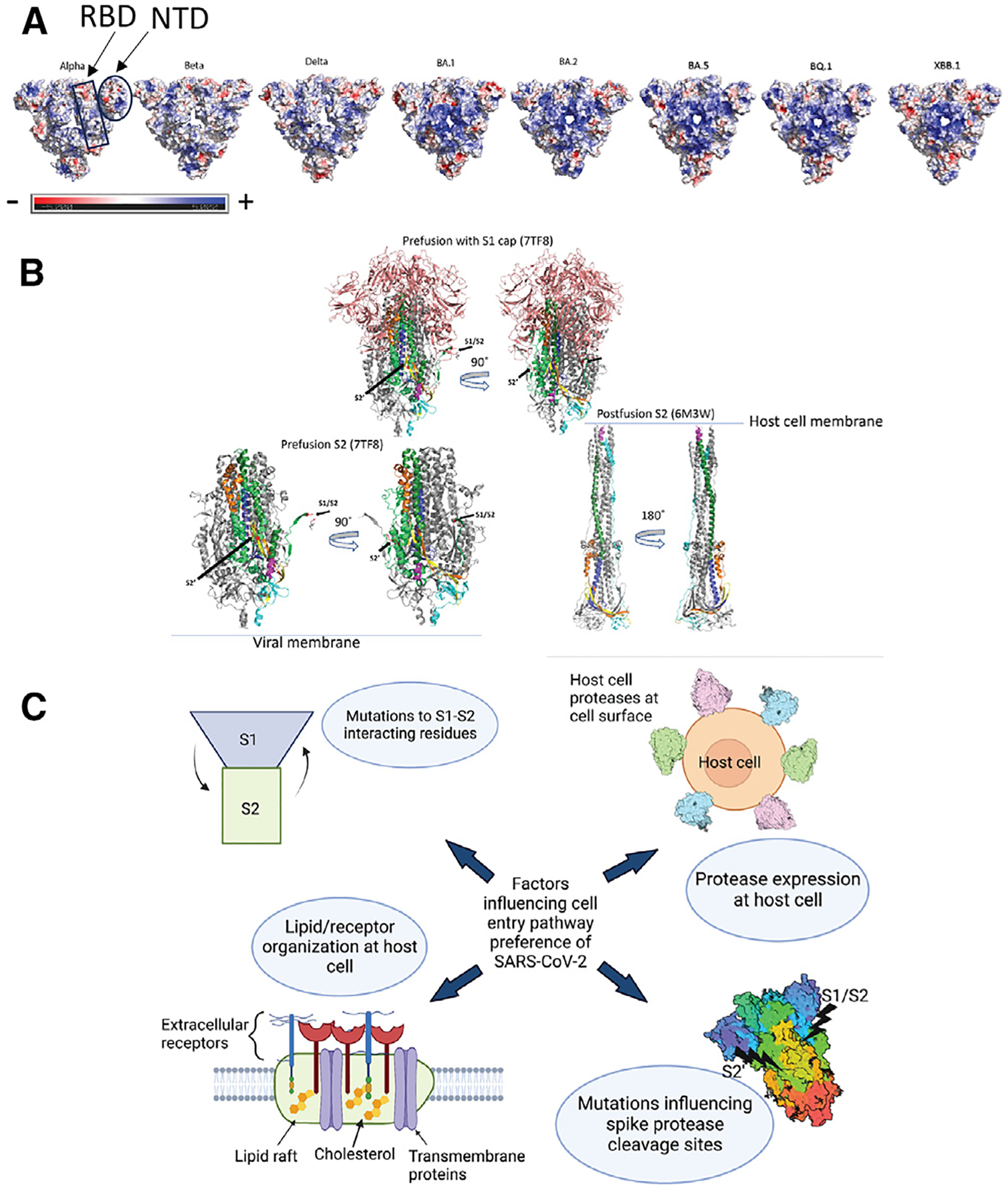
(A) Top surface views of S protein ectodomain colored by electrostatic potential. PDB IDs: 7TOU (Delta), 7TL1 (BA.1), 7UB6 (BA.2), 8IOS (XBB.1). Mutations for BA.5 and BQ.1 are modeled on the BA.2 structure. Beta and Gamma have one RBD in the up position in these structures. The rest of the structures have 3 RBDs in the down position.
(B) Top: pre-fusion Omicron BA.1 (PDB: 7TF8) showing the S1 cap (salmon) covering the S2 subunit. Bottom left: pre-fusion S2 shown without the S1 cap. Bottom right: postfusion S2. Coloring for (A)–(C): gray, monomers 2 and 3 (coloring is only shown on 1 monomer); salmon: S1 cap; forest green: HR2; yellow: CD; TV blue, CH; lime green: regions in S2 of pre-fusion structure but not shown in postfusion because the residues are not resolved; magenta: postfusion structure: residues 901–909, pre-fusion structure: 919–927; cyan: C-terminal. Postfusion: 1069–1178. Pre-fusion: 1087–1147. Red: S1/S2 and S2′ cleavage sites as labeled on pre-fusion structure. Orange: region after S1/S2.
(C) Diagram showing possible factors involved in SARS-CoV-2 cell entry pathway selection.
The role of heparan sulfate binding to the S and its interplay with ACE2 (and other receptors) binding have yet to be thoroughly evaluated. Different cell types express different lengths and sulfation patterns of heparan sulfate,86 and the SARS-CoV-2 S protein has been shown to bind heparan sulfate in a length-dependent manner.84 Although no structures exist of heparan sulfate bound to the SARS-CoV-2 S protein, computational methods predict that heparan sulfate may bind to a long positively charged polyanion binding site extending from the RBD and running between the RBD and the NTD down to the furin cleavage site.13,49,87 Four mutations in the Omicron S increase the positive charge in this putative binding channel,85,88 indicating that the S may be utilizing heparan sulfate to enhance its ability to bind to the host cell surface. The NTD typically had a neutral charge in pre-Omicron variants until Omicron BA.1, which obtained a Glu-Pro-Glu insertion at position 214, making it net negative. The Glu-Pro-Glu insertion was reversed in further Omicron variants, but the XBB variant NTD also has a net negative charge due to the H146Q and Q183E substitutions89 (Figure 4A). Additional studies are needed to determine whether the change in charge on the NTD affects host cell receptor or glycan binding.
Current gaps in our understanding of the Omicron S protein mutational effects
The S1 subunit, consisting of the NTD, RBD, SD1, and SD2, forms a “cap” over the S2 subunit, which contains the FP (Figure 4B). S1 must detach from S2 after receptor binding and cleavage by furin and other host cell proteases such as TMPRSS2 at their respective cleavage sites.90 After cleavage at the furin cleavage site (S1/S2 site), although residues 1–681 are no longer covalently linked to the S2 subunit, in the absence of receptor interactions or cleavage by additional proteases, S1 remains attached to S2.16,91 After cleavage at the S2′site (TMPRSS2 cleavage site), residues 681–815 can then detach, with residue 815 becoming the new N terminus (starting with the FP) of the detached S2 subunit. If S1 does not detach efficiently and completely from S2, cell entry by the virus could be diminished. If the method of entry allowed by TMPRSS2 cleavage is interfered with, then the virus will evolve to use other mechanisms to enter cells. Mutations at the S1–S2 interface found in the Omicron variants could play a role in determining the extent of attachment between the two subunits. Mutations that create or abrogate hydrogen bonding or electrostatic interactions at the S1–S2 interface could either increase or decrease S1–S2 interaction. Decreasing the S1–S2 interaction could lead to premature triggering or incorrect folding of the pre-fusion S protein. Increasing the S1–S2 interaction could lead to S1 becoming “stuck” to S2 and not allowing the helices of the pre-fusion S to elongate into the postfusion form (Figure 4B). The Omicron variant harbors several S2 mutations at the interface between S1 and S2, including N764K, N856K, Q954H, N969K, and L981F in BA.1, with N856K and L981F reverted in later Omicron variants. Further studies of the dynamics of S1 detachment would inform our understanding of the transition between the pre- and postfusion states of the S protein. It has recently been determined that additional proteases, including matrix metalloproteinases (MMPs), at the host cell surface can cleave the S protein at a different cleavage site than TMPRSS2.92 BA.1 demonstrated increased efficiency of membrane-type matrix MMP (MT-MMP) usage compared to previous and further evolved variants.92 MMP cleavage has been observed at or near the S2′cleavage site and not at the S1/S2 cleavage site.However, the putative cleavage site of MMPs is different from serine protease cleavage sites93 and should be further studied within the context of the SARS-CoV-2 S protein.
Epistatic effects of different mutations influence the fitness of the S, and the functions of individual mutations are connected to those of other mutations through the interconnected network of the S protein structure. In addition to the in vitro data discussed here, extensive structure-based vector analysis and molecular dynamics simulations have been employed to understand S-protein mobility and interdomain allostery, indicating extensive crosstalk between the domains and potential for mutations to impact more than one domain.94,95 As the number of mutations grows through viral evolution, the network of interactions becomes more complex. Understanding how the mutations interconnect becomes a daunting task that cannot be ignored as further variants are detected. The Omicron variant mutation epistatic interactions have demonstrated the necessity for high-throughput analysis of how groups of mutations influence antibody binding and viral fitness. One solution is presented in technology by the Bloom group that allows for high-throughput deep mutational scanning using yeast display53 and pseudotyped lentiviruses.96 These studies have allowed the mapping of mutations that escape monoclonal antibodies targeting multiple epitopes on the S as well as how mutations affect pseudovirus infection.96
Concluding remarks
The SARS-CoV-2 Omicron variants have emerged and spread in a global environment where an increasing number of individuals are vaccinated against or have been infected by SARS-CoV-2. The shifts in Omicron’s immune-evasion strategies are dramatic compared to previous variants. As the ongoing evolution of Omicron is occurring in settings where a large proportion of the global population has acquired some level of immunity against SARS-CoV-2, either through natural infection or induced by vaccination, immune evasion is a primary driver for Omicron evolution.
The SARS-CoV-2 Omicron S protein differs from previous variants in the increased interactions between the down-state RBDs, the changes to host protease usage, and the accumulation of positively charged amino acids at its host cell-interacting surface, which leads to increased interactions with the negatively charged host cell glycocalyx. Between Omicron variant subtypes, additional differences exist, such as altered packing of S2 central helices and rearrangements at the FPPR and N2R regions, that demonstrate the continued evolution and optimization of the structure of the S protein as new versions of the Omicron variant emerge and sweep local and global populations. The altered characteristics of the Omicron S protein enable the virus to evade many of the antibody therapeutics that have been used to treat SARS-CoV-2 in the past. Several factors influence the cell entry pathway of SARS-CoV-2 and should be further studied and monitored, including mutations to S1–S2-interacting residues and mutations that influence the S protein protease cleavage sites. Lipid/receptor organization at the host cell surface and host cell protease expression levels that vary between cell types could be considered influencing factors for the preference of certain SARS-CoV-2 variants for certain cell types (Figure 4C). The SARS-CoV-2 S protein will likely continue to mutate as it evolves over time, and optimized viral detection and monitoring is necessary.
With the relaxation of the use of countermeasures such as masking and social distancing, continued surveillance and early detection of new variants will be key for staying ahead of the evolving virus and for identifying emergence of potentially concerning variants. Initiatives such as wastewater viral sampling, which involves viral detection and classification from wastewater samples from the community and PCR-based genotyping of the viruses found in the water over time, can provide such information. This method has been used to identify emerging variants up to 14 days earlier than clinical genomic surveillance97 in multiple studies around the world.97–99 Recent technological advances in nucleic acid sequencing and computational tools allow sequencing with near 95% genome coverage of viruses in wastewater samples,97 while only 40% coverage was possible in previous studies.100–102 Full-length SARS-CoV-2 variant genomic sequence data generated by wastewater sampling provides a comprehensive picture of all viral sequences present in a population, including emerging variants, as opposed to clinical samples that typically only give data on a single virus variant from a single individual. Given that wastewater samples are distributed evenly throughout the total wastewater pool that a community shares, wastewater sampling has the advantage of being less biased than clinical genomic surveillance generated by nasal swab methods. Moreover, this sampling method primarily accounts for human waste and not animal waste, which open water in rivers and other such water bodies would contain. Several programs at the NIH and CDC work to monitor changes in viral sequence. The NIH SARS-CoV-2 Assessment of Viral Evolution (SAVE) Program brings together three teams of scientists. The first evaluates viral sequencing data from the population and identifies variants. The second characterizes variants for antibody escape, antigenic landscaping, and replication kinetics. The third team performs in vivo characterization using animal models to estimate virulence changes in emerging variants (https://www.niaid.nih.gov/research/sars-cov-2-assessment-viral-evolution-program). The NIH Accelerating COVID-19 Therapeutic Interventions and Vaccines: Tracking Resistance and Coronavirus Evolution (ACTIV TRACE) program focuses on identifying emerging variants and prioritizes studying viral variants that provide information for vaccines and therapeutics (https://www.nih.gov/research-training/medical-research-initiatives/activ/tracking-resistance-coronavirus-evolution-trace). In addition, the CDC’s National SARS-CoV-2 Strain Surveillance (NS3) nationally requests specimens from state and local public health agencies, sequences them, and characterizes new viral variants (https://www.cdc.gov/coronavirus/2019-ncov/variants/cdc-role-surveillance.html). Close partnership between wastewater surveillance, sequencing, and in vitro/in vivo viral characterization could enable prediction of upcoming VOCs so that safety protocols can be put in place in communities where these variants are identified.
While the S protein is the best studied, it is not the only predictor of SARS-CoV-2 infectivity. This was prominently exemplified by Omicron BA.5, which overtook BA.4 despite both variants having identical S protein sequences. SARS-CoV-2 genomic RNA is made up of 14 open reading frames (ORFs), 2/3 of which encode non-structural proteins important in viral replication and 1/3 of which encode proteins important for the viral capsid structure.103 While the influence of some S mutations on SARS-CoV-2 infectivity are somewhat understood, the other SARS-CoV-2 proteins are less well studied, and further research into the influence of their mutations is needed before sequencing-based surveillance methods can be used to predict the effect of mutations in these proteins.
The unique structural and conformational features of the SARS-CoV-2 Omicron S protein modulate its ability to bind cell surface receptors and infect cells, to alter its conformation upon receptor interactions and as it is processed by cellular proteases, and to evade immune attack by hiding antibody-binding epitopes. During its evolution, the Omicron S protein has experimented with and improved upon all three of these strategies both relative to previous variants and within its own subvariants as it has continued to evolve. Changes to viral entry mechanisms such as protease usage signal potential changes in host cell tropism. The utilization of new host cell proteases indicates the identification of new sites of cell vulnerability by the viral evolution process. Future mutations may optimize the viral utilization of these new pathways and allow the virus to enter the cells in ways that therapeutics do not currently target and vaccines do not protect against. Identification of cleavage sites targeted by newly identified host proteases and monitoring of mutations that change the presentation of these parts of the protein will be essential in future studies. Studying Omicron S protein structural evolution unveils a tale of compensation between immune evasion and S function. Overcompensation in favor of immune evasion, compromising S stability as well as cell entry abilities that occurred initially in BA.1, was followed quickly by a reversion to a more stable and functional S as Omicron continued to evolve.
ACKNOWLEDGMENTS
This work was supported by NIH R01 grant AI165947. Thank you to all the members of the Acharya lab who provided comments and edits for this review. Special thanks to Alexandria Calloway, Susan Fetics, Aaron May, Ellie Zhang, Logan Scott, and Ki Song. Many images in this review came from use of Pymol (http://www.pymol.org/pymol) and ChimeraX (https://www.cgl.ucsf.edu/chimerax/).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Syed AM, Ciling A, Taha TY, Chen IP, Khalid MM, Sreekumar B, Chen PY, Kumar GR, Suryawanshi R, Silva I, et al. (2022). Omicron mutations enhance infectivity and reduce antibody neutralization of SARS-CoV-2 virus-like particles. Proc. Natl. Acad. Sci. USA 119, e2200592119. 10.1073/pnas.2200592119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou Y, Zhi H, and Teng Y (2023). The outbreak of SARS-CoV-2 Omicron lineages, immune escape, and vaccine effectivity. J. Med. Virol 95, e28138. 10.1002/jmv.28138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boson B, Legros V, Zhou B, Siret E, Mathieu C, Cosset FL, Lavillette D, and Denolly S (2021). The SARS-CoV-2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus-like particles. J. Biol. Chem 296, 100111. 10.1074/jbc.RA120.016175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mannar D, Saville JW, Zhu X, Srivastava SS, Berezuk AM, Tuttle KS, Marquez AC, Sekirov I, and Subramaniam S (2022). SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein-ACE2 complex. Science 375, 760–764. 10.1126/science.abn7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sternberg A, and Naujokat C (2020). Structural features of coronavirus SARS-CoV-2 spike protein: Targets for vaccination. Life Sci. 257, 118056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang Y, Yang C, Xu X. f., Xu W, and Liu S. w. (2020). Structural and functional properties of SARS-CoV-2 spike protein: potential anti-virus drug development for COVID-19. Acta Pharmacol. Sin 41, 1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson CB, Farzan M, Chen B, and Choe H (2022). Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol 23, 3–20. 10.1038/s41580-021-00418-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, and Li F (2020). Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 117, 11727–11734. 10.1073/pnas.2003138117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hikmet F, Méar L, Edvinsson Å, Micke P, Uhlén M, and Lindskog C (2020). The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol 16, e9610. 10.15252/msb.20209610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gobeil SMC, Henderson R, Stalls V, Janowska K, Huang X, May A, Speakman M, Beaudoin E, Manne K, Li D, et al. (2022). Structural diversity of the SARS-CoV-2 Omicron spike. Mol. Cell 82, 2050–2068.e6. 10.1016/j.molcel.2022.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang MY, Zhao R, Gao LJ, Gao XF, Wang DP, and Cao JM (2020). SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. Front. Cell. Infect. Microbiol 10, 587269. 10.3389/fcimb.2020.587269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baggen J, Jacquemyn M, Persoons L, Vanstreels E, Pye VE, Wrobel AG, Calvaresi V, Martin SR, Roustan C, Cronin NB, et al. (2023). TMEM106B is a receptor mediating ACE2-independent SARSCoV-2 cell entry. Cell 186, 3427–3442.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clausen TM, Sandoval DR, Spliid CB, Pihl J, Perrett HR, Painter CD, Narayanan A, Majowicz SA, Kwong EM, McVicar RN, et al. (2020). SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 183, 1043–1057.e15. 10.1016/j.cell.2020.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, van der Meer F, Kallio K, Kaya T, Anastasina M, et al. (2020). Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 370, 856–860. 10.1126/science.abd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, and Veesler D (2020). Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 183, 1735. 10.1016/j.cell.2020.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gobeil SMC, Janowska K, McDowell S, Mansouri K, Parks R, Manne K, Stalls V, Kopp MF, Henderson R, Edwards RJ, et al. (2021). D614G Mutation Alters SARS-CoV-2 Spike Conformation and Enhances Protease Cleavage at the S1/S2 Junction. Cell Rep. 34, 108630. 10.1016/j.celrep.2020.108630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gobeil SMC, Janowska K, McDowell S, Mansouri K, Parks R, Stalls V, Kopp MF, Manne K, Li D, Wiehe K, et al. (2021). Effect of natural mutations of SARS-CoV-2 on spike structure, conformation, and antigenicity. Science 373, eabi6226. 10.1126/science.abi6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt F, Muecksch F, Weisblum Y, Da Silva J, Bednarski E, Cho A, Wang Z, Gaebler C, Caskey M, Nussenzweig MC, et al. (2022). Plasma Neutralization of the SARS-CoV-2 Omicron Variant. N. Engl. J. Med 386, 599–601. 10.1056/NEJMc2119641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sievers BL, Chakraborty S, Xue Y, Gelbart T, Gonzalez JC, Cassidy AG, Golan Y, Prahl M, Gaw SL, Arunachalam PS, et al. (2022). Antibodies elicited by SARS-CoV-2 infection or mRNA vaccines have reduced neutralizing activity against Beta and Omicron pseudoviruses. Sci. Transl. Med 14, eabn7842. 10.1126/scitranslmed.abn7842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qu P, Evans JP, Faraone JN, Zheng YM, Carlin C, Anghelina M, Stevens P, Fernandez S, Jones D, Lozanski G, et al. (2023). Enhanced neutralization resistance of SARS-CoV-2 Omicron subvariants BQ.1, BQ.1.1, BA.4.6, BF.7, and BA.2.75.2. Cell Host Microbe 31, 9–17.e3. 10.1016/j.chom.2022.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Chen X, Tan J, Yue S, Zhou R, Xu Y, Lin Y, Yang Y, Zhou Y, Deng K, et al. (2022). 35B5 antibody potently neutralizes SARS-CoV-2 Omicron by disrupting the N-glycan switch via a conserved spike epitope. Cell Host Microbe 30, 887–895.e4. 10.1016/j.chom.2022.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carreño JM, Alshammary H, Tcheou J, Singh G, Raskin AJ, Kawabata H, Sominsky LA, Clark JJ, Adelsberg DC, Bielak DA, et al. (2022). Activity of convalescent and vaccine serum against SARS-CoV-2 Omicron. Nature 602, 682–688. 10.1038/s41586-022-04399-5. [DOI] [PubMed] [Google Scholar]
- 23.Tao K, Tzou PL, Kosakovsky Pond SL, Ioannidis JPA, and Shafer RW (2022). Susceptibility of SARS-CoV-2 Omicron Variants to Therapeutic Monoclonal Antibodies: Systematic Review and Meta-analysis. Microbiol. Spectr 10, e0092622. 10.1128/spectrum.00926-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffmann M, Kruger N, Schulz S, Cossmann A, Rocha C, Kempf A, Nehlmeier I, Graichen L, Moldenhauer AS, Winkler MS, et al. (2022). The Omicron variant is highly resistant against antibody-mediated neutralization: Implications for control of the COVID-19 pandemic. Cell 185, 447–456.e411. 10.1016/j.cell.2021.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cameroni E, Bowen JE, Rosen LE, Saliba C, Zepeda SK, Culap K, Pinto D, VanBlargan LA, De Marco A, di Iulio J, et al. (2022). Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature 602, 664–670. 10.1038/s41586-021-04386-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Javanmardi K, Segall-Shapiro TH, Chou CW, Boutz DR, Olsen RJ, Xie X, Xia H, Shi PY, Johnson CD, Annapareddy A, et al. (2022). Antibody escape and cryptic cross-domain stabilization in the SARS-CoV-2 Omicron spike protein. Cell Host Microbe 30, 1242–1254.e6. 10.1016/j.chom.2022.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tada T, Zhou H, Dcosta BM, Samanovic MI, Chivukula V, Herati RS, Hubbard SR, Mulligan MJ, and Landau NR (2022). Increased resistance of SARS-CoV-2 Omicron variant to neutralization by vaccine-elicited and therapeutic antibodies. EBioMedicine 78, 103944. 10.1016/j.ebiom.2022.103944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meng B, Abdullahi A, Ferreira IATM, Goonawardane N, Saito A, Kimura I, Yamasoba D, Gerber PP, Fatihi S, Rathore S, et al. (2022). Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 603, 706–714. 10.1038/s41586-022-04474-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Planas D, Saunders N, Maes P, Guivel-Benhassine F, Planchais C, Buchrieser J, Bolland W-H, Porrot F, Staropoli I, Lemoine F, et al. (2022). Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature 602, 671–675. 10.1038/s41586-021-04389-z. [DOI] [PubMed] [Google Scholar]
- 30.Imai M, Ito M, Kiso M, Yamayoshi S, Uraki R, Fukushi S, Watanabe S, Suzuki T, Maeda K, Sakai-Tagawa Y, et al. (2023). Efficacy of Antiviral Agents against Omicron Subvariants BQ.1.1 and XBB. N. Engl. J. Med 388, 89–91. 10.1056/nejmc2214302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Q, Iketani S, Li Z, Liu L, Guo Y, Huang Y, Bowen AD, Liu M, Wang M, Yu J, et al. (2023). Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants. Cell 186, 279–286.e8. 10.1016/j.cell.2022.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stalls V, Lindenberger J, Gobeil SMC, Henderson R, Parks R, Barr M, Deyton M, Martin M, Janowska K, Huang X, et al. (2022). Cryo-EM structures of SARS-CoV-2 Omicron BA.2 spike. Cell Rep. 39, 111009. 10.1016/j.celrep.2022.111009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe Y, Allen JD, Wrapp D, McLellan JS, and Crispin M (2020). Site-specific glycan analysis of the SARS-CoV-2 spike. Science 369, 330–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsieh C-L, Goldsmith JA, Schaub JM, DiVenere AM, Kuo H-C, Javanmardi K, Le KC, Wrapp D, Lee AG, Liu Y, et al. (2020). Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 369, 1501–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhan W, Tian X, Zhang X, Xing S, Song W, Liu Q, Hao A, Hu Y, Zhang M, Ying T, et al. (2022). Structural study of SARS-CoV-2 antibodies identifies a broad-spectrum antibody that neutralizes the omicron variant by disassembling the spike trimer. J. Virol 96. e00480222–e100422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye G, Liu B, and Li F (2022). Cryo-EM structure of a SARS-CoV-2 omicron spike protein ectodomain. Nat. Commun 13, 1214. 10.1038/s41467-022-28882-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ni D, Turelli P, Beckert B, Nazarov S, Uchikawa E, Myasnikov A, Pojer F, Trono D, Stahlberg H, and Lau K (2023). Cryo-EM structures and binding of mouse and human ACE2 to SARS-CoV-2 variants of concern indicate that mutations enabling immune escape could expand host range. PLoS Pathog. 19, e1011206. 10.1371/journal.ppat.1011206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cerutti G, Guo Y, Liu L, Liu L, Zhang Z, Luo Y, Huang Y, Wang HH, Ho DD, Sheng Z, and Shapiro L (2022). Cryo-EM structure of the SARS-CoV-2 Omicron spike. Cell Rep. 38, 110428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Cai Y, Lavine CL, Peng H, Zhu H, Anand K, Tong P, Gautam A, Mayer ML, Rits-Volloch S, et al. (2022). Structural and functional impact by SARS-CoV-2 Omicron spike mutations. Cell Rep. 39, 110729. 10.1016/j.celrep.2022.110729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J, Tang W, Gao H, Lavine CL, Shi W, Peng H, Zhu H, Anand K, Kosikova M, Kwon HJ, et al. (2023). Structural and functional characteristics of the SARS-CoV-2 Omicron subvariant BA.2 spike protein. Nat. Struct. Mol. Biol 30, 980–990. 10.1038/s41594-023-01023-6. [DOI] [PubMed] [Google Scholar]
- 41.Wieczór M, Tang PK, Orozco M, and Cossio P (2023). Omicron mutations increase interdomain interactions and reduce epitope exposure in the SARS-CoV-2 spike. iScience 26, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cui Z, Liu P, Wang N, Wang L, Fan K, Zhu Q, Wang K, Chen R, Feng R, Jia Z, et al. (2022). Structural and functional characterizations of infectivity and immune evasion of SARS-CoV-2 Omicron. Cell 185, 860–871.e13. 10.1016/j.cell.2022.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao Z, Zhou J, Tian M, Huang M, Liu S, Xie Y, Han P, Bai C, Han P, Zheng A, et al. (2022). Omicron SARS-CoV-2 mutations stabilize spike up-RBD conformation and lead to a non-RBM-binding monoclonal antibody escape. Nat. Commun 13, 4958. 10.1038/s41467-022-32665-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tamura T, Ito J, Uriu K, Zahradnik J, Kida I, Anraku Y, Nasser H, Shofa M, Oda Y, Lytras S, et al. (2023). Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nat. Commun 14, 2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang K, Pan Y, Wang D, Yuan Y, Li M, Chen Y, Bi L, and Zhang X-E (2023). Altered hACE2 binding affinity and S1/S2 cleavage efficiency of SARS-CoV-2 spike protein mutants affect viral cell entry. Virol. Sin 38, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mahalingam G, Arjunan P, Periyasami Y, Dhyani AK, Devaraju N, Rajendiran V, Christopher AC, Kt RD, Dhanasingh I, Thangavel S, et al. (2023). Correlating the differences in the receptor binding domain of SARS-CoV-2 spike variants on their interactions with human ACE2 receptor. Sci. Rep 13, 8743. 10.1038/s41598-023-35070-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang X, Wu S, Wu B, Yang Q, Chen A, Li Y, Zhang Y, Pan T, Zhang H, and He X (2021). SARS-CoV-2 Omicron strain exhibits potent capabilities for immune evasion and viral entrance. Signal Transduct. Targeted Ther 6, 430. 10.1038/s41392-021-00852-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim S, Liu Y, Ziarnik M, Seo S, Cao Y, Zhang XF, and Im W (2023). Binding of human ACE2 and RBD of Omicron enhanced by unique interaction patterns among SARS-CoV-2 variants of concern. J. Comput. Chem 44, 594–601. 10.1002/jcc.27025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mycroft-West CJ, Su D, Pagani I, Rudd TR, Elli S, Gandhi NS, Guimond SE, Miller GJ, Meneghetti MCZ, Nader HB, et al. (2020). Heparin Inhibits Cellular Invasion by SARS-CoV-2: Structural Dependence of the Interaction of the Spike S1 Receptor-Binding Domain with Heparin. Thromb. Haemostasis 120, 1700–1715. 10.1055/s-0040-1721319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu L, Zhou L, Mo M, Liu T, Wu C, Gong C, Lu K, Gong L, Zhu W, and Xu Z (2022). SARS-CoV-2 Omicron RBD shows weaker binding affinity than the currently dominant Delta variant to human ACE2. Signal Transduct. Targeted Ther 7, 8. 10.1038/s41392-021-00863-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schubert M, Bertoglio F, Steinke S, Heine PA, Ynga-Durand MA, Maass H, Sammartino JC, Cassaniti I, Zuo F, Du L, et al. (2022). Human serum from SARS-CoV-2-vaccinated and COVID-19 patients shows reduced binding to the RBD of SARS-CoV-2 Omicron variant. BMC Med. 20, 102. 10.1186/s12916-022-02312-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Da Costa CHS, De Freitas CAB, Alves CN, and Lameira J (2022). Assessment of mutations on RBD in the Spike protein of SARS-CoV-2 Alpha, Delta and Omicron variants. Sci. Rep 12, 8540. 10.1038/s41598-022-12479-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KHD, Dingens AS, Navarro MJ, Bowen JE, Tortorici MA, Walls AC, et al. (2020). Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 182, 1295–1310.e20. 10.1016/j.cell.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li L, Liao H, Meng Y, Li W, Han P, Liu K, Wang Q, Li D, Zhang Y, Wang L, et al. (2022). Structural basis of human ACE2 higher binding affinity to currently circulating Omicron SARS-CoV-2 sub-variants BA. 2 and BA. 1.1. Cell 185, 2952–2960.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCallum M, Czudnochowski N, Rosen LE, Zepeda SK, Bowen JE, Walls AC, Hauser K, Joshi A, Stewart C, Dillen JR, et al. (2022). Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 375, 864–868. 10.1126/science.abn8652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tuekprakhon A, Nutalai R, Dijokaite-Guraliuc A, Zhou D, Ginn HM, Selvaraj M, Liu C, Mentzer AJ, Supasa P, Duyvesteyn HME, et al. (2022). Antibody escape of SARS-CoV-2 Omicron BA. 4 and BA. 5 from vaccine and BA. 1 serum. Cell 185, 2422–2433.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cao Y, Yisimayi A, Jian F, Song W, Xiao T, Wang L, Du S, Wang J, Li Q, Chen X, et al. (2022). BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 608, 593–602. 10.1038/s41586-022-04980-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Q, Guo Y, Iketani S, Nair MS, Li Z, Mohri H, Wang M, Yu J, Bowen AD, Chang JY, et al. (2022). Antibody evasion by SARSCoV-2 Omicron subvariants BA.2.12.1, BA.4 and BA.5. Nature 608, 603–608. 10.1038/s41586-022-05053-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taylor AL, and Starr TN (2023). Deep mutational scans of XBB.1.5 and BQ.1.1 reveal ongoing epistatic drift during SARS-CoV-2 evolution. Preprint at bioRxiv. 10.1101/2023.09.11.557279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qu P, Xu K, Faraone JN, Goodarzi N, Zheng Y-M, Carlin C, Bednash JS, Horowitz JC, Mallampalli RK, and Saif LJ (2023). Immune Evasion, Infectivity, and Fusogenicity of SARS-CoV-2 Omicron BA. 2.86 and FLip Variants. Preprint at bioRxiv. 10.1101/2023.09.11.557206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jian F, Yang S, Yu Y, Song W, Yisimayi A, Chen X, Xu Y, Wang P, Yu L, and Wang J (2023). Convergent evolution of SARS-CoV-2 XBB lineages on receptor-binding domain 455–456 enhances antibody evasion and ACE2 binding. Preprint at bioRxiv. 10.1101/2023.08.30.555211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou T, Wang L, Misasi J, Pegu A, Zhang Y, Harris DR, Olia AS, Talana CA, Yang ES, Chen M, et al. (2022). Structural basis for potent antibody neutralization of SARS-CoV-2 variants including B. 1.1. 529. Science 376, eabn8897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guo H, Gao Y, Li T, Li T, Lu Y, Zheng L, Liu Y, Yang T, Luo F, Song S, et al. (2022). Structures of Omicron spike complexes and implications for neutralizing antibody development. Cell Rep. 39, 110770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin S, Chen Z, Zhang X, Wen A, Yuan X, Yu C, Yang J, He B, Cao Y, and Lu G (2022). Characterization of SARS-CoV-2 Omicron spike RBD reveals significantly decreased stability, severe evasion of neutralizing-antibody recognition but unaffected engagement by decoy ACE2 modified for enhanced RBD binding. Signal Transduct. Targeted Ther 7, 56. 10.1038/s41392-022-00914-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klinakis A, Cournia Z, and Rampias T (2021). N-terminal domain mutations of the spike protein are structurally implicated in epitope recognition in emerging SARS-CoV-2 strains. Comput. Struct. Biotechnol. J 19, 5556–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mannar D, Saville JW, Sun Z, Zhu X, Marti MM, Srivastava SS, Berezuk AM, Zhou S, Tuttle KS, Sobolewski MD, et al. (2022). SARS-CoV-2 variants of concern: spike protein mutational analysis and epitope for broad neutralization. Nat. Commun 13, 4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang J, Xiao T, Cai Y, and Chen B (2021). Structure of SARS-CoV-2 spike protein. Current opinion in virology 50, 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Unione L, Moure MJ, Lenza MP, Oyenarte I, Ereño-Orbea J, Ardá A, and Jiménez-Barbero J (2022). The SARS-CoV-2 spike glycoprotein directly binds exogeneous sialic acids: A NMR view. Angew. Chem 61, e202201432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berkowitz RL, and Ostrov DA (2022). The Elusive Coreceptors for the SARS-CoV-2 Spike Protein. Viruses 15, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li F (2016). Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol 3, 237–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qing E, Kicmal T, Kumar B, Hawkins GM, Timm E, Perlman S, and Gallagher T (2021). Dynamics of SARS-CoV-2 spike proteins in cell entry: control elements in the amino-terminal domains. mBio 12. e01590211–e201521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu L, Wang P, Nair MS, Yu J, Rapp M, Wang Q, Luo Y, Chan JF-W, Sahi V, Figueroa A, et al. (2020). Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 584, 450–456. [DOI] [PubMed] [Google Scholar]
- 73.Liu L, Iketani S, Guo Y, Chan JFW, Wang M, Liu L, Luo Y, Chu H, Huang Y, Nair MS, et al. (2022). Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature 602, 676–681. 10.1038/s41586-021-04388-0. [DOI] [PubMed] [Google Scholar]
- 74.Saville JW, Mannar D, Zhu X, Berezuk AM, Cholak S, Tuttle KS, Vahdatihassani F, and Subramaniam S (2023). Structural analysis of receptor engagement and antigenic drift within the BA. 2 spike protein. Cell Rep. 42, 111964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kumar S, Delipan R, Chakraborty D, Kanjo K, Singh R, Singh N, Siddiqui S, Tyagi A, Jha S, Thakur KG, et al. (2023). Mutations in S2 subunit of SARS-CoV-2 Omicron spike strongly influence its conformation, fusogenicity and neutralization sensitivity. Online ahead of Print. J. Virol e0092223. 10.1128/jvi.00922-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peacock TP, Brown JC, Zhou J, Thakur N, Sukhova K, Newman J, Kugathasan R, Yan AW, Furnon W, De Lorenzo G, et al. (2022). The altered entry pathway and antigenic distance of the SARS-CoV-2 Omicron variant map to separate domains of spike protein. Preprint at bioRxiv. 10.1101/2021.12.31.474653. [DOI] [Google Scholar]
- 77.Yamamoto M, Tomita K, Hirayama Y, Inoue J-I, Kawaguchi Y, and Gohda J (2022). SARS-CoV-2 Omicron spike H655Y mutation is responsible for enhancement of the endosomal entry pathway and reduction of cell surface entry pathways. Preprint at bioRxiv. 10.1101/2022.03.21.485084. [DOI] [Google Scholar]
- 78.Pastorio C, Zech F, Noettger S, Jung C, Jacob T, Sanderson T, Sparrer KMJ, and Kirchhoff F (2022). Determinants of Spike infectivity, processing, and neutralization in SARS-CoV-2 Omicron subvariants BA.1 and BA.2. Cell Host Microbe 30, 1255–1268.e5. 10.1016/j.chom.2022.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hu B, Chan JFW, Liu H, Liu Y, Chai Y, Shi J, Shuai H, Hou Y, Huang X, Yuen TTT, et al. (2022). Spike mutations contributing to the altered entry preference of SARS-CoV-2 omicron BA.1 and BA.2. Emerg. Microb. Infect 11, 2275–2287. 10.1080/22221751.2022.2117098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Qu P, Evans JP, Kurhade C, Zeng C, Zheng Y-M, Xu K, Shi P-Y, Xie X, and Liu S-L (2023). Determinants and Mechanisms of the Low Fusogenicity and High Dependence on Endosomal Entry of Omicron Subvariants. mBio 14. e03176222–e403122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aggarwal A, Akerman A, Milogiannakis V, Silva MR, Walker G, Stella AO, Kindinger A, Angelovich T, Waring E, Amatayakul-Chantler S, et al. (2022). SARS-CoV-2 Omicron BA.5: Evolving tropism and evasion of potent humoral responses and resistance to clinical immunotherapeutics relative to viral variants of concern. EBioMedicine 84, 104270. 10.1016/j.ebiom.2022.104270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim SH, Kearns FL, Rosenfeld MA, Votapka L, Casalino L, Papanikolas M, Amaro RE, and Freeman R (2023). SARS-CoV-2 evolved variants optimize binding to cellular glycocalyx. Cell Rep. Phys. Sci 4, 101346. 10.1016/j.xcrp.2023.101346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tarbell JM, and Cancel LM (2016). The glycocalyx and its significance in human medicine. J. Intern. Med 280, 97–113. 10.1111/joim.12465. [DOI] [PubMed] [Google Scholar]
- 84.Liu L, Chopra P, Li X, Bouwman KM, Tompkins SM, Wolfert MA, De Vries RP, and Boons G-J (2021). Heparan Sulfate Proteoglycans as Attachment Factor for SARS-CoV-2. ACS Cent. Sci 7, 1009–1018. 10.1021/acscentsci.1c00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kearns FL, Sandoval DR, Casalino L, Clausen TM, Rosenfeld MA, Spliid CB, Amaro RE, and Esko JD (2022). Spike-heparan sulfate interactions in SARS-CoV-2 infection. Curr. Opin. Struct. Biol 76, 102439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ledin J, Staatz W, Li J-P, Götte M, Selleck S, Kjellén L, and Spill-mann D (2004). Heparan Sulfate Structure in Mice with Genetically Modified Heparan Sulfate Production. J. Biol. Chem 279, 42732–42741. 10.1074/jbc.m405382200. [DOI] [PubMed] [Google Scholar]
- 87.Kim SY, Jin W, Sood A, Montgomery DW, Grant OC, Fuster MM, Fu L, Dordick JS, Woods RJ, Zhang F, and Linhardt RJ (2020). Characterization of heparin and severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) spike glycoprotein binding interactions. Antivir. Res 181, 104873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Paiardi G, Richter S, Oreste P, Urbinati C, Rusnati M, and Wade RC (2022). The binding of heparin to spike glycoprotein inhibits SARS-CoV-2 infection by three mechanisms. J. Biol. Chem 298, 101507. 10.1016/j.jbc.2021.101507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ciccozzi M, and Pascarella S (2023). Two sides of the same coin: the N-terminal and the receptor-binding domains of SARS-CoV-2 Spike. Future Virol. 18, 75–78. 10.2217/fvl-2022-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bestle D, Heindl MR, Limburg H, Van Lam van T, Pilgram O, Moulton H, Stein DA, Hardes K, Eickmann M, Dolnik O, et al. (2020). TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 3, e202000786. 10.26508/lsa.202000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wrobel AG, Benton DJ, Xu P, Roustan C, Martin SR, Rosenthal PB, Skehel JJ, and Gamblin SJ (2020). SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol 27, 763–767. 10.1038/s41594-020-0468-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chan JFW, Huang X, Hu B, Chai Y, Shi H, Zhu T, Yuen TTT, Liu Y, Liu H, Shi J, et al. (2023). Altered host protease determinants for SARS-CoV-2 Omicron. Sci. Adv 9, eadd3867. 10.1126/sciadv.add3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Eckhard U, Huesgen PF, Schilling O, Bellac CL, Butler GS, Cox JH, Dufour A, Goebeler V, Kappelhoff R, Keller U.A.d., et al. (2016). Active site specificity profiling of the matrix metalloproteinase family: Proteomic identification of 4300 cleavage sites by nine MMPs explored with structural and synthetic peptide cleavage analyses. Matrix Biol. 49, 37–60. [DOI] [PubMed] [Google Scholar]
- 94.Manrique PD, Chakraborty S, Henderson R, Edwards RJ, Mansbach R, Nguyen K, Stalls V, Saunders C, Mansouri K, Acharya P, et al. (2023). Network analysis uncovers the communication structure of SARS-CoV-2 spike protein identifying sites for immunogen design. iS-cience 26, 105855. 10.1016/j.isci.2022.105855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Henderson R, Edwards RJ, Mansouri K, Janowska K, Stalls V, Gobeil SMC, Kopp M, Li D, Parks R, Hsu AL, et al. (2020). Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol 27, 925–933. 10.1038/s41594-020-0479-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dadonaite B, Crawford KHD, Radford CE, Farrell AG, Yu TC, Hannon WW, Zhou P, Andrabi R, Burton DR, Liu L, et al. (2023). A pseudovirus system enables deep mutational scanning of the full SARS-CoV-2 spike. Cell 186, 1263–1278.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Karthikeyan S, Levy JI, De Hoff P, Humphrey G, Birmingham A, Jepsen K, Farmer S, Tubb HM, Valles T, Tribelhorn CE, et al. (2022). Wastewater sequencing reveals early cryptic SARS-CoV-2 variant transmission. Nature 609, 101–108. 10.1038/s41586-022-05049-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kirby AE, Welsh RM, Marsh ZA, Yu AT, Vugia DJ, Boehm AB, Wolfe MK, White BJ, Matzinger SR, Wheeler A, et al. (2022). Notes from the Field: Early Evidence of the SARS-CoV-2 B.1.1.529 (Omicron) Variant in Community Wastewater — United States, November–December 2021. MMWR Morb. Mortal. Wkly. Rep 71, 103–105. 10.15585/mmwr.mm7103a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhao L, Zou Y, Li Y, Miyani B, Spooner M, Gentry Z, Jacobi S, David RE, Withington S, McFarlane S, et al. (2022). Five-week warning of COVID-19 peaks prior to the Omicron surge in Detroit, Michigan using wastewater surveillance. Sci. Total Environ 844, 157040. 10.1016/j.scitotenv.2022.157040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Baaijens JA, Zulli A, Ott IM, Nika I, van der Lugt MJ, Petrone ME, Alpert T, Fauver JR, Kalinich CC, Vogels CBF, et al. (2022). Lineage abundance estimation for SARS-CoV-2 in wastewater using transcriptome quantification techniques. Genome biology 23, 236. 10.1186/s13059-022-02805-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Amman F, Markt R, Endler L, Hupfauf S, Agerer B, Schedl A, Richter L, Zechmeister M, Bicher M, Heiler G, et al. (2022). Viral variant-resolved wastewater surveillance of SARS-CoV-2 at national scale. Nat. Biotechnol 40, 1814–1822. 10.1038/s41587-022-01387-y. [DOI] [PubMed] [Google Scholar]
- 102.Crits-Christoph A, Kantor RS, Olm MR, Whitney ON, Al-Shayeb B, Lou YC, Flamholz A, Kennedy LC, Greenwald H, Hinkle A, et al. (2021). Genome Sequencing of Sewage Detects Regionally Prevalent SARS-CoV-2 Variants. mBio 12, 10–1128. 10.1128/mBio.02703-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Arya R, Kumari S, Pandey B, Mistry H, Bihani SC, Das A, Prashar V, Gupta GD, Panicker L, and Kumar M (2021). Structural insights into SARS-CoV-2 proteins. J. Mol. Biol 433, 166725. [DOI] [PMC free article] [PubMed] [Google Scholar]